
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of triblock copolymer polydopamine-polyacrylic-polyoxyethylene with excellent performance as a binder for silicon anode lithium-ion batteries
Lei Lüab,
Hongming Loua,
Yinglin Xiaob,
Guangzhao Zhang c,
Chaoyang Wang*c and
Yonghong Deng*b
c,
Chaoyang Wang*c and
Yonghong Deng*b
aSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou, China 516640. E-mail: cehmlou@scut.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, China 518055. E-mail: yhdeng08@163.com
cResearch Institute of Materials Science, South China University of Technology Guangzhou, China 510640
First published on 25th January 2018
Abstract
Triblock copolymer polydopamine-polyacrylic-polyoxyethylene (PDA-PAA-PEO) with excellent performance as a binder for silicon anodes was synthesized. Its structure was confirmed by 1H-NMR, FTIR and UV-vis spectroscopy. Results of electrochemical measurements indicated that a silicon anode based on PDA-PAA-PEO binder exhibited excellent cycle performance with a reversible capacity of 1597 mA h g−1 after 200 cycles at a current density of 0.5 C, much better than that of an electrode based on a polyvinylidene-fluoride binder. Improvement of the cycle performance and reversible capacity for silicon anodes could be attributed to the strong adhesive and high ion conductivity of PDA-PAA-PEO.
Introduction
Lithium-ion batteries (LIBs) are very promising electrochemical energy-storage devices.1 As an anode material for LIBs, silicon (Si) has been considered to be an intriguing candidate for next-generation, high-energy-density anode materials for LIBs due to its high theoretical capacity (4200 mA h g−1) and natural abundance.2,3 However, the drastic volume changes of Si during charging–discharging result in pulverization and rapid capacity fade of Si-based electrodes, which hinders its practical application.4 Numerous efforts have been proposed to address this problem, including nanostructuring of Si, formation of Si-based alloys and design of alternative binder polymers.5–9 Extensive research has demonstrated that the binder plays an important part in maintaining electrode integrity, which affects the electrochemical performance of Si-based electrodes directly.10–12 Polyvinylidene fluoride (PVDF), as a commercial binder for graphite anodes, cannot accommodate the drastic volume expansion of Si anodes effectively due to the weak van der Waals' force between PVDF and active particles. Furthermore, poisonous N-methyl pyrrolidines (NMPs), which are expensive, are usually employed as the solvent for PVDF binders, which has limited their application.13,14 Studies have shown that carboxyl-containing polymers such as carboxymethyl cellulose (CMC) and poly(acrylic acid) (PAA) are excellent binders owing to the formation of hydrogen bonds or covalent bonds between the carboxyl groups of binders and hydroxyl groups on the Si surface, which greatly improves the cycle stability of Si-based electrodes.15 Extensive research has focused on the modification of carboxyl-containing binders such as PAA-PBI,16 PAA-PVA,17 c-PAA-CMC.18Recently, mussels have attracted much attention due to their extraordinary adhesion to almost all types of surfaces, which has been attributed to a catechol functional group.19–21 Research suggests that catechol-modified polymers with strong adhesion possess could be applied extensively.21–25 Poly(ethylene oxide) (PEO) is a well-known as solid-state electrolyte in LIBs owing to its high conduction of lithium ions, which is ascribed to the lone-pair electrons on the oxygen of the ether groups.26,27 PEO-based polymers have been applied as cathode binders to enhance the transport of lithium ions in active materials.28–30 Besides, the ether moiety can increase the interface adhesion and ductility of binder polymers.31–34
Herein, we prepared a triblock copolymer containing the characteristics of ethylene oxide moieties as well as carboxyl and catechol groups, and used it as binder for Si anodes in LIBs. Compared with the Si/PVDF electrode, the Si/PDA-PAA-PEO electrode exhibited more stable cyclic performance and lower electrochemical impedance due to the strong adhesion and high ionic conductivity of PDA-PAA-PEO. Moreover, our results showed that incorporation of catechol groups in the binders could improve the capacities of the electrodes. Furthermore, compared with oil-solvent binding of PVDF, water-solvent PDA-PAA-PEO had much better application prospects because it is environmentally friendly.
Experimental
Materials
2-Methyl-2-[(dodecylsulfanylthiocarbonyl) sulfanyl] propanoic acid (CTA) and the macro-RAFT agent CTA-PEO-CTA were synthesized following a procedure reported previously.35,36 Poly(ethylene oxide) (molecular weight = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), trifluoroacetic acid (TFA), EDC·3HCl and dopamine hydrochloride were purchased from Aladdin. Dichloromethane (DCM), diethyl ether, 1,4-dioxane were obtained from Shanghai Lingfeng Chemicals. tert-Butyl acrylate (AR; Aladdin) was purified through chromatography by flow through an alkaline aluminum oxide column to remove inhibitors. Azodiisobutyronitrile (99%; Aladdin) was recrystallized twice from methanol. Phosphate-buffered saline (PBS) was obtained by mixing potassium dihydrogen phosphate (KH2PO4) with disodium hydrogen phosphate (Na2HPO4). Si nanopowder (50 nm, 99.9%), super P and other battery accessories were purchased from Shenzhen Kejing. Electrolytes were obtained from Capchem.
000), trifluoroacetic acid (TFA), EDC·3HCl and dopamine hydrochloride were purchased from Aladdin. Dichloromethane (DCM), diethyl ether, 1,4-dioxane were obtained from Shanghai Lingfeng Chemicals. tert-Butyl acrylate (AR; Aladdin) was purified through chromatography by flow through an alkaline aluminum oxide column to remove inhibitors. Azodiisobutyronitrile (99%; Aladdin) was recrystallized twice from methanol. Phosphate-buffered saline (PBS) was obtained by mixing potassium dihydrogen phosphate (KH2PO4) with disodium hydrogen phosphate (Na2HPO4). Si nanopowder (50 nm, 99.9%), super P and other battery accessories were purchased from Shenzhen Kejing. Electrolytes were obtained from Capchem.
Preparation of the triblock copolymer binder PDA-PAA-PEO
Characterization and measurements
:20:20 were mixed and stirred for 12 h. Then, the slurries were cast onto copper foils. After the cast films had been dried in a vacuum oven for 12 h at 120 °C, they were cut into wafers of diameter 1.2 cm and then dried under vacuum for 12 h at 120 °C. The mass loading of all active materials was ≈0.4 mg cm−2. 2025-type coin cells were assembled in an argon-filled glove box using lithium foil as counter electrode and Celgard 2400 as a separator. The electrolyte consisted of a solution of 1 M of LiPF6 in a mixture of ethylene carbonate, diethyl carbonate and dimethyl carbonate (EC:DEC:DMC, 1:1:1 by volume). The cycle performance was tested on a LAND battery test system (Wuhan) at 0.01–1 V. Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were employed on an electrochemical workstation (Solartron Analytical).Results and discussion
PDA-PAA-PEO was obtained by a three-step method. First, PtBA-PEO-PtBA was synthesized through RAFT polymerization using tBA as a monomer and CTA-PEO-CTA as a chain-transfer agent. Second, PAA-PEO-PAA was obtained by the hydrolysis of PtBA-PEO-PtBA with excess TFA. Third, –COOH groups in PAA-PEO-PAA reacted with primary amines in dopamine through coupling reactions with EDC·3HCl as a catalytic agent, and PDA-PAA-PEO was obtained (Fig. 1).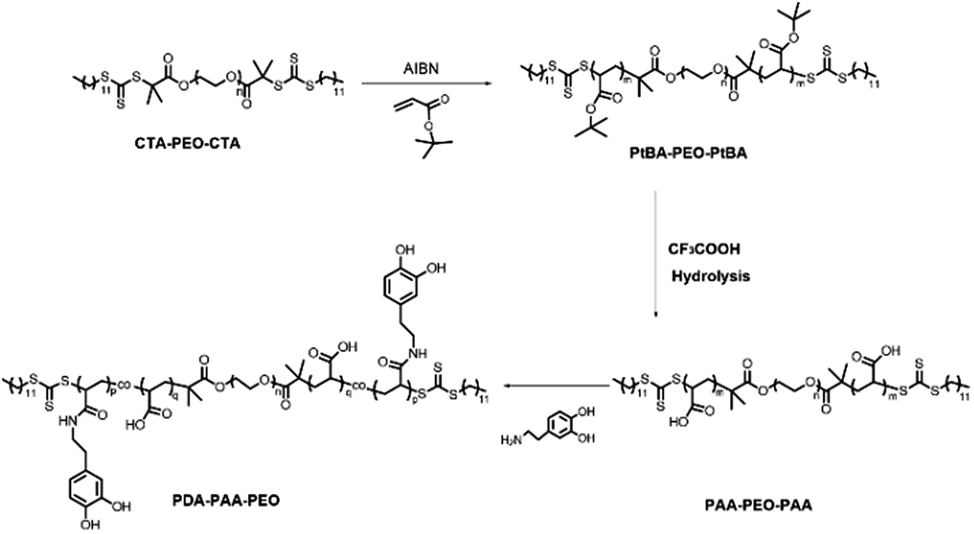 | ||
| Fig. 1 Synthesis of PDA-PAA-PEO triblock copolymers. | ||
The structures of PtBA-PEO-PtBA and PAA-PEO-PAA were confirmed by the 1H-NMR spectra (Fig. 2a). The 1H-NMR spectra of CTA-PEO-CTA showed a singlet at ≈3.6 ppm assigned to –CH2CH2O– in PEO blocks. The singlet at ≈1.45 ppm corresponding to tert-butyl groups confirmed the structure of PtBA-PEO-PtBA. The singlet at ≈1.45 ppm disappeared when PtBA-PEO-PtBA was hydrolyzed to PAA-PEO-PAA. The spectra of PAA-PEO-PAA showed a wide peak at ≈12 ppm that was assigned to –COOH. The structure of PDA-PAA-PEO was verified by a UV spectrum (Fig. 2b). The absorption band at 280 nm was related to the catechol group. According to the standard curve of dopamine (inset in Fig. 2b), the content of dopamine in polymers was ≈13.6%, which corresponded to the results of elemental analysis shown in Table 1. According to GPC, the weight-average molecular weight of PDA-PAA-PEO was 70795 g mol−1.
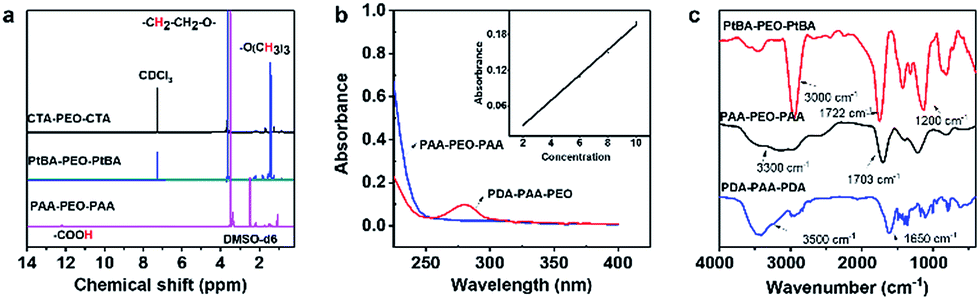 | ||
| Fig. 2 Structural characteristics of PtBA-PEO-PtBA, PAA-PEO-PAA and PDA-PAA-PEO. (a) 1H-NMR spectra (b) UV spectrum and (c) FTIR spectra. | ||
| N% | C% | H% | S% | |
|---|---|---|---|---|
| PtBA-PEO-PtBA | 0 | 64.76 | 10.373 | 0 |
| PAA-PEO-PAA | 0 | 47.48 | 6.874 | 0.284 |
| PDA-AA-EO | 1.31 | 40.76 | 6.08 | 0 |
All of these structures were verified further by FTIR spectroscopy (Fig. 2c). In the spectra of PtBA-PEO-PtBA, a pointed peak at ≈3000 cm−1 was observed, which was assigned to the stretching vibrations of C–H in PtBA blocks. The peaks at 1722 cm−1 and 1200 cm−1 were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of ester groups (COOR) and C–O–C stretching of ethylene oxide moieties, respectively. FTIR spectroscopy of PAA-PEO-PAA showed a shift in the position of the CO stretching band from 1722 cm−1 to 1703 cm−1 due to the hydrolysis of COOR to COOH, and the pointed peak at ≈3000 cm−1 disappeared, which signified the loss of a tert-butyl group. A new broad peak at 3300 cm−1 was assigned to the stretching vibrations of O–H, which confirmed the existence of –COOH. In the spectra of PDA-PAA-PEO, the peaks at ≈1650 cm−1 and ≈3500 cm−1 were assigned to the CO stretching band and N–H stretching band, respectively, which indicated that the amino groups of dopamine had reacted with the carboxyl group, and that amide groups (CONH) were obtained.
O stretching of ester groups (COOR) and C–O–C stretching of ethylene oxide moieties, respectively. FTIR spectroscopy of PAA-PEO-PAA showed a shift in the position of the CO stretching band from 1722 cm−1 to 1703 cm−1 due to the hydrolysis of COOR to COOH, and the pointed peak at ≈3000 cm−1 disappeared, which signified the loss of a tert-butyl group. A new broad peak at 3300 cm−1 was assigned to the stretching vibrations of O–H, which confirmed the existence of –COOH. In the spectra of PDA-PAA-PEO, the peaks at ≈1650 cm−1 and ≈3500 cm−1 were assigned to the CO stretching band and N–H stretching band, respectively, which indicated that the amino groups of dopamine had reacted with the carboxyl group, and that amide groups (CONH) were obtained.
These characteristic peaks demonstrated that PDA-PAA-PEO had been synthesized.
We conducted a series of electrochemical measurements to investigate the performance of PDA-PAA-PEO as a binder for Si anodes in LIBs using PAA-PEO-PAA and PVDF binders as reference. CV curves were measured in a potential range between 0.01 V and 2.5 V at a scan rate of 0.05 mV s−1 for three cycles (Fig. 3a–c). In the first cycle, a cathodic peak at 1.5 V may have corresponded to formation of a solid electrolyte interphase, which disappeared at the second and third cycles, demonstrating no formation of a new solid electrolyte interphase (SEI) layer on Si surfaces.37,38 For the CV curve of Si/PDA-PAA-PEO (Fig. 3a), a cathodic peak at 0.1 V was assigned to the formation of LixSi in the first discharge process; the corresponding charge curve showed peaks at 0.35 V and 0.5 V assigned to the delithiation of LixSi.39 The second and third cycles showed the same cathodic and anodic peaks, implying that the lithiation and delithiation reactions were highly reversible.40,41 A cathodic peak at 2.0 V could be ascribed to the electron transfer between carbonyl groups and lithium ions, a finding that is similar to previous reports.42 Fig. 3b shows the similar CV curve except that the anodic peak at 0.35 V is inconspicuous. For the CV curve of Si/PVDF, the anodic peak at 0.35 V disappeared, which indicated that the lithiation and delithiation reactions were irreversible. For all of the electrodes, the peaks at 0.25 V which appeared at the third cycle were assigned to a crystal-to-amorphous transition.43
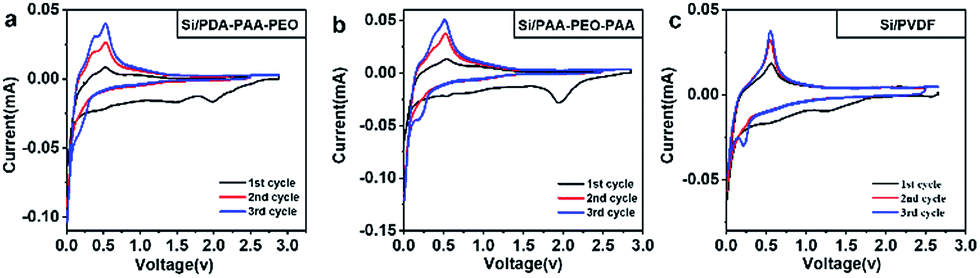 | ||
| Fig. 3 Cyclic voltammograms of Si electrodes made with PDA-PAA-PEO (a), PAA-PEO-PAA (b), PVDF (c) binders. | ||
Fig. 4a shows the first discharge–charge profiles of Si electrodes using PVDF, PAA-PEO-PAA and PDA-PAA-PEO as binders at 0.01–1.0 V at a current density of 0.05 C. The discharge curve displayed a plateau profile at 0.1–0.01 V, consistent with the behavior of lithium-ion insertion.44 The initial discharge capacities of Si/PDA-PAA-PEO and Si/PAA-PEO-PAA were 2983 and 3344 mA h g−1 with coulombic efficiencies (CEs) of 74% and 80% (Table 2), respectively; however, the Si/PVDF electrode delivered a discharge capacity of 2777 mA h g−1 with a CE of 68%. Moreover, the CEs of Si/PDA-PAA-PEO and Si/PAA-PEO-PAA electrodes increased to 98% at the fifth cycle, but the Si/PVDF electrode showed a CE of 92% at the fifth cycle (Fig. 4b). The higher CEs of Si/PDA-PAA-PEO and Si/PAA-PEO-PAA electrodes could be ascribed to the formation of a relatively stable SEI layer on the Si surface at the first lithium insertion/extraction.45 Fig. 4c shows the cyclic charge–discharge curves of Si/PVDF, Si/PAA-PEO-PAA and Si/PDA-PAA-PEO at a current density of 0.5 C. The Si/PDA-PAA-PEO electrode exhibited quite a high capacity of 1597 mA h g−1 after 200 cycles, which was higher than that of the Si/PAA-PEO-PAA electrode with a capacity of 1205 mA h g−1 over 200 cycles, and the Si/PVDF electrode had a capacity of 40 mA h g−1 after 50 cycles. These results suggested that PDA-PAA-PEO could improve the electrochemical performance of the electrode effectively. The improvement of electrochemical performance could be attributed to the intensive adhesive cohesive force of PDA-PAA-PEO due to the strong interaction between functional groups (carboxyl and catechol) and hydroxyl groups on the SI surface.46–48 Furthermore, the higher ion conductivity of PEO blocks was beneficial for decreasing the interfacial charge transfer impedance of electrodes, which resulted in the high capacity of Si/PDA-PAA-PEO.49,50
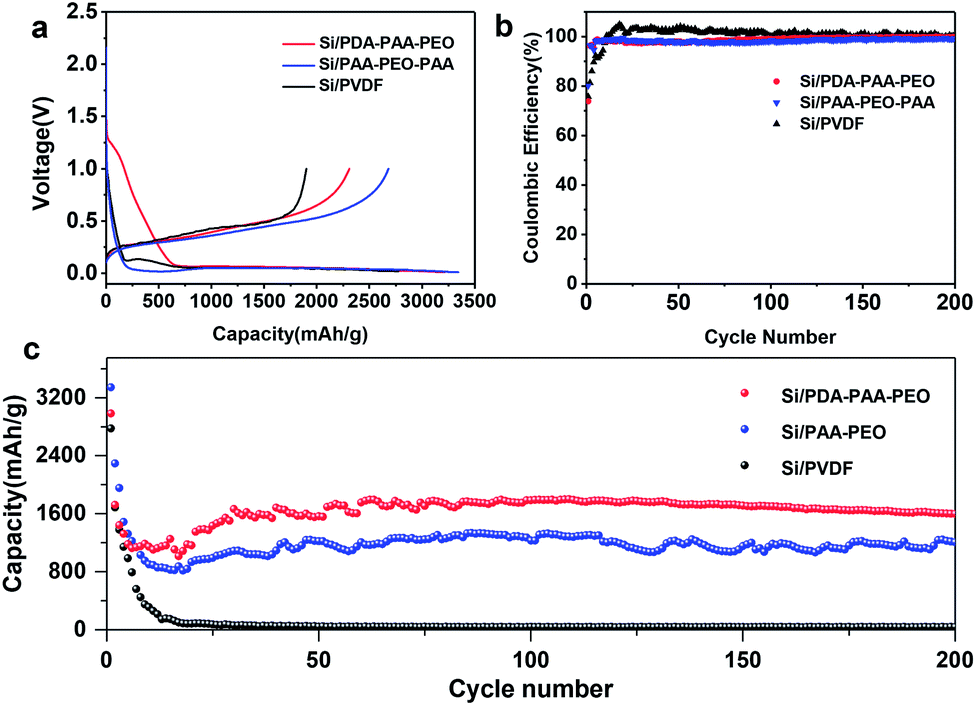 | ||
| Fig. 4 Electrochemical properties of Si electrodes made with PDA-PAA-PEO, PAA-PEO-PAA and PVDF. | ||
| Si/PVDF | Si/PAA-PEO-PAA | Si/PDA-PAA-PEO | |
|---|---|---|---|
| Discharge capacity at 1st cycle (mA h g−1) | 2777 | 3344 | 2983 |
| Discharge capacity at 200th cycle (mA h g−1) | 37 | 1205 | 1597 |
| Initial coulombic efficiency (%) | 68 | 80 | 74 |
To investigate further the electrochemical characteristics of binders, EIS was done between 10 kHz and 10 MHz. Fig. 5 reveals the Nyquist plots of Si nanoparticle electrodes with the three binders after three discharge–charge cycles at a current density of 0.05 C. The diameters of the semicircles in high-frequency and medium-frequency ranges reflect the SEI resistance and interfacial charge transfer impedance, respectively. The slope in the low-frequency range represents the ionic diffusion resistance.40,51 Notably, PVDF showed a relatively high SEI resistance (130 Ω) according to the fitted semicircles, and PDA-PAA-PEO with the lowest resistance (50 Ω). These results substantiate the notion that PDA-PAA-PEO can decrease the interfacial charge transfer impedance of electrodes due to the higher ion conductivity of PEO blocks.51 Moreover, studies have shown that binders have important roles in connecting whole complex electrodes and maintaining the integrity of conductive networks, all of which can decrease the interfacial charge transfer impedance of electrodes effectively.52 The 180° peeling test suggested that PDA-PAA-PEO with a strong cohesion force (Fig. 6) could decrease the interfacial charge transfer impedance of electrodes further.
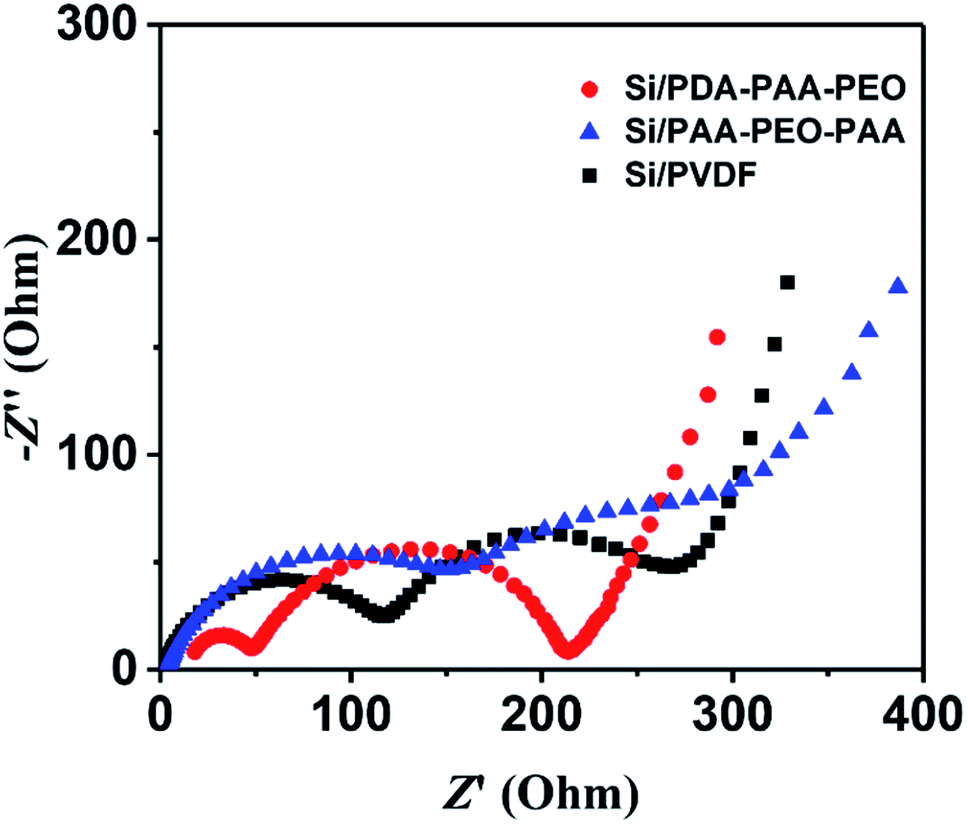 | ||
| Fig. 5 Nyquist plots of Si nanoparticle electrodes with PDA-PAA-PEO, PAA-PEO-PAA and PVDF. | ||
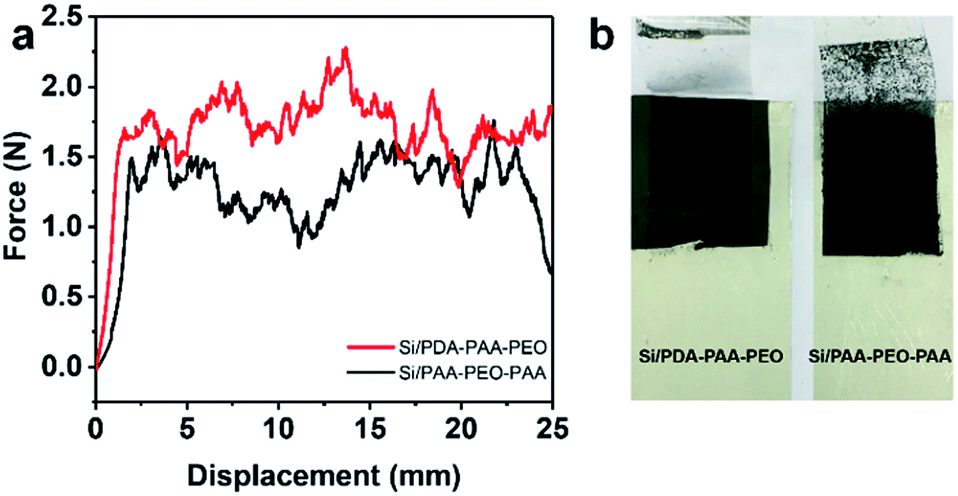 | ||
| Fig. 6 Peeling test of Si/PDA-PAA-PEO and Si/PAA-PEO-PAA laminates: (a) force–displacement curves. (b) Digital photograph of laminates after the peeling test. | ||
Fig. 6a shows the force–displacement curves of the electrodes with PDA-PAA-PEO and PAA-PEO-PAA. We did not need to conduct an adhesion test for PVDF considering its poor electrochemical behavior and reported weak bonding force.52 The average force of PDA-PAA-PEO was 1.76 N, much higher than that for PAA-PEO-PAA (1.31 N). Moreover, it is clearly observed from Fig. 6b that many particles were detached from Si/PAA-PEO-PAA laminates. These results suggest that PDA-PAA-PEO binder could allow the cohesion of Si particles to the Cu current collector. The enhanced adhesive strength for PDA-PAA-PEO could be attributed to the strong interaction between functional groups (carboxyl and catechol) and hydroxyl groups on the Si surface. This is beneficial for decreasing the interfacial charge transfer impedance of electrodes and improves the electrochemical performance of the electrode. The results of the adhesion test were in agreement with that of the charge–discharge test (Fig. 4c). The Si/PDA-PAA-PEO electrodes exhibited ≈400 mA h g−1 larger capacities compared with that of Si/PDA-PAA-PEO, which was attributed to the enhanced adhesion by the catecholic interactions.
Conclusions
Triblock copolymer PDA-PAA-PEO containing the characteristics of ethylene oxide moieties as well as carboxyl and catechol groups was obtained via RAFT polymerization and a coupling reaction. The Si/PDA-PAA-PEO electrode exhibited excellent electrochemical performance with a quite high capacity of 1597 mA h g−1 after 200 cycles. The improvement of electrochemical performance could be attributed to the intensive adhesive cohesive force of PDA-PAA-PEO and higher ion conductivity of PEO blocks. Our research offers an environmentally friendly binder with excellent application prospects for the next generation of LIBs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for financial support from the Natural Science Foundation of Guangdong Province, China (2016A030311031) and Fundamental Research Foundation of Shenzhen (20170410160631170). The authors also acknowledge support from the members of the Advanced Polymeric Energy Materials Laboratory and Department of Materials Science and Engineering of South University of Science and Technology of China.Notes and references
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- H. Tao, L. Z. Fan, W. L. Song, M. Wu, X. He and X. Qu, Nanoscale, 2014, 6, 3138–3142 RSC.
- M.-S. Wang, W.-L. Song, J. Wang and L.-Z. Fan, Carbon, 2015, 82, 337–345 CrossRef CAS.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87–90 CrossRef CAS.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo and H.-K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed.
- M.-S. Wang, W.-L. Song and L.-Z. Fan, J. Mater. Chem. A, 2015, 3, 12709–12717 CAS.
- M.-S. Wang, W.-L. Song and L.-Z. Fan, ChemElectroChem, 2015, 2, 1699–1706 CrossRef CAS.
- M.-S. Wang, Y. Song, W.-L. Song and L.-Z. Fan, ChemElectroChem, 2014, 1, 2124–2130 CrossRef CAS.
- S. L. Chou, Y. Pan, J. Z. Wang, H. K. Liu and S. X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 RSC.
- G. Zhang, Y. Chen, Y. Deng, T. Ngai and C. Wang, ACS Macro Lett., 2017, 6, 641–646 CrossRef CAS.
- D. Mazouzi, Z. Karkar, C. Reale Hernandez, P. Jimenez Manero, D. Guyomard, L. Roué and B. Lestriez, J. Power Sources, 2015, 280, 533–549 CrossRef CAS.
- M. A. Spreafico, P. Cojocaru, L. Magagnin, F. Triulzi and M. Apostolo, Ind. Eng. Chem. Res., 2014, 53, 9094–9100 CrossRef CAS.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CAS.
- S. Lim, H. Chu, K. Lee, T. Yim, Y. J. Kim, J. Mun and T. H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 23545–23553 CAS.
- J. Song, M. Zhou, R. Yi, T. Xu, M. L. Gordin, D. Tang, Z. Yu, M. Regula and D. Wang, Adv. Funct. Mater., 2014, 24, 5904–5910 CrossRef CAS.
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N. S. Choi and J. Cho, Angew. Chem., Int. Ed. Engl., 2012, 51, 8762–8767 CrossRef CAS PubMed.
- Q. Lin, D. Gourdon, C. Sun, N. Holten-Andersen, T. H. Anderson, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3782–3786 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- S. Moulay, Polym. Rev., 2014, 54, 436–513 CrossRef CAS.
- Y. K. Jeong, S. H. Park and J. W. Choi, ACS Appl. Mater. Interfaces, 2017 DOI:10.1021/acsami.7b08495.
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L.-T. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372 CrossRef CAS.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CAS.
- M. H. Ryou, J. Kim, I. Lee, S. Kim, Y. K. Jeong, S. Hong, J. H. Ryu, T. S. Kim, J. K. Park, H. Lee and J. W. Choi, Adv. Mater., 2013, 25, 1571–1576 CrossRef CAS PubMed.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- R. Kido, K. Ueno, K. Iwata, Y. Kitazawa, S. Imaizumi, T. Mandai, K. Dokko and M. Watanabe, Electrochim. Acta, 2015, 175, 5–12 CrossRef CAS.
- C.-H. Tsao, C.-H. Hsu and P.-L. Kuo, Electrochim. Acta, 2016, 196, 41–47 CrossRef CAS.
- T. Nakazawa, A. Ikoma, R. Kido, K. Ueno, K. Dokko and M. Watanabe, J. Power Sources, 2016, 307, 746–752 CrossRef CAS.
- Y. H. Kwon, K. Minnici, M. M. Huie, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Reichmanis, Chem. Mater., 2016, 28, 6689–6697 CrossRef CAS.
- M. Wu, X. Xiao, N. Vukmirovic, S. Xun, P. K. Das, X. Song, P. Olalde-Velasco, D. Wang, A. Z. Weber, L. W. Wang, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2013, 135, 12048–12056 CrossRef CAS PubMed.
- S. J. Park, H. Zhao, G. Ai, C. Wang, X. Song, N. Yuca, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2015, 137, 2565–2571 CrossRef CAS PubMed.
- N. Yuca, H. Zhao, X. Song, M. F. Dogdu, W. Yuan, Y. Fu, V. S. Battaglia, X. Xiao and G. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 17111–17118 CAS.
- G. Zhang, Y. Chen, Y. Deng and C. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36301–36310 CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- Y. He and T. P. Lodge, Macromolecules, 2008, 41, 167–174 CrossRef CAS.
- J. H. Ryu, J. W. Kim, Y.-E. Sung and S. M. Oh, Electrochem. Solid-State Lett., 2004, 7, A306–A309 CrossRef CAS.
- Y. Jin, S. Li, A. Kushima, X. Zheng, Y. Sun, J. Xie, J. Sun, W. Xue, G. Zhou, J. Wu, F. Shi, R. Zhang, Z. Zhu, K. So, Y. Cui and J. Li, Energy Environ. Sci., 2017, 10, 580–592 CAS.
- B. Key, M. Morcrette, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2010, 133, 503–512 CrossRef PubMed.
- N. Yuca and Ü. Çolak, Electrochim. Acta, 2016, 222, 1538–1544 CrossRef CAS.
- J. Yoon, D. X. Oh, C. Jo, J. Lee and D. S. Hwang, Phys. Chem. Chem. Phys., 2014, 16, 25628–25635 RSC.
- K. Liu, J. Zheng, G. Zhong and Y. Yang, J. Mater. Chem., 2011, 21, 4125 RSC.
- M. Ge, J. Rong, X. Fang and C. Zhou, Nano Lett., 2012, 12, 2318–2323 CrossRef CAS PubMed.
- J. Liu, Q. Zhang, T. Zhang, J.-T. Li, L. Huang and S.-G. Sun, Adv. Funct. Mater., 2015, 25, 3599–3605 CrossRef CAS.
- S. P. V. Nadimpalli, V. A. Sethuraman, S. Dalavi, B. Lucht, M. J. Chon, V. B. Shenoy and P. R. Guduru, J. Power Sources, 2012, 215, 145–151 CrossRef CAS.
- J. I. Lee, H. Kang, K. H. Park, M. Shin, D. Hong, H. J. Cho, N. R. Kang, J. Lee, S. M. Lee, J. Y. Kim, C. K. Kim, H. Park, N. S. Choi, S. Park and C. Yang, Small, 2016, 12, 3119–3127 CrossRef CAS PubMed.
- H.-K. Park, B.-S. Kong and E.-S. Oh, Electrochem. Commun., 2011, 13, 1051–1053 CrossRef CAS.
- Y. Shi, X. Zhou and G. Yu, Acc. Chem. Res., 2017, 50, 2642–2652 CrossRef CAS PubMed.
- S. Lim, K. Lee, I. Shin, A. Tron, J. Mun, T. Yim and T.-H. Kim, J. Power Sources, 2017, 360, 585–592 CrossRef CAS.
- M. Ling, J. Qiu, S. Li, C. Yan, M. J. Kiefel, G. Liu and S. Zhang, Nano Lett., 2015, 15, 4440–4447 CrossRef CAS PubMed.
- J. Guo, A. Sun, X. Chen, C. Wang and A. Manivannan, Electrochim. Acta, 2011, 56, 3981–3987 CrossRef CAS.
- L. Zhang, L. Zhang, L. Chai, P. Xue, W. Hao and H. Zheng, J. Mater. Chem. A, 2014, 2, 19036–19045 CAS.
| This journal is © The Royal Society of Chemistry 2018 |