
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported cobalt catalysts for the selective hydrogenation of ethyl levulinate to various chemicals†
Youliang Cen ab,
Shanhui Zhu*a,
Jing Guoab,
Jiachun Chaiab,
Weiyong Jiaoa,
Jianguo Wanga and
Weibin Fan*a
ab,
Shanhui Zhu*a,
Jing Guoab,
Jiachun Chaiab,
Weiyong Jiaoa,
Jianguo Wanga and
Weibin Fan*a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, PR China. E-mail: zhushanhui@sxicc.ac.cn; fanwb@sxicc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 1st March 2018
Abstract
A highly active and selective cobalt catalyst was developed for the hydrogenation of biomass-derived ethyl levulinate (EL) to γ-valerolactone (GVL), ethyl 4-hydroxypentanoate (EHP), 1,4-pentanediol (1,4-PDO) and 2-methyltetrahydrofuran (2-MTHF), which are considered to be value-added chemicals and important biofuels. The effects of reaction time, reaction temperature, catalyst amount and solvent on its catalytic performance were investigated. In addition, the reaction pathway was studied as well. It was found that the selectivity of GVL, 1,4-PDO and 2-MTHF on Co/ZrO2 can be easily tuned by changing reaction conditions, and can reach as high as 94%, 78% and 77%, respectively. The product selectivity is also significantly affected by the catalyst support. With SBA-15 as the support, the selectivity of EHP can reach 90%. Moreover, Co/ZrO2 gave an extraordinarily high GVL productivity of 1.50 mol gmetal−1 h−1 and displayed excellent stability and reusability. Interestingly, coke has a positive effect on the enhancement of GVL yield. AL dimers and trimers were identified as the coke species in the hydrogenation of EL. As far as we know, this is the first work conducting the flexible transformation of EL on cobalt catalysts.
1. Introduction
With the diminishment of fossil fuel reserves and growing concerns about global warming, great efforts are being made to search for alternatives.1 Lignocellulosic biomass, derived from terrestrial plants, waste biomass and energy crops, represents one of the most viable substitutions for fossil fuels.2 In recent years, many research studies have been devoted to the development of new catalytic routes for the conversion of raw biomass and biomass-based platform molecules into biofuels and value-added chemicals.2b,3 One of the typical chemicals is γ-valerolactone (GVL), which could be used as fuel additive, food ingredient, renewable solvent, and intermediate for the production of chemicals and high-grade alkenes.4GVL can be synthesized by hydrogenation of levulinic acid (LA) and its esters, which are important platform chemicals and have already been efficiently produced from lignocellulosic biomass on a large scale.5 Ethyl levulinate (EL), like LA, is formed by ethanolysis of chloromethyl furfural, 5-hydroxymethyl furfural, furfuryl alcohol or direct alcoholysis of monosaccharide and polysaccharide.6 Compared to LA, the conversion of EL is less strict for the catalyst. The strong acidity of LA will severely destroy the support such as zeolites and leach the active metal sites.7 In addition, it also heavily corrodes equipment. EL yielded from direct alcoholysis is obviously higher than that of LA from hydrolysis due to substantial suppression of humins formation, and EL is easier to be separated in alcohol medium.8
Several metal catalysts have been reported to be active for the hydrogenation of LA and its ester to GVL. Homogeneous catalysts such as Ru(acac)3 in combination with tris(3-sulfonatophenylphosphine) (TPPTS) or PBu3, iridium complexes and Shvo catalyst give very high conversion and GVL yield.9 However, the complexity in the ligand synthesis, together with the difficulty of catalyst recovery and recycling, greatly lowers the application potential of homogeneous catalysts in commercial applications. To solve these problems, heterogeneous catalysts have been successfully developed, and a series of supported noble metal catalysts such as Ru/C, Ru/SiO2, Ru/Al2O3, Ru/TiO2, RuCs/Al2O3, RuRe/C, Au/ZrO2, AuPd/TiO2 and PdNb/AC exhibit excellent performance.4b,10 However, the high cost and limited reserve of noble metals necessitate the development of abundant non-noble metal catalysts.
In this aspect, RANEY® Ni, Hf-ATMP, Zr-MOFs, Zr-beta and ZrO2 have been demonstrated to be effective for the transformation of LA and its esters to GVL through catalytic transfer hydrogenation process.11 In addition, supported non-noble metal catalysts (Cu, Co and Ni) and reduced Co3O4 also present moderate ability in activating LA and its esters.7a,12 Nevertheless, the GVL formation rate is rather low, and the highest rate reported by Chia et al.11b reached only 6.5 mmolGVL gcat−1 h−1 over ZrO2. Particularly, relatively higher reaction temperature is required to obtain high activity over non-noble metals than over noble ones.
The intermediate for hydrogenation EL to GVL has been identified as ethyl 4-hydroxypentanoate (EHP), which is a kind of chiral γ-hydroxy ester and an important bioactive molecule. It is currently made by fermenting and chemoenzymatic protocols.13 GVL can be further hydrogenated to highly valuable 1,4-pentanediol (1,4-PDO) and 2-methyltetrahydrofuran (2-MTHF). Biogenic diols are interesting building blocks for biodegradable polyesters and other polymeric materials.14 2-MTHF is advocated as a bioderived “green” solvent and a component of P-series fuels in future.15 Geilen et al.14a have demonstrated that homogeneous Ru(acac)3 in combination with phosphine ligands and acidic additives can selectively convert LA into GVL, 1,4-PDO and 2-MTHF. Despite its high activity, this catalyst system is highly expensive and shows severe limitation of homogeneous catalysts, and hence, possibly influencing product quality. Thus, various homogeneous catalysts were integrated with heterogeneous ones,16 and most of researches devote to production of one type of chemical. Concerning preparation of 1,4-PDO, supported noble metal catalysts were generally employed, including Ir–MoOx/SiO2, Rh–MoOx/SiO2, RuRe/C and Pt–Mo/HAP.17 With respect to production of 2-MTHF, Pt–Mo/H-β and Ru–N–triphos complexes are only reported to be active.15,18 Recently, it is very interesting that Yang et al.19 achieve transfer hydrogenation of methyl levulinate into GVL, 1,4-PDO and 1-pentanol (1-PAO) over Cu/ZrO2 although their selectivities only get to 75%, 39% and 13.8%, respectively, under optimal reaction conditions, because it shows the possibility for controllable transformation of LA and its ester over non-noble metal heterogeneous catalyst.
In this context, we prepare a cobalt catalyst of Co/ZrO2 that can flexibly hydrogenate EL in 1,4-dioxane solvent to GVL, 1,4-PDO and 2-MTHF with selectivity of 94%, 78% and 77%, respectively. A change of the support to SBA-15 leads to formation of 90% EHP. We demonstrate for the first time that the efficient non-noble metal cobalt catalyst for the flexible transformation of EL. It is interesting that deposition of appropriate amounts of coke species has a positive effect on the formation of GVL.
2. Experimental section
2.1 Materials
Co(NO3)2·6H2O, methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 1-pentanol, cyclohexane, benzene and tetrahydrofuran were supplied by Sinopharm Chem. Reagent Co., Ltd. 1,4-Dioxane, ethyl levulinate, α-angelica lactone and bis(2-methoxyethyl)ether were all purchased from Aladdin. 1,4-Pentanediol and γ-valerolactone was bought from Sigma-Aldrich and Damas-beta respectively. The above-mentioned chemicals were all of analytical grade and used as received without further purification. ZrO2 (Jiangsu Qianye Co., Ltd, China), TiO2 (Degussa P25), γ-Al2O3 (Shandong aluminum Co., Ltd, China), SBA-15 (Nanjing XFNANO Mater. Technol. Co., Ltd.), and montmorillonite (MMT) K-10 (Aladdin) were used as supports.2.2 Catalyst preparation
All the supported Co catalysts were prepared by the wetness impregnation method. Prior to the impregnation, the support was vacuum dried at 120 °C overnight. The dried support was dispersed in cobalt nitrate aqueous solution under vigorous stirring condition for 24 h. This is followed by directly drying at 100 °C for 10 h, and further calcining at 500 °C for 4 h in air.2.3 Catalyst characterization
N2 adsorption–desorption isotherms were measured at −196 °C on a Micromeritics TriStar 3000 instrument. BET surface area and BJH pore size distribution were calculated based on the desorption branch of the isotherms.The temperature-programmed desorption of ammonia (NH3-TPD) was performed on a Micromeritics AutoChem II 2920. Typically, 0.1 g sample was first pretreated at 400 °C for 30 min in Ar flow. Then, it was flushed with NH3 up to adsorptive saturation after being cooled to 100 °C. After that, the sample was heated to 600 °C at a rate of 10 °C min, and the desorbed NH3 was monitored by a thermal conductivity detector (TCD). The H2-TPR experiment was carried out in the same apparatus as that used for NH3-TPD. The sample was first pretreated at 500 °C for 30 min in Ar flow. Then, it was exposed to 10% H2–Ar flow after being cooled down to 40 °C. Finally, the temperature was raised to 800 °C at a rate of 5 °C min−1, and the amount of consumed H2 was monitored with a TCD. X-ray photoelectron spectra (XPS) were measured on a Kratos Axis Ultra DLD spectrometer equipped with a monochromated AlKα radiation source and a multichannel detector. All the binding energies were calibrated with adventitious C 1s peak at 284.8 eV.
The cobalt content was determined by an inductively coupled plasma-atomic emission spectrometer (ICP-AES, Thermo iCAP 6300). Powder X-ray diffraction (XRD) patterns were collected on a Rigaku MiniFlex II desktop X-ray diffractometer with monochromated CuKα radiation (0.154 nm, 30 kV, and 15 mA) at a scanning speed of 4° min−1. Transmission electron microscopy (TEM) images were taken on a field-emission transmission electron microscope (JEM-2100F) at an acceleration voltage of 200 kV. Prior to the measurements, the sample powders were dispersed into ethanol by ultrasonicating for 15 min, and dropped onto copper grids. Co/SBA-15 sample was embedded in a polymeric resin and polished to 20 nm thick in the middle of the sample using precision ion polishing system (Gatan 691). The average Co particle sizes were determined by counting more than 200 particles from several TEM images.
Thermal gravimetric analyses (TGA) of used catalysts were carried out on a Rigaku Thermo plus Evo TG 8120 instrument. The sample was initially heated to 160 °C at a rate of 10 °C min−1 and kept for 30 min. Then, it was further heated to 600 °C at the same rate in air flow (30 mL min−1). The deposited carbonaceous species extracted with methyl isobutyl ketone (MIBK) after destroying the catalyst structure with HF (40 wt%) were analyzed by the positive-ion ESI mass spectroscopy (Bruker micrOTOF-Q III). The IR spectra in the range of 400–4000 cm−1 were recorded on a Bruker Vertex 70 FTIR spectrometer using the conventional KBr (99 wt%) pellet method.
2.4 Catalyst test
All the reactions were carried out in a 50 mL high-pressure autoclave equipped with a magnetic stirrer. Before reaction, 0.1 g catalyst was reduced at 500 °C for 2 h in 10 vol% H2–Ar flow. It was quickly added into the autoclave together with 17.4 mmol ethyl levulinate and 15 mL solvent. The autoclave was purged with H2 for three times, and charged of 4.0 MPa H2. The reaction was stopped by quickly cooling the autoclave in an ice water bath. The catalyst stability was investigated by the regeneration method. The spent sample was magnetically separated from the reaction mixture, thoroughly washed with corresponding solvent, and directly reused under identical reaction conditions. The catalyst was regenerated by calcining at 500 or 550 °C for 2 h in static air and reducing at 500 °C for 2 h in 10 vol% H2–Ar.The reaction products were analyzed by a Shimadzu GC-2014C gas chromatograph equipped with a DB-WAX column (30 m × 0.53 mm × 0.25 μm) and a flame ionization detector (FID). The unknown products were identified with GC-MS (Shimadzu QP2010 Ultra) by comparing with authentic chemicals. The products were also qualitatively analyzed by the positive-ion ESI mass spectroscopy (Bruker micrOTOF-Q III) and NMR spectroscopy (Bruker AV-III 400 MHz NMR spectrometer). The product amount was quantitatively determined by calibrated area normalization. In the experiments for investigating the effect of solvents, the EL conversion and GVL yield were calculated on the basis of GC analysis results obtained with bis(2-methoxyethyl)ether as internal standard.
3. Results and discussion
3.1 Characterization of the catalysts and hydrogenation activity
First, the effect of support on the catalytic performance was investigated. Fig. S1† shows that all the samples contain sole crystalline Co phase, viz. spinel Co3O4 (JCPDS 42-1467). Table 1 shows the Co3O4 particle size, as calculated by the Scherrer equation from the most intense peak at 2θ = 36.8°, of different samples. The Co size calculated by the equation of dCo = 0.75dCo3O4 is consistent with that of TEM results20 (Fig. 1). The metal dispersions in the calcined catalysts are estimated on the basis of cobalt size.20 The formation of Co species in the reduced samples, e.g. Co/ZrO2, can be verified by its lattice planes of Co (200) and (111) (Fig. 1h and i). Fig. 1a–d shows that Co nanoparticles are highly dispersed on γ-Al2O3 and well confined in the mesopores of SBA-15,21 while much larger Co particles are present in the other supports such as TiO2, MMT and ZrO2. Thus, Co/γ-Al2O3 and Co/SBA-15 have higher dispersions than the other samples. Table 1 shows that no distinct relationship exists between BET surface area and Co dispersion, indicating that Co dispersion is not significantly affected by the BET surface area of catalysts.| Sample | Co loadinga (wt%) | SBET (m2 g−1) | dpore (nm) | Co3O4 particle sizeb (nm) | Cobalt dispersionc | NH3 uptaked mmol g−1 |
|---|---|---|---|---|---|---|
| a Determined by ICP.b Estimated by Scherrer equation, according to the (311) reflection of Co3O4.c Co dispersion is calculated by D = 96/d, where d is the cobalt size determined by the relative molar volume correction: dCo = 0.75dCo3O4.d Determined by NH3-TPD. | ||||||
| Co/γ-Al2O3 | 9.0 | 127.7 | 8.4 | 9.1 | 14.1 | 0.31 |
| Co/SBA-15 | 9.2 | 472.5 | 7.8 | 10.5 | 12.2 | 0.06 |
| Co/TiO2 | 9.2 | 31.0 | 37.4 | 14.8 | 8.6 | 0.17 |
| Co/MMT | 8.8 | 80.2 | 7.8 | 15.8 | 8.1 | 0.13 |
| Co/ZrO2 | 9.6 | 38.1 | 8.4 | 19.9 | 6.4 | 0.18 |
 | ||
| Fig. 1 TEM and HRTEM images of reduced Co/SBA-15 (a and b), Co/γ-Al2O3 (c and d), Co/MMT (e), Co/TiO2 (f), and Co/ZrO2 (g–i). | ||
Fig. 2a shows the H2-TPR profiles of different catalysts. Two peaks were observed for all the samples. The peak between 200 and 350 °C is attributed to the reduction of Co3O4 to CoO, and that in the range of 300–550 °C is due to the reduction of CoO to metallic Co. As for the reduction peaks observed at temperature higher than 550 °C in the profiles of Co/Al2O3, Co/SBA-15 and Co/MMT can be assigned to the reduction of cobalt species strongly interacted with support.22 Metallic Co0 was proved to be the active species in the hydrogenation reaction.12d Clearly, the Co3O4 species in the Co/ZrO2 and Co/TiO2 are easily reduced, consequently giving larger amounts of active Co species. Fig. 2b shows the XRD spectra of the reduced catalysts. The characteristic peaks of Co3O4 at 36.8°, 59.4° and 65.2° nearly cannot be detected on Co/ZrO2 and Co/TiO2, and the peaks of Co at 41.7° and 44.7° (JCPDS 05-0727) can be seen clearly. There is no significant change of Co/Al2O3 after reduction and the lattice planes of Co3O4 (111) can be observed in Fig. 1f. These results are in good consistent with the TPR profiles.
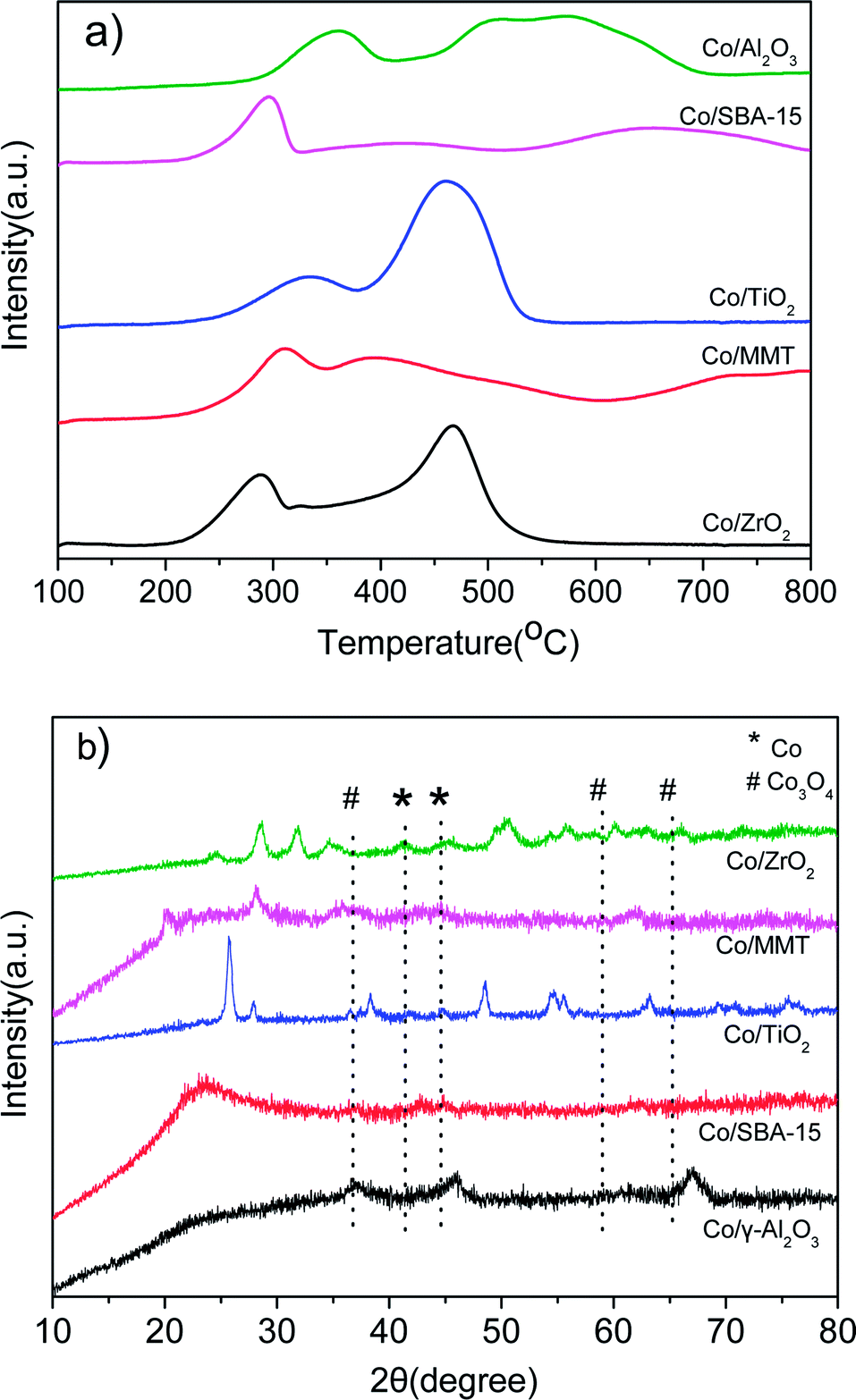 | ||
| Fig. 2 (a) H2-TPR curves of the as-prepared Co-based catalysts; (b) XRD patterns of the reduced Co-based catalysts. | ||
Fig. 3 shows that all the supported Co catalysts enabled a nearly total conversion of EL except that Co/γ-Al2O3 obtained only 31.9% conversion. Interestingly, Co/ZrO2 gave a GVL yield as high as 83.5%, whereas Co/SBA-15 catalyst exhibited the highest EHP yield of 57.1%. This is probably due to their different acidity and reducibility of Co3O4 species. Co/ZrO2 shows high reducibility of Co3O4 species and moderate acidity in dealcoholization process (Table 1), while Co/SBA-15 exhibits weak acidity. It has been reported that acid sites are beneficial to increase GVL yield.23 It should be noted that EHP was generated as byproducts, but it has not been definitely identified yet although it is a value-added product.11d,24 We unambiguously confirmed the formation of EHP with 1H and 13C NMR as well as mass spectroscopy (Fig. S2–S4†), and found that its yield was increased by lowering reaction temperature. At 160 °C, the EHP yield obtained over Co/SBA-15 reached 90%. To the best of our knowledge, this value is much higher than the reported results.13 Such a high EHP yield is due to the high dispersion of Co species on Co/SBA-15 and weak acidity of SBA-15.
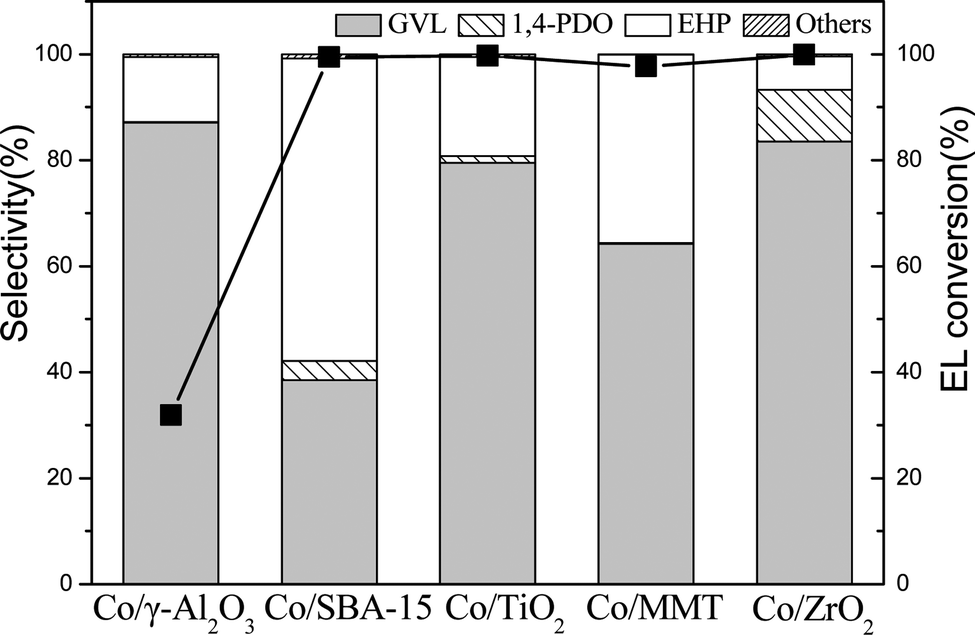 | ||
| Fig. 3 Catalytic results for hydrogenation of EL in 1,4-dioxane over different catalysts (reaction conditions: 17.4 mmol EL, 15 mL 1,4-dioxane, 190 °C, 4 MPa H2, 2 h and 0.1 g catalyst. Others mainly include 2-MTHF, 1-PAO, and 2-PAO). | ||
3.2 Effect of reaction conditions
The influence of reaction conditions on the catalytic performance was investigated with Co/ZrO2 as model catalysts as it gave the highest GVL yield. Fig. 4a shows that the selectivity of EHP drastically declined with the reaction time, while 1,4-PDO and 2-MTHF selectivity gradually increased to 23.2% and 11.8% respectively at 24 h. The GVL selectivity quickly increased to 81.5% within one 1 h, and then, maintained in the range of 83.5–86.7% between 2 and 5 h. After that, it monotonically decreased with increasing reaction time.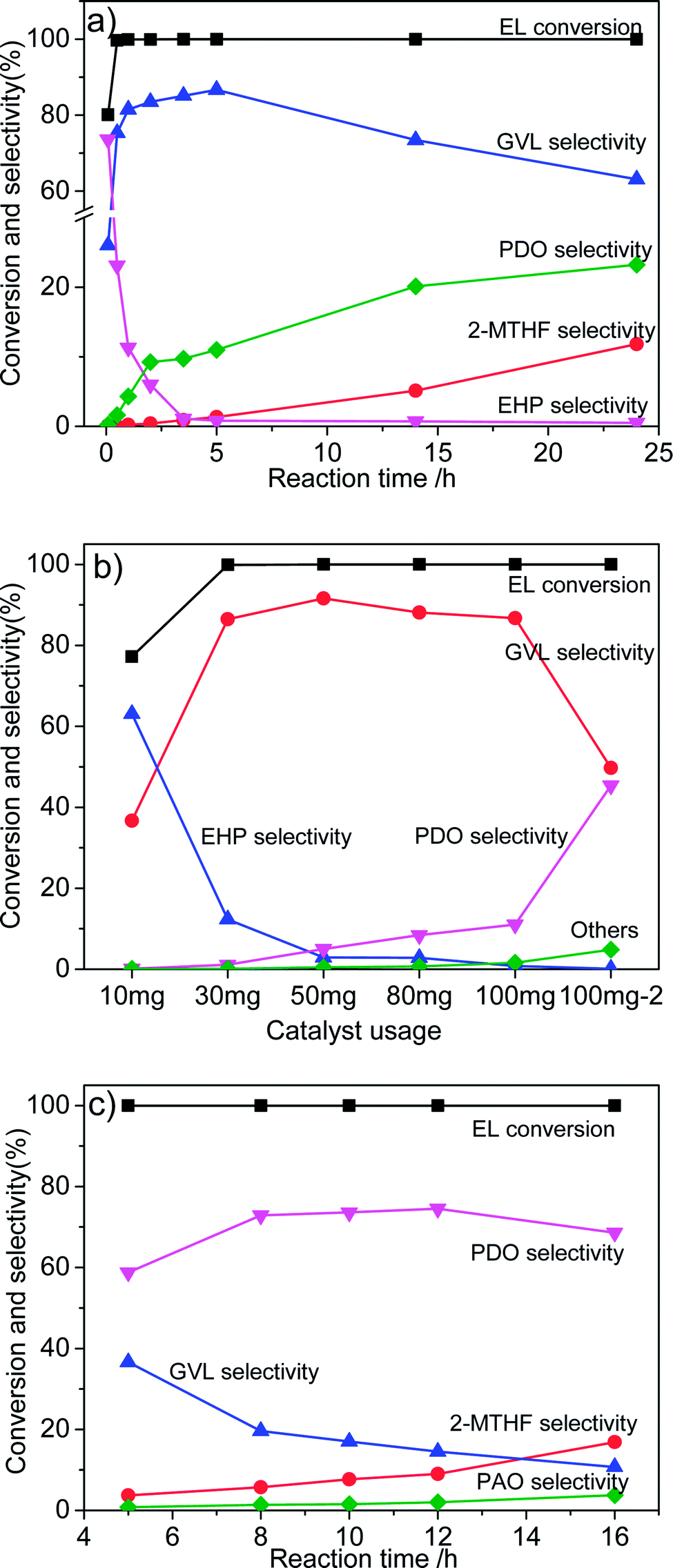 | ||
| Fig. 4 Effect of reaction conditions on the catalytic performance of Co/ZrO2 for hydrogenation of EL. (a) 17.4 mmol EL, 15 mL 1,4-dioxane, 190 °C, 4 MPa H2 and 0.1 g Co/ZrO2; (b) 17.4 mmol EL (the 100 mg−2 in the horizontal ordinate represents addition of 3.47 mmol EL), 15 mL 1,4-dioxane, 190 °C, 5 h and 4 MPa H2; (c) 3.47 mmol EL, 15 mL 1,4-dioxane, 190 °C, 6 MPa H2 and 0.1 g Co/ZrO2. | ||
Fig. 4b shows that 1,4-PDO selectivity linearly increases with increasing catalyst amount from 10 to 100 mg. This suggests that introduction of more amount of active Co species in the reaction system leads to formation of more 1,4-PDO. To confirm this hypothesis, 3.47 mmol EL (one fifth of normally added amount) was added into the reaction system (named as 100 mg−2). As expected, a very high 1,4-PDO yield (45.4%) was obtained. However, it is strange that a further increase in the catalyst weight to 300 mg contrarily decreased 1,4-PDO yield to 26.1%, and large amounts of 2-MTHF was detected.
Thus, a new way needs to be developed for further enhancing the production of 1,4-PDO. It was found that the 1,4-PDO yield considerably increased to 59% with increasing reaction pressure to 6 MPa (Fig. 4c), and could be further increased to nearly 74% by prolonging reaction time to 8 h. It reached as high as 78% at 8 h when the H2 pressure was increased to 8 MPa.
It is worth pointing out that Co/ZrO2 can selectively catalyze hydrogenolysis of EL into not only GVL and 1,4-PDO but also 2-MTHF. Table 2 shows that the increase of reaction temperature to 230 °C enhanced 2-MTHF selectivity to 76.7% (entry 1) due to promotion of the dehydration of diol to cyclic ether.25
| Entry | T (°C) | Substrate | Time (h) | Conv. (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|---|---|
| GVL | 1,4-PDO | MTHF | PAO | |||||
| a Reaction conditions: 15 mL 1,4-dioxane, 4 MPa H2 and 0.1 g Co/ZrO2.b 1-PAO selectivity.c 2-PAO selectivity.d 44.2% 1-PAO and 55.8% 2-PAO, respectively. | ||||||||
| 1 | 230 | 3.4 mmol EL | 8 | 100 | 2.0 | 0.6 | 76.7 | 20.6 |
| 2 | 190 | 5 mmol GVL | 5 | 23.9 | — | 79.6 | 17.8 | 2.7 |
| 3 | 230 | 1 mmol GVL | 2 | 97.0 | — | 1.9 | 74.4 | 23.7 |
| 4 | 230 | 1 mmol 1,4-PDO | 2 | 99.3 | 0.2 | — | 75.1 | 24.7 |
| 5 | 230 | 1 mmol 2-PAO | 8 | 4.1 | n.d. | n.d. | 91.5 | 8.5b |
| 6 | 230 | 1 mmol 1-PAO | 8 | 8.2 | n.d. | n.d. | 91.3 | 8.7c |
| 7 | 230 | 1 mmol 2-MTHF | 3 | 4.0 | n.d. | n.d. | — | 100d |
3.3 Reaction route for the conversion of EL
In order to illustrate the reaction process of EL hydrogenation, conversion of GVL, 1,4-PDO, 1-PAO, 2-pentanol (2-PAO) and 2-MTHF over Co/ZrO2 in the presence of H2 were explored under the same conditions as hydrogenation of EL. Table 2 shows that 1,4-PDO was mainly produced from GVL at 190 °C with a selectivity of nearly 80% (entry 2). This is different from the results attained on Ru/H-ZSM-5, Ru/SBA-15-SO3H, Pt/MFI, and Co@ZSM-5 that petanoic acid (PA) is primarily generated via ring opening of GVL7b,26 due to the weaker acidity of ZrO2 than of zeolites and sulfonated SBA-15 supports.High temperature promotes the dehydration of 1,4-PDO to 2-MTHF. Table 2 shows that MTHF selectivity reached 74.4% at 230 °C (entry 3). Although 1,4-PDO, like all diols, can also convert into 1-PAO and 2-PAO through consecutive hydrogenolysis reaction,27 the PAO selectivity is just about 1/3 of that 2-MTHF, and it seems to be independent of the substrates (Table 2, entries 1, 3, and 4). This indicates that PAO and 2-MTHF are both thermodynamically stable products with a molar ratio of ≈1/3 at 230 °C. This is confirmed by the mutual transformation of PAO and 2-MTHF in the presence of H2 with a very high selectivity. Table 2 shows that both 1-PAO and 2-PAO can dehydrogenate to 2-MTHF with a selectivity >91%, while 2-MTHF almost completely hydrogenate to PAO, although their conversions were <8.5% (entries 5–7).
Based on the above experimental results, a possible reaction pathway for the hydrogenation of EL on Co/ZrO2 catalyst, as shown in Scheme 1, is proposed in combination with previous researches.25,28 First, EL quickly hydrogenates to EHP on active Co species. Then, the acid sites on the ZrO2 support catalyze dealcoholization of EHP to GVL, which is further hydrogenated to 1,4-PDO. Finally, 2-MTHF is formed through the dehydration of two hydroxyl species of 1,4-PDO. With respect to PAO, it is generated by hydrogenolysis of 1,4-PDO, and reaches a thermodynamic balance with 2-MTHF.
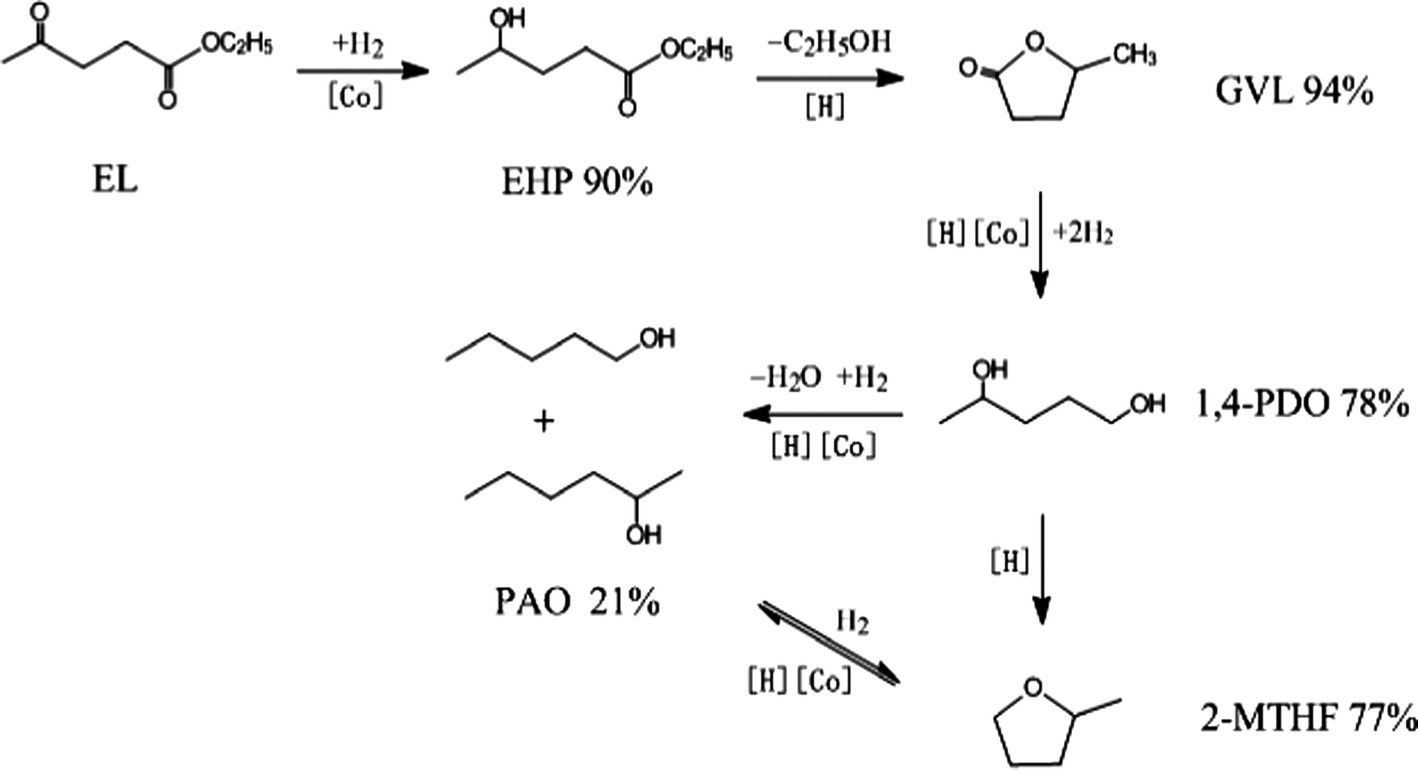 | ||
| Scheme 1 Possible reaction pathway and maximum yields for the selective conversion of EL into GVL, 1,4-PDO, PAO and 2-MTHF in the presence of 1,4-dioxane. | ||
3.4 Effect of solvent on EL hydrogenation to GVL
The choice of suitable solvent is very important from viewpoints of catalytic performance, environment and upscaling.29 Table 3 shows the reaction results of solvent effect and Co leaching percentage occurred in the hydrogenation of EL to GVL over Co/ZrO2 under optimal conditions. Clearly, water as solvent gave not only high EL conversion of 97.4% but also high GVL yield of 91.8%. In contrast, benzene caused the catalyst nearly inactive. Ethanol, 1-propanol, 1-butanol, cyclohexane and 1,4-dioxane exhibited a GVL yield of 80–87% despite that EL was nearly completely converted. Concerning methanol, 2-propanol and 1-pentanol solvents, both moderate EL conversion and GVL yield were obtained. This is partially due to the different solubility of H2 in different solvents. The Henry's law constants (kh)‡ of H2 in methanol, ethanol, 1-propanol and 1-butanol are 596, 452, 408 and 358 MPa, respectively,30 consequently leading to these alcohol solvents with a declined H2 concentration under the same H2 pressure. Similar result was also obtained in the LA hydrogenation with Ru/C as catalyst in different alcohols.31 However, the Co leaching in water is severe. It was found that about 28.7% of Co leached in the reaction solution, making its color turning into pink as a result of formation of soluble metal carboxylate complex between Co2+ and generated levulinic acid. Similar phenomenon was observed by Hengne et al.7a in the hydrogenolysis of LA over Cu/ZrO2 in water medium. The leached copper species led to formation of blue reaction solution. The negligible activity with benzene as solvent is due to its strong adsorption on the active Co species. In contrast, the alcohol solvent has a great potential, especially when combining with the upstream processes such as production of EL from monosaccharide, polysaccharides, furfuryl alcohol, 5-hydroxymethylfurfural in ethanol32 for the abatement of separation process. The utilization of alcohols has proved to be advantageous, as such solvents arguably have a relatively low net environmental impact and can be derived from biomass.33 Nonetheless, the possible etherification of alcohols with the product of 1,4-PDO make 1,4-dioxane be an appropriate solvent.| Entry | Solvent | EL conv. (%) | GVL yield (%) | Co leachingb (%) |
|---|---|---|---|---|
| a Reaction conditions: 17.4 mmol EL, 15 mL solvent, 190 °C, 4 MPa H2, 2 h and 0.1 g Co/ZrO2.b Determined by ICP. | ||||
| 1 | Water | 97.4 | 91.8 | 28.4 |
| 2 | Methanol | 97.3 | 58.7 | 0.8 |
| 3 | Ethanol | >99.5 | 84.9 | 0.8 |
| 4 | 2-Propanol | 63.2 | 53.1 | 0.4 |
| 5 | 1-Propanol | >99.5 | 84.9 | 0.2 |
| 6 | 1-Butanol | >99.5 | 86.7 | 0.6 |
| 7 | 1-Pentanol | 78.7 | 64.2 | 0.5 |
| 8 | Cyclohexane | >99.5 | 80.0 | DNT |
| 9 | Benzene | 1.0 | 0.8 | DNT |
| 10 | 1,4-Dioxane | >99.5 | 83.5 | 0.46 |
3.5 Reusability of the Co/ZrO2 catalyst
Reusability is of great significance for heterogeneous catalysts. After reaction, Co/ZrO2 can be simply separated by a magnet (Fig. S5†). Co/ZrO2 shows high catalytic stability. It gave a GVL yield higher than 83.5% within 12 repeated runs with regeneration before starting the fifth recycle by calcining in air and reducing with H2 at 500 °C for 2 h respectively (Fig. 5). An increase in the calcination temperature to 550 °C led to a decrease of GVL yield to about 83%, but it gradually increased to 94% again, and maintained for four recycles, while 1,4-PDO yield gradually declined and approached zero.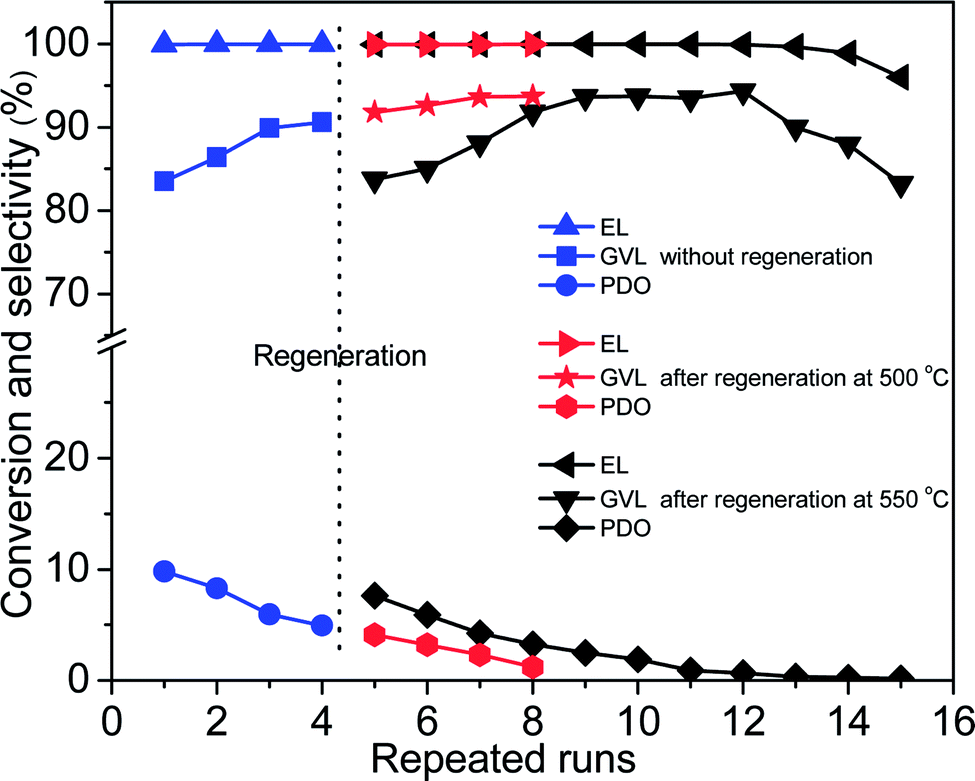 | ||
| Fig. 5 Reusability tests of Co/ZrO2 for hydrogenation of EL with regeneration after four repeated runs (reaction conditions: 17.4 mmol EL, 15 mL 1,4-dioxane, 190 °C; 4 MPa H2, 2 h and 0.1 g Co/ZrO2). | ||
To investigate the reason inducing the change of GVL yield in the repeated runs, the reduced and used catalysts were comprehensively characterized with TEM, XPS, TG and IR techniques. Fig. S6† shows that the morphology and particle size of Co/ZrO2 were kept intact during the reduction and the first three recycles. The peak at 781.5 eV in the XPS is attributed to Co2+/3+ 2p3/2 configuration, while that at 797 eV is assigned to Co2+/3+ 2p1/2. The weak ones at 786.8 eV and 803.5 eV are the shake-up peaks of Co2+/3+ 2p3/2 and Co2+/3+ 2p1/2. The Co0 2p3/2 and Co0 2p1/2 are characterized by the peaks at 779.8 and 795.4 eV respectively.12d,34 The Co0/Co ratio was kept constant in all the runs (Fig. S7a†). The Zr 3d spectrum (Zr 3d5/2 at 181.8 eV and Zr 3d3/2 at 184.2 eV with a splitting of 2.4 eV)35 did not significantly change in the spectral feature and the peak intensity after reduction and the first three recycles, revealing that ZrO2 was not reduced in the reaction system (Fig. S7b†), which is supported by the unchange of O 1s peak (Fig. S7c†). The major component of O(I) corresponds to ZrO2 (529.9 eV), and the components O(II) (531.6 eV) and O(III) (533.3 eV) are related to the organic or hydroxylic O and adsorbed water, respectively.35 The high stability of ZrO2 support was also observed in the hydrogenation of LA over Ru/ZrO2.35
Fig. 6A shows that the weight loss of Co/ZrO2 in 450–600 °C is around 0.44%, 1.41% and 4.13% after one, two and three repeated runs, respectively. This is accompanied by the gradual increase in the GVL yield and the decrease in the 1,4-PDO yield although the EL conversion is still higher than 99%. When the sample was regenerated by calcining at 550 °C for 2 h in static air and reducing with H2 at 500 °C for 2 h after four repeated runs, the catalytic performance was completely recovered, and showed similar changes with increasing repeated runs (Fig. 5, repeated runs 5–10).
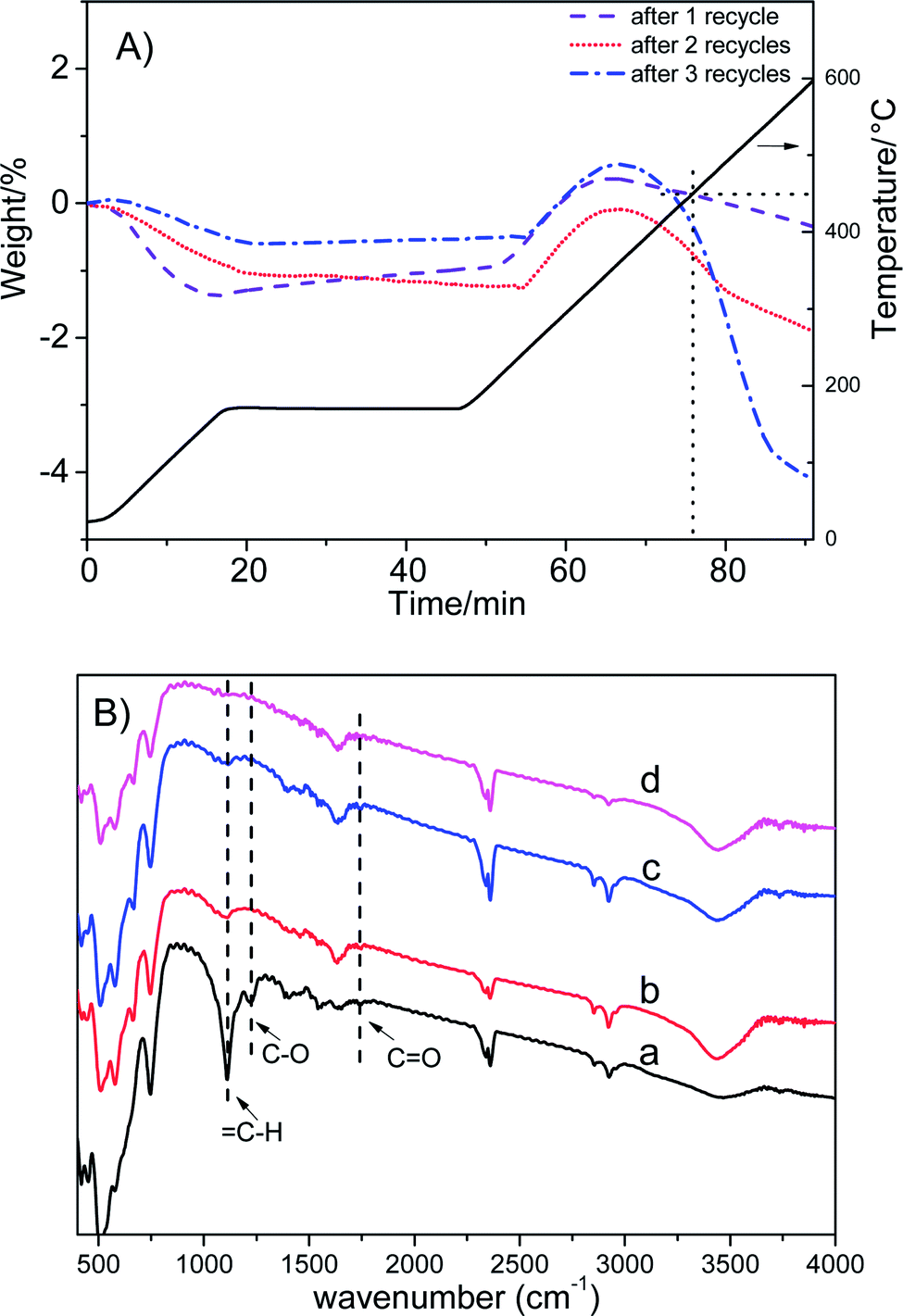 | ||
| Fig. 6 (A) TG profiles of Co/ZrO2 used for 1–3 recycles; (B) FTIR spectra of spent Co/ZrO2 (a), Co/ZrO2 regenerated at 500 °C in air (b), Co/ZrO2 regenerated at 550 °C in air (c), reduced Co/ZrO2 (d). | ||
Interestingly, when the reaction temperature was lowered down to 140 °C, the EL conversion almost linearly decreased with increasing repeated runs although the product selectivity was kept nearly the same (Fig. S8†). This shows that the hydrogenation activity of Co/ZrO2 declines with the recycles maybe due to deposition of coke species on some active Co sites. Consequently, the over-hydrogenation of GVL to 1,4-PDO was suppressed, thus increasing GVL selectivity. It is unexpected that regeneration of the sample by calcining at 500 °C in static air gave a GVL yield same as that attained over the sample at 4th recycles, being about 94%. This reveals that the deposited coke species cannot be completely removed by calcining at 500 °C. Thus, the decrease in both EL conversion and GVL selectivity (Fig. 5, repeated runs 13–15) can be accounted for by deposition of too many coke species on the Co/ZrO2, which results in a significant decrease of acid sites. More EHP was not converted in the reaction system (a selectivity up to 16% after 15 repeat runs), and it resulted in a decline of GVL yield.
Weckhuysen et al. reported that angelica lactone (AL) might be involved in the formation of coke species in the hydrogenation of levulinic acid.36 However, no AL was detected in the products and the extracted organic species from used catalyst. Zhang et al.37 found that AL dimers and trimers were generated through C–C bond coupling in the presence of moderately strong alkalis such as K2CO3 and Na2CO3. As we know, ZrO2 is an acid–base amphoteric oxide.38 Thus, it is possible that dimerization and/or trimerization of AL may occur. To prove this point, the organic species occluded in used catalyst were extracted with MIBK and analyzed by the negative-ion ESI mass spectroscopy. Indeed, the mass signals typical of AL dimers and trimers were observed (Fig. S9†). This is supported by the appearance of vibration bands attributed to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H, C–O and CO groups in the FT-IR spectrum (Fig. 6B).39 Another evidence was obtained by the following experiment. First, a very small amount of α-AL (3.5 mg) was added in the reactor together with Co/ZrO2 and 1,4-dioxane, and reacted for 1 h under the conditions (2 MPa N2, 80 °C) typical for formation of dimers and trimers.35 Then, 2.51 g EL was added after cooling to room temperature and reacted under the above normal conditions, which gave a GVL yield of 87.8%, being similar to the GVL yield in the third recycle (Fig. 5). Thus, it can be deduced that AL dimers and trimers are the coke species deposited on the Co/ZrO2 in the hydrogenation of EL.
C–H, C–O and CO groups in the FT-IR spectrum (Fig. 6B).39 Another evidence was obtained by the following experiment. First, a very small amount of α-AL (3.5 mg) was added in the reactor together with Co/ZrO2 and 1,4-dioxane, and reacted for 1 h under the conditions (2 MPa N2, 80 °C) typical for formation of dimers and trimers.35 Then, 2.51 g EL was added after cooling to room temperature and reacted under the above normal conditions, which gave a GVL yield of 87.8%, being similar to the GVL yield in the third recycle (Fig. 5). Thus, it can be deduced that AL dimers and trimers are the coke species deposited on the Co/ZrO2 in the hydrogenation of EL.
Table S1† compares the catalytic results for the hydrogenation of LA and its ester to GVL over Co/ZrO2 and reported catalysts. The commercial Ru/C shows the best catalytic performance for the hydrogenation of LA.31 In contrast, Cu-based and Co-based catalysts and RANEY® Ni exhibit a GVL selectivity of 80–90% except for metallic Co and 4Co/Al2O3 prepared by calcining corresponding hydrotalcite at 700 °C, on which about 94% of EL and >99% of LA were hydrogenated into GVL respectively.7a,11b,12c,12d,40 Regardless of this, the GVL productivity of Co/ZrO2 reaches 1.5 mol gmetal−1 h−1, which is much higher than that obtained over the reported non-noble metal catalysts. In addition, the GVL selectivity could be significantly increased to 94.3% with increasing repeated runs to 12 after regeneration by calcining at 550 °C for 2 h and reducing at 500 °C for 2 h before the 5th recycle.
4. Conclusions
Co/ZrO2 shows high activity and selectivity to GVL, 1,4-PDO or 2-MTHF in the reaction of EL hydrogenation. The GVL, 1,4-PDO and 2-MTHF reach 94%, 78% and 77%, respectively. It is interesting that a change of the ZrO2 support to SBA-15 selectively convert EL into EHP (selectivity > 90%) as a result of decreasing acid sites in catalyst. In addition, Co/ZrO2 shows very high catalytic stability, and can be reused with regeneration by calcining at 550 °C in air. No significant leaching and coordination state changes of Co species were observed. The Co/ZrO2 also exhibits the highest GVL productivity, being 1.50 mol gmetal−1 h−1, among the reported non-noble metal catalysts. The reaction conditions, including temperature, H2 pressure, time, catalysts amount and solvent, have great effect on the catalytic performance. It was found that the deposited coke species are AL dimers and trimers, which decreases the hydrogenation activity, but favors formation of GVL yield due to cover of some active Co species. It's envisaged that such an active, selective and robust catalyst will hold remarkable advantages in the EL flexible transformation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21403269 and 21603254), the Natural Science Foundation of Shanxi Province (2016021033), the Science Foundation for Youth Scholars of State Key Laboratory of Coal Conversion (2016BWZ002), the Youth Innovation Promotion Association CAS (2015140) and the CAS/SAFEA International Partnership Program for Creative Research Teams (2015YC901).Notes and references
- P. N. Vennestrom, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef CAS PubMed.
- (a) C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC; (b) C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- (a) A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (c) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (d) D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2013, 6, 76–80 RSC; (e) Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- (a) S. Zhu, Y. Xue, J. Guo, Y. Cen, J. Wang and W. Fan, ACS Catal., 2016, 6, 2035–2042 CrossRef CAS; (b) W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed; (c) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- (a) H. Chen, B. Yu and S. Jin, Bioresour. Technol., 2011, 102, 3568–3570 CrossRef CAS PubMed; (b) E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed; (c) F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Green Chem., 2016, 18, 1694–1705 RSC.
- (a) E. Ahmad, M. I. Alam, K. K. Pant and M. A. Haider, Green Chem., 2016, 18, 4804–4823 RSC; (b) S. Zhu, Y. Cen, J. Guo, J. Chai, J. Wang and W. Fan, Green Chem., 2016, 18, 5667–5675 RSC.
- (a) A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC; (b) W. H. Luo, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2014, 320, 33–41 CrossRef CAS.
- (a) X. Hu and C. Z. Li, Green Chem., 2011, 13, 1676–1679 RSC; (b) S. Zhu, J. Guo, X. Wang, J. Wang and W. Fan, ChemSusChem, 2017, 10, 2547–2559 CrossRef CAS PubMed.
- (a) A. D. Chowdhury, R. Jackstell and M. Beller, ChemCatChem, 2014, 6, 3360–3365 CrossRef; (b) U. Omoruyi, S. Page, J. Hallett and P. W. Miller, ChemSusChem, 2016, 9, 2037–2047 CrossRef CAS PubMed.
- (a) W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed; (b) S. Cao, J. R. Monnier and J. R. Regalbuto, J. Catal., 2017, 347, 72–78 CrossRef CAS.
- (a) C. Xie, J. L. Song, B. W. Zhou, J. Y. Hu, Z. R. Zhang, P. Zhang, Z. W. Jiang and B. X. Han, ACS Sustainable Chem. Eng., 2016, 4, 6231–6236 CrossRef CAS; (b) M. Chia and J. A. Dumesic, Chem. Comm., 2011, 47, 12233–12235 RSC; (c) Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Comm., 2013, 49, 5328–5330 RSC; (d) A. H. Valekar, K. H. Cho, S. K. Chitale, D.-Y. Hong, G. Y. Cha, U. H. Lee, D. W. Hwang, C. Serre, J. S. Chang and Y. K. Hwang, Green Chem., 2016, 18, 4542–4552 RSCJ. Wang, S. Jaenicke and G. K. Chuah, RSC Adv., 2014, 4, 13481–13489 RSC.
- (a) V. Mohan, V. Venkateshwarlu, C. V. Pramod, B. D. Raju and K. S. R. Rao, Catal.Sci. Technol., 2014, 4, 1253–1259 RSC; (b) K. Jiang, D. Sheng, Z. H. Zhang, J. Fu, Z. Y. Hou and X. Y. Liu, Catal. Today, 2016, 274, 55–59 CrossRef CAS; (c) K. Yan and A. C. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS; (d) H. C. Zhou, J. L. Song, H. L. Fan, B. B. Zhang, Y. Y. Yang, J. Y. Hu, Q. G. Zhu and B. X. Han, Green Chem., 2014, 16, 3870–3875 RSC.
- (a) A. Diaz-Rodriguez, W. Borzecka, I. Lavandera and V. Gotor, ACS Catal., 2014, 4, 386–393 CrossRef CAS; (b) A. Manzocchi, R. Casati, A. Fiecchi and E. Santaniello, J. Chem. Soc., Perkin Trans. 1, 1987, 2753–2757 RSC.
- (a) F. M. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed; (b) M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. DellaPina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed; (c) Y. Shen, B. B. Yao, G. Yu, Y. Fu, F. S. Liu and Z. B. Li, Green Chem., 2017, 19, 4930–4938 RSC.
- A. Phanopoulos, A. J. P. White, N. J. Long and P. W. Miller, ACS Catal., 2015, 5, 2500–2512 CrossRef CAS.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- (a) Z. Wang, G. Li, X. Liu, Y. Huang, A. Wang, W. Chu, X. Wang and N. Li, Catal. Commun., 2014, 43, 38–41 CrossRef CAS; (b) M. X. Li, G. Y. Li, N. Li, A. Q. Wang, W. J. Dong, X. D. Wang and Y. Cong, Chem. Comm., 2014, 50, 1414–1416 RSC; (c) L. Corbel-Demailly, B. K. Ly, D. P. Minh, B. Tapin, C. Especel, F. Epron, A. Cabiac, E. Guillon, M. Besson and C. Pinel, ChemSusChem, 2013, 6, 2388–2395 CrossRef CAS PubMed; (d) T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC.
- T. Mizugaki, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustainable Chem. Eng., 2016, 4, 682–685 CrossRef CAS.
- Y. Yang, X. Xu, W. Zou, H. Yue, G. Tian and S. Feng, Catal. Commun., 2016, 76, 50–53 CrossRef CAS.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486–499 CrossRef.
- (a) M. Audemar, C. Ciotonea, K. De Oliveira Vigier, S. Royer, A. Ungureanu, B. Dragoi, E. Dumitriu and F. Jérôme, ChemSusChem, 2015, 8, 1885–1891 CrossRef CAS PubMed; (b) A. Ungureanu, B. Dragoi, A. Chirieac, S. Royer, D. Duprez and E. Dumitriu, J. Mater. Chem., 2011, 21, 12529–12541 RSC.
- K. Cheng, V. Subramanian, A. Carvalho, V. V. Ordomsky, Y. Wang and A. Y. Khodakov, J. Catal., 2016, 337, 260–271 CrossRef CAS.
- J. M. Nadgeri, N. Hiyoshi, A. Yamaguchi, O. Sato and M. Shirai, Appl. Catal., A, 2014, 470, 215–220 CrossRef CAS.
- F. Ye, D. Zhang, T. Xue, Y. Wang and Y. Guan, Green Chem., 2014, 16, 3951–3957 RSC.
- X. Du, Q. Bi, Y. Liu, Y. Cao, H. He and K. Fan, Green Chem., 2012, 14, 935–939 RSC.
- (a) P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 4136–4142 CrossRef CAS; (b) K. Kon, W. Onodera and K. Shimizu, Catal. Sci. Technol., 2014, 4, 3227–3234 RSC; (c) T. Pan, J. Deng, Q. Xu, Y. Xu, Q. X. Guo and Y. Fu, Green Chem., 2013, 15, 2967–2974 RSC.
- S. Zhu, X. Gao, Y. Zhu, Y. Zhu, X. Xiang, C. Hu and Y. Li, Appl. Catal., B, 2013, 140, 60–67 CrossRef.
- (a) A. Villa, M. Schiavoni, C. E. Chan-Thaw, P. F. Fulvio, R. T. Mayes, S. Dai, K. L. More, G. M. Veith and L. Prati, ChemSusChem, 2015, 8, 2520–2528 CrossRef CAS PubMed; (b) T. Pan, J. Deng, Q. Xu, Y. Xu, Q. X. Guo and Y. Fu, Green Chem., 2013, 15, 2967–2974 RSC; (c) D. Zhang, F. Ye, Y. Guan, Y. Wang and E. J. M. Hensen, RSC Adv., 2014, 4, 39558–39564 RSC.
- (a) J. G. Zhang and N. Yan, ChemCatChem, 2017, 9, 2790–2796 CrossRef CAS; (b) X. Hu, R. J. M. Westerhof, D. H. Dong, L. P. Wu and C. Z. Li, ACS Sustainable Chem. Eng., 2014, 2, 2562–2575 CrossRef CAS.
- (a) Purwanto, R. M. Deshpande, R. V. Chaudhari and H. Delmas, J. Chem. Eng. Data, 1996, 41, 1414–1417 CrossRef CAS; (b) M. S. Wainwright, T. Ahn, D. L. Trimm and N. W. Cant, J. Chem. Eng. Data, 1987, 32, 22–24 CrossRef CAS.
- M. G. Al-Shaal, W. R. H. Wright and R. Palkovits, Green Chem., 2012, 14, 1260–1263 RSC.
- (a) G. Pasquale, P. Vázquez, G. Romanelli and G. Baronetti, Catal. Commun., 2012, 18, 115–120 CrossRef CAS; (b) S. Saravanamurugan and A. Riisager, ChemCatChem, 2013, 5, 1754–1757 CrossRef CAS; (c) R. Liu, J. Chen, X. Huang, L. Chen, L. Ma and X. Li, Green Chem., 2013, 15, 2895–2903 RSC; (d) B. Lu, S. An, D. Song, F. Su, X. Yang and Y. Guo, Green Chem., 2015, 17, 1767–1778 RSC.
- M. Fatih Demirbas, M. Balat and H. Balat, Energy Convers. Manage., 2011, 52, 1815–1828 CrossRef CAS.
- (a) B. Ernst, S. Libs, P. Chaumette and A. Kiennemann, Appl. Catal., A, 1999, 186, 145–168 CrossRef CAS; (b) T. Cai, H. Huang, W. Deng, Q. Dai, W. Liu and X. Wang, Appl. Catal., B, 2015, 166, 393–405 CrossRef.
- J. Ftouni, A. Muñoz-Murillo, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, L. Lu, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Catal., 2016, 6, 5462–5472 CrossRef CAS.
- W. H. Luo, U. Deka, A. M. Beale, E. R. H. van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- J. Xin, S. Zhang, D. Yan, O. Ayodele, X. Lu and J. Wang, Green Chem., 2014, 16, 3589–3595 RSC.
- K. Tanabe and T. Yamaguchi, Catal. Today, 1994, 20, 185–198 CrossRef CAS.
- T. Chen, Z. Qin, Y. Qi, T. Deng, X. Ge, J. Wang and X. Hou, Polym. Chem., 2011, 2, 1190–1194 RSC.
- (a) Q. Xu, X. L. Li, T. Pan, C. G. Yu, J. Deng, Q. X. Guo and Y. Fu, Green Chem., 2016, 18, 1287–1294 RSC; (b) X. D. Long, P. Sun, Z. L. Li, R. Lang, C. G. Xia and F. W. Li, Chin. J. Catal., 2015, 36, 1512–1518 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01316k |
| ‡ Henry's law constant, kh = P(H2)/χ(H2), where P is H2 partial pressure and χ is the mole fraction of H2 in a given solution. |
| This journal is © The Royal Society of Chemistry 2018 |