
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ahmpatinin iBu, a new HIV-1 protease inhibitor, from Streptomyces sp. CPCC 202950†
Ming-Hua Chen‡
 ac,
Shan-Shan Chang‡ab,
Biao Donga,
Li-Yan Yua,
Ye-Xiang Wua,
Ren-Zhong Wanga,
Wei Jianga,
Zeng-Ping Gao*b and
Shu-Yi Si*a
ac,
Shan-Shan Chang‡ab,
Biao Donga,
Li-Yan Yua,
Ye-Xiang Wua,
Ren-Zhong Wanga,
Wei Jianga,
Zeng-Ping Gao*b and
Shu-Yi Si*a
aInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: sisyimb@hotmail.com
bSchool of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing 100102, China. E-mail: gaozp@bucm.edu.cn
cKey Laboratory for Uighur Medicine, Institute of Materia Medica of Xinjiang, Urumqi 830004, China
First published on 30th January 2018
Abstract
Ahmpatinin iBu (1) and statinin iBu (2), two new linear peptides, a novel pyrrolidine derivative, (−)-(S)-2-[3-(6-methylheptanamido)-2-oxopyrrolidin-1-yl] acetic acid (3), and three known pepstatin derivatives (4–6) along with their corresponding methanolysis artifacts (7–9) were isolated from Streptomyces sp. CPCC 202950. Their structures were elucidated on the basis of extensive spectroscopic data using Marfey's analysis, chiral-phase HPLC, and ECD and OR calculation to determine the absolute configurations. Compound 1 contains an unusual amino acid, 4-amino-3-hydroxy-5-(4-methoxyphenyl)pentanoic acid (Ahmppa), and 3 is the first natural product with a 2-(3-amino-2-oxopyrrolidin-1-yl)acetic acid system. Compounds 1, 2, and 4–9 are HIV-1 protease inhibitors. In particular, ahmpatinin iBu (1) exhibits significant inhibitory activity against HIV-1 protease with an IC50 value of 1.79 nM. A preliminary structure–activity relationship is discussed.
Introduction
The most effective treatment for HIV infection, HAART (Highly Active AntiRetroviral Therapy) that including several HIV-1 protease inhibitors combination with reverse transcriptase inhibitors, and other inhibitors with integrase, membrane fusion, and viral attachment, have significantly reduced the mortality of AIDS patients and improved the quality of life of those infected with HIV.1–4 However, according to World Health Organization and Joint United Nations Program on HIV/AIDS data, in 2016, there were still 36.7 million people living with HIV, 1.8 million newly infected and 1.0 million HIV-related deaths.5,6 Therefore, AIDS is still a substantial threat to global public health. HIV-1 protease (HIV-1 PR) is an essential enzyme in the life cycle of HIV and has been used as a promising target for AIDS therapy.7–9 Although 11 HIV-1 PR inhibitors such as amprenavir, indinavir, nelfinavir, ritonavir, saquinavir, and tipranavir have been approved by the FDA and applied extensively in clinical evaluations,10,11 drug toxicity, resistance, and drug–drug interactions remain serious problems for HIV/AIDS treatments,1,12–14 prompting us to discover new anti-HIV drugs.In our previous report, a new HIV-1 protease inhibitor named 4862F15 and 11 known compounds16 were isolated from the liquid fermentation of the strain Streptomyces sp. CPCC 202950. Unexpectedly, the crude extract of the rice culture of Streptomyces sp. CPCC 202950 also significantly inhibited HIV-1 protease activity with an IC50 value of 20.0 ng ml−1. Extensive investigation of the secondary metabolites of the rice culture extract of the strain resulted in the isolation of two new linear peptides, ahmpatinin iBu (1) and statinin iBu (2), a new pyrrolidine derivative, (−)-(S)-2-[3-(6-methylheptanamido)-2-oxopyrrolidin-1-yl]acetic acid (3), and three known pepstatin derivatives along with their corresponding methanolysis artifacts (4–9) (Fig. 1). Among them, compound 1 contains an unusual amino acid, 4-amino-3-hydroxy-5-(4-methoxyphenyl)pentanoic acid. Compound 3 represents the first natural product with a 2-(3-amino-2-oxopyrrolidin-1-yl)acetic acid system, which was synthesized in a previous study.17 Herein, we report the isolation, structural elucidation, and biological activities of these compounds.
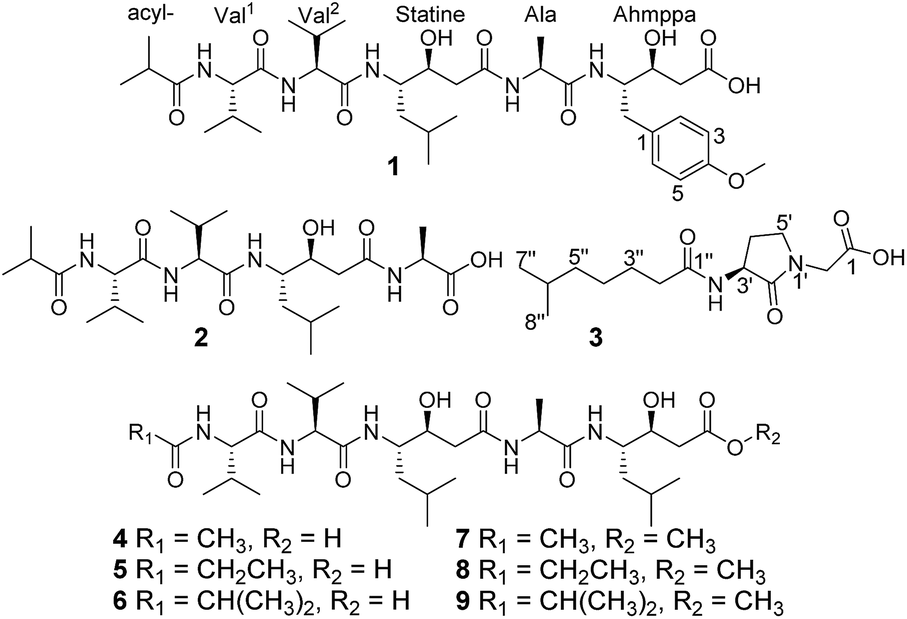 | ||
| Fig. 1 Chemical structures of compounds 1–9. | ||
Results and discussion
Ahmpatinin iBu (1) was isolated as a white powder and was determined to be C37H61N5O10 on the basis of positive HRESIMS, indicating 10 degrees of unsaturation. The 1H NMR spectrum of 1 (Table 1) in DMSO-d6 exhibited characteristics of a typical peptide, with five NH protons (δH 7.4–7.9) and five amino acid α-protons (δH 3.8–4.3). Furthermore, para-disubstituted aromatic ring protons [δH 6.80 (2H, d, H-3 and H-5) and 7.12 (2H, d, H-2 and H-6)], two oxygenated methine protons (δH 3.82 and 3.85), four methylene protons, four methine protons, and a methoxyl singlet [δH (3.70, s, 3H)] along with nine additional methyl doublets (δH 0.7–1.2) were also observed in the 1H NMR spectrum. Six carbonyl signals (δC 170–176) and five α-amino acid carbon signals (δC 48–58) were observed in the 13C NMR spectrum, further indicating that 1 was a peptide. Analysis of the DEPT and 13C NMR spectra revealed additional 1,4-disubstitued benzene ring groups (δC 113–158), ten methyl groups, four methylenes, and six sp3 methines.| No. | 1 | 2 | |||
|---|---|---|---|---|---|
| δH | δC, type | δH | δC, type | ||
| a NMR data (δ) were measured at 600 MHz for 1H and at 150 MHz for 13C in DMSO-d6. Proton coupling constants (J) in Hz are given in parentheses. The assignments were based on 1H–1H COSY, HSQC, and HMBC experiments.b The J-value was not determined due to overlapping signals. | |||||
| Val1 | CO | 171.1, C | 171.0, C | ||
| α | 4.14, dd (7.8, 8.4) | 57.7, CH | 4.14, ddb | 57.7, CH | |
| β | 1.96, m | 30.1, CH | 1.94, m | 30.1, CH | |
| γ | 0.81, d(6.6) | 18.4, CH3 | 0.81, d(6.6) | 18.2, CH3 | |
| γ′ | 0.81, d(6.6) | 19.8, CH3 | 0.81, d(6.6) | 19.8, CH3 | |
| NH | 7.79, d(8.4) | 7.78, d(9) | |||
| Val2 | CO | 170.6, C | 170.6, C | ||
| α | 4.14, dd(7.8, 8.4) | 57.8, CH | 4.14, ddb | 57.8, CH | |
| β | 1.96, m | 30.4, CH | 1.94, m | 30.5, CH | |
| γ | 0.81, d(6.6) | 18.2, CH3 | 0.81, d(6.6) | 18.1, CH3 | |
| γ′ | 0.83, d(6.6) | 19.4, CH3 | 0.83, d(6.6) | 19.4, CH3 | |
| NH | 7.71, d(9) | 7.68, d(9) | |||
| Sta | CO | 170.7, C | 170.8, C | ||
| α | 2.11, m | 39.2, CH2 | 2.10, m | 39.5, CH2 | |
| β | 3.82, m | 69.0, CH | 3.81, m | 69.0, CH | |
| γ | 3.82, m | 50.7, CH | 3.85, m | 50.7, CH | |
| δ | 1.34, m | 38.7, CH2 | 1.32, m | 38.5, CH2 | |
| 1.25, m | 1.25, m | ||||
| ε | 1.52, m | 24.2, CH | 1.52, m | 24.2, CH | |
| ζ | 0.78, d(6.6) | 21.6, CH3 | 0.78, d(6.6) | 21.6, CH3 | |
| ζ′ | 0.83, d(6.6) | 23.4, CH3 | 0.83, d(6.6) | 23.5, CH3 | |
| NH | 7.47, d(9) | 7.50, d(9) | |||
| Ala | CO | 172.1, C | 174.2, C | ||
| α | 4.22, m | 48.2, CH | 4.17, dqb | 47.4, CH | |
| β | 1.12, d(7.2) | 18.0, CH3 | 1.23, d(7.8) | 17.2, CH3 | |
| NH | 7.85, d(7.2) | 8.07, d(7.2) | |||
| Ahmppa | CO | 172.8, C | |||
| α | 2.23, dd(15.6, 3.6) | 38.8, CH2 | |||
| 2.16, dd(15.6, 8.4) | |||||
| β | 3.85, m | 67.2, CH | |||
| γ | 3.83, m | 54.4, CH | |||
| δ | 2.75, dd(13.8, 6) | 35.4, CH2 | |||
| 2.56, dd(13.8, 8.4) | |||||
| 1 | 130.9, C | ||||
| 2 | 7.12, d(8.4) | 130.0, CH | |||
| 3 | 6.80, d(8.4) | 113.5, CH | |||
| 4 | 157.5, C | ||||
| 5 | 6.80, d(8.4) | 113.5, CH | |||
| 6 | 7.12, d(8.4) | 130.0, CH | |||
| NH | 7.53, d(9) | ||||
| OMe-4 | 3.70, s | 54.9, CH3 | |||
| Acyl | CO | 176.2, C | 176.2, C | ||
| α | 2.51, m | 33.6, CH | 2.52, m | 33.6, CH | |
| β | 0.95, d(6.6) | 19.2, CH3 | 0.95, d(6.6) | 19.2, CH3 | |
| β′ | 0.98, d(6.6) | 19.3, CH3 | 0.98, d(6.6) | 19.3, CH3 | |
A comparison of the NMR spectra of compounds 1 and ahpatinin iBu18 revealed that the mono-substituted benzene moiety in ahpatinin iBu was replaced with a para-disubstituted aromatic ring group in 1. This was confirmed by 2D NMR (Fig. S6–S9†). HMBC correlations (Fig. 2) of H-4-OMe and C-4, δ-H-Ahmppa/C-1, C-2, C-6, C-β-Ahmppa and C-γ-Ahmppa, α-H-Ahmppa/C-β-Ahmppa, C-γ-Ahmppa and C-CO-Ahmppa, β-H-Ahmppa and C-CO-Ahmppa, and NH-Ahmppa and C-γ-Ahmppa indicated the presence of a 4-amino-3-hydroxy-5-(4-methoxyphenyl)pentanoic acid (Ahmppa) residue. Integrated 2D NMR (1H NMR, COSY, HSQC, and HMBC) data were used to assign five partial structures consisting of the amino acids alanine, two valines, statine (Sta), and an isobutyl (iBu) group. HMBC correlations from α-H-Val1 (δH 4.14) and NH-Val1 (δH 7.79) to iBu-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (δC 176.2), α-H-Val2 (δH 4.14) to Val1-CO (δC 171.1), γ-H-Sta (δH 3.82) to Val2-CO (δC 170.6), α-H-Ala (δH 4.22) to Sta-CO (δC 170.7), and γ-H-Ahmppa (δH 3.83) to Ala-CO (δC 172.1) established the sequence iBu–Val1–Val2–Sta–Ala–Ahmppa, which was verified by NOESY correlations (Fig. 3 and S9†) from NH-Val1 (δH 7.79) to α-H-iBu (δH 2.51), NH-Val2 (δH 7.71) to α-H-Val1 (δH 4.14), NH-Sta (δH 7.47) to α-H-Val2 (δH 4.14), NH-Ala (δH 7.85) to α-H-Sta (δH 2.11), and NH-Ahmppa (δH 7.53) and α-H-Ala (δH 4.22).
O (δC 176.2), α-H-Val2 (δH 4.14) to Val1-CO (δC 171.1), γ-H-Sta (δH 3.82) to Val2-CO (δC 170.6), α-H-Ala (δH 4.22) to Sta-CO (δC 170.7), and γ-H-Ahmppa (δH 3.83) to Ala-CO (δC 172.1) established the sequence iBu–Val1–Val2–Sta–Ala–Ahmppa, which was verified by NOESY correlations (Fig. 3 and S9†) from NH-Val1 (δH 7.79) to α-H-iBu (δH 2.51), NH-Val2 (δH 7.71) to α-H-Val1 (δH 4.14), NH-Sta (δH 7.47) to α-H-Val2 (δH 4.14), NH-Ala (δH 7.85) to α-H-Sta (δH 2.11), and NH-Ahmppa (δH 7.53) and α-H-Ala (δH 4.22).
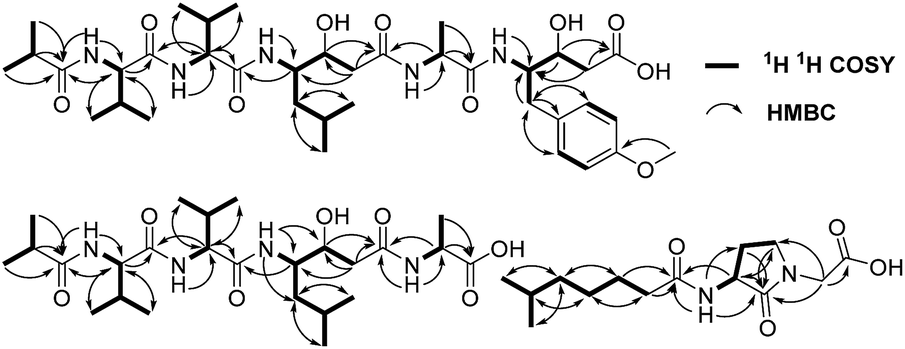 | ||
| Fig. 2 The key 1H–1H COSY and HMBC correlations of 1–3. | ||
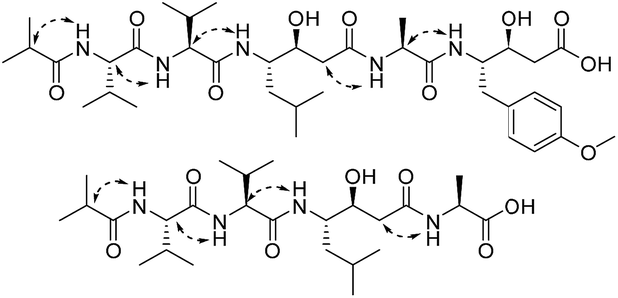 | ||
| Fig. 3 The key NOESY correlations of 1 and 2. | ||
The absolute configurations of Ala and Val in 1 were determined to be L using Marfey's method (Fig. S27† and Experimental section),19,20 whereas the Ahmppa residue was established to be 3S, 4S by chiral-phase HPLC after acid hydrolysis in comparison to synthetic standards (Fig. S28† and Experimental section). In addition, the acid hydrolysis of 1 followed by HPLC purification gave statine, and its 1H NMR data agreed with the synthetic standard of (3S,4S)-statine. Meanwhile, the specific rotation {[α]20D = −19.5 (c 0.05, H2O)} of the statine from the hydrolysis of 1 was consistent with that of (3S,4S)-statine {[α]20D = −20.4 (c 0.502, H2O)}.21 Therefore, the final structure of 1 was determined to be iBu-L-Val1-L-Val2-(3S,4S)-Sta-L-Ala-(3S,4S)-Ahmppa and was named ahmpatinin iBu.
Compound 2 was obtained as a white amorphous powder, and the molecular formula was assigned as C25H46N4O7 by HRESIMS based on a [M − H]− ion at 513.3275 (calc. 513.3283). The MS data for 2 indicated the absence of the Ahmppa moiety relative to compound 1. Extensive analysis of 2D NMR (1H–1H COSY, HSQC, HMBC, and NOESY) spectroscopic data (Fig. S15–S18†) revealed the sequence of iBu–Val1–Val2–Sta–Ala. The absolute configurations of the amino acid residues and the statine unit were also confirmed to be L-Ala, L-Val and (3S,4S)-statine using the same protocol as 1. Hence, compound 2 was deduced to be iBu-L-Val1-L-Val2-(3S,4S)-Sta-L-Ala and was named statinin iBu.
Compound 3 was isolated as a white powder. Its molecular formula C14H24N2O4, requiring four degrees of unsaturation, was established from the HRESIMS data (m/z 285.1802 [M + H]+, calcd for C14H25N2O4, 285.1808). The IR spectrum of 3 displayed the presence of hydroxy and/or amino (3287 cm−1), carboxyl (1700 cm−1) and amide (1645 cm−1) functionalities. Analysis of the 1H NMR, 13C NMR and HSQC spectra (Table 2) of 3 revealed two doublet methyls, seven sp3 methylenes, two sp3 methines, and three carbonyls (δC 170.0, 172.3, and 172.4). The 1H–1H COSY cross-peaks of H2-2′′/H2-3′′/H2-4′′/H2-5′′/H-6′′/H3-7′′(H3-8′′) and HMBC correlations of H3-7′′(H3-8′′)/C-6′′ and C-5′′; H-6′′/C-4′′, C-5′′, C-7′′ and C-8′′; H2-5′′/C-3′′, C-4′′, C-6′′, C-7′′ and C-8′′; H2-4′′/C-2′′, C-3′′, C-5′′ and C-6′′; H2-3′′/C-1′′, C-2′′, C-4′′ and C-5′′; and H2-2′′/C-1′′, C-3′′ and C-4′′ indicated the presence of a 6-methylheptanoyl moiety in 3. Meanwhile, in conjunction with the degrees of unsaturation and the molecular composition, 1H–1H COSY correlations of NH/H-3′/H-4′/H-5′ along with HMBC correlations of NH/C-2′, C-3′ and C-4′; H-3′/C-2′ and C-4′; H2-4′/C-2′, C-3′ and C-5′; H2-5′/C-2′, C-3′ and C-4′; and H2-2/C-1, C-2′ and C-5′ demonstrated the presence of a 2-(3-amino-2-oxopyrrolidin-1-yl)acetic acid moiety.17 Finally, the HMBC correlation from H-3′ and NH to C-1′′ suggested that the two moieties were connected via an amide bond. Thus, compound 3 was confirmed to be 2-(3-(6-methylheptanamido)-2-oxopyrrolidin-1-yl)acetic acid. The absolute configuration at C-3′ was determined by comparing the experimental circular dichroism (CD) spectrum and optical rotation (OR) data with the computed ECD and OR data. A comparison of the experimental CD spectrum with the calculated ECD spectrum of 3 (Fig. 4) demonstrated that compound 3 possessed an S configuration, which was verified by the computed OR, which was −41.5 for (S)-3. The experimental OR value was −100.5 in acetonitrile. Therefore, compound 3 was elucidated to be (−)-(S)-2-[3-(6-methylheptanamido)-2-oxopyrrolidin-1-yl]acetic acid.
| No. | 3 | |
|---|---|---|
| δH | δC, type | |
| a NMR data (δ) were measured at 600 MHz for 1H and at 150 MHz for 13C in DMSO-d6. Proton coupling constants (J) in Hz are given in parentheses. The assignments were based on 1H–1H COSY, HSQC, and HMBC experiments. | ||
| 1 | 170.0, C | |
| 2 | 3.99, d(17.4) | 44.1, CH2 |
| 3.89, d(17.4) | ||
| 2′ | 172.4, C | |
| 3′ | 4.41, m | 49.5, CH |
| 4′ | 2.29, m | 26.2, CH2 |
| 1.76, m | ||
| 5′ | 3.33, m | 43.8, CH2 |
| 1′′ | 172.3, C | |
| 2′′ | 2.10, t(7.2) | 35.3, CH2 |
| 3′′ | 1.47, m | 25.5, CH2 |
| 4′′ | 1.25, m | 26.4, CH2 |
| 5′ | 1.14, m | 38.2, CH2 |
| 6′′ | 1.51, m | 27.3, CH |
| 7′′ | 0.84, d(6.6) | 22.5, CH3 |
| 8′′ | 0.84, d(6.6) | 22.5, CH3 |
| NH | 8.13, d(8.4) | |
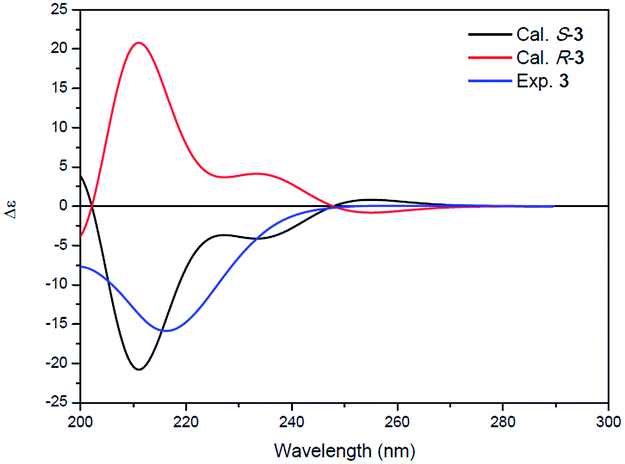 | ||
| Fig. 4 Experimental ECD spectrum of 3 (blue) and the calculated ECD spectra of (R)-3 (red) and (S)-3 (black). | ||
In addition to compounds 1–3, the known pepstatin Ac (4),18,22 pepstatin Pr (5),18,22 pepsinostreptin (6),18 pepstatin Ac methyl ester (7), pepstatin Pr methyl ester (8), and pepsinostreptin methyl ester (9) were also isolated and identified from the strain Streptomyces sp. CPCC 202950. Compounds 4–6 incubated with methanol showed the presence of corresponding methyl esters 7–9, respectively. These results indicated that 7–9 were indeed artifacts derived from methanolysis during the isolation process. Furthermore, a new benzamide analog and seven known compounds were isolated from the inactive fractions of the crude extract in our previous report.23
Pepstatin and ahpatinin derivatives have been shown to exhibit significant inhibitory activity against aspartic protease.18,24–28 HIV-1 PR is a 22 kDa dimeric aspartyl protease. As expected, compounds 1 and 2 along with pepstatin derivatives 4–9 from the active fractions showed significant inhibitory activity against HIV-1 protease with IC50 values of 1.79 nM to 3.19 μM, while compound 3 was inactive (the positive control indinavir gave an IC50 value of 1.82 nM) in preliminary in vitro assays (Table 3). A comparison of the activities of 4–6 and 7–9 demonstrated that the exposed terminal carboxyl group was very important for the HIV-1 protease inhibitory activity. Analysis of the inhibitory abilities of 1 and 4–9 indicated that the Ahmppa unit was better than the statine moiety. Compound 2 showed weaker inhibitory ability than 1 and 4–9, suggesting that the presence of the statine or Ahmppa unit could enhance the HIV-1 protease inhibitory activity. In addition, all of the compounds were not active at toward Hela, HepG2, and U2OS tumor cells at 100 μM in in vitro tests.
| Compd | IC50 (nM) | Compd | IC50 (nM) |
|---|---|---|---|
| 1 | 1.79 | 6 | 9.90 |
| 2 | 3190.0 | 7 | 203.30 |
| 3 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
8 | 138.60 |
| 4 | 92.10 | 9 | 782.10 |
| 5 | 22.27 | Indinavir | 1.82 |
Experimental
General experimental methods
ORs were measured using a Rudolph Research Autopol III automatic polarimeter. UV, CD, and IR spectra were recorded using a Cary 300 spectrometer, a JASCO J-815 CD spectrometer, and a Nicolet 5700 FT-IR spectrometer (FT-IR microscope transmission), respectively. 1H and 13C NMR spectra were obtained at 600 and 150 MHz, respectively, using a Bruker-AVIIIHD-600 spectrometer with solvent peaks used as references. HR-ESIMS data were measured using a Micromass Autospec-Ultima ETOF spectrometer and Thermo LTQ Orbitrap XL mass spectrometer. Column chromatography was performed using silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China) and MCI gel CHP20P (Mitsubishi Chemical Corporation, Tokyo, Japan). HPLC separation was performed using an Agilent 1200 series (quaternary pump, autosampler, diode array detector) with a Shiseido Capcell-Pak C18 MGII column (5 μm, 250 × 10 mm). TLC was performed using glass-precoated silica gel GF254 plates (Qingdao Marine Chemical Inc., Qingdao, China). Cervical cancer (Hela), hepatocellular carcinoma (HepG2), and human osteosarcoma (U2OS) cell lines were obtained from the National Infrastructure of Cell Line Resource (Beijing, China).Microorganism and fermentation
Strain CPCC 202950 was identified as a member of the genus Streptomyces on the basis of 16S rRNA sequence analysis and deposited at the China Pharmaceutical Culture Collection (Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, no. CPCC 202950).The strain was cultured on slants of YM (0.4% yeast extract, 1% malt extract, 0.4% glucose, and 1.2% agar) at 28 °C for 7 d. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions and 5 of these pieces were used to inoculate in three Erlenmeyer flasks (500 ml), each containing 100 ml of media (glucose 0.5%, yeast extract 0.5%, soluble starch 2.0%, soybean meal 1%, peptone 0.5%, beef extract 0.5%, corn steep liquor 0.4%, CaCO3 0.4%, and CoCl2·6H2O 0.002%), and the final pH was adjusted to 7.2. After sterilization, three flasks of the inoculated media were incubated at 28 °C on a rotary shaker at 220 rpm for two days to prepare the seed culture. Spore inoculum was prepared by suspending the seed culture in sterile, distilled H2O to give a final spore/cell suspension of 1 × 106/ml. Fermentation was carried out in 30 Fernbach flasks (500 ml), each containing 80 g of rice. Distilled H2O (120 ml) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 10.0 ml of the spore inoculum and incubated at 28 °C for 30 day.
Extraction and isolation
The fermented material was ultrasonicated with 95% EtOH (2 × 8.0 l × 40 min), and the EtOH extracts were combined and evaporated under reduced pressure to yield an aqueous suspension (2.0 l). The suspension was partitioned with EtOAc (4 × 2.0 l). The EtOAc extract (15.0 g) was chromatographed over a silica column using a gradient elution of increasing MeOH (0–100%) in CH2Cl2 to give 10 fractions (F1–F10). The fraction of F6 was also subjected to silica gel CC using a gradient elution of increasing MeOH (0–100%) in CH2Cl2 to give six parts (F6-1–F6-6). Further purification of F6-1 and F6-3 with reversed-phase semipreparative HPLC (Capcell PAK C18-MGII 5 μm, 10 mm × 250 mm, 1.5 ml min−1, 37% MeCN in 0.1% trifluoroacetic acid) yielded 2 (13.5 mg) and 1 (8.6 mg), respectively. The aqueous phase was chromatographed over MCI gel (CHP20P, 1 l) with successive elution using H2O, 10% EtOH, 20% EtOH, 35% EtOH, 50% EtOH, and 70% EtOH to afford fractions M1–M5. M4 was further separated by reversed-phase semipreparative HPLC (Capcell PAK C18-MGII 5 μm, 10 mm × 250 mm, 1.5 ml min−1, 35% MeCN in 0.1% trifluoroacetic acid) to afford 3 (102.0 mg).Physical–chemical properties of 1–3
:H2O 1:1); UV (MeOH) λmax (logε) 223 (4.44), 275 (3.56) nm; IR νmax 3285, 2961, 2925, 2873, 2851, 1635, 1547, 1515, 1468, 1390, 1249, 1183, 1148, 1099, 1038, 722 cm−1; 1H NMR (DMSO-d6, 600 MHz) data and 13C NMR (DMSO-d6, 150 MHz) data, see Table 1. (+)-HR-ESIMS m/z 736.4474 [M + H]+ (calcd for C37H62N5O10, 736.4439).:H2O 1:1); IR νmax 3285, 3080, 2964, 2874, 1727, 1636, 1545, 1466, 1389, 1284, 1229, 1150, 1098, 1053, 721 cm−1; 1H NMR (DMSO-d6, 600 MHz) data and 13C NMR (DMSO-d6, 150 MHz) data, see Table 1. (−)-HR-ESIMS m/z 513.3275 [M − H]− (calcd for C25H45N4O7, 513.3283).![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ); (3R,4S)-isomer (minor product) δ 0.84 (3H, d, J = 6.6 Hz, ζ-CH3), 0.89 (3H, d, J = 6.6 Hz, ζ′-CH3), 1.27 (1H, m, δ-Ha), 1.40 (1H, m, δ-Hb), 1.64 (1H, m, ε-H), 2.29 (1H, dd, J = 8.4, 15.6 Hz, α-Ha), 2.43 (1H, dd, J = 4.8, 15.6 Hz, α-Hb), 3.13 (1H, m, β-H), 4.11 (1H, m, γ-H), 5.50 (1H, br s, β-OH), 7.74 (2H, br s, γ-NH2), 12.32 (1H, br s, –COO).); (3R,4S) and (3S,4R)-isomer (minor product) δ 2.29 (1H, dd, J = 9.0, 15.6 Hz, α-Ha), 2.54 (1H, dd, J = 3.6, 15.6 Hz, α-Hb), 2.70 (1H, dd, J = 7.8, 14.4 Hz, δ-Ha), 2.80 (1H, dd, J = 6.6, 14.4 Hz, δ-Hb), 3.38 (1H, m, β-H), 3.73 (3H, s, 4-OCH3), 4.10 (1H, m, γ-H), 6.89 (2H, d, J = 8.4 Hz, H-2 and H-6), 7.19 (2H, d, J = 8.4 Hz, H-3 and H-5), 5.56 (1H, br s, β-OH), 7.80 (2H, br s, γ-NH2), 12.31 (1H, br s, –COO).).
); (3R,4S)-isomer (minor product) δ 0.84 (3H, d, J = 6.6 Hz, ζ-CH3), 0.89 (3H, d, J = 6.6 Hz, ζ′-CH3), 1.27 (1H, m, δ-Ha), 1.40 (1H, m, δ-Hb), 1.64 (1H, m, ε-H), 2.29 (1H, dd, J = 8.4, 15.6 Hz, α-Ha), 2.43 (1H, dd, J = 4.8, 15.6 Hz, α-Hb), 3.13 (1H, m, β-H), 4.11 (1H, m, γ-H), 5.50 (1H, br s, β-OH), 7.74 (2H, br s, γ-NH2), 12.32 (1H, br s, –COO).); (3R,4S) and (3S,4R)-isomer (minor product) δ 2.29 (1H, dd, J = 9.0, 15.6 Hz, α-Ha), 2.54 (1H, dd, J = 3.6, 15.6 Hz, α-Hb), 2.70 (1H, dd, J = 7.8, 14.4 Hz, δ-Ha), 2.80 (1H, dd, J = 6.6, 14.4 Hz, δ-Hb), 3.38 (1H, m, β-H), 3.73 (3H, s, 4-OCH3), 4.10 (1H, m, γ-H), 6.89 (2H, d, J = 8.4 Hz, H-2 and H-6), 7.19 (2H, d, J = 8.4 Hz, H-3 and H-5), 5.56 (1H, br s, β-OH), 7.80 (2H, br s, γ-NH2), 12.31 (1H, br s, –COO).).Conclusions
In summary, we have isolated two new linear peptides, ahmpatinin iBu (1) and statinin iBu (2), and a novel pyrrolidine anolog, (−)-(S)-2-[3-(6-methylheptanamido)-2-oxopyrrolidin-1-yl]acetic acid (3), from a Streptomyces sp. strain. The unusual amino acid, 4-amino-3-hydroxy-5-(4-methoxyphenyl)pentanoic acid (Ahmppa) of ahmpatinin iBu was identified for the first time from a natural source, and 3 represents the first reported instance of a natural pyrrolidine possessing a 2-(3-amino-2-oxopyrrolidin-1-yl)acetic acid system. In addition, ahmpatinin iBu (1) displayed significant inhibitory activity against HIV-1 protease.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (NNSFC, 81302675), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-2-002), PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (3332015133), Natural Science Foundation of Beijing Municipality (7144224), and National Infrastructure of Microbial Resources (NIMR 2017-3) is acknowledged.Notes and references
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172–5208 CrossRef CAS PubMed.
- Y. Mehellou and E. D. Clercq, J. Med. Chem., 2010, 53, 521–538 CrossRef CAS PubMed.
- B. Gaucher, M. Rouquayrol, D. Roche, J. Greiner, A. M. Aubertin and P. Vierling, Org. Biomol. Chem., 2004, 2, 345–357 CAS.
- P. Piot, S. S. A. Karim, R. Hecht, H. Legido-Quigley, K. Buse, J. Stover, S. Resch, T. Ryckman, S. MØgedal, M. Dybul, E. Goosby, C. Watts, N. Kilonzo, J. Mcmanus and M. Sidibé, Lancet, 2015, 386, 171–218 CrossRef.
- World Health Organization, Global Health Observatory (GHO) data, http://www.who.int/gho/hiv/en/, accessed Dec 2, 2017.
- Joint United Nations Programme on HIV/AIDS, Latest statistics on the status of the AIDS epidemic, http://www.unaids.org/en/resources/fact-sheet, accessed Dec 2, 2017.
- A. Brik and C. H. Wong, Org. Biomol. Chem., 2003, 1, 5–14 CAS.
- N. E. Kohl, E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. F. Dixon, E. M. Scolnick and I. S. Sigal, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4686–4690 CrossRef CAS.
- A. G. Tomasselli and R. L. Heinrikson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1477, 189–214 CrossRef CAS.
- E. J. Arts and D. J. Hazuda, Cold Spring Harbor Perspect. Med., 2012, 2, a007161 Search PubMed.
- J. Chauhan, S. E. Chen, K. J. Fenstermacher, A. Naser-Tavakolian, T. Reingewertz, R. Salmo, C. Lee, E. Williams, M. Raje, E. Sundberg, J. J. DeStefano, E. Freire and S. Fletcher, Bioorg. Med. Chem., 2015, 23, 7095–7109 CrossRef CAS PubMed.
- HIV Drug Resistance Report 2017, World Health Organization, Geneva, 2017 Search PubMed.
- L. Palmisano and S. Vella, Ann. Ist. Super. Sanita, 2011, 47, 44–48 CAS.
- R. L. Hamers, C. L. Wallis, C. Kityo, M. Siwale, K. Mandaliya, F. Conradie, M. E. Botes, M. Wellington, A. Osibogun, K. C. E. Sigaloff, I. Nankya, R. Schuurman, F. W. Wit, W. S. Stevens, M. van Vugt and T. F. R. de Wit, Lancet Infect. Dis., 2011, 11, 750–759 CrossRef CAS PubMed.
- X. Liu, M. Gan, B. Dong, T. Zhang, Y. Li, Y. Zhang, X. Fan, Y. Wu, S. Bai, M. Chen, L. Yu, P. Tao, W. Jiang and S. Si, Molecules, 2013, 18, 236–243 CrossRef CAS PubMed.
- M. H. Chen, Y. X. Wu, B. Dong, X. Y. Fan, L. Y. Yu, W. Jiang and S. Y. Si, Zhongguo Zhongyao Zazhi, 2015, 40, 1320–1324 CAS.
- S. Hecht, L. Dedkoval, R. Maini, C. S. Roy and R. Paul, WO 2016118877 A1, 2016.
- Y. Sun, K. Takada, Y. Nogi, S. Okada and S. Matsunaga, J. Nat. Prod., 2014, 77, 1749–1752 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- M. H. Chen, Y. X. Wu, Y. N. Xu, P. Liu, L. Y. Yu, B. Hong, W. Jiang and S. Y. Si, J. Asian Nat. Prod. Res., 2015, 17, 676–682 CrossRef CAS PubMed.
- Y. Takemoto, T. Matsumoto, Y. Ito and S. Terashima, Tetrahedron Lett., 1990, 31, 217–218 CrossRef CAS.
- T. Aoyagi, Y. Yagisawa, M. Kumagai, M. Hamada, H. Morishima, T. Takeuchi and H. Umezawa, J. Antibiot., 1973, 26, 539–541 CrossRef CAS PubMed.
- S. S. Chang, M. H. Chen, R. Z. Wang, Y. X. Wu, B. Dong, L. Y. Yu, Z. P. Gao and S. Y. Si, Zhongguo Zhongyao Zazhi, 2017, 42, 2097–2101 Search PubMed.
- F. M. Tumminello, R. J. Bernacki, N. Gebbia and G. Leto, Med. Res. Rev., 1993, 13, 199–208 CrossRef CAS PubMed.
- H. Umezawa, T. Aoyagi, H. Morishima, M. Matsuzaki, M. Hamada and T. Takeuchi, J. Antibiot., 1970, 23, 259–262 CrossRef CAS PubMed.
- S. Kunimoto, T. Aoyagi, H. Morishima, T. Takeuchi and H. Umezawa, J. Antibiot., 1972, 25, 251–255 CrossRef CAS PubMed.
- S. Omura, N. Imamura, K. Kawakita, Y. Mori, Y. Yamazaki, R. Masuma, Y. Takahashi, H. Tanaka, L. Y. Huang and H. B. Woodruff, J. Antibiot., 1986, 39, 1079–1085 CrossRef CAS PubMed.
- T. Sato, M. Shibazaki, H. Yamaguchi, K. Abe, H. Matsumoto and M. Shimizu, J. Antibiot., 1994, 47, 588–590 CrossRef CAS PubMed.
- K. X. Zhan, W. H. Jiao, F. Yang, J. Li, S. P. Wang, Y. S. Li, B. N. Han and H. W. Lin, J. Nat. Prod., 2014, 77, 2678–2684 CrossRef CAS PubMed.
- Y. Song, Q. Li, X. Liu, Y. Chen, Y. Zhang, A. Sun, W. Zhang, J. Zhang and J. Ju, J. Nat. Prod., 2014, 77, 1937–1941 CrossRef CAS PubMed.
- Spartan 10, Wavefunction, Inc., Irvine, CA Search PubMed.
- M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Y. Nakajima, O. Honda, H. Kitao, T. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, Gausssum 2.25, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- M. Chen, Y. Wu, Y. He, Y. Xu, Y. Li, D. Li, T. Feng, L. Yu, B. Hong, W. Jiang and S. Si, Bioorg. Med. Chem. Lett., 2015, 25, 562–565 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D, 2D NMR, HRESIMS and IR spectra for compounds 1–3. See DOI: 10.1039/c7ra13241g |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |