
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the cycling stability of lithium–sulfur batteries by hollow dual-shell coating†
Jianhua Zhang,
Rujia Zou *,
Qian Liu,
Shu-ang He,
Kaibing Xu and
Junqing Hu*
*,
Qian Liu,
Shu-ang He,
Kaibing Xu and
Junqing Hu*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: rjzou@dhu.edu.cn; hu.junqing@dhu.edu.cn
First published on 1st March 2018
Abstract
Herein, a novel hybrid S@MnO2@C nanosphere, comprising sulfur nanoparticles encapsulated by a MnO2@C hollow dual-shell, is reported. Benefiting from a conductive C outer layer, the S@MnO2@C hybrid nanosphere provided highly efficient pathways for fast electron/ion transfer and sufficient free space for the expansion of the encapsulated sulfur nanoparticles. Moreover, the dual-shell composed of a MnO2 inner layer and a C outer layer coating on S not only improved the efficacious encapsulation of sulfur, but also significantly suppressed the dissolution of polysulfides during cycling. As a result, the S@MnO2@C electrode shows high capacity, high coulombic efficiency and excellent cycling stability. The S@MnO2@C cathode delivered a discharge capacity of 593 mA h g−1 in the fourth cycle and was able to maintain 573 mA h g−1 after 100 charge–discharge cycles at 1.0C, corresponding to a capacity retention of 96.6%.
1. Introduction
Rechargeable lithium–sulfur (Li–S) batteries show promising potential for next-generation high-energy-storage technology due to the high capacities of both the sulfur cathode and the Li metal anode.1,2 The sulfur cathode has a high theoretical specific capacity of 1672 mA h g−1, which is much higher than that of the conventional lithium-ion battery cathodes (e.g., 140 mA h g−1 for LiCoO2, 148 mA h g−1 for LiMn2O4, and 170 mA h g−1 for LiFePO4)4–6 and corresponds to a specific energy of around 2600 W h kg−1.3 It should also be noted that sulfur is a low-cost material because it is one of the most abundant element on earth and hence, it is favorable for inexpensive large-scale storage devices.7 However, Li–S batteries suffer from poor cycling stabilities, low capacities, and low coulombic efficiencies primarily due to the defects described as follows.8,9 First, sulfur is reduced to high-order polysulfides and then to low-order polysulfides during the discharge process (Li2Sx, x = 1–8); both sulfur and low-order polysulfides show poor ionic and electronic conductivities. Second, intermediate polysulfide ions formed at the cathode are soluble in liquid electrolytes, resulting in active-material loss. Moreover, the dissolved polysulfides travel between the electrodes during cycling and give rise to the deposition of insulating Li2S2 or Li2S on the Li anode via a reoxidized process, which results in poor cycle stability and low coulombic efficiencies. Third, large volumetric expansion and shrinkage (∼80%) of sulfur gradually decrease the mechanical integrity and the stability of S-composite structures during charge and discharge processes.Currently, most efforts to tackle these issues are based on S-composite cathode architectures in Li–S batteries. A commonly employed strategy is using high-surface-area carbon materials, such as hollow carbon structures, porous carbon, and graphene/graphene oxides.10–12 Though recognized as materials with great potential for improving electrochemical performance, C/S composite structures cannot sustain prolonged-life cycling due to the surface polarity difference between carbon and lithium polysulfides. Recently, polar materials, metal oxides and metal sulfides were employed to effectively suppress the polysulfides-shuttle via creating a relatively strong chemical interaction with dissolvable polysulfides, thus effectively confining them within the S-composite cathode, which significantly improved the cycling stability.13,14 However, despite the significant advances in the use of these polar materials, S/polar materials' composite structures cannot quickly transfer electrons and ions in the electrodes due to the increase in internal resistance of the battery, which causes rate capability decay and low specific capacity, particularly under large current densities. Recently, a new way to solve these problems of Li–S batteries using S/C/polar materials-based composites was put forward, which could result in a more stable performance.15–18 These S/C/polar materials-based composites not only confine the shuttle effect, but also increase the conductivity of the entire cathode, easily prolonging the cycle life of Li–S batteries. However, S/C/polar composites were always imperfectly designed as pores or open structures cannot effectively prevent the loss of polysulfides into the electrolyte, which could help obtain an improved but diminished cycle.15–17,19 Moreover, the outermost coating layer of polar materials often has high resistance compared with carbon materials, which can block the transport of electrons among active materials.9,20 Therefore, it is still a great challenge to design innovative nanostructures for efficacious encapsulation of sulfur to control polysulfide dissolution and obtain long life-cycling stability at high current densities.
Herein, a rational design and hybrid structure with sulfur nanoparticles filled in MnO2@C hollow dual-shell spheres to form S@MnO2@C hybrid nanospheres is reported. This S@MnO2@C hybrid nanosphere has several apparent advantages. First, hollow C nanospheres with high conductivity provide highly efficient pathways for fast electron/ion transfer during long-term cycling. Second, the dual-shell composed of an internal MnO2 coating layer and an external hollow C coating layer can provide chemical adsorption to polysulfides and effectively prevent polysulfides from rapidly dissolving into the electrolyte. Third, the additional void after sulfur introduction provides sufficient free space for the expansion of encapsulated sulfur nanoparticles. The special material composition and structure design endows the S@MnO2@C electrode with a much improved capacity, high coulombic efficiency and an excellent cycling stability.
2. Experimental
2.1 Materials synthesis
All chemical agents used in this experiment were commercially purchased and used without further purification.2.2 Materials characterization
The morphologies and microstructures were characterized by FE-SEM (Field Emission Scanning Electron Microscope, Hitachi S-4800 equipped with an energy-dispersive X-ray spectrometer) and FE-TEM (Field Emission Scanning Electron Microscope, JEM-2100F). The spectrum recorded using an X-ray Diffractometer (D/MAX-2550VB+) was used to confirm the ingredients and the phase of the compositions. XPS data were obtained using an EALA Lab MKII spectrometer with an excitation source of Mg-Kα radiation. The weight ratio of sulfur was examined by TGA (Thermal Gravimetric Analyzer TG 209 F1) under nitrogen flow with a heating rate of 20 °C min−1. The carbon content of the MnO2@C hollow sphere was evaluated by Elemental Analysis (Vario EL III).2.3 Electrochemical measurements
Powder composed of active material (S@MnO2@C, MnO2/S), carbon black and poly(vinylidene fluoride) (PVDF) binder with a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 was sufficiently mixed in an appropriate amount of N-methyl-2-pyrrolidone (NMP) to form a homogeneous slurry. The typical loading density of active materials of S@MnO2@C was 1.2–1.4 mg cm−2. The slurry was then coated onto the aluminum foil uniformly before drying in a vacuum oven of 60 °C for 12 h to form the working electrode. In an argon-filled glove box, 2032-type coin cells were assembled and lithium foil was used as the counter electrode. Then, 1 M LiPF6 dissolved in 1:1 v/v DOL and DME containing 0.5 M LiNO3 was used as the electrolyte and Celgard 2400 was the separator. Galvanostatic discharge–charge tests were carried out on a LANDHE Measurement System (Landhe, Wuhan, China) between cutoff voltages of 2.8 V and 1.7 V (vs. Li/Li+). Current densities and specific capacities were both calculated based on the sulfur loading on the working electrode (1C = 1672 mA h g−1). Electrochemical impedance spectroscopy was conducted by testing at 5 mV ac oscillation amplitude over the frequency range of 100 kHz to 100 mHz. CV measurements were conducted at the scan rate of 0.1 mV s−1 over the potential range of 1.7–2.8 V on an electrochemical workstation (Metrohm Autolab).
:2:1 was sufficiently mixed in an appropriate amount of N-methyl-2-pyrrolidone (NMP) to form a homogeneous slurry. The typical loading density of active materials of S@MnO2@C was 1.2–1.4 mg cm−2. The slurry was then coated onto the aluminum foil uniformly before drying in a vacuum oven of 60 °C for 12 h to form the working electrode. In an argon-filled glove box, 2032-type coin cells were assembled and lithium foil was used as the counter electrode. Then, 1 M LiPF6 dissolved in 1:1 v/v DOL and DME containing 0.5 M LiNO3 was used as the electrolyte and Celgard 2400 was the separator. Galvanostatic discharge–charge tests were carried out on a LANDHE Measurement System (Landhe, Wuhan, China) between cutoff voltages of 2.8 V and 1.7 V (vs. Li/Li+). Current densities and specific capacities were both calculated based on the sulfur loading on the working electrode (1C = 1672 mA h g−1). Electrochemical impedance spectroscopy was conducted by testing at 5 mV ac oscillation amplitude over the frequency range of 100 kHz to 100 mHz. CV measurements were conducted at the scan rate of 0.1 mV s−1 over the potential range of 1.7–2.8 V on an electrochemical workstation (Metrohm Autolab).
3. Results and discussion
The synthesis of the S@MnO2@C hybrid nanosphere is illustrated in Fig. 1a (see the Experimental section for the details). Uniform SiO2 nanospheres were prepared by the hydrolysis of TEOS with the assistance of (NH3)·H2O. The SiO2 nanospheres were coated with a thin MnO2 layer, which acts as a chemical adsorption layer and prevents the polysulfides from diffusing into the electrolyte. Then, a thin PDA layer was coated on SiO2@MnO2 nanospheres as the carbon precursor. After carbonization under H2 atmosphere and removal of SiO2 with NaOH, hollow MnO2 nanospheres coated with a hollow carbon layer to form MnO2@C dual-shell hollow spheres were obtained. Finally, sulfur was introduced into the MnO2@C via a melt-diffusion strategy to fabricate the S@MnO2@C hybrid nanosphere. As shown in Fig. 1b, SiO2 nanospheres were obtained and with uniform size of ∼310 nm. After coating with MnO2 layer as a polar chemical adsorption layer, the average diameter of the SiO2@MnO2 hybrid nanosphere increased to ∼330 nm (Fig. 1c), indicating that a thin layer of MnO2 was formed. The SiO2@MnO2@PDA maintained its spherical shape after PDA-layer coating as shown in Fig. 1d. After carbonization and SiO2 etching by NaOH, the hollow dual-shell MnO2@C still retained their spherical morphology (Fig. 1e). The elemental analysis suggests that the carbon content in MnO2@C was ∼43.78%. The TEM image also reveals that the diameter of the MnO2@C hollow sphere is ∼370 nm, which can provide sufficient free-space for high loading of sulfur. Sulfur was introduced into the MnO2@C to form S@MnO2@C hybrid nanospheres through a traditional melt-diffusion method as shown in Fig. 1f and S1.† The S@MnO2@C hybrid nanosphere effectively retained its morphology after the melt-diffusion reaction, exhibiting the firmness of the structure (Fig. S1†). The apparent brightness contrast observed in Fig. 1f demonstrates the extra space available after the infiltration of S, which effectively buffers the volume expansion of sulfur during lithiation and delithiation.21 Thermogravimetric analysis curve is presented in Fig. S2,† which shows high sulfur loading (∼60%) in the S@MnO2@C hybrid nanosphere. The high magnification TEM image further uncovers the detailed structure and clearly distinguishes uniform hollow dual-shell of carbon and MnO2 layer coated on the S-core for the S@MnO2@C hybrid nanosphere (Fig. 1g). The elemental distribution of the S@MnO2@C hybrid nanosphere was detected through X-ray mapping with a matching TEM image, which revealed that C, Mn, O and S are uniformly distributed in the core (S) and the shell (C, Mn, O) regions, which is in accordance with the TEM image (Fig. S3†). The thickness of the dual-shell layer is ∼20 nm. As shown in Fig. 1h, the high resolution transmission electron microscope (HRTEM) image of the MnO2 layer shows a lattice spacing of 0.22 nm, which can be indexed to the (201) planes of birnessite δ-MnO2.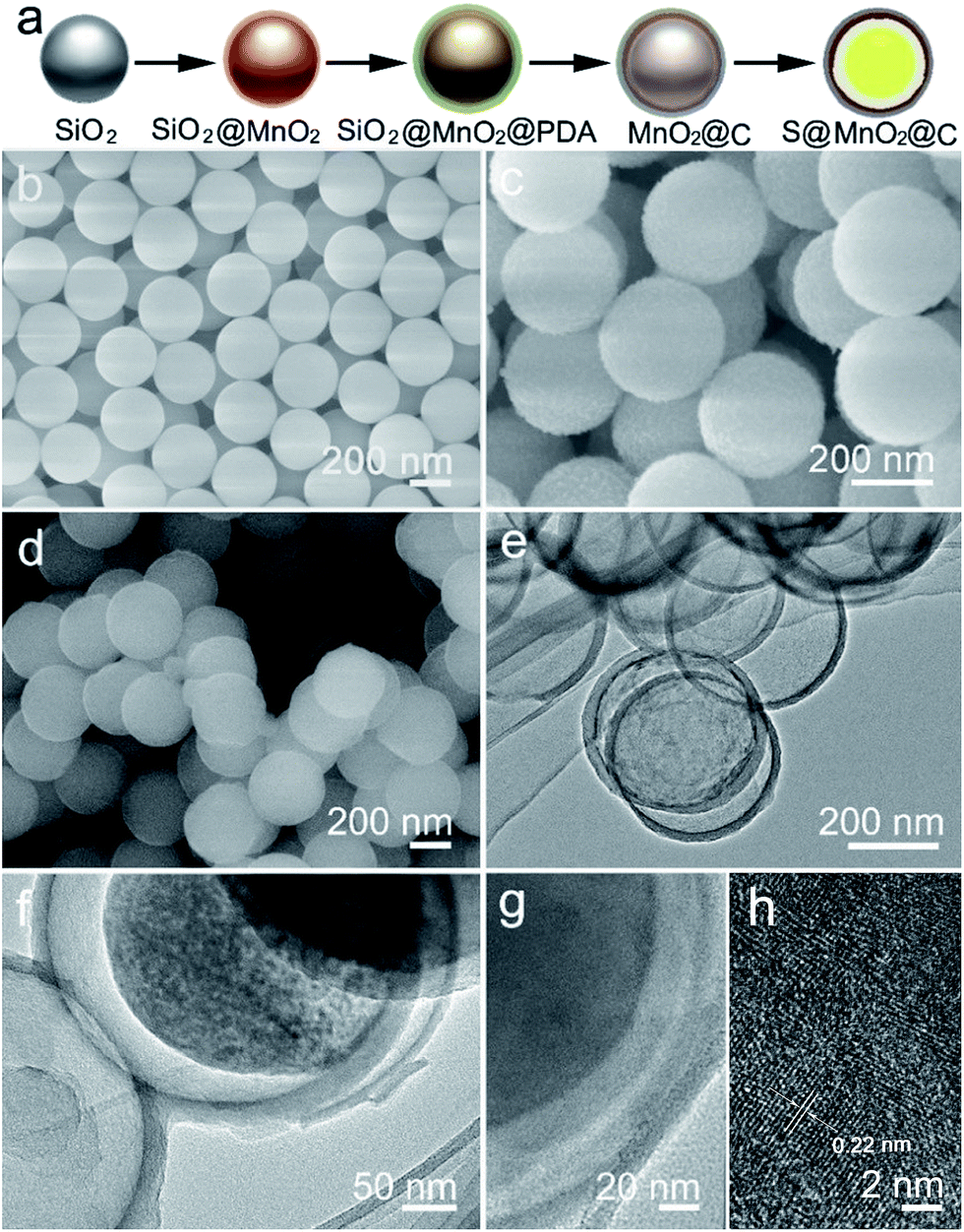 | ||
| Fig. 1 (a) Schematic illustrations showing the fabrication of the S@MnO2@C hybrid nanosphere. (b) SEM image of SiO2 nanospheres. (c) SEM image of the SiO2@MnO2 hybrid nanosphere. (d) SEM image of the SiO2@MnO2@PDA hybrid nanospheres. (e) TEM image of the MnO2@C hybrid nanospheres. (f) TEM image of the S@MnO2@C hybrid nanospheres. (g) High magnification TEM image of the S@MnO2@C hybrid nanospheres. (h) HRTEM image of the S@MnO2@C hybrid nanospheres. | ||
In order to further understand the structure characterization, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were utilized to analyze the synthesized hybrid nanospheres. As shown in Fig. 2a, the XRD pattern of the as-prepared MnO2@C hierarchical microspheres is characterized (higher panel of Fig. 2a). Three sharp and intense peaks at around 35.1°, 40.9° and 59.1° can be respectively well-indexed to the (111), (200) and (220) planes of birnessite MnO2 (JCPDS-43-1456, lower panel of Fig. 2a). No impure phase of other manganese oxides was detected, indicating that phase pure birnessite MnO2 was obtained. The absence of carbon diffraction peaks may result from its low content and crystallization structure.16,22 As shown in Fig. 2b, diffused sulfur in the S@MnO2@C hybrid nanospheres (higher panel of Fig. 2b) showed the same crystal structure of sulfur indexed in JCPDS-53-1109 as displayed in the XRD pattern (lower panel of Fig. 2b). XPS survey assured the existence of Mn (Fig. 2b) and C (Fig. 2c). Binding energies at 643.9 eV and 641.8 eV were ascribed to the chemical state of Mn4+ and Mn3+, respectively.23,24 In addition, a peak appeared in the lower binding energy region, which can be attributed to the Mn–S bonds.25 There is no new peak in the C 1s spectra, indicating no interaction between C and S.
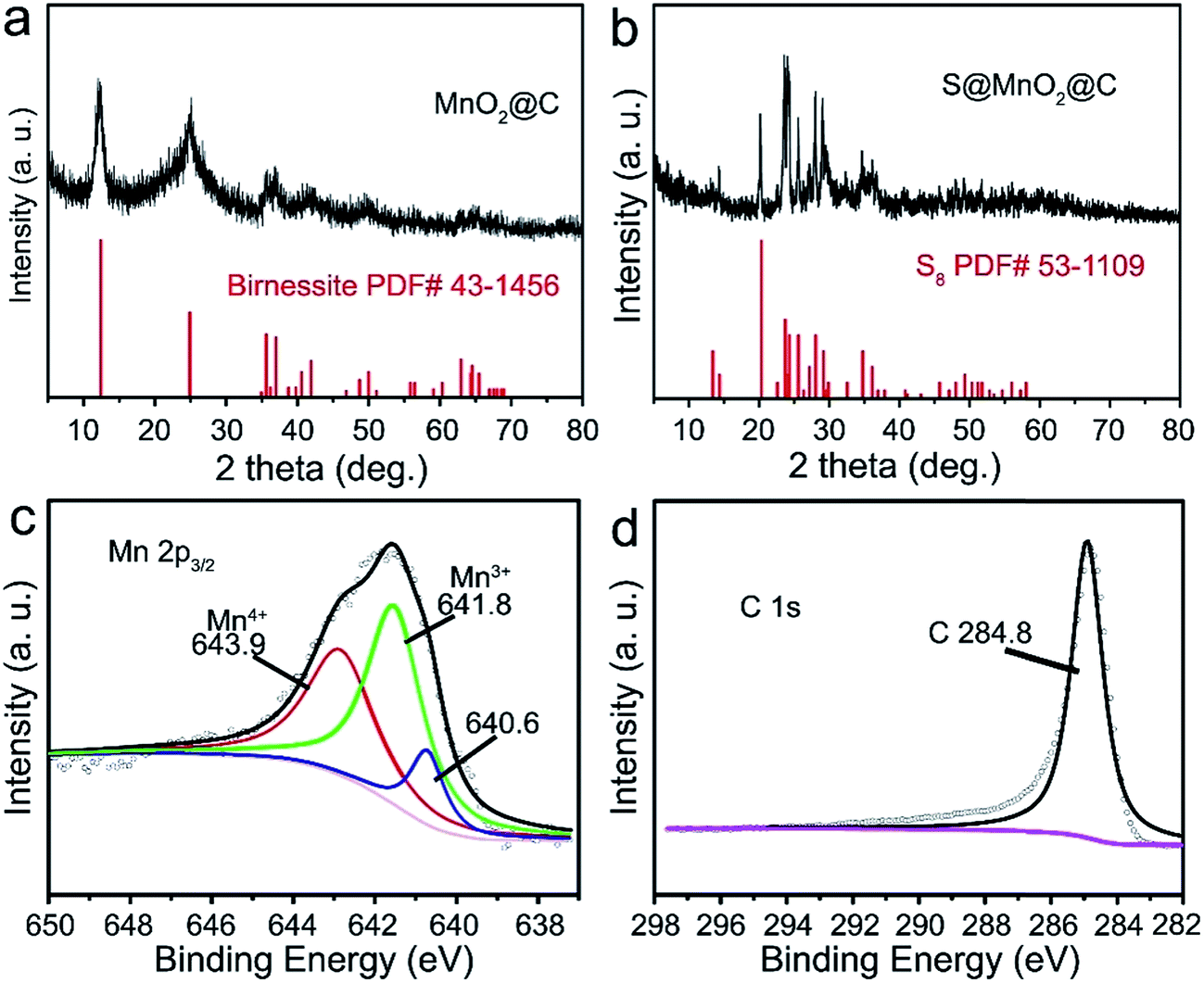 | ||
| Fig. 2 (a) XRD patterns of MnO2@C hybrid nanospheres (upper panel) and JCPDS card (MnO2, no. 43-1456) (lower panel). (b) XRD patterns of S@MnO2@C hybrid nanospheres (upper panel) and JCPDS card (S8, no. 53-1109) (lower panel). XPS spectra of S@MnO2@C hybrid nanospheres: (c) Mn 2p3/2 and (d) C 1s. | ||
The electrochemical test of the electrodes was performed in a coin-type cell and the lithium foil was used as the counter electrode. The cell was tested between the cut-off voltage of 1.7–2.8 V (vs. Li/Li+). Fig. 3a shows the cyclic voltammetry (CV) curves of the S@MnO2@C cathode for the first five cycles at 0.1 mV s−1. The CV curves show two reduction peaks appearing at 2.3 and 2.05 V. The broad reduction peak at 2.3 V is attributed to the reduction of S8 to intermediate polysulfides (Li2Sx, 4 ≤ x ≤ 8) and further reduction to insoluble Li2S2 or Li2S.26,27 A strong oxidation peak at 2.38 V corresponds to the delithiation process from Li2S/Li2S2 to Li2Sx and eventually to S8.28,29 In the following cycles, the intensities of reduction and oxidation peaks remain almost unchanged, suggesting an excellent reversibility of the cycling process. Moreover, compared with the S@C cathode in previous reports,29–32 the CV curve of the S@MnO2@C cathode shows a distinct positive shift in the reduction peaks and a negative shift in the oxidation peaks. This implies a strong electrocatalytic effect and an improved polysulfide redox kinetics resulting from the MnO2 layer inside the S@MnO2@C hybrid nanospheres,33,34 indicating the successful creation of relatively strong chemical interactions with Li2Sx (x = 4–8). Fig. 3b shows the first cycle discharge–charge voltage profile of the S@MnO2@C cathode at the current density of 0.2C. In the discharge–charge cycle, two potential plateaus are presented in the discharge process and one potential plateau is observed in the charge process. The charge/discharge profiles show similar features and are well-consistent with the CV curves.
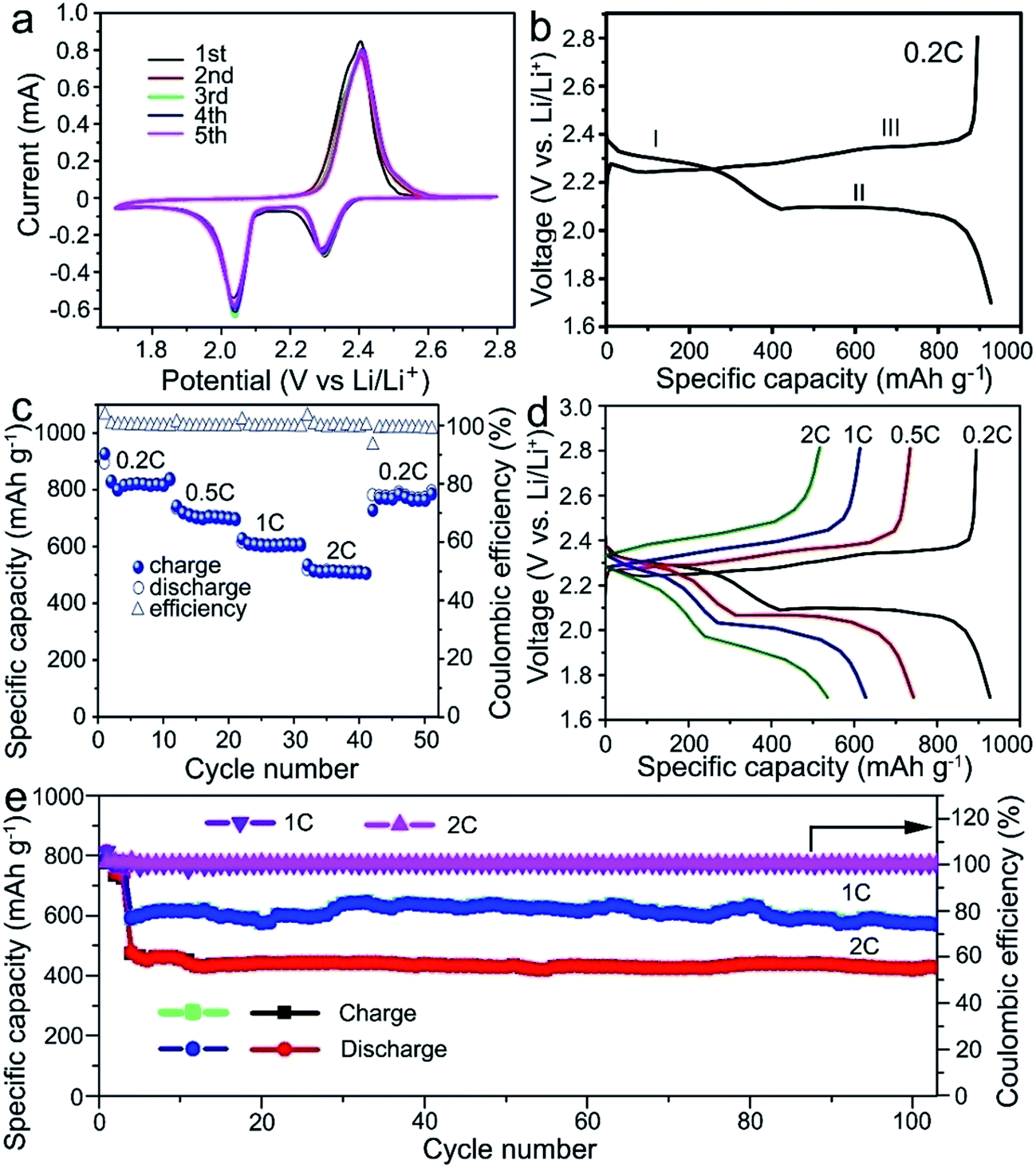 | ||
| Fig. 3 (a) CV profiles of the S@MnO2@C electrode between the cut-off voltages of 1.7 V and 2.8 V. (b) Charge and discharge curves of the S@MnO2@C electrode under the current density of 0.2C. (c) Rating performance the S@MnO2@C electrode under different current rates. (d) Charge and discharge curves of the S@MnO2@C electrode under different current rates. (e) Cycling performance of the S@MnO2@C electrode under the current density of 1.0C and 2.0C. | ||
Fig. 3c and d show the rate performances of the S@MnO2@C cathode under various current rates. The capacities decreased from ∼820.2 to 703.3, 602.3, and 510.1 mA h g−1 when the current density was increased from 0.2 to 0.5, 1.0, and 2.0C, respectively. It should be noted that a capacity of ∼510.1 mA h g−1 was still retained at a high current density of to 2.0C, which is clearly higher than that of the S@MnO2 and S@C cathodes reported previously.35,36 After cycling at the high current density of 2.0C for 10 cycles, the capacity of about 784.8 mA h g−1 was recovered as the current density was switched back to 0.2C, corresponding to a capacity retention of ∼95.7%, which indicates a high stability as well as an excellent reversibility of the electrode. Moreover, a distinct discharging plateau was still obtained at high rates (Fig. 3d), indicating the facile mass transport and reaction kinetics of the S@MnO2@C cathode. The cycling stabilities of the S@MnO2@C cathodes were tested by carrying out the discharge–charge measurements up to 100 cycles as shown in Fig. 3e. It should be noted that the test was performed at a low rate of 0.2C in the initial three cycles for the slow activation of S@MnO2@C. As shown in Fig. 3e, the S@MnO2@C cathode delivered the discharge capacity of 593 mA h g−1 for the fourth cycle and it was able to maintain 574.1 mA h g−1 in the 100th charge–discharge cycle at 1.0C, corresponding to a capacity retention of 96.8%. In particular, at a high current density of 2.0C, the discharge capacity of the S@MnO2@C cathode was 476.7 mA h g−1 in the first cycle and it was able to maintain 426.9 mA h g−1 after 100 charge–discharge cycles at 2.0C. On average, the S@MnO2@C cathode showed a capacity fading rate of only 0.1% per cycle, which is clearly smaller than those of previously reported S@MnO2 or S@C-based cathodes.37,38 During the entire cycling at 2.0C, the coulombic efficiency was higher than 99.7%. As a control sample, S/MnO2 composite cathodes were synthesized (Fig. S4†) and the rate performance and cyclic stability were also tested. The capacity of the S/MnO2 composite decreased from 651.5 to 515.5, 444.6, and 382.4 mA h g−1 when the current density was increased from 0.2 to 0.5, 1.0, and 2.0C, respectively, which is significantly lower than those for the S@MnO2@C cathode. After cycling at the high current density of 2.0C, the capacity recovered to about 531 mA h g−1 as the current density was switched back to 0.2C, corresponding to a capacity retention of ∼81.5%, which indicates low reversibility of the electrode. In a sharp contrast to the S@MnO2@C composite cathode, the capacity of the S/MnO2 electrode declined drastically from ∼497.6 mA h g−1 in the fourth cycle to 381.6 mA h g−1 in the 100th cycle, corresponding to a capacity retention of ∼76.6%. Moreover, the coulombic efficiency of the cell was only 99.1–99.6%, indicating its low electrochemical reversibility. The electrochemical results of the two electrodes clearly revealed that only the interaction of polysulfides with MnO2 nanomaterials is not enough to improve the sulfur cathode performance.18,39,40 Furthermore, MnO2 hollow spheres and carbon hollow spheres were synthesized (Fig. S5†) and S@MnO2 and S@C cathodes were formed to demonstrate the advantage of S@MnO2@C. The sulfur contents of the two samples were almost the same as that of the S@MnO2@C electrode (Fig. S6†). In addition, the cycling performance of the S@MnO2 and S@C electrodes was also investigated (Fig. S7†). The capacities of the S@MnO2 and S@C electrodes declined drastically from ∼415.7 mA h g−1 and ∼584.9 mA h g−1 in the fourth cycle to 373.2 mA h g−1 and 311.6 mA h g−1 in the 100th cycle under the current density of 1.0C, respectively (Fig. S7†). The performances of both S@MnO2 and S@C electrodes were worse than that of S@MnO2@C.
To understand the significant increase in the electrochemical performance, the electrochemical impedance spectra (EIS) of the S@MnO2@C electrode was first collected before and after 100 cycles as shown in Fig. 4a. The Nyquist plots of both samples depict a semicircle in the high-to-medium frequency region and an inclined line at the low frequency region. The semicircle is attributed to the charge transfer process at the electrode/electrolyte interface and the inclined line corresponds to the lithium ion-diffusion process into the bulk of the electrode, which is known as the Warburg diffusion.41 The impedance parameter of the S@MnO2@C electrode shows a minimum change between values before and after 100 cycles, indicating good electrochemical kinetics of these composite electrodes. Such minor change in charge transfer resistance upon cycling indicated a relatively stable electrode–electrolyte interface and provided significant advantages of rapid electron transport and fast faradic reaction during the electrochemical lithium insertion/extraction processes, which is favorable for increased capacity and good cycle performance. In addition, the morphologies of the S@MnO2@C hybrid nanospheres before and after 100 cycles at 2.0C were also examined by SEM (Fig. 4b and S8†). It was found that the S@MnO2@C electrode was well preserved without any apparent destruction, crack and joint points as compared to that before cycling, demonstrating the advantages of rapid charge transfer among active materials, conductive carbon and binders during the electrochemical lithium insertion/extraction processes as shown in Fig. S8.†42 Moreover, it was also found that the S@MnO2@C hybrid nanospheres were well preserved without any apparent destruction as indicated by the high magnification SEM image (Fig. 4b), indicating that the insertion and extraction of the lithium ion had no distinct impact on the composite structures because sufficient free space was available for the expansion of the loaded S nanoparticles inside the S@MnO2@C hybrid nanospheres. Finally, the thin MnO2 layer between the S nanoparticles and C hollow spheres is an efficient sulfur adsorption layer, which can provide a much larger contact area for the mitigation of polysulfide dissolution owing to the strong interactions between MnO2 and the polysulfides and promote stable redox activity to suppress the shuttle effect during the cycling. In addition, encapsulation of sulfur of the porous carbon layer also prevents intermediate product polysulfides from outflowing into the electrolyte. Therefore, the hollow dual-shell sphere of MnO2 layer and C layer spheres increases the effectiveness of preventing polysulfide dissolution, accommodates volumetric expansion in discharge cycling and also facilitates ion and electron transport, which improves cycle stability. The unique material composition, morphology and structure of the S@MnO2@C hybrid nanospheres-based electrode resulted in its good specific capacity, excellent cycling stability and high rate performance.
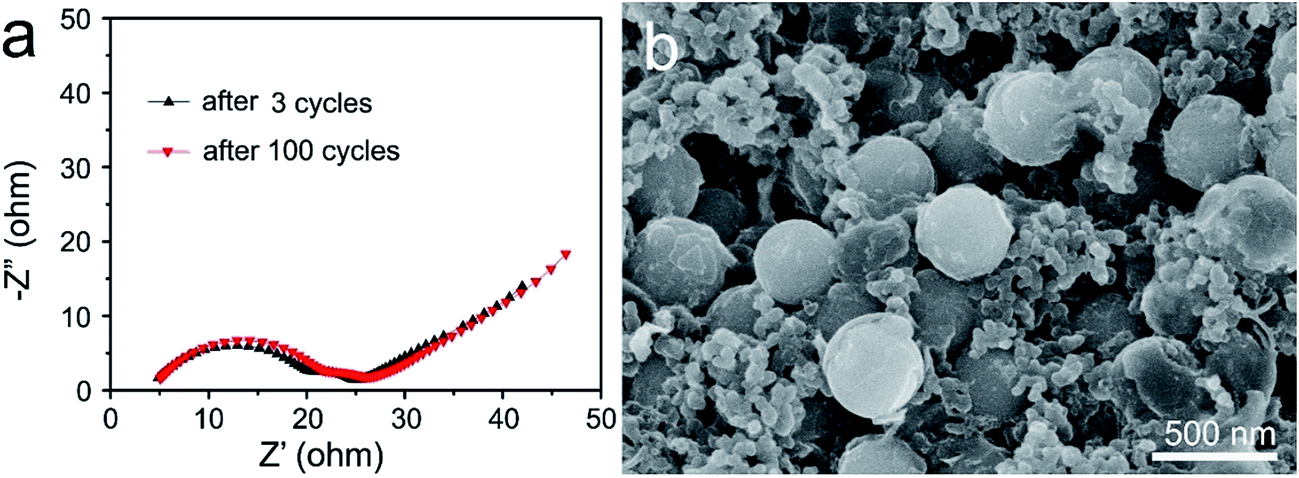 | ||
| Fig. 4 (a) Nyquist plots of the S@MnO2@C hybrid nanospheres electrode measured at an amplitude of 5 mV over a frequency range from 100 kHz to 0.01 Hz after 3 cycles and 100 cycles. (b) SEM image of the S@MnO2@C hybrid nanospheres-based electrode after 100 cycles at 2.0C. | ||
4. Conclusion
In summary, we demonstrated a facile and effective method for the synthesis of S@MnO2@C hybrid nanospheres. The hollow C nanospheres not only provide highly efficient pathways for fast electron/ion transfer, but also prevent rapid dissolution of polysulfides into the electrolyte. The MnO2 layer offers chemical adsorption to polysulfides and restricts the polysulfides from dissolving into the electrolyte. Moreover, the sufficient voids provide extra space for the expansion of the encapsulated sulfur nanoparticles. With this advanced design, the S@MnO2@C electrode shows high capacity, high coulombic efficiency and an excellent cycling stability. At a high current density of 1.0C, the capacity of the S@MnO2@C cathode was 593 mA h g−1 in the fourth cycle and it was able to maintain 574.1 mA h g−1 in the 100th cycle at 1.0C, corresponding to a capacity retention of 96.6%. The obtained results of the S@MnO2@C cathode make it a promising cathode material for lithium–sulfur batteries for power-intensive energy storage applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 51672044, 51741203 and 51472049), the Shanghai Pujiang Program (Grant No. 16PJ1400200), the Shanghai Sailing Program (Grant No. 17YF1400300), the Program for Innovative Research Team in University of Ministry of Education of China (IRT_16R13), the DHU Distinguished Young Professor Program, and the Fundamental Research Funds for the Central Universities.References
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- Y. Wang and G. Cao, Adv. Mater., 2008, 20, 2251 CrossRef CAS.
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. Zhou, Nano Lett., 2009, 9, 1045 CrossRef CAS PubMed.
- L.-X. Yuan, Z.-H. Wang, W.-X. Zhang, X.-L. Hu, J.-T. Chen, Y.-H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269 CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135 CrossRef CAS PubMed.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 9592 CrossRef CAS PubMed.
- G. He, X. Ji and L. Nazar, Energy Environ. Sci., 2011, 4, 2878 CAS.
- J. Zhang, C.-P. Yang, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Adv. Mater., 2016, 28, 9539 CrossRef CAS PubMed.
- X. Tao, J. Wang, Z. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288 CrossRef CAS PubMed.
- Q. Pang, D. Kundu and L. F. Nazar, Mater. Horiz., 2016, 3, 130 RSC.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- S. Rehman, T. Tang, Z. Ali, X. Huang and Y. Hou, Small, 2017, 13, 1700087 CrossRef PubMed.
- T. Chen, L. Ma, B. Cheng, R. Chen, Y. Hu, G. Zhu, Y. Wang, J. Liang, Z. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 38, 239 CrossRef CAS.
- J. Shen, J. Liu, Z. Liu, R. Hu, J. Liu and M. Zhu, Chem.–Eur. J., 2018, 24, 1 CrossRef.
- X. Liang and L. F. Nazar, ACS Nano, 2016, 10, 4192 CrossRef CAS PubMed.
- Z. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605 RSC.
- H. Kim, J. T. Lee, A. Magasinski, K. Zhao, Y. Liu and G. Yushin, Adv. Energy Mater., 2015, 5, 1501306 CrossRef.
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, J. Mater. Chem., 2011, 21, 6251 RSC.
- Y. Umezawa and C. N. Reilley, Anal. Chem., 1978, 50, 1290 CrossRef CAS.
- V. Di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117 CrossRef CAS.
- T. Ji, Q. Liu, R. Zou, Y. Sun, K. Xu, L. Sang, M. Liao, Y. Koide, L. Yu and J. Hu, Adv. Funct. Mater., 2016, 26, 1400 CrossRef CAS.
- S.-R. Chen, Y.-P. Zhai, G.-L. Xu, Y.-X. Jiang, D.-Y. Zhao, J.-T. Li, L. Huang and S.-G. Sun, Electrochim. Acta, 2011, 56, 9549 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2013, 231, 153 CrossRef CAS.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347 CAS.
- M.-Q. Zhao, Q. Zhang, J.-Q. Huang, G.-L. Tian, J.-Q. Nie, H.-J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410 Search PubMed.
- G. C. Li, J. J. Hu, G. R. Li, S. H. Ye and X. P. Gao, J. Power Sources, 2013, 240, 598 CrossRef CAS.
- W. Deng, A. Hu, X. Chen, S. Zhang, Q. Tang, Z. Liu, B. Fan and K. Xiao, J. Power Sources, 2016, 322, 138 CrossRef CAS.
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367 CrossRef CAS PubMed.
- S. Xin, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2014, 26, 1261 CrossRef PubMed.
- Y. Qiu, W. Li, G. Li, Y. Hou, L. Zhou, H. Li, M. Liu, F. Ye, X. Yang and Y. Zhang, Nano Res., 2014, 7, 1355 CrossRef CAS.
- S. A. Ahad, P. Ragupathy, S. Ryu, H.-W. Lee and D. K. Kim, Chem. Commun., 2017, 53, 8782 RSC.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., 2012, 124, 9730 CrossRef.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- L. Ni, Z. Wu, G. Zhao, C. Sun, C. Zhou, X. Gong and G. Diao, Small, 2017, 13, 1603466 CrossRef PubMed.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- S. Rehman, T. Tang, Z. Ali, X. Huang and Y. Hou, Small, 2017, 13, 1700087 CrossRef PubMed.
- Z. Deng, Z. Zhang, Y. Lai, J. Liu, J. Li and Y. Liu, J. Electrochem. Soc., 2013, 160, A553 CrossRef CAS.
- P. Gao, J. Fu, J. Yang, R. Lv, J. Wang, Y. Nuli and X. Tang, Phys. Chem. Chem. Phys., 2009, 11, 11101 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13235b |
| This journal is © The Royal Society of Chemistry 2018 |