
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A highly sensitive and selective spectrofluorimetric method for the determination of manganese at nanotrace levels in some real, environmental, biological, soil, food and pharmaceutical samples using 2-(α-pyridyl)-thioquinaldinamide
M. Jamaluddin Ahmed *,
M. Tazul Islam and
Faisal Hossain
*,
M. Tazul Islam and
Faisal Hossain
Laboratory of Analytical Chemistry, Department of Chemistry, University of Chittagong, Chittagong-4331, Bangladesh. E-mail: pmjahmed55@gmail.com
First published on 1st February 2018
Abstract
A very simple, ultra-sensitive and highly selective non-extractive spectrofluorimetric method is presented for the determination of manganese at nano-trace levels using 2-(α-pyridyl)-thioquinaldinamide (PTQA). PTQA has been proposed as a new analytical reagent for the direct non-extractive spectrofluorimetric determination of manganese(VII). This novel fluorimetric reagent, PTQA becomes oxidized in a slightly acidic (0.0125–0.05 M H2SO4) solution with manganese(VII) in absolute ethanol to produce a highly fluorescent oxidized product (λex = 319 nm; λem = 373 nm). Constant and maximum fluorescence intensities were observed over a wide range of acidity (0.0125–0.05 M H2SO4) for the period between 5 min and 24 h. Linear calibration graphs were obtained for 0.01–800 μg L−1 of Mn, having a detection limit of 1 ng L−1; the quantification limit of the reaction system was found to be 10 ng L−1 and the RSD was 0–2%. A large excess of over 60 cations, anions and complexing agents do not interfere in the determination. The developed method was successfully used in the determination of manganese in several standard reference materials (alloys, steels, hair and sediments) as well as in some environmental waters (potable and polluted), biological samples (human blood, urine and hair), soil samples, food samples (vegetables, fruits, tea, rice, and wheat), fertilizer samples and pharmaceutical samples (multivitamin-mineral tablets and syrup), solutions containing both manganese(II) and manganese(VII) speciation and complex synthetic mixtures. The results of the proposed method for assessing biological, food and vegetables samples were comparable with AAS and ICP-MS and were found to be in excellent agreement.
Introduction
Manganese is an essential trace element in the metabolism of all living organisms, including plants and bacteria, and is found in all tissues of man. But manganese is also a potential carcinogen.1 Among the inorganic chemicals, manganese, chromium, and selenium and certain organometallic derivatives of these metals have been found to be carcinogenic under special conditions and are also considered potential inorganic genotoxic agents.2 The hazards of manganese have been known for a long time. The accurate determination of manganese in environmental and biological materials is of considerable importance for both metabolic and toxicological studies in humans and animals because of its dose-dependent harmful and beneficial roles. Until recently, analytical techniques of satisfactory sensitivity and accuracy have not been widely available and attempts by many researchers to use unsatisfactory procedures have led to the publication of much data and also to biochemical conclusions of dubious value.3 Clearly, major problems which have given rise to this situation are the very low concentrations of manganese in biological samples, and a lack of awareness of the need to exercise control over the extraneous contamination and interfering of foreign ions during all the steps of an analytical procedure. The present method that is being recommended over the existing methods almost in every respect in their own terms-selectivity, range of determination, accuracy, sensitivity, simplicity and rapidity of the operation, stability of the fluorescent system and acidity of wide variation, etc.In expanding analytical fields such as environmental, biological and material monitoring of trace metals, there is an increasing need to develop the simple, sensitive and selective analytical techniques that do not use expensive or complicated test equipment. Many sophisticated technique, such as pulse polarography, NAA, HPLC, spectrophotometry, spectrofluorimetry, AAS, FAAS, ICP-OES and ICP-MS have been widely applied to the determination of manganese. However, the spectrofluorimetric method still has the advantages of being simple and without requiring expensive or complicated test equipment. For this reason, a wide variety of spectrofluorimetric methods for determination of manganese has been developed. Several authors have reported on the extractive spectrofluorimetric determination of manganese using complexes formed variety of reagents.4–40 In most of the methods4–40 cited above, manganese forms soluble or insoluble complexes with reagents with various organic solvents for spectrofluorimetric determination. Most of these reagents are expensive and non-recoverable. Most of the organic solvents which were used are carcinogenic.
The aim of this study was to develop a simpler direct spectrofluorimetric method for the nano-trace determination of manganese. In the search for a more sensitive reagent, in this work a new reagent was synthesized according to the method of Porter41 and a oxidation reaction of 2-(α-pyridyl)-thioquinaldinamide (PTQA); with Mn(VII) and forms an intensely fluorescent oxidized product. The method possesses distinct advantages over existing methods4–40 with respect to sensitivity, selectivity, range of determination, simplicity, speed, pH/acidity range, thermal stability, accuracy, precision and ease of operation. The method is based on the oxidative reaction of non-fluorescent PTQA in a slightly acidic (0.0125–0.05 M H2SO4) solution with Mn(VII) in presence of ethanol to produce a highly fluorescent oxidized product, followed by a direct measurement of the fluorescence intensity in an aqueous solution at room temperature. Oxidation is very rapid and no extraction is required. With suitable masking, the reaction can be made to be highly selective and the reagent blank solutions do not show any fluorescence.
Experimental section
Apparatus
A Shimadzu (Kyoto, Japan) (Model-RF-5301PC) Spectrofluorophotometer and a Jenway (England, UK) (Model-3010) pH meter with combination of electrodes were used for measurements of the fluorescence intensity and pH, respectively. The calibration and linearity of the instrument were frequently checked with standard quinine sulphate (10 μg L−1). A Thermo Fisher Scientific (Model-iCE 3000, origin USA) atomic absorption spectrophotometer equipped with a microcomputer-controlled nitrous oxide-acetylene flame and an Inductively Coupled Plasma-Mass Spectrometer (ICP-MS), Model – ELAN DRC-II, Perkin Elmer (Toronto, Canada) were used to compare of the results. The Elemental Analyzer (Exeter Analytical Inc. Model: CE 440) equipped with supersensitive thermal conductivity detector for simultaneous determination of CHN was used. Infrared spectrum was recorded with FTIR Spectrophotometer, Shimadzu (Model-IR Prestige 21, Detector-DTGS KBr) in the range 7500–350 cm−1 and model: JEOL 500SS, magnetic field strength: 500 MHz, solvent used: DMSO D6, standard: TMS, four channel NMR spectrometer with signal-to-noise ratio of ∼5000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for proton were used for characterization of the ligand.
:1 for proton were used for characterization of the ligand.
Synthesis and characterization of the reagent
:1:1.5 were mixed and refluxed for 6 hours in 250 mL round bottom flask fitted with bulb condenser under controlled temperature (140–150) °C at 1 atm. Pressure over oil bath. The reaction mixture was kept overnight. The thiocompound was filtered and crystallized using petroleum ether (60–80)° C to give a bright yellow crystalline (needle shaped) solid. The compound recrystallized from lime-distilled ethanol and was kept under vacuum (0.1 mm of Hg) for 24 hours. Yield of the product was 70%. The structure of the reagent is shown in Scheme 1.
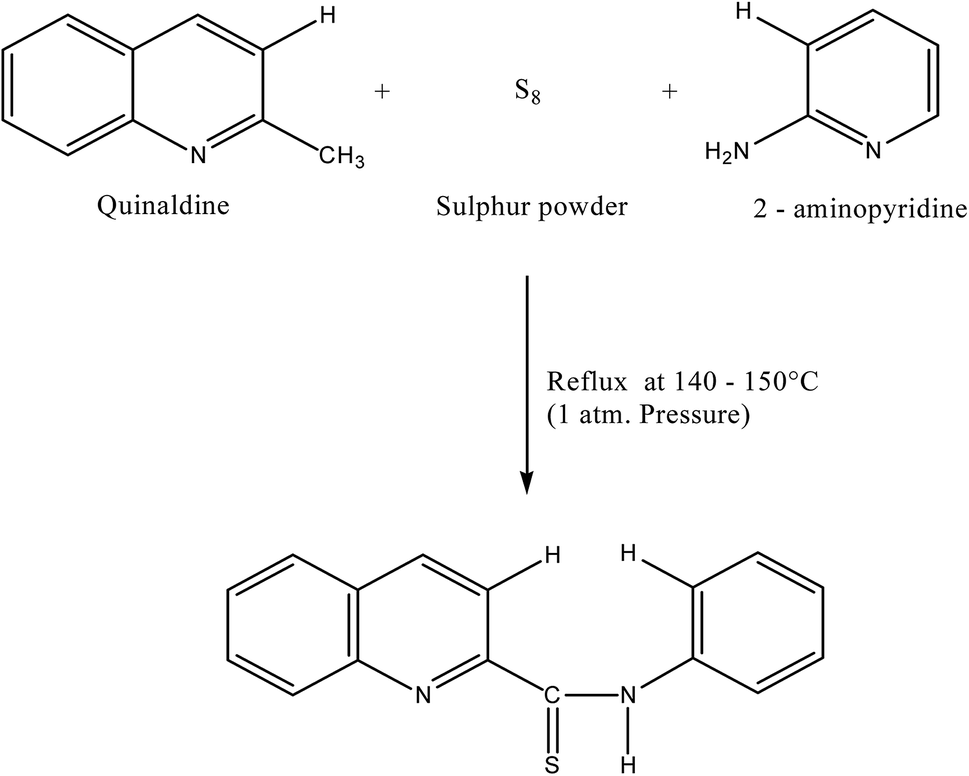 | ||
| Scheme 1 Reaction scheme of 2-(α-pyridyl)-thioquinaldinamide (PTQA). | ||
The results elemental analysis (C = 72.25, N = 13.35 and H = 4.25%) of the reagent are very in good agreement with the calculated values (C = 72.43, N = 13.55 and H = 4.55%). The FTIR spectrum of prepared reagent (PTQA) is shown in Fig. 1. The presence of FTIR peak at 1126.43 cm−1 in Fig. 1 was due to the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond peak (νCS, 1050–1200 cm−1)41 of the reagent indicating the formation of PTQA. Both FTIR spectral and elemental analysis data indicated the formation of the reagent PTQA. The formation of the reagent also tested by 1HNMR spectrum is shown in Fig. 2. The steadiness of the thermogravimetric curve obtained for about 1 g of the reagent at 80–90 °C indicated that the reagent did not contain any moisture.
S double bond peak (νCS, 1050–1200 cm−1)41 of the reagent indicating the formation of PTQA. Both FTIR spectral and elemental analysis data indicated the formation of the reagent PTQA. The formation of the reagent also tested by 1HNMR spectrum is shown in Fig. 2. The steadiness of the thermogravimetric curve obtained for about 1 g of the reagent at 80–90 °C indicated that the reagent did not contain any moisture.
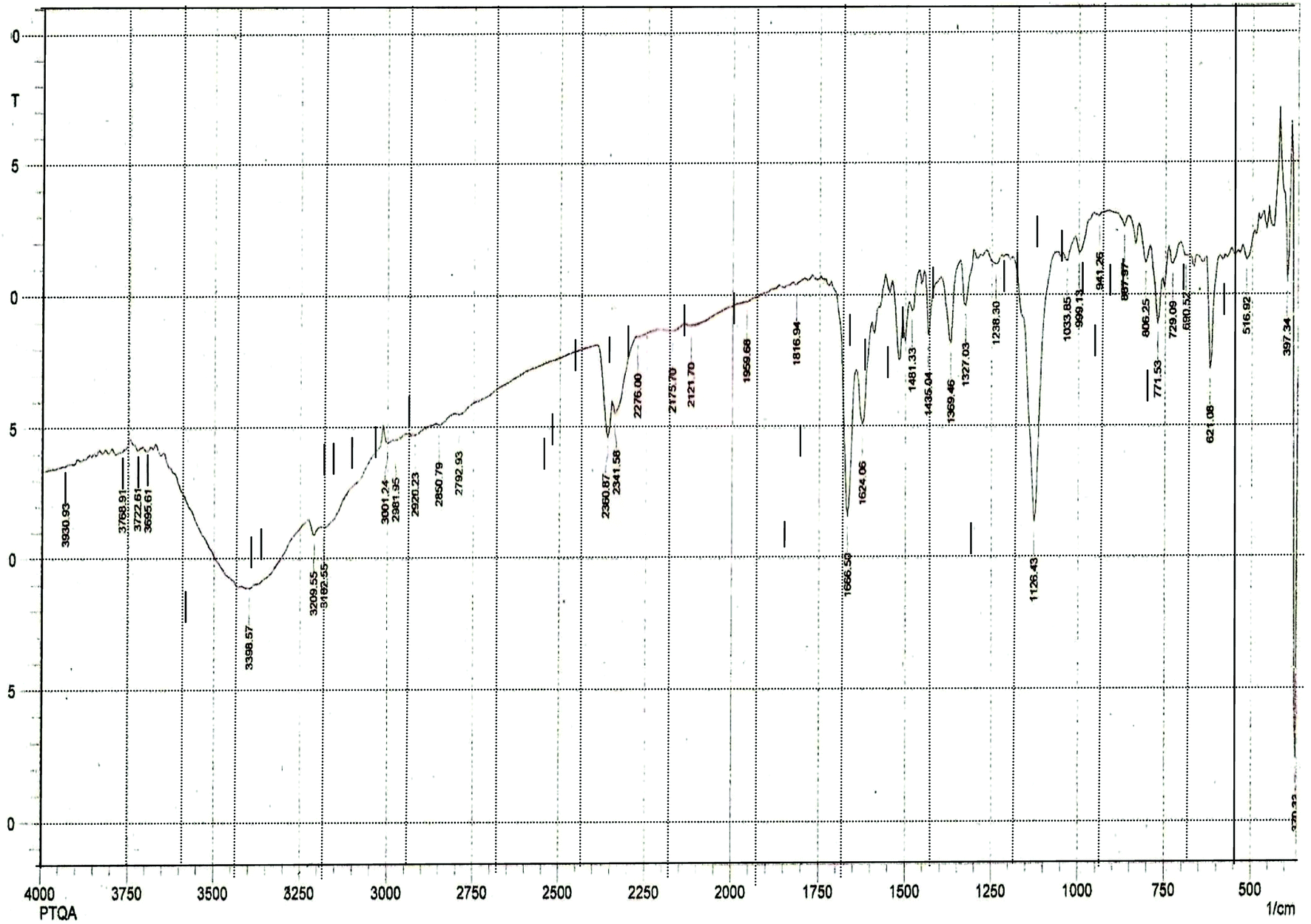 | ||
| Fig. 1 FTIR spectrum of 2-(α-pyridyl) thioquinaldinamide. | ||
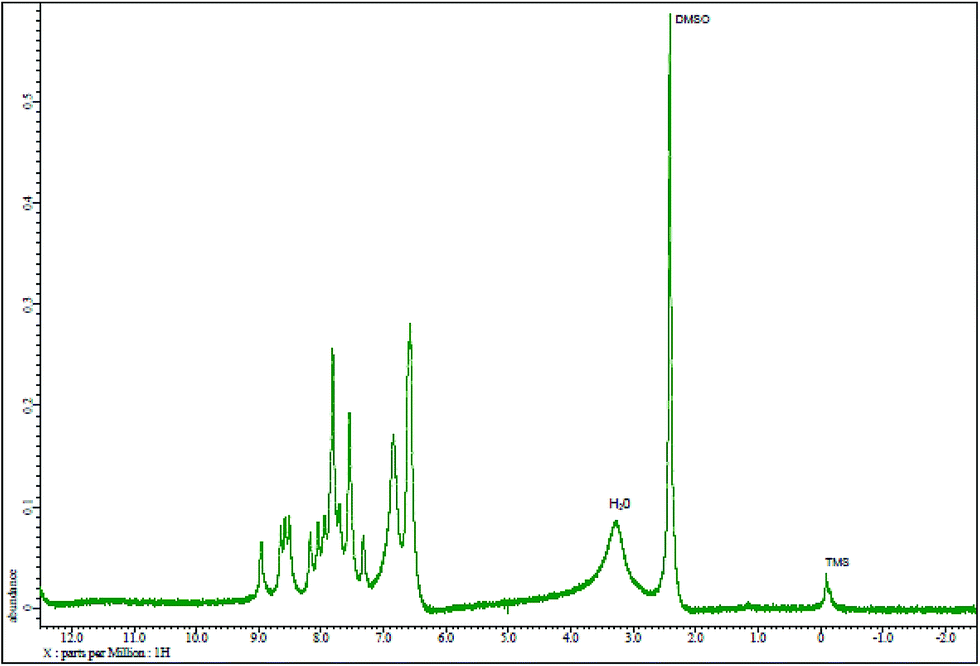 | ||
| Fig. 2 1HNMR spectrum of 2-(α-pyridyl)-thioquinaldinamide. | ||
The elemental analysis were performed by the National Center of Excellence in Analytical Chemistry, University of Sindh, Pakistan and FTIR spectra was recorded with FTIR spectrophotometer, Shimadzu (Model-IR Prestige 21, Detector-DTGS KBR) in the range 7500–350 cm−1 from our laboratory and 1HNMR spectrum was recorded with 1HNMR spectrophotometer model: JEOL 500SS from Kanazawa University, Japan.
Reagents and solutions
All the chemicals used were of analytical reagent grade of the highest purity available. High-purity absolute ethanol and high-purity de-ionized water were used throughout. High-purity water was obtained by passing tap water through cellulose absorbent and to mixed-bed ion exchange columns, followed by distillation in a corning AG-11 unit. Glass vessel were cleaned by soaking in acidified solutions of KMnO4 or K2Cr2O7 followed by washing with concentrated HNO3 and rinsed several times with high purity de-ionized water. Stock solutions and environmental water sample (1000 mL each) were kept in polypropylene bottles containing 1 mL concentrated HNO3. More rigorous contamination control was used when the manganese levels in the specimens were low.PTQA solution (1 × 10−3 M)
The reagent solution was prepared by dissolving the requisite amount (0.0024 g) of PTQA, in a known volume (10 mL) of absolute ethanol. A freshly prepared reagent solution (10−4 M) was used whenever required.Manganese(VII) standard solution (1.82 × 10−2 M)
A 100 mL amount of stock solution (1 mg mL−1) of heptavalent manganese was prepared by dissolving 287.66 mg of potassium permanganate (KMnO4) (Aldrich A.C.S. grade) in doubly distilled de-ionized water. Aliquots of this solution were standardized with oxalic acid.42 More dilute standard solutions were prepared by appropriate dilution of aliquots from the stock solution with de-ionized water when required. A freshly standardized solution was always used.Mn(II) standard solution (1.82 × 10−2 M)
A 100 mL amount of stock solution (1 mg L−1) of divalent manganese was prepared by dissolving 307.7 mg of purified-grade (E Merck proanalysis grade) monohydrated manganese sulphate (MnSO4·H2O) (super special grade J. T. Baker) in doubly distilled de-ionized water. Aliquots of this solution were standardized titrimetrically with disodiumdihydrogenethylenediaminetetraacetate (Na2H2EDTA) using Eriochrome Black T.42 More dilute standard solutions were prepared by appropriate dilution of aliquots from the stock solution with de-ionized water as and when required.Potassium dichromate solution
A 100 mL amount of stock solution (0.1 N) was prepared by dissolving 500 mg of finely powdered K2Cr2O7 (Merck) in 100 mL de-ionized water.Ammonium persuphate solution
Ammonium persuphate solution (2% w/v) (A.C.S.-grade 99% pure) was freshly prepared by dissolving 2 g in 100 mL of de-ionized water.Tartrate solution
A 100 mL stock solution of tartrate (0.01% w/v) was prepared by dissolving 10 mg of A.C.S.-grade (99%) potassium sodium tartrate tetrahydrate in (100 mL) de-ionized water.Aqueous ammonia solution
A 100 mL solution of an aqueous ammonia solution was prepared by diluting 10 mL concentrated NH4OH (28–30%, A.C.S.-grade) to 100 mL with de-ionized water. The solution was stored in a polypropylene bottle.EDTA solution
A 100 mL stock solution of EDTA (0.01% w/v) was prepared by dissolving 10 mg A.C.S.-grade (≥99%) ethylenediaminetetraacetic acid as disodium salt dihydrate in (100 mL) de-ionized water.Other solutions
Solutions of a large number of inorganic ions and complexing agents were prepared from their AnalaR grade or equivalent grade water-soluble salts (or the oxides and carbonates in hydrochloric acid); those of niobium, tantalum, titanium, zirconium and hafnium were specially prepared from their corresponding oxides (Specpure, Johnson Matthey) according to the recommended procedures of Mukharjee.43In the case of insoluble substances, special dissolution methods were adopted.44Procedure
To 0.1–1.0 mL of a neutral aqueous solution containing 0.1–8000 ng of manganese(VII) in a 10 mL calibrated flask was mixed with a 1:70–1:300 fold molar excess (preferably 1 mL of 1 × 10−4 M) of the 2-(α-pyridyl) thioquinaldinamide (PTQA) reagent solution followed by the addition of 0.5–2 mL (preferably 1 mL) of 0.025 M of sulfuric acid. The solution was mixed well and allowed to stand for 5 min after which 2 mL of absolute ethanol was added and the mixture was diluted to the mark with de-ionized water. The fluorescence intensity of the system was measured at 373 nm against a corresponding reagent blank, prepared concurrently, keeping the excitation wavelength maximum at 319 nm and the instrument setting the same. The manganese content in an unknown sample was determined using a concurrently prepared calibration graph.
Sample collection and preservation
Results and discussion
Factors affecting the fluorescence intensity
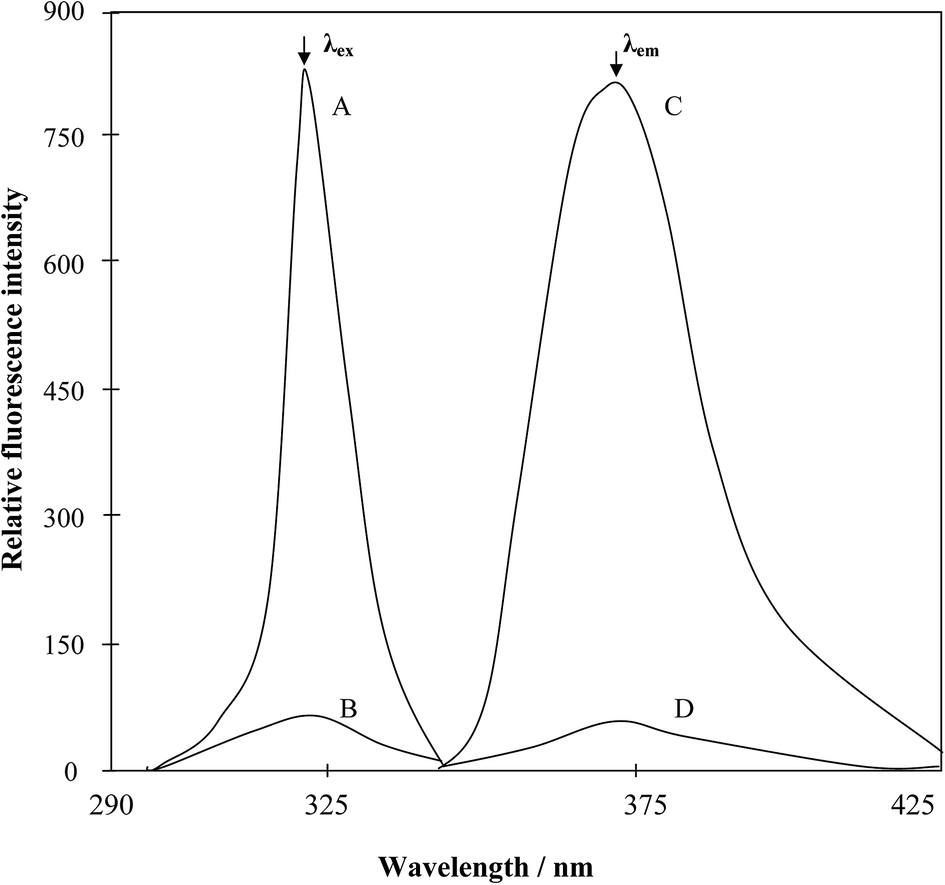 | ||
| Fig. 3 (A and B), Excitation spectra of the MnVII–PTQA system and the reagent blank (λem = 373 nm); and (C and D) are the corresponding emission spectra (λex = 319 nm). | ||
Optimization of some parameters on the fluorescence intensity
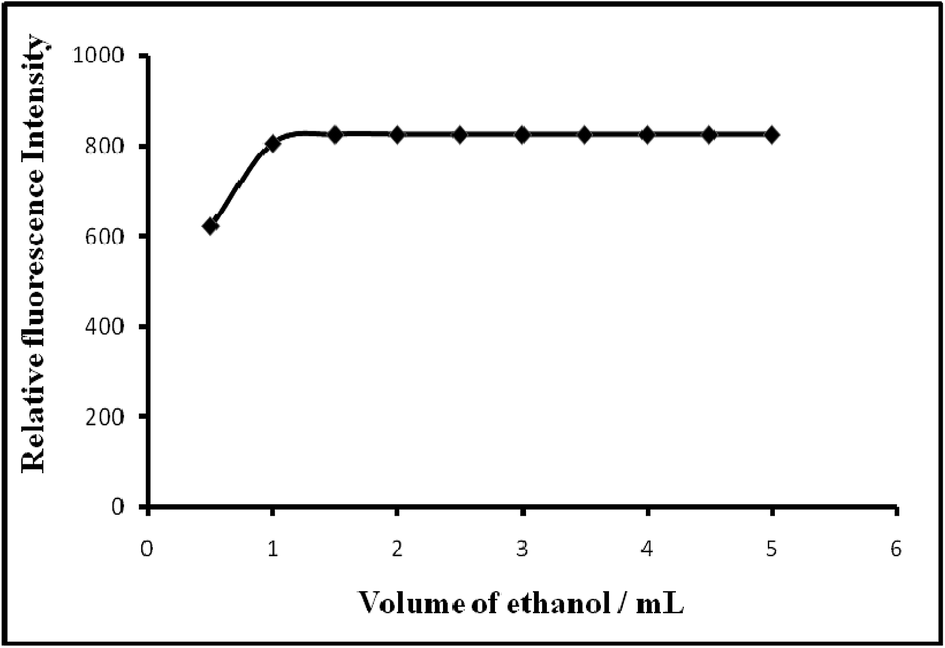 | ||
| Fig. 4 Effect of solvent (ethanol) on the fluorescence of Mn(VII)–PTQA system. | ||
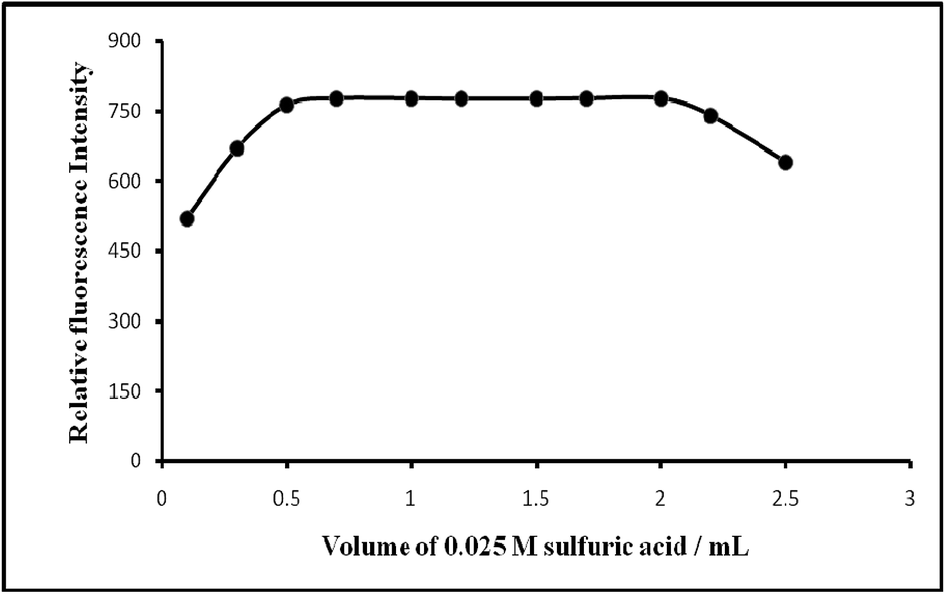 | ||
| Fig. 5 Effect of acidity on the fluorescence of Mn(VII)–PTQA system. | ||
:70–1:300 produced a constant fluorescence intensity of the oxidized product. Outside this range of reagent, the fluorescence intensity decreased (Fig. 6). At different manganese(VII) concentrations (0.5 and 1 μg L−1), the effect of varying the reagent concentration was similar. For all subsequent measurements 1 mL of 1 × 10−4 M PTQA reagent was added.
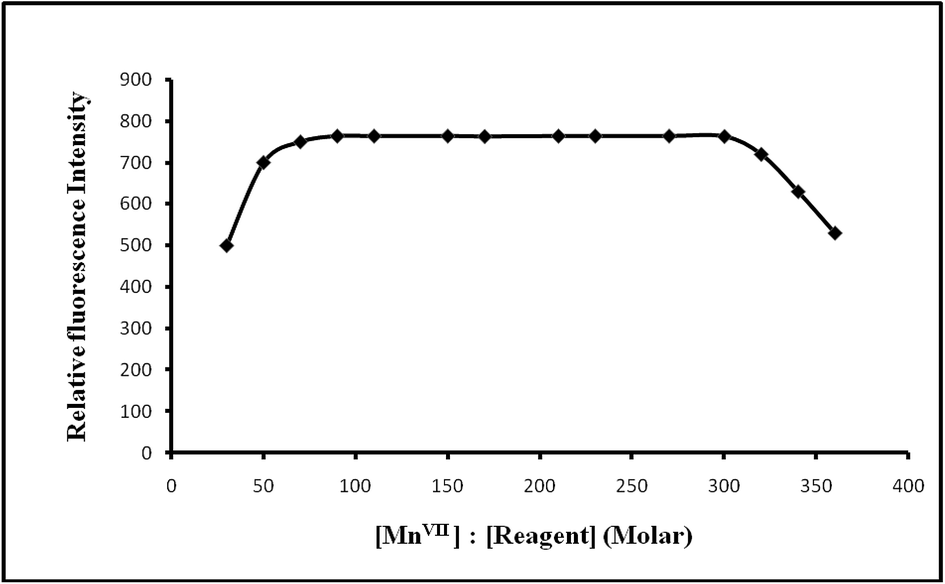 | ||
| Fig. 6 Effect of reagent on the fluorescence of Mn(VII)–PTQA system. | ||
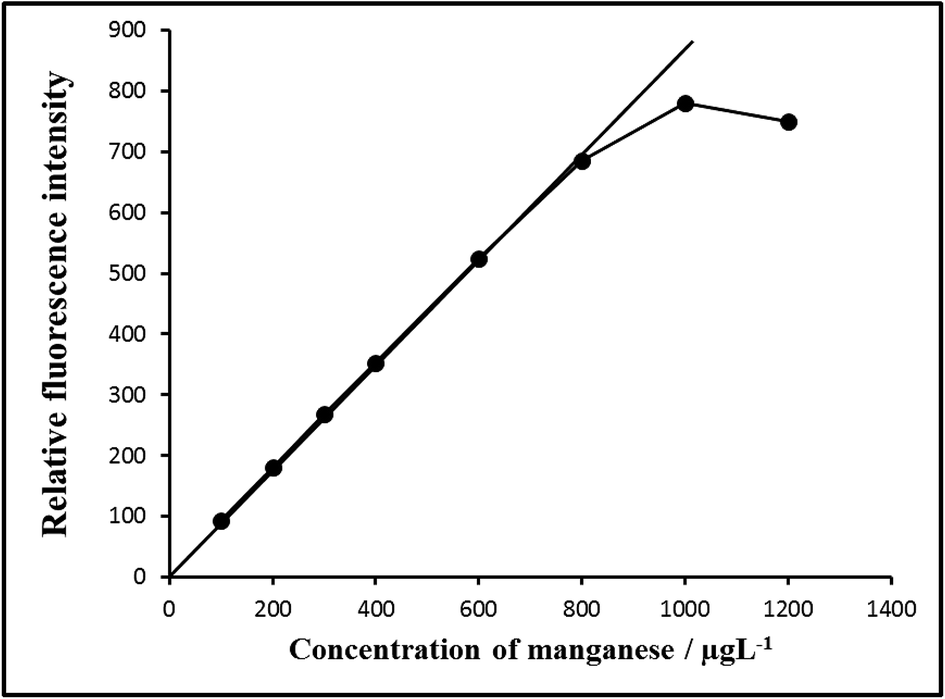 | ||
| Fig. 7 Calibration graph E: 100–800 μg L−1 of manganese(VII). Bandwidth: Ex. slit-1.5, Em. slit-1.5, sensitivity: high. | ||
| Parameters | Studied range | Selected value |
|---|---|---|
| Excitation wavelength maximum/λex (nm) | 200–700 | 319 |
| Emission wavelength maximum/λem (nm) | 200–700 | 373 |
| Solvent/amount of absolute ethanol/mL | 0–6 | 1–5 (preferably 2) |
| Acidity/M H2SO4 | 0.001–0.075 | 0.0125–0.05 (preferably 0.025) |
| pH | 3.69–0.69 | 3.0–1.0 (preferably 1.3) |
| Time/h | 0–72 | 1 min–24 h (preferably 5 min) |
| Temperature/°C | 10–70 | 15–40 (preferably 25 ± 5) |
| Reagent (fold molar excess, M:R) |
1:1–1:500 |
1:70–1:300 (preferably 1:100) |
| Linear range/μg L−1 | 0.001–1000 | 0.01–800 |
| Limit of quantification/ng L−1 | 1–100 | 10 |
| Detection limit/ng L−1 | 0.01–10 | 1 |
| Reproducibility (% RSD) | 0–10 | 0–2% |
| Regression co-efficient (R2) | 0.9992–0.9999 | 0.9998 |
| Species x | Tolerance ratio x/Mn (w/w) | Species x | Tolerance ratio x/Mn (w/w) |
|---|---|---|---|
| a Tolerance limit was defined as ratio that causes less than ±5 percent interference.b With 10 mg L−1 tartrate.c With 10 mg L−1 EDTA. | |||
| Ammonium(I) | 1000 | Lithium | 1000 |
| Arsenic(III) | 500b | Lead(II) | 200 |
| Arsenic(V) | 500 | Magnesium | 1000 |
| Aluminum | 200 | Manganese(II), manganese(IV) | 1000, 500 |
| Azide | 200 | Mercury(II) | 200 |
| Ascorbic acid | 1000 | Molybdenum(VI) | 500 |
| Antimony | 500 | Nitrate | 1000 |
| Bromide | 100 | Nickel | 500 |
| Bismuth(III) | 500 | Oxalate | 1000 |
| Beryllium(II) | 1000 | Potassium | 1000 |
| Calcium | 1000 | Phosphate | 1000 |
| Chloride | 1000 | Selenium(VI) | 500 |
| Cobalt(II) | 200c | Selenium(IV) | 50c |
| Cobalt(III) | 500 | Silver | 500 |
| Chromium(III) | 1000 | Sodium | 1000 |
| Chromium(VI) | 100c | Strontium | 500 |
| Cadmium | 1000 | Sulfate | 500 |
| Carbonate | 1000 | Titanium(IV) | 100 |
| Cesium | 1000 | Tellurium(IV) | 500 |
| Citrate | 1000 | Tartrate | 1000 |
| Cerium(III) | 500 | Copper(II) | 500 |
| Cerium(IV) | 500b | Thiocyanate | 1000 |
| Cyanide | 200 | Thiourea | 100 |
| EDTA | 1000 | Tungsten(VI) | 1000 |
| Fluoride | 1000 | Tin(II) | 50b |
| Iodide | 1000 | Tin(IV) | 50b |
| Iron(II) | 200 | Vanadium(V) | 1000 |
| Iron(III) | 200 | Zinc | 1000 |
The greater tolerance limits for these ions can be achieved by using several masking agents. In order to eliminate the interference of Se(IV), Cr(VI) and Ce(IV) ions, EDTA and tartrate can be used as masking agents, respectively.46 A 50, 100 and 500-fold excess of Se(IV), Cr(VI) and Ce(IV) ions could be masked with EDTA and tartrate, respectively. During the interference studies, if a precipitate was formed, it was removed by centrifugation. The amount mentioned is not the tolerance limit but the actual amount studied. However, for those ions whose tolerance limits have been studied, their tolerance ratios are mentioned in Table 2.
Strong reducing agents such as tin(II), chloride, iron(II), sulphate, hydroxylamine, hydrochloride and sodium azide, which would otherwise reduce manganese(VII), undergo oxidation during the treatment of manganese(II) solution with persulphate and hence are not a problem.
| Sample | Certified reference materials (composition, %) | Manganese (%) | ||
|---|---|---|---|---|
| In CRM sample | Found (n = 5) | RSDa | ||
| a The measure of precision is the relative standard deviation (RSD).b Values in μg g−1.c Values in mg kg−1. | ||||
| 1 | BCS-CRM 163, unalloyed steel (C = 1.2, Mn = 0.472, Ni = 0.608, Si = 0.18, Cu = 0.05, Cr = 0.03) | 0.472 | 0.47 | 1.5 |
| 2 | BAS-CRM-10g, high-speed brass (Cu = 60.8, Fe = 1.56, Pb = 0.23, Ni = 0.16, Sn = 0.21, Al = 3.34, Zn = 32.0, Mn = 1.36) | 1.36 | 1.35 | 1.2 |
| 3 | BCS-CRM-238, unalloyed steel (C = 0.2, Si = 0.06, S = 0.05, Mn = 0.51, Ni = 0.06, Cr = 0.06 and Cu = 0.05) | 0.51 | 0.50 | 2.0 |
| 4 | BAS-CRM-1d, mild steel (C = 0.19, Si = 0.06, S = 0.04, Mn = 0.52, P = 0.04) | 0.52 | 0.518 | 1.8 |
| 5 | BAS-CRM-33b, alloy cast iron (Mn = 0.64, Cr = 0.61, Mo = 0.04, Ni = 2.24) | 0.64 | 0.625 | 1.6 |
| 6 | CRM-BCR-397, human hair | 11.2 ± 0.3b | 11.15 ± 0.5 | 1.0 |
| 7 | NIST-SRM-1646, estuarian sediment | 234.5c | 232.0 | 1.5 |
| Sample | Manganese/μg L−1 | Recovery ± s (%) | srb (%) | ||
|---|---|---|---|---|---|
| Added | Founda (n = 5) | ||||
| a Average of five replicate determinations of each sample.b The measure precision is the relative standard deviation(sr).c T. S. P. Complex Ltd., Patenga, Chittagong.d PHP Glass factory, Chittagong.e Bangladesh Steel Re-rolling Mills Ltd. (BSRM), Baizid Bosthami, Chittagong.f Karnaphuly Paper Mills, Chandraghona, Chittagong.g Berger Paints Bangladesh Limited, Kalurghat, Chittagong. | |||||
| Tap water | 0, 10, 50 | 55.0, 65.0, 108.0 | 100.0 ± 0.0, 103.0 ± 0.5 | 0.00, 0.25 | |
| Well water | 0, 10, 50 | 35.0, 45.0, 85.0 | 100 ± 0.0, 100 ± 0.0 | 0.00, 0.00 | |
| River water | Karnaphully (upper) | 0, 10, 50 | 75.0, 88.0, 125.0 | 104.0 ± 06, 100.0 ± 0.0 | 0.35, 0.00 |
| Karnaphully (lower) | 0, 10, 50 | 80.5, 90.0, 135.0 | 99.5 ± 0.5, 103.0 ± 0.8 | 0.21, 0.39 | |
| Halda (upper) | 0, 10, 50 | 45.0, 55.0, 98.0 | 100.0 ± 0.00, 103.0 ± 0.6 | 0.00, 0.29 | |
| Halda (lower) | 0, 10, 50 | 53.0, 65.0, 105.0 | 103.0 ± 0.8, 102.0 ± 0.5 | 0.36, 0.33 | |
| Sea water | Bay of Bengal (upper) | 0, 10, 50 | 25.0, 35.0, 80.0 | 100.0 ± 0.0, 106.0 ± 1.0 | 0.00, 0.45 |
| Bay of Bengal (lower) | 0, 10, 50 | 35.0, 45.0, 90.0 | 100.0 ± 0.0, 105.8 ± 1.0 | 0.00, 0.38 | |
| Drain water | T. S. P. Complexc | 0, 100, 500 | 130.0, 235.0, 630.0 | 102.0 ± 0.8, 100.2 ± 0.0 | 0.18, 0.00 |
| PHPd | 0, 100, 500 | 160.0, 260.0, 665.0 | 100.0 ± 0.0, 100.7 ± 0.5 | 0.00, 0.25 | |
| BSRMe | 0, 100, 500 | 245.0, 350.0, 750.0 | 101.0 ± 0.8, 100.6 ± 1.0 | 0.29, 0.35 | |
| K. P. M. waterf | 0, 100, 500 | 188.0, 288.0, 690.0 | 100.0 ± 0.8, 100.2 ± 0.5 | 0.00, 0.42 | |
| Berger paintsg | 0, 100, 500 | 270.0, 370.0, 775.0 | 100.0 ± 0.0, 100.6 ± 1.2 | 0.00, 0.55 | |
| Serial no. | Sample | Manganese/μg L−1 | Sample sourcea | |
|---|---|---|---|---|
| AAS (n = 5) | Proposed method (n = 5) | |||
| a Samples were from Chittagong Medical College Hospital.b Values in ng g−1. | ||||
| 1 | Blood, urine | 800 ± 1.0, 210.00 ± 1.5 | 798.5 ± 1.3, 201.5 ± 1.5 | Manganism (damage of central nervous system or neurological disorder) patient (female) |
| 2 | Blood, urine | 460.5 ± 1.3, 120.6 ± 1.5 | 465.8 ± 1.5, 121.5 ± 1.7 | Leucopenia patient (female) |
| 3 | Blood, urine | 385.8 ± 1.3, 106.5 ± 1.8 | 390.5 ± 1.5, 112.2 ± 2.0 | Liver cirrhosis patient (male) |
| 4 | Blood, urine | 370.6 ± 1.5, 93.5 ± 1.8 | 372.5 ± 1.8, 98.5 ± 2.2 | Pneumonitis (manganic pneumonia) patient (male) |
| 5 | Blood, urine | 230.8 ± 1.6, 85.6 ± 2.0 | 232.5 ± 2.0, 85.6 ± 1.8 | Hypertension patient (male) |
| 6 | Blood, urine | 110.0 ± 2.1, 30.5 ± 1.9 | 115.0 ± 2.0, 32.8 ± 2.5 | Asthma patient (male) |
| 7 | Blood, urine | 15.5 ± 2.0, 3.85 ± 1.5 | 15.8 ± 1.5, 3.82 ± 1.8 | Normal adult (male) |
| 8 | Hairb | 790.5 ± 1.8 | 795.8 ± 1.5 | Normal human hair (male) |
Nature of the fluorescent species
The non fluorescent reagent, PTQA, produced the same spectral characteristics with excitation and emission wavelengths almost invariably around 319 nm and 373 nm, with manganese(VII), chromium(VI), selenium(IV) and cerium(IV) and with persulphate, hydrogen peroxide, and triiodide in acidic media. This indicates that the fluorescence species is an oxidized product of the reagent itself and not a chelate. Similar oxidative fluorescent reactions have been utilized previously.47 The PTQA reagent produced here has so many potential reaction sites that the structure of the oxidized fluorescent species is difficult to predict. Given that ring closure can lead to intense fluoresce in some circumstances, it seems likely that photo-oxidative cyclization takes place, leading to the formation of structure (A) in resonance with structure (B) (Fig. 8).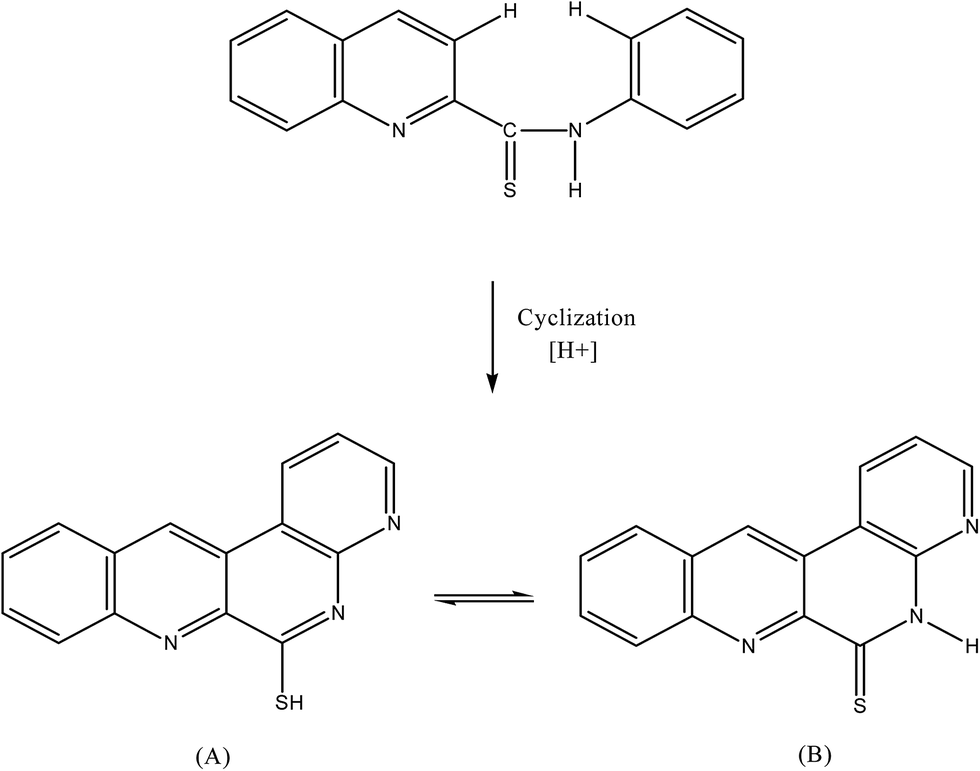 | ||
| Fig. 8 Mechanism of oxidative cyclization reaction of 2-(α-pyridyl)-thioquinaldinamide (PTQA). | ||
Applications
The present method was successfully applied to the determination of manganese(VII) in a series of synthetic mixtures of various compositions and also in a number of real samples e.g. several Certified Reference Materials (CRM) (Table 3). The method was also extended to the determination of manganese in a number of environmental, biological, soil, food, tea, fertilizer, vegetable, fruit and pharmaceutical samples. In view of the unknown composition of environmental water samples, the same equivalent portions of each such sample were analyzed for manganese content; the recoveries in both the “spiked” (added to the samples before the mineralization or dissolution) and the “unspiked” samples are in excellent agreement (Table 4). The results of biological analyses by spectrofluorimetric method were found to be in excellent agreement with those obtained by AAS (Table 5). The results of soil analyses by the spectrofluorimetric method are shown in Table 6. The results of pharmaceutical samples by the spectrofluorimetric method are shown in Table 7. The results of vegetable and fruit samples by the spectrofluorimetric method were found to be in excellent agreement with that of found by AAS (Table 8). The results of food analyses by spectrofluorimetric method were also found to be in good agreement with those obtained by ICP-MS (Table 9). The results of speciation of manganese(II) and manganese(VII) in mixtures are shown in Table 10. Statistical comparison of proposed method with reference methods are shown in Table 11.| Serial no. | Manganesea (mg kg−1) (n = 5) | RSD (%) | Sample sourceb |
|---|---|---|---|
| a Average of five analyses of each sample.b Composition of the soil samples: C, N, P, K, Na, Ca, Mg, Cu, Mo, Fe, Pb, V, Zn, Mn, Co, NO3, SO4 etc. | |||
| S1b | 3.5 ± 0.5 | 1.8 | Agriculture soil (Chittagong University Campus) |
| S2 | 1.05 ± 0.3 | 1.5 | Marine soil (Bay of Bengal) |
| S3 | 21.5 ± 1.0 | 1.8 | Traffic soil (Kadamtali Bus Terminal) |
| S4 | 95.6 ± 1.5 | 2.2 | Industrial soil (T. S. P. Complex, Chittagong) |
| S5 | 78.5 ± 1.0 | 2.3 | Industrial soil (Eastern refinery, Chittagong, Bangladesh) |
| S6 | 130.5 ± 1.3 | 2.5 | Industrial soil (Eastern Cables) |
| S7 | 145.0 ± 1.0 | 2.0 | Industrial soil (P·H·P glass) |
| S8 | 7.85 ± 0.8 | 2.1 | Road side soil (Dhaka-Chittagong Highway) |
| S9 | 117.0 ± 1.5 | 2.0 | Paint soil (Berger paint, Chittagong) |
| S10 | 7.5 ± 0.8 | 1.8 | River soil (River Karnaphuly, Chittagong) |
| Pharmaceutical samples | Brand name | Trade name | Manganese/μg g−1 or mg L−1 | RSD (%) | |
|---|---|---|---|---|---|
| Reported (claimed) value | Found (n = 5) | ||||
| Tablet | Square Pharmaceuticals Ltd. | Mulvit plus (multivitamin-mineral)/μg g−1 | 101.5 | 102.5 | 2.0 |
| Eskayef Bangladesh Ltd (SK + F) | Ostocal M (calcium, vitamin D & minerals)/μg g−1 | 185.5 | 184.2 | 1.5 | |
| Beximco Pharmaceuticals Ltd (BPL) | Bextram gold (A to Z) (multivitamin-mineral)/μg g−1 | 201.5 | 205.0 | 2.0 | |
| Syrup | The ACME Laboratory | Menuvit syrup (multivitamin-mineral)/mg L−1 | 155.0 | 153 | 2.3 |
| Finecure Pharmaceuticals Ltd. | Menuvit syrup (multivitamin-mineral)/mg L−1 | 160.0 | 158.8 | 1.8 | |
| Serial no. | Sample | Manganese/mg kg−1 Founda ± s (n = 5) | Sample source | |||
|---|---|---|---|---|---|---|
| AAS (n = 5) | Proposed method | |||||
| Found | RSDb | Found | RSDb | |||
| a Average of five replicate analyses of each sample.b The measure of precision is the relative standard deviation (RSD). | ||||||
| 1 | White cabbage (Brassica oleraceacupitata) | 20.5 | 1.0 | 21.8 | 1.2 | Local market, Chittagong |
| 2 | Radish (Raphanus sativus) | 29.5 | 1.5 | 30.0 | 2.0 | Local market, Chittagong |
| 3 | Tomato (Lycopersicon esculentum) | 15.75 | 1.8 | 17.5 | 2.5 | Local market, Chittagong |
| 4 | Cauliflower (Brassica oleracea) | 45.5 | 1.6 | 46.5 | 1.5 | Local market, Chittagong |
| 5 | Onion (Allium cepa) | 75.5 | 1.8 | 78.0 | 2.0 | Local market, Chittagong |
| 6 | Coriander seeds (Coriandrum sativum) | 40.5 | 2.0 | 41.5 | 1.8 | Local market, Chittagong |
| 7 | Potato (Solanum tuberosum) | 7.50 | 1.4 | 8.50 | 1.5 | Local market, Chittagong |
| 8 | Garlic (Allium sativum) | 49.5 | 1.5 | 50.4 | 1.9 | Local market, Chittagong |
| Samplea | Manganese/μg g−1 | ||||
|---|---|---|---|---|---|
| ICP-MS (n = 5) | Proposed method (n = 5) | ||||
| Found | RSDb (%) | Found | RSDb (%) | ||
| a Samples were from Local market, Chittagong.b The measure of precision is the relative standard deviation (RSD). | |||||
| Tea (Camellia sinensis) | Proyash tea | 285.8 | 1.8 | 289.5 | 1.5 |
| Ispahani tea | 282.4 | 1.9 | 285.0 | 1.7 | |
| Taza tea | 188.8 | 2.0 | 185.5 | 1.8 | |
| Seylon tea | 297.6 | 2.1 | 299.3 | 1.5 | |
| Green tea | 295.5 | 2.0 | 198.2 | 1.8 | |
| Rice (Oryza sativa) | 1.95 | 1.0 | 1.98 | 1.2 | |
| Wheat (Triticum aestivum) | 2.88 | 1.6 | 2.91 | 1.6 | |
| Serial no. | Mn(VII): Mn(II) |
Mn, taken (μg L−1) | Mn, found (μg L−1) | Error (μg L−1) | |||
|---|---|---|---|---|---|---|---|
| Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | ||
| 1 | 1:1 |
10 | 10 | 9.98 | 9.99 | 0.02 | 0.01 |
| 1 | 1:1 |
10 | 10 | 10.00 | 10.02 | 0.00 | 0.02 |
| 1 | 1:1 |
10 | 10 | 9.97 | 9.98 | 0.03 | 0.02 |
| Mean error: Mn(VII) = ±0.017; Mn(II) = ±0.017 | |||||||
| Standard deviation: Mn(VII) = ±0.012; Mn(II) = ±0.011 | |||||||
| Serial no. | Mn(VII):Mn(II) |
Mn, taken (μg L−1) | Mn, found (μg L−1) | Error (μg L−1) | |||
|---|---|---|---|---|---|---|---|
| Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | ||
| 1 | 1:5 |
10 | 50 | 9.98 | 49.80 | 0.02 | 0.20 |
| 1 | 1:5 |
10 | 50 | 9.99 | 49.70 | 0.01 | 0.30 |
| 1 | 1:5 |
10 | 50 | 9.98 | 49.80 | 0.02 | 0.20 |
| Mean error: Mn(VII) = ±0.017; Mn(II) = ±0.234 | |||||||
| Standard deviation: Mn(VII) = ±0.0015; Mn(II) = ±0.0018 | |||||||
| Serial no. | Mn(VII):Mn(II) |
Mn, taken (μg L−1) | Mn, found (μg L−1) | Error (μg L−1) | |||
|---|---|---|---|---|---|---|---|
| Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | Mn(VII) | Mn(II) | ||
| 1 | 1:10 |
10 | 100 | 9.99 | 99.8 | 0.01 | 0.2 |
| 1 | 1:10 |
10 | 100 | 9.98 | 99.9 | 0.02 | 0.1 |
| 1 | 1:10 |
10 | 100 | 9.97 | 99.8 | 0.03 | 0.2 |
| Mean error: Mn(VII) = ±0.02; Mn(II) = ±0.17 | |||||||
| Standard deviation: Mn(VII) = ±0.0019; Mn(II) = ±0.0018 | |||||||
| Samples | F – test resultsa | ||||
|---|---|---|---|---|---|
| (s12/s22)15 | (s12/s32)26 | (s12/s42)31 | (s12/s52)33 | (s12/s62)28 | |
| a Tabulated F-value for (5, 5) degrees of freedom at P(0.98) is 5.72. s1 = standard deviation of proposed method, s2 = standard deviation of reference method,15 s3 = standard deviation of reference method,26 s4 = standard deviation of reference method,31 s5 = standard deviation of reference method,33 s6 = standard deviation of reference method.28 | |||||
| Water | 0.127 | 0.58 | 0.23 | 0.0019 | |
| Water | 0.0516 | 0.063 | 0.028 | 0.37 | |
| Water | 0.219 | 0.100 | 0.273 | ||
| Water | 0.119 | ||||
| Blood | 0.95 | ||||
| Blood | 0.52 | ||||
| Blood | 0.73 | ||||
| Soil | 0.32 | 0.0002 | 0.0004 | ||
| Soil | 0.95 | 0.0003 | 0.0007 | ||
| Soil | 1.0 | 0.0001 | 0.0001 | ||
| Alloy | 0.074 | ||||
| Alloy | 0.12 | ||||
| Synthetic mixture | 0.13 | ||||
| Synthetic mixture | 0.52 | ||||
| Synthetic mixture | 0.72 | ||||
| Food | 0.79 | ||||
| Food | 0.68 | ||||
| Urine | 1.02 | ||||
| Urine | 0.84 | ||||
| Tea | 0.98 | ||||
| Wheat | 0.76 | ||||
Determination of manganese in synthetic mixtures
Several synthetic mixtures of varying compositions containing manganese(VII) and diverse ions of known concentrations were determined by the present method using EDTA as masking agent. The results were found to be highly reproducible. Accurate recoveries were achieved in all solutions in the range 98 ± 0.5 to 100 ± 0.0%. The reliability of our manganese–PTQA procedure was approved by quantitative recovery of manganese(VII) spiked in several synthetic mixtures containing manganese(VII) and diverse ions. The method has high precision and accuracy (s = ±0.01 for 0.5 μg L−1).Determination of manganese in certified reference materials
A 0.1 g amount of an alloy or steel sample containing 0.472–1.36% of manganese was weighed accurately and placed in a 50 mL Erlenmeyer flask in presence of excess oxidizing agent following a method recommended by Parker.48 To it, 10 mL of 20% (w/v) sulfuric acid was added and the mixture was heated gently. The residual carbides were decomposed by gently simmering with 1 mL of concentrated H3PO4 and 10 mL of concentrated HNO3. Then a further 2 mL of 1 + 1 H2SO4 and 2 mL 2% w/v freshly prepared persulphate were added and the solution was evaporated carefully to dense white fumes of sulphur trioxide, then cooled to room temperature (25 ± 5 °C). After suitable dilution with de-ionized water, the contents of the Erlenmeyer flask were warmed so as to dilute the soluble salts. The solution was then cooled and neutralized with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) EDTA solution. The resulting solution was filtered if necessary, through a Whatman no. 40 filter paper into a 100 mL calibrated flask. The residue (silica tungstanic acid) was washed with a small volume of hot 1 + 99 H2SO4, followed by water; the volume was made up to mark with de-ionized water.A suitable aliquot (1–2 mL) of the above-mentioned solution was taken into a 10 mL calibrated flask and the manganese(VII) content was determined; as described under procedure using tartrate or EDTA as masking agent. The proposed procedure for the spectrofluorimetric determination of manganese was applied to the analysis of estuarine sediment (NIST-SRM-1646) and human hair (CRM-BCR-397) CRMs obtained from the National Resource Council of Canada using tartrate or EDTA as a masking agent, following a method recommended by Sun et al.49 Based on five replicate analyses, average manganese concentration determined by spectrofluorimetric method was in excellent agreement with the certified values. The results are given in Table 3.
Determination of manganese in environmental water samples
Each filtered (with Whatman no. 40) environmental sample (25 mL) contained in a 50 mL Pyrex beaker were added 1 mL of concentrated H2SO4, 1 mL of concentrated H3PO4 and 2 mL of concentrated HNO3 and the mixture was heated on a hot plate until white fumes of sulphur trioxide; in the presence of freshly prepared 2 mL of 2% (w/v) persulphate solution in a fume cupboard to oxidize manganese(II) to manganese(VII) following a method recommended by Greenberg et al.50An aliquot (1–2 mL) of this water sample was pipetted into a 10 mL calibrated flask and the manganese content was determined as described under the general procedure using tartrate or EDTA as masking agent. The results of analyses of environmental water samples from various sources for manganese are shown in Table 4.
Spectrofluorimetric methods for determination of manganese in natural and sea-water does not require any preconcentration of manganese.51 The concentration of manganese in natural and sea water is a few μg L−1 in developed countries.52 The mean concentration of manganese found in U.S. drinking water is greater than 20 μg L−1.52
Determination of manganese in biological samples
Human blood (1–2 mL) or urine (5–10 mL) or hair (1–2 g) sample was taken into a 100 mL micro-Kjeldahl flask. A glass bead and 10 mL of concentrated nitric acid were added, and the flask was placed on the digester under gentle heating. The sample was digested in the presence of an excess persulphate solution according to the method recommended by Stahr.53 As the heating process continued 1 mL of H2SO4 is added and heated for about 0.5 hour to dense white fumes of sulphur trioxide.When the initial brisk reaction was completed, the solution was removed and cooled at room temperature. A 1 mL volume of 3.33 M H3PO4 was carefully added, followed by the addition of 2 mL of 2% (w/v) freshly prepared ammonium persulphate solution to oxidize Mn(II) to Mn(VII) and heating was continued to dense white fumes, while repeating the nitric acid addition, if necessary. The solution was then cooled and neutralized with a dilute NH4OH. The final solution is made up to the mark with de ionized water.
A suitable aliquot (1–2 mL) of the final solution was pipetted out into a 10 mL calibrated flask and the manganese content was determined as described under procedure using EDTA or tartrate as masking agent. The results of biological analyses by the spectrofluorimetric method were found to be in excellent agreement with those obtained by AAS. The results are shown in Table 5.
The abnormally high value for the manganism patient is probably due to the involvement of high manganese concentrations with As and Zn. The occurrence of such high manganese contents are also reported in manganism patient from some developed countries.54
Determination of manganese in soil samples
An air-dried homogenized soil sample (10 g) was accurately weighed and placed in a 100 mL micro-Kjeldahl flask. The sample was digested in the presence of an oxidizing agent (2 mL of 2% freshly prepared ammonium persulphate solution) following method recommended by Jackson.55 The solution was cooled and in the resulting solution 1 mL of 3.33 M H3PO4, 2 mL of 2% (w/v) freshly prepared ammonium persulphate was added. The solution was heated for 5–10 min to affect complete oxidation from manganese(II) to manganese(VII). The solution is reduced in volume by evaporation to eliminate excess persulphate.The content of the flask was filtered through a Whatman no. 40 filter paper into 25 mL calibrated flask and neutralized with dilute NH4OH solution in the presence of 1–2 mL of 0.01% (w/v) EDTA solution. The resulting solution was then diluted up to the mark with de-ionized water.
A suitable aliquot (1 mL) of the final solution was pipetted out into a 10 mL calibrated flask and the manganese content was determined as described under procedure using tartrate or EDTA as masking agent. The manganese content was then determined by the above procedure and quantified from a calibration graph prepared concurrently. The results are shown in Table 6. The average value of manganese in Bangladesh surface soil was found to be 43.29 mg kg−1.56
Determination of manganese in pharmaceutical samples
Finished pharmaceutical samples (each Mn containing 1 mg tablet or 5 mL syrup or required weight) were quantitatively taken in a 50 mL beaker and digested in the presence of excess oxidizing agent following a method recommended by Ahmed et al.57 10 mL of concentrated nitric acid is added and heated to dryness and then added 10 mL of 20% (v/v) of H2SO4 and heated to white fume. In the resulting solution 1 mL of 3.33 M H3PO4, 2 mL of 2% (w/v) freshly prepared ammonium persulphate is added. The solution is heated for 5–10 min to effect complete oxidation of manganese(II) to manganese(VII). The solution is reduced in volume by evaporation to eliminate excess persulphate. The solution was then cooled and neutralized with dilute NH4OH in the presence of 1–2 mL of 0.01% (w/v) EDTA solution. The resulting solution was then filtered and quantitatively transferred into a 25 mL calibrated flask and made up to the mark with de-ionized water.An aliquot (1–2 mL) of this digested sample was pipetted into a 10 mL calibrated flask and then manganese content was determined as described under the general procedure using tartrate or EDTA as a masking agent. The results of some pharmaceutical analyses are in excellent agreement with the reported values. The analyses of pharmaceutical samples from several Pharmaceutical Companies for manganese are given in Table 7.
Determination of manganese in vegetable and fruit samples
The vegetable and fruit samples collected prior to the determination were pretreated in the following way: edible portion of samples was first washed clean with tap water followed by rewashing with de-ionized water. After removing de-ionized water from the surface of vegetables and fruits, the samples were cut into small pieces and dried at 65 °C in oven. An air dried vegetables and fruits samples (10 gm) were taken in a 100 mL micro-Kjeldahl flask in presence of oxidizing agent and digested following a method recommended by Stahr53 and 10 mL of concentrated nitric acid were added and the flask was placed on the digester under gentle heating. When the initial brisk reaction was over, the solution was removed and cooled at room temperature. 1 mL volume of concentrated sulfuric acid was added carefully, followed by the addition of 2 mL of concentrated HF, and heating was continued for at least 1/2 h and then cooled. In the resulting solution 1 mL of 3.33 M H3PO4, 2 mL of 2% (w/v) freshly prepared ammonium persulphate is added. The mixture of each foodstuff was heated below the boiling point for 5–10 min to oxidize manganese(II) to manganese(VII). The resulting solution was then cooled and neutralized with dilute NH4OH in presence of 1–2 mL of (w/v) EDTA solution. The resulting solution was filtered and quantitatively transferred into a 25 mL calibrated flask and mixed well and made up to the mark with de ionized water.A suitable aliquot (1–2 mL) of the final solution was pipetted into a 10 mL calibrated flask and the manganese content was determined as described under the procedure using tartrate as masking agent. High value of manganese for Allium cepa (onion) is probably due to the involvement of high manganese concentration in the soil.58 The results of vegetable and fruit analysis by spectrofluorimetric method were also found to be in excellent agreement with those obtained by AAS. The results are shown in Table 8.
Determination of manganese in food and tea samples
The food samples used were rice, wheat and tea and these were used under dry conditions. Each sample was first ground in a mortar. Tea samples (0.1 g) or rice and wheat samples (1.0 g) were weighed accurately and placed in a porcelain crucible and charred in an electric furnace; the sample was ashed at 555 °C in a muffle furnace in presence of excess oxidizing agent following a method recommended by Stahr.53 2 mL of HCl and 10 mL of water were added to the ash. The mixture of each foodstuff was heated below the boiling point for a moment. In the resulting solution 1 mL of 3.33 M H3PO4, 2 mL of 2% (w/v) freshly prepared ammonium persulphate is added. The solution is heated for 5–10 min to effect complete oxidation from Mn(II) to Mn(VII). The solution was then cooled and neutralized with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) EDTA solution and filtered. The resulting solution was quantitatively transferred transferred into a 25 mL calibrated flask and mixed well and made up to the mark with de-ionized water.A suitable aliquot (1–2 mL) of the final solution was pipetted out into a 10 mL calibrated flask and the manganese content was determined as described under procedure using tartrate as masking agent. The results of food analyses by the spectrofluorimetric method were also found to be in excellent agreement with those obtained by ICP-MS. The results are shown in Table 9.
High value of manganese for Camellia sinensis (green tea) is probably due to the involvement of high manganese concentration in the soil.
Determination of manganese(II) and manganese(VII) speciation in mixtures
Suitable aliquots (1–2 mL) of manganese (VII + II) mixtures (preferably 1:1, 1:5, 1:10) were taken in a 25 mL Pyrex beaker. In this solution 1 mL of 3.33 M H3PO4, 1 mL of 10−4 M silver nitrate and 2 mL of 2% (w/v) freshly prepared ammonium persulphate is added. The solution (4–5 mL) is heated for 5–10 min to effect complete oxidation to convert all Mn(II) into Mn(VII). The solution is reduced in volume by evaporation to half of its initial volume to eliminate excess persulphate. The solution was then cooled and neutralized with dilute NH4OH. The solution is taken quantitatively in to 10 mL volumetric flux and 1 mL of 1 × 10−4 M PTQA reagent solution was added followed by the addition of 1 mL of 0.025 M H2SO4 and 2 mL ethanol. It was made up to the mark with de-ionized water. The fluorescence intensity was measured after 5 min at 373 nm when excited at 319 nm, against a reagent blank. The total manganese content was calculated with the help of a calibration graph prepared concurrently.58,59
An equal aliquot (1–2 mL) of the above manganese (VII + II) mixture was taken into a 25 mL beaker. Neutralize the solution with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) tartrate solution. After, the content of the beaker was transferred quantitatively into a 10 mL volumetric flask, 1 mL of 1 × 10−4 M PTQA reagent solution was added, followed by the addition of 1 mL of 0.025 M H2SO4 and 2 mL ethanol. It was made up to the mark with de-ionized water. After 5 min the fluorescence intensity was measured at 373 nm when excited at 319 nm against a reagent blank, as before. The manganese concentration was calculated in μg L−1 or ng L−1 with the aid of a calibration graph. This gives a measure of manganese(VII) originally present in the mixture. This value was subtracted from that of the total manganese to get the manganese(II) present in the mixture. The results were found to be highly reproducible. The occurrence of such reproducible results is also reported for different oxidation states of manganese.60 The results of a set of determination are given in Table 10.
The present method was compared with some reported methods15,26,28,31,33 statistically. It was found that present method is much superior those of the reported methods. The results are shown in Table 11.
Conclusions
A new simple, ultra sensitive, highly selective and inexpensive spectrofluorimetric method with the manganese(VII)–PTQA system was developed for the determination of manganese in some real, environmental, biological, soil, food, vegetable, fruit and pharmaceutical samples, for continuous monitoring to establish the nano-trace levels of manganese in different samples matrices. Compared with other methods4–40 in the literature, the proposed method has several remarkable analytical characteristics. Firstly, the proposed method is highly sensitive that amount of ng L−1 of manganese can be determined without preconcentration. Secondly, the proposed method is very simple, rapid, and stable. The reaction of manganese(VII) with PTQA is completed rapidly in 1 min at room temperature so it does not involve any stringent reaction conditions and offer the advantages of stability of fluorescence intensity (24 h). Thirdly, the method has added the advantage of determining individual amounts of Mn(VII) and Mn(II). With suitable masking agents, the reaction can be made highly selective. The proposed method using PTQA in aqueous solutions not only is one of the most sensitive methods for the determination of manganese but also is excellent in terms of selectivity and simplicity. Therefore, this method will be successfully applied to the monitoring of nano-trace amounts of manganese in real, environmental, biological, soil, fruit, vegetables, food and pharmaceutical samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the authorities of Chittagong Medical College Hospital for their generous help in supplying biological samples. We are especially indebted to the authorities of BCSIR Laboratories, Chittagong for analyzing the biological and food samples by AAS and ICP-MS, respectively. We are also thankful to the Director of the Center of Excellence in Analytical Chemistry, University of Sindh, Jamshro, Pakistan for elemental analysis of our prepared reagent. We are also thankful to the department of Chemistry, University of Kanazawa, Japan for 1NMR analysis of our prepared reagent.References
- www.en.wikipedia/org/wiki/manganese.com.
- www.lenntech.com/periodic/elements/mn.htm.
- www.chemistry.about.com/od/elementfacts/a/manganese.htm.
- G. G. Guilbault, Anal. Chem., 1968, 40, 459–471 CrossRef.
- A. Navas and F. S. Rojas, Talanta, 1984, 31, 437–449 CrossRef CAS PubMed.
- B. K. Pal, M. Jamaluddin Ahmed and A. K. Chakrabarti, Analyst, 1990, 115, 439–443 RSC.
- W. Ruiz, Talanta, 1984, 31, 29–32 CrossRef PubMed.
- S. Rubio, G. H. Agustina and M. Valcárcel, Analyst, 1984, 109, 715–717 Search PubMed.
- F. J. L. Benet, F. Hernández, J. M. Escriche and R. M. Saez, Analyst, 1986, 111, 1325–1331 RSC.
- D. P. Bendito, J. Peinado and F. Toribio, Analyst, 1984, 109, 1497–1502 RSC.
- Z. T. Jiang and R. Li, Chem. Anal., 2008, 53, 647–659 Search PubMed.
- F. Lazaro, M. D. Luque De Castrto and M. Valcarcel, Anal. Chim. Acta, 1985, 169, 141–147 CrossRef CAS.
- M. Y. Fujita, K. Ikuta, Y. Nakahashi and K. Kato, Fresenius Z. Anal. Chem., 1989, 334(3), 252–259 Search PubMed.
- S. Huiming, T. Bo and Y. Yiqiong, Chin. J. Anal. Chem., 1992, 20, 1043–1048 Search PubMed.
- Y. Yu, C. Hong, R. Wang and J. Huang, Chin. J. Anal. Chem., 1998, 26, 410–414 Search PubMed.
- Z. G. Cheng, Phys. Test. Chem. Anal. Part A Phys. Test., 2004, 12, 1–45 Search PubMed.
- V. L. Biddle and E. L. Wehry, Am. J. Anal. Chem., 1978, 50, 865–867 CrossRef.
- J. Juhui, Z. Luqing, C. Dingxi and Z. Gui’en, Anal. Lab., 1999, 01, 47–53 Search PubMed.
- Z. Shuxian, A. Gaowa, S. Gaowa, S. Yin and Y. Wenbin, Chin. J. Anal. Chem., 1995, 23, 393–398 Search PubMed.
- G. C Ragos, M. A. Demertzis and P. B. Issopoulos, Farmaco, 1997, 52, 199–203 Search PubMed.
- W. Kunihiro, R. Kazuhiro and I. Masayuki, Bunseki Kagaku, 2009, 44, 929–933 Search PubMed.
- F. Yang, G. J. X. Zhao, L. J. Qia, L. I. M. Jing and C. U. I. X. Yu, Chin. J. Spectrosc. Lab., 2013, 30, 2365–2371 Search PubMed.
- C. S. jie, L. I. Ying-jiea and Q. Cheng-lib, Chem. Reagents, 2010, 3, 87 Search PubMed.
- B. Tang and F. Han, Anal. Lett., 2001, 34, 1353–1368 CrossRef CAS.
- Z. Zhiqi, X. Fenghe and G. Xiaodan, Chin. J. Anal. Chem., 1989, 17, 749–753 Search PubMed.
- A. Moreno, M. Silva and D. Perez-Bendito, Anal. Chim. Acta, 1984, 159, 319–328 CrossRef CAS.
- T. Xiu-lan, M. A. Yong-jun, Z. Min, P. Yu-ying and Y. Hai-qing, Chem. Reagents, 2009, 8, 16–31 Search PubMed.
- G. S. guang, Z. Li-na, D. Ping, C. Xiao-liang and Y. Jing-hua, Chin. J. Anal. Lab., 2008, 11, 39–44 Search PubMed.
- H. Shuhui, H. Youying and C. Lanhua, J. Huaibei Ind. Teach. Coll., Nat. Sci. Ed., 1993, 03, 3–10 Search PubMed.
- Z. Zhaosheng and L. Tong, Chin. J. Anal. Chem., 1990, 18, 1071–1079 Search PubMed.
- Q. Shan-bao and Y. Hai-yan, Stud. Trace Elem. Health, 2004, 1, 11–30 Search PubMed.
- Z. Lina, G. Shenguang, Z. Yan and L. Haiyun, Phys. Test. Chem. Anal., 2008, 9, 1–20 Search PubMed.
- Z. Guien, C. Dingxi and F. Suling, Chin. J. Anal. Chem., 1993, 8, 148–153 Search PubMed.
- P. Zhigong, W. Baoqi and W. Congying, Anal. Lab., 1994, 01, 11–15 Search PubMed.
- L. Verne, E. Biddle and L. Wehry, Anal. Chem., 1978, 50, 867–870 CrossRef.
- A. Moreno, M. Silva and D. Perez-Bendito, Anal. Chim. Acta, 1984, 159, 319–328 CrossRef CAS.
- A. D. Sherry and G. L. Cottam, Arch. Biochem. Biophys., 1973, 156, 665–672 CrossRef CAS PubMed.
- A. D. Gaowa, S. Yin, Y. Jinlong and Z. A. Shuxian, Anal. Lab., 1995, 3, 23–30 Search PubMed.
- X. Yu-sheng and Q. Shan-bao, J. South. Yangtze Univ., Nat. Sci. Ed., 2004, 6, 187–196 Search PubMed.
- Y. Ying, H. Jianfeng and D. Feng, Chin. J. Anal. Chem., 1997, 5, 17–26 Search PubMed.
- H. D. Porter, J. Am. Chem. Soc., 1954, 76, 127–131 CrossRef CAS.
- G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denney, Text-book of Quantitative Inorganic Analysis, John Wiley and Sons Inc, 5th edn, New York, 1989, pp. 309–368 Search PubMed.
- A. K. Mukharjee, Analytical Chemistry of Zirconium and Hafnium, Pergamon Press, 1st edn, New York, 1970, pp. 12–37 Search PubMed.
- B. K. Pal and B. Chowdhury, Mikrochim. Acta, 1984, 11, 121–127 CrossRef.
- B. K. Pal and D. E. Ryan, Anal. Chim. Acta, 1969, 47, 227–231 CrossRef.
- A. Pierre, D. Karine, D. Franck and C. Gwenaelle, Chem. Geol., 2005, 218, 265–279 CrossRef.
- B. K. Pal, F. Toneguzzo, A. Corsini and D. E. Ryan, Anal. Chim. Acta, 1977, 88, 353–361 CrossRef CAS.
- G. A. Parker, Analytical Chemistry of Molybdenum, Springer-Verlag, Berlin, 1983 Search PubMed.
- C. Sun, J. Y. Yang and S. R. Tzeng, Analyst, 1999, 124, 421–429 RSC.
- Standard Methods for the Examination of Water and Wastewater, ed. E. A. Greenberg, S. L. Clesceri and D. A. Eaton, American Public Health Association, 18th edn, Washington D. C., 1992, p. 53 Search PubMed.
- C. Ojeda, A. Garcia de Torres, F. Sanchez Rojas and J. M. Cano Pavon, Analyst, 1987, 112, 1499–1502 RSC.
- Manganese in Drinking Water, World Health Organization, 1992, pp. 1–15 Search PubMed.
- H. M. Stahr, Analytical Methods in Toxicology, John Wiley and Sons, 3rd edn, New York, 1991, pp. 75–85 Search PubMed.
- American Conference of Governmental Industrial Hygienists (ACGIH), Threshold limit values for chemical substances and physical agents and biological exposure indices for 1995/96, ACGIH, Cincinnati, OH, 1996 Search PubMed.
- M. L. Jackson, Soil Chemical Analysis, Prentice Hall, Englewood Cliffs, 1965, pp. 326–337 Search PubMed.
- A. L. Page, A. C. Chang and M. El-Amany, in: Lead, Mercury, Cadmium, Manganese and Arsenic in the Environment, ed. T. C. Hutchinsm and K. M. Mecma, John Wiley and Sons Ltd., New York, 1987, p. 119 Search PubMed.
- M. Jamaluddin Ahmed, M. Reazul Houqe, A. S. M. Shahed Hossain Khan and S. C. Bhattacharjee, Eurasian J. Anal. Chem., 2010, 5, 1–15 Search PubMed.
- H. P. Beck, D. Kostova and B. Zhang, Agron. Res., 2006, 4(2), 493–498 Search PubMed.
- L. F. Delgadillo, G. H. Silva, R. A. Martinez and M. M. Vermeerch, Rev. Int. Contam. Ambient., 1992, 8, 37–46 Search PubMed.
- A. Pierre, D. Karine, D. Franck and C. Gwenalle, Chem. Geol., 2005, 218, 265–279 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |