
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal coordination in the high-temperature leaching of roasted NdFeB magnets with the ionic liquid betainium bis(trifluoromethylsulfonyl)imide†
Martina Orefice ,
Koen Binnemans* and
Tom Vander Hoogerstraete
,
Koen Binnemans* and
Tom Vander Hoogerstraete
KU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. Box 2404, B-3001 Heverlee, Belgium. E-mail: koen.binnemans@kuleuven.be
First published on 5th March 2018
Abstract
Ionic liquids are largely used to leach metals from primary (ores) and secondary sources (end-of-life products). However, dry ionic liquids with a carboxylic function on the cation have not yet been used to leach metals at temperature above 100 °C and under atmospheric pressure. The ionic liquid betainium bis(trifluoromethylsulfonyl)imide, [Hbet][Tf2N], was used in the dry state to recover neodymium, dysprosium and cobalt from NdFeB magnets and NdFeB production scrap. The magnets and the scrap were crushed, milled and roasted before being leached above 100 °C. Recovery efficiencies below 10% and a lack of selectivity for all the parameters tested pointed to the importance of water in the dissolution process. The influence of the viscosity of the ionic liquid and the composition of the metal oxides after roasting was investigated as well. Although the dissolution of pure metal oxides was faster than the dissolution of the magnets, the low leaching efficiencies could not be attributed to the composition and crystal structure of the samples, since magnets roasted with the same protocol have already been successfully leached in the past, albeit in the presence of water. The role of water on the mass transfer and on the coordination of the metals was studied by viscometry and by spectroscopic methods, respectively. It is shown that for leaching of rare earths with [Hbet][Tf2N], the presence of ligands such as water is mandatory to saturate the first coordination sphere of the dissolved rare-earth ions. This paper provides new insights in the dissolution mechanism of metal oxides by [Hbet][Tf2N] at leaching temperatures higher than those typically used in hydrometallurgical leaching processes.
Introduction
Manufacturers of high-tech devices strongly rely on metal supply and availability. The supply also has a strong effect on the materials' price. In the last report (2017), the European Union confirmed the criticality of the rare-earth elements (REEs) because of their high demand and supply risk.1 Among many other applications, rare earths are mainly used in neodymium–iron–boron (NdFeB) permanent magnets, which also contain dysprosium (to improve the thermal stability), cobalt (to increase the coercivity) and praseodymium (as an impurity of neodymium), in addition to neodymium in the alloy with iron. NdFeB permanent magnets are present in the motors of electric cars and electric bikes, computer hard-disk drives, wind turbines and scanners for magnetic resonance imaging (MRI).2 Rare-earth resources are limited and three options are proposed to mitigate their supply risk in Europe: (1) opening new mines, with the most promising project taking place in Greenland; (2) substitution of rare-earths by less critical metals; or (3) recycling and/or recovering of rare earths from end-of-life products and industrial waste streams.3,4 The recovery of metals from urban or industrial waste is crucial when one wants to move from the consumption economy towards the circular economy.5,6 Recycling technologies are traditionally classified as hydro-, pyro- and electrometallurgy. Hydrometallurgy refers to the group of processes based on the use of aqueous solutions to extract metals from solid species. Pyrometallurgical processes use high temperatures to melt and transform the solids of interest. In electrometallurgy, a series of electrical and electrolytic techniques are employed to extract metals from solutions.7,8 However, a combination of these three groups rather than the implementation of only one of them is frequently used. In this classification, a new branch and not much yet explored metallurgical field was recently added: solvometallurgy.9 The prefix “solvo” indicates solvent and refers to a pool of techniques based on non-aqueous solvents. The even more specific term “ionometallurgy” is adopted in case the non-aqueous solvent is an ionic liquid (IL) or a deep-eutectic solvent (DES).10–15 Ionic liquids are solvents that consist entirely of ions and usually melt below 100 °C. Ionic liquids are mainly implemented in processes between room temperature (RT) and 100 °C.16–21 Ionic liquids have a low volatility and a low flammability. Very few studies have reported on the dissolution of metals or metal oxides in ionic liquids above 100 °C. Davris et al. carried out studies on the use of the ionic liquids [EMIM][HSO4] and [Hbet][Tf2N] to leach rare earths from bauxite residues.22–26 In particular for the [Hbet][Tf2N] they worked at temperatures between 60 °C and 180 °C in an autoclave reactor. They investigated, among the other parameters, the role of water on the leaching both for solid waste and for natural ore, and they found an optimum at 40% v/v content of water in [Hbet][Tf2N]. They also found that the used mixture is selective against Fe, as Dupont and Binnemans reported as well.17 Molten pyridinium chloride (m. p. = 115 °C) was used to dissolve metals, metal oxides and metal salts in the works of Audrieth and Edwards (1936),27 Starke (1950)28 and Hollebone and Wiles (1967).29 Metals and metal oxides dissolved faster in molten pyridinium chloride than they did in metal salts. However, it must be pointed out that all these authors did not report detailed quantitative analysis of their tests. Hollebone and Wiles observed that grinding the mineral has a positive influence on the dissolution efficiency, while Audrieth and Edwards observed the crucial role of water on the dissolution of oxides in molten “onium” salts.27 According to Starke, the high dissociating power of water, which is often introduced in combination with the solvent, is a driving force for the dissolution of the material but it can lead to undesired dissociation reactions involving the solvent.28 Molecular compounds, such as pyridinium chloride, offer an alternative to mineral acids and do not introduce water as diluent in the system. In fact, Hollebone and Wiles commented that the reactions of inorganic compounds in HCl in pyridine are very similar to those of HCl in aqueous solutions.29In this paper, the ionic liquid betainium bis(trifluoromethylsulfonyl)imide, [Hbet][Tf2N], without addition of water is used to leach NdFeB magnets and NdFeB production scrap at atmospheric pressure at a temperature of 175 °C. Exclusion of water has the advantage of allowing to work at temperatures above 100 °C without the use of an autoclave and consequently to increase the reaction kinetics. Dupont and Binnemans (2015) were able to dissolve NdFeB magnets in water-saturated [Hbet][Tf2N] and to selectively separate Nd and Dy from Fe and Co, but the dissolution process was slow.17 At the same time, they showed that the leaching process was efficient only in the presence of water. In the present paper, specific attention is paid to the leaching mechanism and the coordination chemistry of the metals in dry [Hbet][Tf2N]. The role of water or coordinating anions such as chloride in the dissolution process is highlighted. Although a dry system was developed, the presence of water could not be excluded a priori, since water is produced by the reaction between the metal oxide and [Hbet][Tf2N]. The water produced might not evaporate if it coordinates to the metal:
| M2O3 + 6[Hbet][Tf2N] → 2[M(bet)3][Tf2N]3 + 3H2O | (1) |
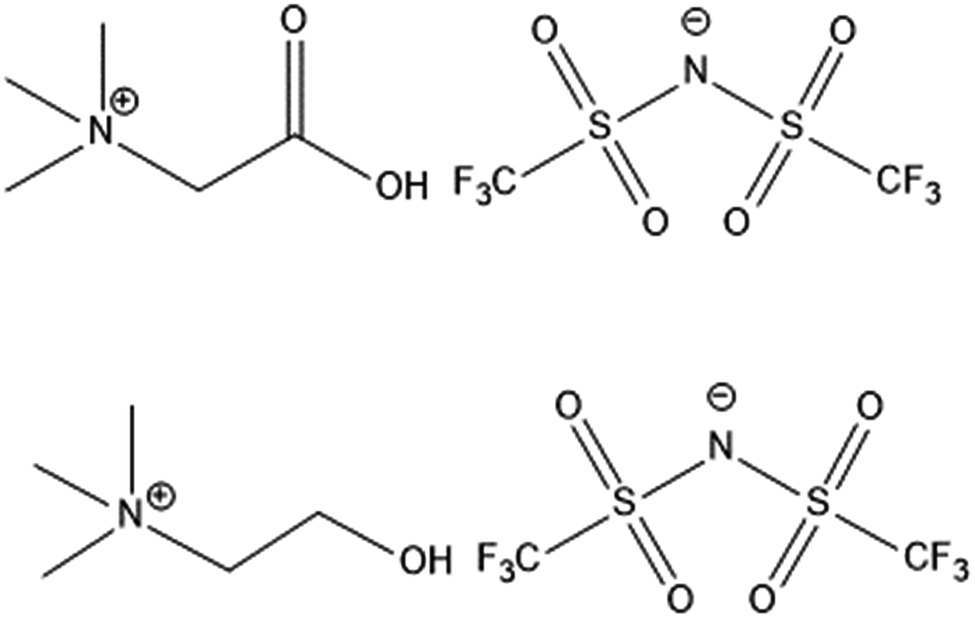 | ||
| Fig. 1 Structure of [Hbet][Tf2N] (top) and of [Chol][Tf2N] (bottom). | ||
Experimental
Chemicals
Betainium chloride and choline chloride were purchased by Acros Organic (Geel, Belgium) and LiTf2N from Iolitec (Heilbronn, Germany). Iron, neodymium, boron, dysprosium, praseodymium, cobalt, copper, gallium, aluminium, holmium, rhodium and strontium standards (1000 μg mL−1) were obtained from Chem-Lab (Zedelgem, Belgium). Nitric acid (65%), D2O (99.99%), deuterated dimethyl sulfoxide (99.9% atom D), Fe2O3 (95%) and calcium chloride dihydrate (99% ultrapure) were obtained from Sigma-Aldrich (Diegem, Belgium); acetic acid (glacial AnalR NORMAPUR, 96%) and CoO (99.995%) were purchased from VWR Chemicals (Leuven, Belgium); absolute ethanol and HCl (∼37%) from Fisher Scientific (Loughborough, United Kingdom). Nd2O3 (99.99%), was obtained from ReactonTM (Widnes, United Kingdom), Dy2O3 (99.9%) from Strem chemicals (Newbury Port, USA), Pr2O3 (99.9%) from Janssen Chimica (Geel, Belgium) and Eu2O3 (99.995%) from Inframat Advanced Materials (Manchester, USA). The silicone solution in isopropanol was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). The chemicals were used without further purification, with the exception of the rare-earth oxides and the CaCl2·2H2O. The rare-earth oxides were calcined before use at a heating rate of 20 °C min−1 from 150 °C to 950 °C and left at 950 °C for 15 h, in order to decompose all rare-earth carbonate impurities to the corresponding oxides. The CaCl2·2H2O was dried overnight under vacuum on the Schlenk line.Synthesis and characterization of [Hbet][Tf2N] and [Chol][Tf2N]
The ionic liquid [Hbet][Tf2N] was prepared following the one-step metathesis reaction between HbetCl and LiTf2N described in the literature.30 Afterwards, the ionic liquid was washed three times with ice-cold water to remove chloride impurities. A AgNO3 test was performed to check the presence of remaining chloride impurities. [Hbet][Tf2N] was dried for 3 hours under vacuum on a rotary evaporator and overnight on the Schlenk line at 60 °C.34 A 1H NMR spectrum of the ionic liquid was taken and compared with literature data. The water-saturated ionic liquid was prepared by equilibrating the dried ionic liquid with demineralized water.The 1H NMR spectrum was recorded on a Bruker Avance 300 spectrometer operating at 300 MHz. A small amount of sample was dissolved in d6-DMSO. The data were analysed with TopSpin 3.5 software. The viscosity of the dry ionic liquid between 60 and 175 °C was determined with an Anton Paar MCR 501 rheometer (geometry: cone–couette, shear rate ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) = 10 s−1, measurement time = 10 s). The thermal stability of the ionic liquid was investigated by dynamic and isothermal thermo-gravimetric analysis (TGA) on a TA Instruments T500 (heating rate: 5 °C min−1, temperature range: from 25 to 300 °C for the dynamic analysis, 175 °C for 1000 min for the isothermal analysis; sample gas: air) and by Fourier Transform Infrared – Attenuated Total Reflectance (FTIR). As for the TGA, solutions of the metal oxides, separately dissolved in dry [Hbet][Tf2N] for 24 h at 90 °C were used to study the possible influence of Fe, Nd and Co on the decomposition rate of the ionic liquid. The amount of metal oxides, preventively calcined, was calculated to reproduce the concentration of metals in the leaching experiments. More precisely Fe2O3, Nd2O3 and CoO were dissolved so that the final concentrations in the ionic liquid were: Fe(III) = 4.19 mg g−1; Nd(III) = 1.57 mg g−1; Co(II) = 0.21 mg g−1. A IR spectrum of the pure ionic liquid and of the leachates with the two powder fractions (vide infra) after 48 h were taken using a FTIR-ATR Vertex 70 by Bruker (operating at room temperature in a wavelength range of 400–4500 cm−1), the spectra were analysed by the OPUS software. [Chol][Tf2N] has been synthetized following the same procedure as used for [Hbet][Tf2N],35 but with [Chol][Cl] instead of [Hbet][Cl] as one of the starting product.
= 10 s−1, measurement time = 10 s). The thermal stability of the ionic liquid was investigated by dynamic and isothermal thermo-gravimetric analysis (TGA) on a TA Instruments T500 (heating rate: 5 °C min−1, temperature range: from 25 to 300 °C for the dynamic analysis, 175 °C for 1000 min for the isothermal analysis; sample gas: air) and by Fourier Transform Infrared – Attenuated Total Reflectance (FTIR). As for the TGA, solutions of the metal oxides, separately dissolved in dry [Hbet][Tf2N] for 24 h at 90 °C were used to study the possible influence of Fe, Nd and Co on the decomposition rate of the ionic liquid. The amount of metal oxides, preventively calcined, was calculated to reproduce the concentration of metals in the leaching experiments. More precisely Fe2O3, Nd2O3 and CoO were dissolved so that the final concentrations in the ionic liquid were: Fe(III) = 4.19 mg g−1; Nd(III) = 1.57 mg g−1; Co(II) = 0.21 mg g−1. A IR spectrum of the pure ionic liquid and of the leachates with the two powder fractions (vide infra) after 48 h were taken using a FTIR-ATR Vertex 70 by Bruker (operating at room temperature in a wavelength range of 400–4500 cm−1), the spectra were analysed by the OPUS software. [Chol][Tf2N] has been synthetized following the same procedure as used for [Hbet][Tf2N],35 but with [Chol][Cl] instead of [Hbet][Cl] as one of the starting product.
Characterization and pre-treatment of the solid materials
Non-coated and demagnetized NdFeB magnets and NdFeB production scrap with a high Dy content were kindly provided by Magneti Ljubljana (Ljubljana, Slovenia) (Fig. 2). | ||
| Fig. 2 NdFeB magnets (left) and NdFeB production scrap (right). | ||
The composition of the different materials was determined by a Perkin Elmer Optima 8300 inductively coupled plasma optical emission spectrometer (ICP-OES). For both the magnets and the scrap, 50 mg of milled, non-roasted solid was dissolved in 10 mL of 37% HCl at 70 °C for 1 h. The solutions were diluted 100 times in 2% v/v HNO3. Three calibration standards were prepared containing 0.5–5–50 mg L−1 Fe, Nd, B, Dy, Pr, Co, Cu, Ga and Al in 2% v/v HNO3. Rh was selected as internal standard for Fe, Nd and Dy; Sr was selected as internal standard for B, Pr, Co, Cu, Ga and Al. According to the composition provided by the supplier, minor elements were neglected and the composition was normalised to 100%.
Pre-treatment of the solids includes three steps: (1) crushing the solids into fragments smaller than 5 mm by using a hydraulic press and a hammer; (2) further milling the obtained powder in a planetary ball mill (Fritsch Planetary Mill Pulverisette 7 with tempered steel balls Ø 15 mm) for 2 min at 700 rpm, and for 5 min at 500 rpm with a ball-to-powder weight ratio of 1.3; (3) sieving the powder using two sieves with pore sizes of 400 μm and 45 μm to obtain three fractions: a fraction with a diameter >400 μm, a fraction with a diameter between 45 and 400 μm, and a fraction with a diameter <45 μm. The fraction above 400 μm was discarded because it was too large. The fraction between 45 and 400 μm and that below <45 μm were named, respectively, S400/M400 and S45/M45, where S and M stay respectively for “scrap” and “magnet” and 400 or 45 stay for the upper limit of the particle size fraction (see ESI†).
The size distribution of the fractions between 45 and 400 μm and <45 μm were measured by a laser diffractometer (Malvern Masterizer 3000) operating in liquid mode (suspending fluid: water; incident wavelength: λ = 470 nm and 632.8 nm). A muffle furnace operating at 950 °C for 15 h was used to roast the sample.36 Scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra of the roasted samples were taken with a Philips XL 30 FEG microscope and data were analysed with the software EDAX Genesis. The crystalline structure of the non-roasted and roasted solids was studied with quantitative powder X-ray diffraction (XRD) analysis using a Bruker D2 Phaser diffractometer. Homogenisation of the sample powder before analysis was done by milling 2.7 g of non-roasted or roasted scrap with 0.3 g of ZnO standard for 10 min using a McCrone micronizing mill (grinding solvent: heptane 99.9%; grinding elements: alumina pellets). The powders were then dried for 24 h at 40 °C. Diffractograms were recorded in the measurement range of 10–70° 2θ using CuKα radiation and applying an acceleration voltage of 45 kV, a current of 30 mA, a step size of 0.020° and a counting time of 2.5 s per step. The obtained data were evaluated with EVA V.3.1 (Bruker AXS) and processed with X'pert HighScore Plus PANalytical and Topas-Academic V.5 software matching the data with the standard present in the ICSD database.37
Proof-of-concept tests with CaCl2 in dry [Hbet][Tf2N] to coordinate the metal ion by chloride ions instead of water were carried out with NdFeB dross from strip casting production, kindly provided by Less Common Metals Ltd. The dross is too oxidized to be remelted and currently it is not recycled at the company, but its similarity with roasted NdFeB magnets made it an interesting material to test. All the steps of crushing, milling (roasting was unnecessary) and the corresponding characterization, i.e. chemical composition, particle size distribution, quantitative XRD, were conducted in the same way as reported for the NdFeB magnets and scrap. The nomenclature in the ESI† also follows the scheme applied for the solids from Magneti Ljubljana, with D stating for dross.
Leaching tests
Leaching tests were performed at 175 °C in 20 mL glass vials in copper blocks standing on a heating plate and open to the air. Polytetrafluoroethylene-coated magnetic stirring bars were used to stir at 600 rpm. A volume of 5 mL of [Hbet][Tf2N] and a mass ratio ionic liquid/solid of 100 were used. After each test, the leachate was filtered with a glass fibre filter with pore size P5. The composition of the leachate was analysed via total reflection X-ray fluorescence spectroscopy (TXRF) with a Bruker S2 PICOFOX TXRF spectrometer equipped with a molybdenum X-ray source.38,39 The data were analysed via the Bruker Spectra PICOFOX v. 7.5.3.0 software. The leachate was diluted before analysing its composition as follows. A weighed amount of sample was dissolved in 8.5 mL of acetic acid (CH3COOH) and 1.25 mL of 65% nitric acid (HNO3) to keep the pH low and to avoid precipitation of iron(III) hydroxide. Holmium was selected as standard, since the best data accuracy is obtained when the X-ray fluorescence energies of the internal standard are as close as possible to the X-ray fluorescence energy of the measured elements, but far enough to avoid an overlap of the peaks.39,40 An aliquot of 0.9 mL of the leachate in the selected solvent was mixed with 0.1 mL of a 1000 μg mL−1 holmium standard. Afterwards 1 μL of this solution was transferred on a quartz carrier, previously coated with 30 μL of silicone–isopropanol solution and dried for 30 min at 60 °C. The carriers were dried again for 30 min at 60 °C after addition of the sample droplet. Finally, the samples were analysed by TXRF spectrometer for 500 s. In preliminary tests, the relative standard deviation (% RSD) was calculated to check if the measuring time was sufficiently long to get accurate results for such a complicated matrix.40 The leaching process was evaluated in terms of recovery efficiency and in terms of selectivity. The recovery efficiency, η (%), is the ratio in percentage of the total moles of metal in the liquid after leaching (nf) over the total moles initially in the solid (n0):
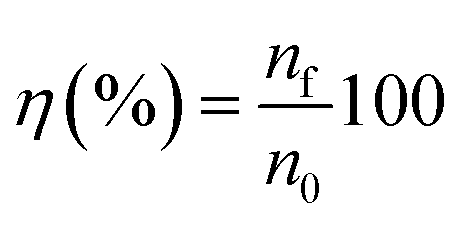 | (2) |
The selectivity, Δ 〈[Nd] + [Dy] + [Co]/[Fe]〉 (—), is defined as the ratio between the concentration of metal values [Nd] + [Dy] + [Co] over the concentration of iron [Fe] in the leachate at final time (tf), divided by the same ratio in the solid at initial time t0 (see solids composition in the Results and discussion section).
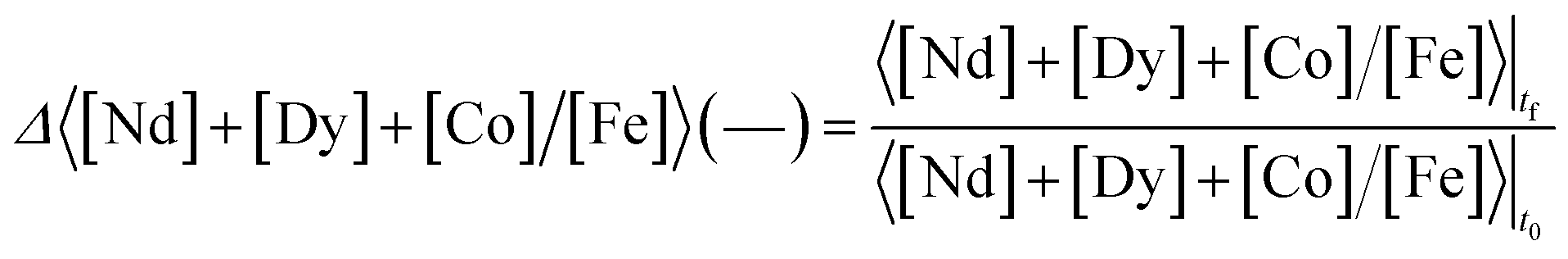 | (3) |
The selectivity indicates how the concentration of the metal values relative to iron changes between the solid phase and the leachate. It has a reference value of 1.0 in case no metal is preferentially dissolved.
The dissolution efficiency of the solids was compared with the dissolution process of the pure metal oxides M2O3 (M = Fe(III), Nd(III) or Dy(III)). The metal oxides were calcined overnight in a muffle furnace at 950 °C to avoid errors in the calculated theoretical concentrations of the metals initially present in the oxides because of a possible presence of carbonates, hydroxides or water impurities. The metal oxides and the pretreated magnets were dissolved separately in about 7.655 g of dry [Hbet][Tf2N] by stirring at 600 rpm for 96 h at 175 °C. An amount of 76.55 mg of magnets was used and the amount of metal oxides to use (Fe2O3, Nd2O3, Dy2O3 and CoO) was calculated to maintain, in solution, the same metal content that is in 76.55 mg of magnets.
The effect of the water content in the ionic liquid L on the metal coordination was investigated by Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy (vide infra), Eu(III) emission spectra and lifetime measurements. The number of coordinating water molecules or, simply, the hydration number q, can be obtained by luminescence analysis only for Eu(III). Therefore, it was used as a model for the lanthanides present in the magnets Dy(III) and Nd(III).
Emission spectra and emission decay curves were recorded on an Edinburgh Instruments F900 spectrofluorimeter (wavelengths of excitation λexc = 394 nm and wavelength of emission λem = 616 nm; Xe lamp, pulsed mode). Quartz cuvettes with 10.0 mm optical path lengths were used. Each measurement was replicated 3 times and the average is reported. Data were elaborated in Origin 8.0 as negative exponentials. τH2O and τD2O, the decay time of Eu(III) in the ionic liquid saturated with H2O and D2O, respectively, were obtained by fitting the curves according to equation:
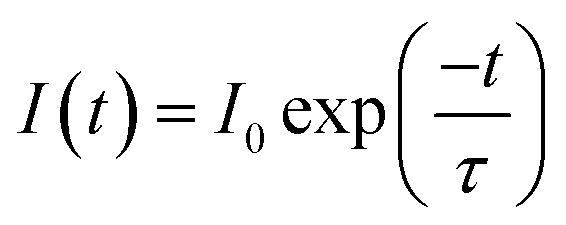 | (4) |
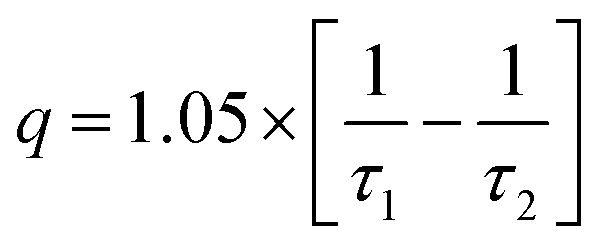 | (5) |
Since H2O is formed as a product of the dissolution reaction between [Hbet][Tf2N] and the metal oxides, it will interfere with the results. To minimize the discrepancies, it was necessary to obtain D2O as a product of the metal oxide dissolution reaction as well. Therefore, the deuterated version of [Hbet][Tf2N] ([Dbet][Tf2N]) was prepared by exchanging the acidic proton of the carboxylic group of [Hbet][Tf2N] with a D+ cation deriving from D2O. [Dbet][Tf2N] was dried or saturated with D2O similarly as done for [Hbet][Tf2N]. Afterwards, 100 mg of Eu2O3 was leached for 96 h at 175 °C with 3.5 g of dry [Hbet][Tf2N] or [Dbet][Tf2N], and for 96 h at 90 °C with 3.5 g of H2O/D2O saturated [Hbet][Tf2N]/[Dbet][Tf2N]. Eu2O3 was preventively calcined to avoid a weighing error due to the presence of carbonate, hydroxide or water impurities.
Leaching tests were carried out dissolving CaCl2·2H2O in the dry ionic liquid to further prove that an efficient leaching system has to contain a dissolving agent and a coordinating agent as well. Although the salt was dried before adding it to the ionic liquid, the possible tiny amount of water introduced was not considered influencing the results since also the water produced by the reaction was proved to evaporate at 175 °C. The assumption that two chloride ions coordinate the metal ion, as is the case for the water molecules, was made and the salt was dissolved in a close to stoichiometric quantity, i.e. 114.66 mg for about 76.55 mg of NdFeB magnets or scrap. The molecular weight considered is that of the dihydrate salt as the corresponding error can only correspond to an excess of CaCl2, beneficial to the system. For convenience, the solid used for the tests was a fraction below 45 μm of NdFeB dross from scrap casting and the leaching time was 24 and 72 hours; the temperature and the stirring speed were 175 °C and 600 rpm, respectively, as in case of the dry [Hbet][Tf2N].
Finally, [Hbet][Tf2N] was partly substituted with [Chol][Tf2N] to lower the viscosity of the system. For the leaching experiments, [Hbet][Tf2N] was mixed with 10, 25 and 50 wt% [Chol][Tf2N]. No other parameters were changed in comparison to the leaching tests described above and only one test was performed for 48 h at 175 °C on the <45 μm magnet fraction.
EXAFS measurements
The chemical environment of Nd(III) in the roasted powder and of rare earths in the leachate was also determined by EXAFS analysis. As for the leachate, Pr(III) dissolved in [Hbet][Tf2N] was investigated since the authors had the opportunity to measure the absorption of X-rays around the Pr(III) K-edge. The K-edge of Nd(III) could not be reached with the used set-up at the beamline, but thanks to the similar properties of the lanthanides, conclusions similar to the results with Pr(III) can be drawn for Nd(III).41 The spectra, of the LIII-edge of Nd(III) (6208 eV) and of the K-edge of Pr(III) (41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 990 eV), were collected at the Dutch–Belgian Beamline (DUBBLE, BM26A) at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). The energy of the X-ray beam was tuned by a double-crystal monochromator operating in fixed-exit mode using a Si(111) crystal pair for Nd and Si(3111) for the Pr. The measurements were done in transmission mode using Ar/He gas filled ionization chambers. The roasted powders were diluted with boron nitride and pressed into pellets. The Pr(III) samples were measured in plastic cuvette holders with a path length of 2.0 mm. Standard procedures were used for pre-edge subtraction and data normalization in order to isolate the EXAFS function (χ). The isolated EXAFS oscillations, accomplished by a smoothing spline as realized in the program Viper,42 were k4 – weighted and Fourier transformed over the k-range from 3 to 7.64 Å−1 using a Kaiser–Bessel window function. The data were fitted using the ab initio code FEFF 7.0,43 which was used to calculate the theoretical phase and amplitude functions that subsequently were used in the non-linear least-squares refinement of the experimental data. Fitting of the data of the roasted powders with the model was performed in R-space between 1.21 and 3.94 Å for the roasted powders, and between 1.26 and 2.51 Å for the Pr(III) dissolved in [Hbet][Tf2N]. Estimated errors on the obtained parameters were calculated by VIPER. S0 was fixed at 0.8. The data were modelled using the VIPER software.42
990 eV), were collected at the Dutch–Belgian Beamline (DUBBLE, BM26A) at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). The energy of the X-ray beam was tuned by a double-crystal monochromator operating in fixed-exit mode using a Si(111) crystal pair for Nd and Si(3111) for the Pr. The measurements were done in transmission mode using Ar/He gas filled ionization chambers. The roasted powders were diluted with boron nitride and pressed into pellets. The Pr(III) samples were measured in plastic cuvette holders with a path length of 2.0 mm. Standard procedures were used for pre-edge subtraction and data normalization in order to isolate the EXAFS function (χ). The isolated EXAFS oscillations, accomplished by a smoothing spline as realized in the program Viper,42 were k4 – weighted and Fourier transformed over the k-range from 3 to 7.64 Å−1 using a Kaiser–Bessel window function. The data were fitted using the ab initio code FEFF 7.0,43 which was used to calculate the theoretical phase and amplitude functions that subsequently were used in the non-linear least-squares refinement of the experimental data. Fitting of the data of the roasted powders with the model was performed in R-space between 1.21 and 3.94 Å for the roasted powders, and between 1.26 and 2.51 Å for the Pr(III) dissolved in [Hbet][Tf2N]. Estimated errors on the obtained parameters were calculated by VIPER. S0 was fixed at 0.8. The data were modelled using the VIPER software.42
Results and discussion
Thermal stability of [Hbet][Tf2N]
The purpose of this study is to investigate the application of [Hbet][Tf2N] at temperatures above 100 °C (implicating dry conditions), because increasing the temperature increases the reaction rate of the leaching process. The highest possible working temperature depends on the ionic liquid decomposition temperature or on the occurrence of undesired reactions that may occur above this temperature. The FTIR spectra are reported in the ESI (Fig. S2†). Two new peaks at about 3200 and 1600 cm−1 appeared after leaching, due to the formation of the metal complexes of [Hbet][Tf2N], whereas the peaks between 1000 and 1500 cm−1 were intensified.A dynamic TGA was performed to find the highest possible working temperature of the ionic liquid. Dynamic TGA allows to detect mass losses of the ionic liquid within a certain temperature range. In Fig. 3, the dynamic TGAs of the ionic liquid and of the ionic liquid containing Fe(III), Nd(III) or Co(II) are shown. The samples were first heated to 110 °C and kept at this temperature for 5 h to remove the remaining water; after which they were cooled to 50 °C and kept at this temperature for 1 h. Afterwards, the temperature was increased every 6 h by 50 °C starting at 150 °C and ending at 300 °C at a heating rate of 5 °C min−1. From 50 to 200 °C, no significant mass losses occurred, neither in the presence of Fe(III), Nd(III) or Co(II). The plots show that about 5 wt% of the sample is lost at 200 °C. Therefore, it was decided to work at 175 °C. An effect of the dissolved metals is noticeable only above 250 °C, where especially Co(II) seems to stabilize the ionic liquid rather than to catalyse any decomposition reaction. The effect of the time on the stability of a compound at a certain temperature cannot be analysed by dynamic TGA but by isothermal TGA. An isothermal TGA measurement was carried out at 175 °C at 1000 min (Fig. 4). Since the dynamic TGA measurements have revealed that the effects of metals are negligible at this temperature, the isothermal test was only performed on the pure [Hbet][Tf2N]. The results show that there is a constant loss of mass equal to 2% of the initial mass of the sample after about 17 hours. In case of decomposition with production of gases a step would be observed in the trend of the remaining mass, while a constant loss of mass can be attributed to the residue water content of the ionic liquid.
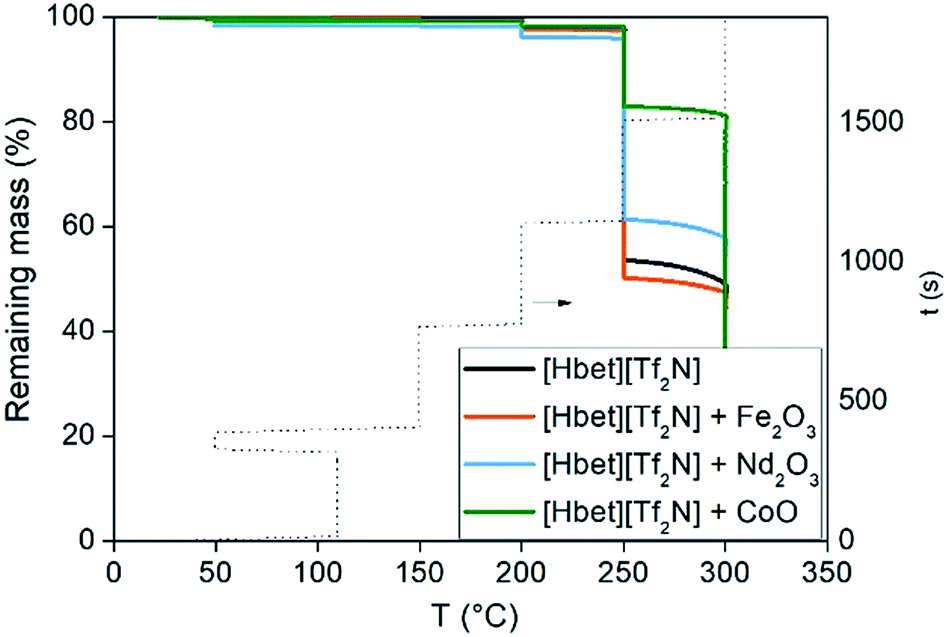 | ||
| Fig. 3 TGA traces of [Hbet][Tf2N] and [Hbet][Tf2N] mixed with Fe2O3, Nd2O3 or CoO as a function of temperature. | ||
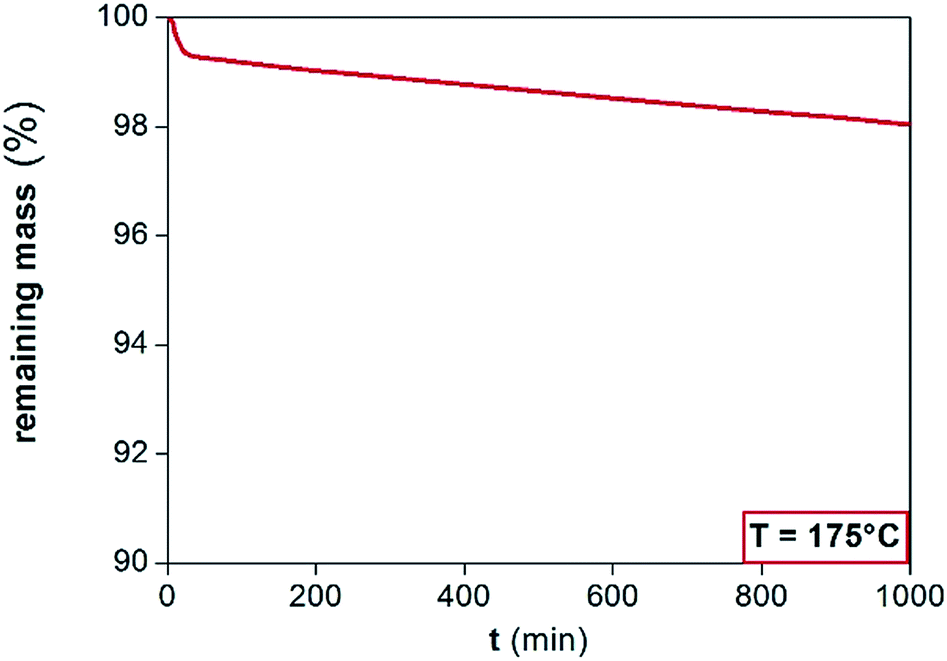 | ||
| Fig. 4 TGA traces of [Hbet][Tf2N] at a fixed temperature of 175 °C as a function of time. | ||
Characterization and pre-treatment of the solid materials
The composition of the magnets and the scrap is reported in Table 1. The samples are rich in dysprosium, (6–9 wt%). The weight ratio ([Nd] + [Dy] + [Co])/[Fe] of the metal values on iron is 0.56, in both the starting materials. In other words, the total content of metal values (Nd(III), Dy(III) and Co(II)) in the solids is approximatively half of the content of Fe(III).| Element | Composition (wt%) | |
|---|---|---|
| Magnet | Scrap | |
| a Compositions normalised to 100%. | ||
| Fe | 64.19 | 62.52 |
| Nd | 20.54 | 24.15 |
| Dy | 8.61 | 6.89 |
| Pr | 1.70 | 1.45 |
| Co | 3.38 | 3.46 |
| Cu | 0.02 | 0.04 |
| Ga | 0.33 | 0.32 |
| Al | 0.20 | 0.11 |
| B | 1.04 | 1.05 |
The solid materials were crushed, milled and separated by sieving into three fractions: below 45 μm, between 45 and 400 μm and above 400 μm. The particle size distribution is described as a probability distribution function (PDF) in Fig. S3† and as a cumulative distribution function (CDF) in Fig. S4.† The diameters d10, d50 and d90, in which 10%, 50% and 90% of the population is included respectively, are reported in Table S1.† The median, d50, is not affected by asymmetries in the curve and is therefore considered as a more reliable parameter to describe a distribution than the mode or the mean.
The distribution functions are very similar for each fraction of the two materials. This means that the scrap and the magnets not only have a similar chemical composition, as shown by the ICP analysis, but also similar hardness. The samples are in fact the output of the same production line since scrap is made of magnets not having the required specification in terms of magnetic properties, size, etc. Once demagnetised and oxidized, there is no difference between production scrap and magnets originating from the same production line in terms of chemical composition and thus of recycling performances. Therefore, the quantitative XRD (before and after roasting) was executed only on the scrap. It shows a clear rearrangement of the alloy Nd2Fe14B into different oxides, such as NdFeO3 and Fe3O4 as summarized in Table 2. There is a risk of partial oxidization of the non-roasted sample during the milling process. In fact, a high amount of energy is transferred to the point of impact between mill balls-material and material–material. This energy turns into a significative punctual increase of temperature and the sample can react with the air in the mill pot with consequent metal oxidization. However, the oxide content was still below the detection limit of the XRD in the non-roasted sample. As for the roasted sample, iron is almost completely converted in hematite (Fe2O3) and partly in the ternary oxide NdFeO3. More than 98% of Nd(III) is incorporated into NdFeO3, rather than in Nd2O3. SEM pictures of the solids before and after roasting are reported in Fig. S5–S7 in the ESI.† They show that the particles are uniformly distributed and that no sintering effects occurred during the milling process. However, smaller particles deposit on the larger ones.
| Compound | Non-roasted (wt%) | Roasted (wt%) |
|---|---|---|
| Nd2Fe14B | 100 | — |
| Nd2O3 | — | 0.44 |
| NdBO3 | — | 3.26 |
| NdFeO3 | — | 25.32 |
| Fe2O3 | — | 70.98 |
The presence of NdFeO3 after roasting of the solids was verified by EXAFS measurements on the fraction of milled and roasted scrap below 45 μm. The structure reported by Streltsov and Ishizawa was used as a model.44 The structure around the central Nd atom is highly disordered. For instance, five different Nd–O distances are found ranging from 2.34 to 2.72 Å. Also the distances from the Nd atom to the Fe atoms are ranging from 3.18 to 3.63 Å. The distances to the next Nd atoms are between 3.82 and 3.98 Å. To reduce the number of free parameters and to deal with the rather short k-range, we limited the number of paths to three and constrained them as found in the crystal structure of NdFeO3: (1) a single scattering path Nd–O; (2) a single scattering path from Nd to the Fe atoms, for which the degeneracy was constrained to be equal to the degeneracy of path (1); (3) a single scattering path between the central Nd atom and the nearest Nd atoms which was constrained to be 0.75 times the degeneracy of path (1). Other paths are excluded from the fit because of their low contribution to the EXAFS signal. The obtained parameters are reported in Table 3 together with the average values found in the crystal structure of NdFeO3.44 All distances found are about 0.1 Å shorter than found in the NdFeO3 crystal structure. The EXAFS spectra of NdFeO3 and Nd2O3 are plotted and compared in Fig. 5. The Fourier Transform (FT) of NdFeO3 present in the sample and of a reference model are given in Fig. 6. The XANES and EXAFS spectrum of Nd2O3 and of the oxidized magnet powder are significantly different as shown in Fig. 5 and 6. This proves that other atoms surround Nd in the roasted NdFeB material in comparison to Nd2O3. The XANES function of the NdFeO3 compared to that of Nd2O3 and the EXAFS spectrum for the NdFeO3, compared to that available from the literature,44 are available in Fig. S8 and S9 in the ESI.† Data for NdFeO3 were obtained from the fraction of the milled and roasted magnet scrap with a particle size smaller than 45 μm. The data in Fig. S8† confirm the results from Fig. 5 that Nd(III) in the roasted magnets and scrap is not present in the form of the binary oxide Nd2O3, but mainly in the form of the ternary oxide NdFeO3. This is further confirmed by the EXAFS data in Fig. S9.† However, the two curves in this latter plot slightly differ from each other because one refers to a real, treated solid, while the other two data sets are taken from the literature. The patterns in Fig. 5 and S8† are both from real, treated solids.
| Path | N (—) | r (Å) | σ (Å2) | r* (Å) |
|---|---|---|---|---|
| a N is the coordination number, dimensionless, r is the interatomic distance (Å), σ is the Debye–Waller (Å2) and r* is the interatomic distance expected from crystal structure.44 The absolute errors on the average values are reported in brackets. | ||||
| Nd–O | 7.8(1) | 2.409(4) | 0.021(1) | 2.52 |
| Nd–Fe | 7.8(1) | 3.250(4) | 0.026(1) | 3.38 |
| Nd–Nd | 5.8(1) | 3.833(1) | 0.006(1) | 3.91 |
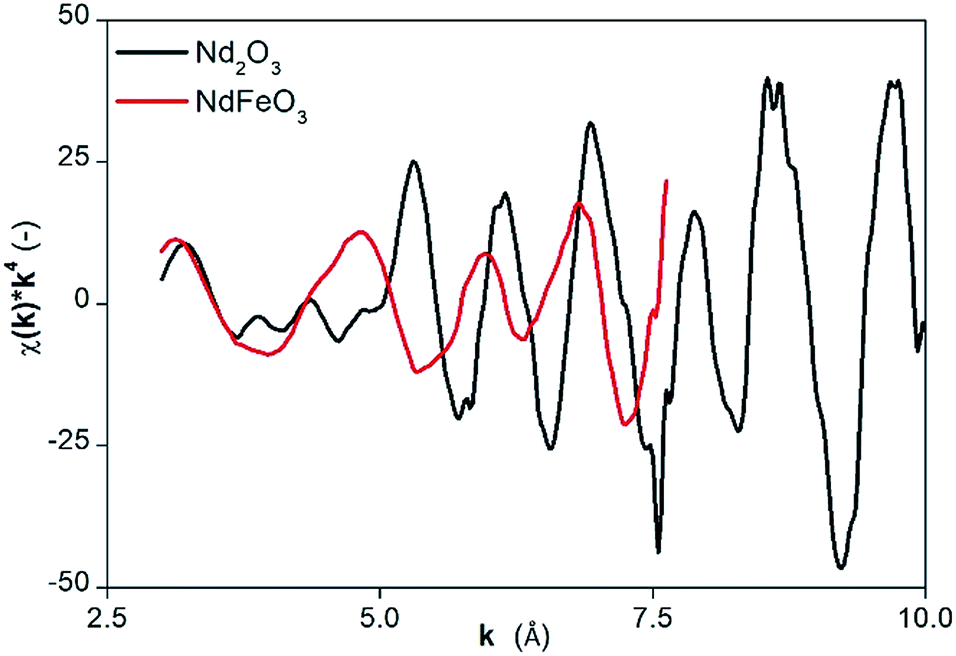 | ||
| Fig. 5 EXAFS function of NdFeO3 compared to that of Nd2O3. | ||
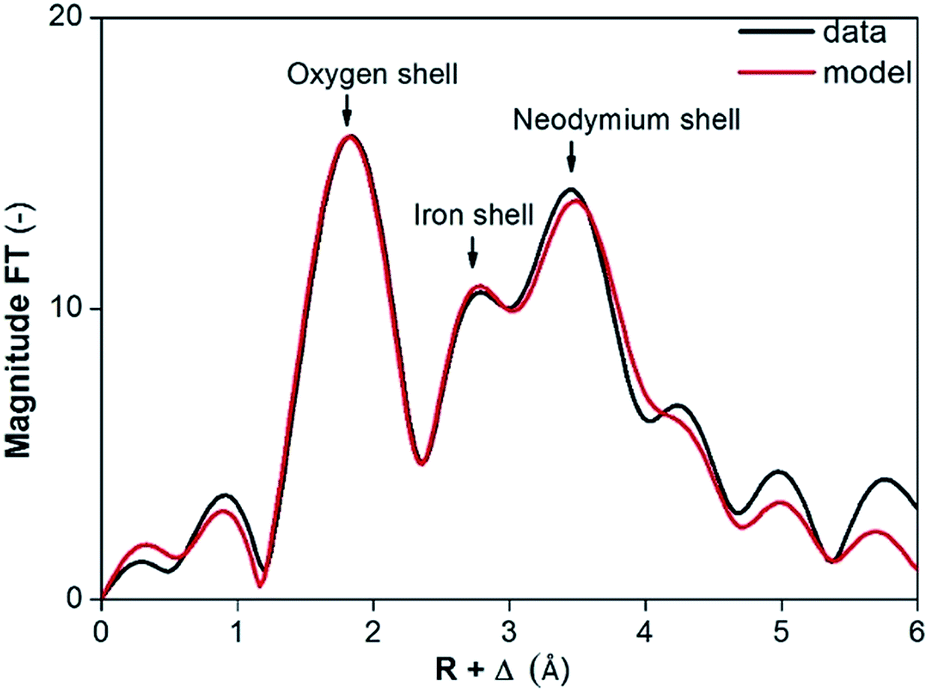 | ||
| Fig. 6 Fourier transform of experimental (black) and model (red) EXAFS function for NdFeO3. | ||
All the results regarding the characterization of the NdFeB dross from scrap casting are reported in the ESI (Tables S2–S4†). As expected, the dross is very comparable to the roasted NdFeB in every aspect: chemical composition, particle size and crystalline structure. Therefore, the conclusions drawn from the tests with the dross are valid also for roasted NdFeB magnets or production scrap.
Leaching tests
Leaching tests with dry [Hbet][Tf2N] were performed on the NdFeB magnet and the scrap material with the aim to recover the (neodymium, dysprosium and cobalt), rather than iron. Our results showed significant differences with the work of Dupont and Binnemans who successfully used water-saturated [Hbet][Tf2N] at 90 °C for recovery of rare earths from a NdFeB magnet.17 The reaction in dry [Hbet][Tf2N] system at 175 °C was less efficient than the reaction in water-saturated [Hbet][Tf2N] at 90 °C. Metal recovery efficiencies and selectivities for the leaching of magnets with dry [Hbet][Tf2N] are reported in Fig. 7 and 8. The leaching results for NdFeB production scrap can be found in Fig. S10 and S11 in the ESI.† The leaching yields is below 10% for every metal at every leaching time or particle size. It is foreseeable from Fig. 7 that there is no preference to leach any of the metals. In fact, the selectivity ratio (Fig. 8) Δ 〈[Nd] + [Dy] + [Co]/[Fe]〉 (—) is around 2 in every case beside than for the fraction between 45 and 400 μm leached for 24 h, where it is around 5. This might anyway not be a case of selective conditions since the concentrations are very small and the error on the ratio can be significant. In other words, the metals concentrations are close to 0 and a slight change, especially one in the denominator, can make the ratio to change significantly. In general, the process is not selective since the amount of iron is still comparable to that of metal values and a next step of extraction will be necessary to separate the metals.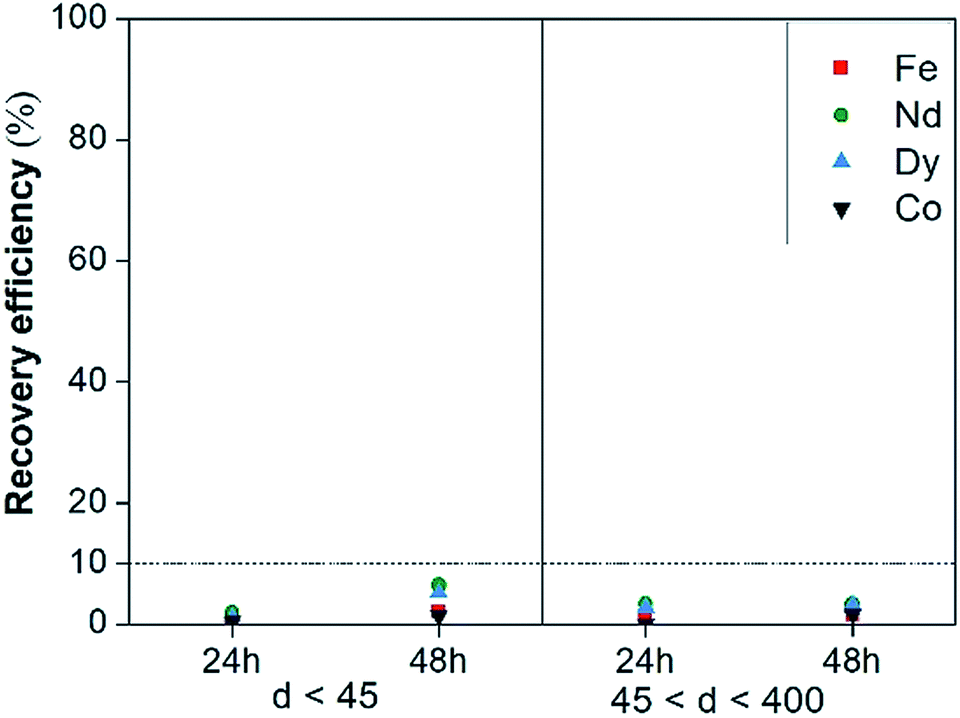 | ||
| Fig. 7 Recovery efficiency of Fe(III), Nd(III), Dy(III) and Co(II) of magnets leached with dry [Hbet][Tf2N] as a function of leaching time and particle size. | ||
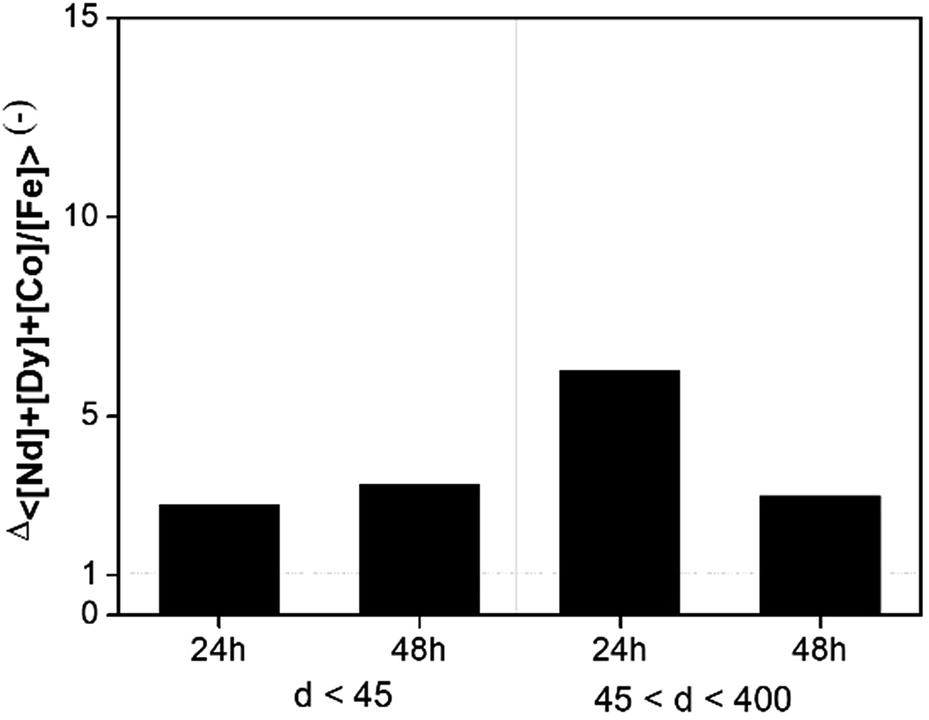 | ||
| Fig. 8 Selectivity of magnets leached with dry [Hbet][Tf2N] as a function of leaching time and particle size. | ||
The influence of the different parameters (crystal structure, water content and coordination chemistry of the metals in the ionic liquid) were investigated to explain the differences in leaching rates.
At first, the influence of the structure in which the metal values are incorporated on the leaching efficiencies was studied by comparing the dissolution efficiencies of Fe2O3, Nd2O3 and Dy2O3 with the dissolution efficiencies of metals from the magnets with dry [Hbet][Tf2N] at 175 °C.
Fig. 9 shows that the dissolution of the pure metal oxides is more efficient than that of the magnet under the same conditions. The structure of the roasted magnets is more stable than that of pure metal oxides. Fe2O3 is known to have a low solubility in [Hbet][Tf2N], whereas Ln2O3 have higher solubilities in [Hbet][Tf2N].35
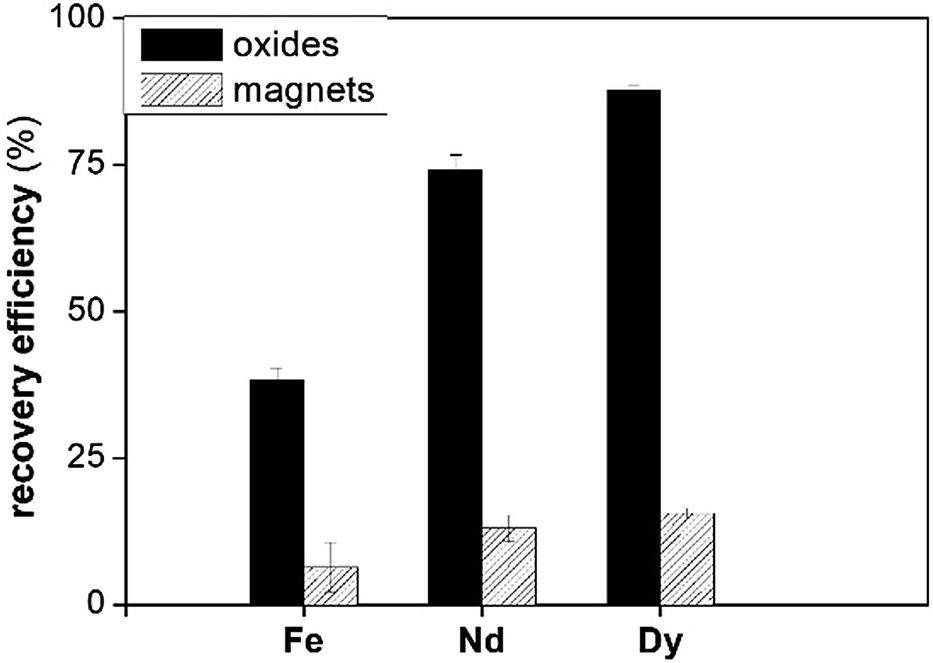 | ||
| Fig. 9 The recovery efficiency (%) of Fe, Dy and Nd from their pure oxides and from the roasted magnet after 96 h at 175 °C. | ||
Dupont and Binnemans were able to completely recover and separate the rare earths from iron at 90 °C with water-saturated [Hbet][Tf2N] after 24 hours; only cobalt showed a behaviour similar to that of iron and could not be separated efficiently. The difference in the yields between magnet leaching in dry and water-saturated [Hbet][Tf2N] is not due to differences in crystal structure as both materials were roasted by the same procedure. The particle size of the powder has an influence on the dissolution of the particles itself because smaller particles dissolve faster than larger ones due to their higher surface-to-volume ratio. Dupont and Binnemans reported that particles with an average diameter of 6 ± 3 μm dissolve faster in water-saturated [Hbet][Tf2N] than particles of 73 ± 40 μm average diameter under the same conditions. The d50 reported in Table S1† is about 28 μm for the fraction <45 μm and 85 μm for the fraction 45–400 μm. Because of the error of ±40 μm, the latter fraction from Dupont's work is comparable with both the fractions reported in the current paper and similar results could be expected. Dupont and Binnemans also obtained higher recovery efficiencies than those obtained in this study with the dry ionic liquid at 175 °C with equal stirring speed, stirring time, solid-to-liquid ratio, and even with an upper particle size of 70 μm, which is greater than the upper particle size of the fraction <45 of this study.
Water might affect the mass transfer in terms of a reduced viscosity or it might be required to saturate the coordination sites of the metals leaching into the ionic liquid. The viscosity of dry [Hbet][Tf2N] measured from 60 °C to 175 °C and of water-saturated [Hbet][Tf2N] is reported in Fig. 10.17 The viscosity of water-saturated [Hbet][Tf2N] above 80 °C cannot be measured because of the formation of water vapour in the ionic liquid. The viscosity of the dry [Hbet][Tf2N] at 175 °C and of the viscosity of water-saturated [Hbet][Tf2N] at 80 °C are 9.1 and 6.0 cP, respectively. The difference between the these values is too small to induce major differences in mass transfer and in leaching efficiencies between dry and water-saturated [Hbet][Tf2N].
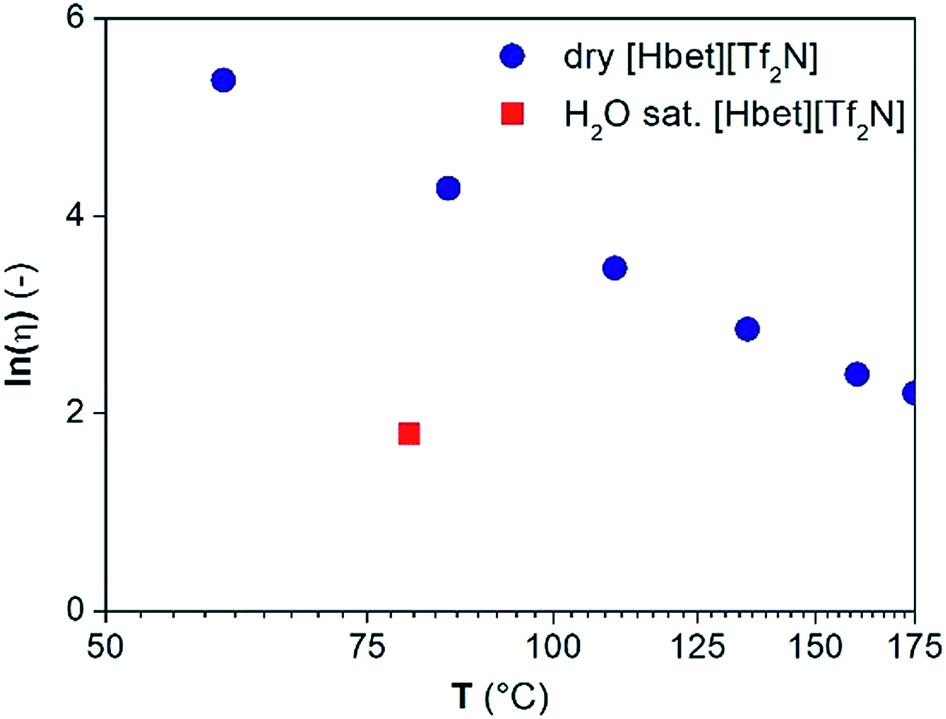 | ||
| Fig. 10 Natural logarithm of the viscosity (ln(μ) (—)) as a function of the temperature (T (°C)) for dry [Hbet][Tf2N] and water-saturated [Hbet][Tf2N] at 80 °C. | ||
The presence of water in the coordination sphere of the leached metals was investigated via EXAFS, Eu(III) luminescence emission spectra and lifetime decay measurements. Although leaching at 175 °C was carried out with dry [Hbet][Tf2N], water is produced during the dissolution reaction which might escape the reaction mixture because of the high temperature, or it might bind to the leached metals. These measurements were performed on Pr(III) or Eu(III) rather than on Nd(III) or Dy(III) for practical reasons (see Experimental section). Nevertheless, the coordination environment of Pr(III) and of Eu(III) is very similar to that of Nd(III) and Dy(III), and to that of all Ln(III), and the results obtained with one Ln(III) may be extrapolated to the other Ln(III).42
The coordination number of Pr(III) dissolved in dry and water-saturated [Hbet][Tf2N] was measured by EXAFS spectroscopy. In case of water-saturated [Hbet][Tf2N], a coordination number for Pr(III) of 7.5(4) was obtained. The corresponding average length of the Pr–O bond was 2.523(5) Å and the Debye–Waller factor 0.010(1) Å2 (Table 4). Whereas, for the Pr(III) dissolved in dry [Hbet][Tf2N] a smaller coordination number 6.2(2) and smaller Debye–Waller factor of 0.009(1) Å2 were obtained. These parameters suggest that the coordination number decreases when Pr(III) leaching is performed without water. However, care should be taken as the data quality is low and the Pr–O bond length is not changing between Pr(III) dissolved in wet (2.523(5) Å) and in dry [Hbet][Tf2N] (2.528(3) Å). The coordination number also depends on the arbitrary value of S0 of the two samples: the consistency of the EXAFS with the luminescence measurements (vide infra) indicates that 0.8 per S0. A stable bond length indicates an equal coordination number, although this might be caused as well by changes in coordination polyhedra, sterical hindrance between the coordinating groups or inefficient coordination bonds. The FT functions of Pr6O11 dissolved in dry and water-saturated [Hbet][Tf2N] are represented in Fig. 11 and 12 and the relative parameters in Table 4, the EXAFS spectra are reported in Fig. S12 and S13 of the ESI.†
| Path | [Hbet][Tf2N] | N (—) | r (Å) | σ (Å2) |
|---|---|---|---|---|
| a N is the coordination number, r is the interatomic distance, (Å), σ is the Debye–Waller (Å2). The error is reported in brackets. | ||||
| Pr–O | Dry | 7.5(4) | 2.523(5) | 0.010(1) |
| Water-saturated | 6.2(2) | 2.528(3) | 0.009(1) | |
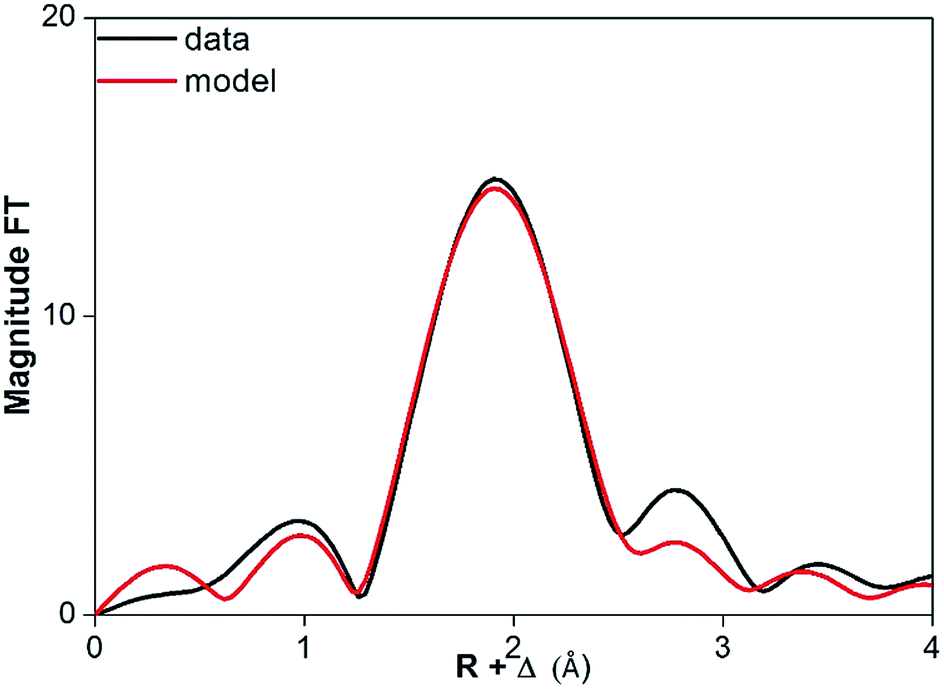 | ||
| Fig. 11 FT function of P6O11 in dry [Hbet][Tf2N], fitted on experimental (black) and theoretical (red) data. | ||
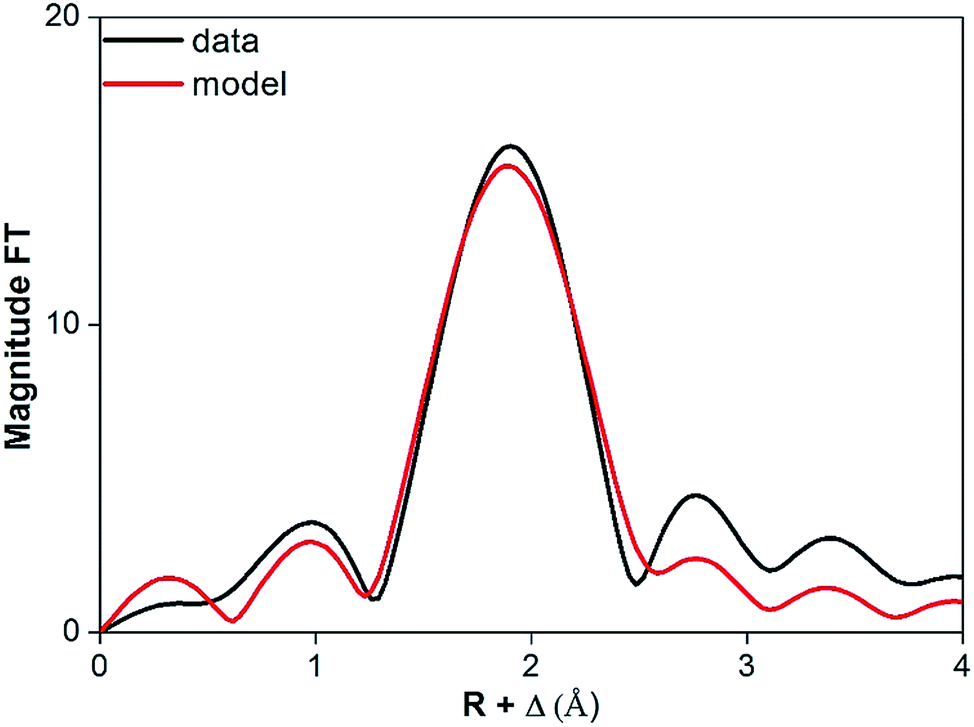 | ||
| Fig. 12 FT function of Pr6O11 in water-saturated [Hbet][Tf2N], fitted on experimental (black) and theoretical (black) data. | ||
Coordination of the lanthanides in the leachate was studied as well by luminescence spectroscopy and, more specifically, via measurements of the emission decay time. Many Ln(III) ions exhibit luminescence, after radiative absorption of energy from UV light or the energy transfer from the triplet state of a ligand. In particular, Eu(III) and Tb(III) show strong metal-ion fluorescence that may be applied to investigate the coordination environment of the ion.41,42 The visible transitions for Eu(III) are 5D0 → 7Fn with n = 4–0. The ground state 7F0 and 5D0 are unsplit. Therefore, each peak seen in the region where the 5D0 → 7F0 transition is expected, to correspond to one europium site. Depopulation of an excited state occurs via both radiative and non-radiative decay. The luminescence lifetime τ is a combination of the probabilities for the two phenomena. In general, the lifetime is defined as the time after which a population decreases by a factor of 1/e compared to its initial value.
Eu(III) emission decay curves are shown in the ESI, Fig. S14 and S15.† The dissolution was carried out simulating the working conditions with dry and water-saturated ionic liquid. The hydration number q is shown in the plot and it was calculated according to eqn (5).41 The calculation of q confirms that no water molecules coordinate the metal ions in dry [Hbet][Tf2N], as occurring in the leaching process at 175 °C, and that there are two water molecules coordinating to Eu(III) in case of water-saturated [Hbet][Tf2N] used in a leaching process at 90 °C. Thijs et al. already reported that REEs dissolved in [Hbet][Tf2N] are coordinated with two molecules of water.35 They prepared several metal complexes of [Hbet][Tf2N] by dissolving the metal oxides in a mixture of [Hbet][Tf2N] and H2O.35 The crystal structure revealed that not all the water evaporated but that dimers with the formula [Ln2(bet)8(H2O)4][Tf2N]6 were formed, in which two water molecules coordinated directly to one of the Ln(III) ions and the other two are forming a bridge between the Ln(III) and the bis(trifluoromethylsulfonyl)imide anion. Nockemann et al. confirmed a similar metals coordination, in an article about the best techniques to study the speciation of REEs in ionic liquids.45 The emission spectra of Eu(III) dissolved in water-saturated [Hbet][Tf2N] and in dry [Hbet][Tf2N] provides information about the symmetry of the metal complexes in solution (Fig. 13). The emission spectrum of Eu(III) dissolved in water-saturated [Hbet][Tf2N] is significantly different from the emission spectrum of Eu(III) dissolved dry [Hbet][Tf2N]. Especially the transition 5D0 → 7F2 which is hypersensitive in terms of symmetry is changing in intensity and shape.41 These results confirm that the lanthanide complex in dry [Hbet][Tf2N] is indeed different from the europium complex dissolved in water-saturated [Hbet][Tf2N] and supports the conclusions drawn from EXAFS measurements and lifetime measurements.
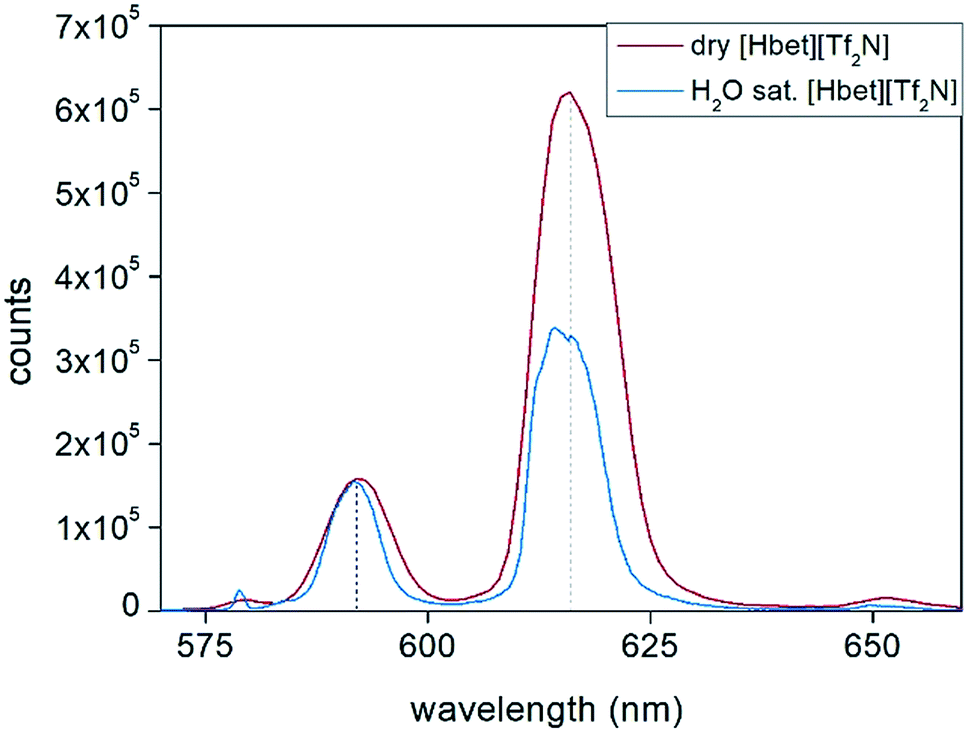 | ||
| Fig. 13 Emission spectra, collected at room temperature, of Eu(III) dissolved at 175 °C in dry and water-saturated [Hbet][Tf2N]. λexc = 394 nm. | ||
NdFeB dross from scrap casting production was leached in [Hbet][Tf2N] with the addition of CaCl2, to investigate the possibility to substitute water with other ligands to keep the process running at 175 °C. The dross was shown to be very similar to the roasted NdFeB magnets so that the results do not depend on the treated solid. Metal recovery efficiencies are reported in Fig. 14, while the selectivity is reported in Fig. S16.† No complete dissolution was achieved neither after 72 h, but the recovery efficiencies significantly increased for all the metals and in particular for Nd, with a leaching yield of more than 60% after 24 hours. This led to the conclusion that a coordinating agent is necessary in the system to stabilise the metal ions in the liquid phase and to promote the dissolution of the metals themselves. However, no selectivity was achieved with the mixture [Hbet][Tf2N]–CaCl2 due to the high iron content in the starting solid, although the recovery of Fe(III) is significantly lower than that of Nd(III) after 24 and 72 hours.
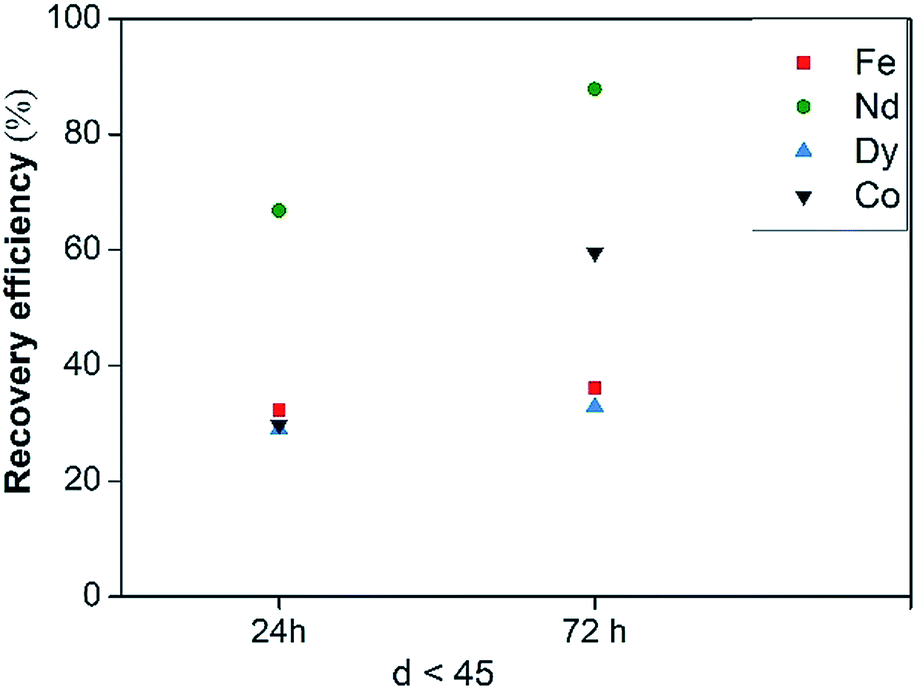 | ||
| Fig. 14 Recovery efficiency of Fe(III), Nd(III), Dy(III) and Co(II) of NdFeB dross leached with dry [Hbet][Tf2N] with addition of CaCl2 as a function of leaching time. | ||
The effect of the coordinating group of the cation on the leaching performance was tested with mixtures of [Hbet][Tf2N] and [Chol][Tf2N], (ESI, Fig. S17 and S18†). The recovery efficiency drops with an increasing percentage of [Chol][Tf2N] and thus with a lower carboxylic acid concentration. On the other side, the selectivity does not depend on the presence of the [Chol][Tf2N] since the capacity of choline to coordinate metals is smaller than the coordination capacity of the betaine cation. The betaine and the choline cation are very similar in composition and differ only in the functional group attached to the nitrogen centre (an R–COOH group in case of betaine and an R–OH group in case of choline).
In conclusion, the water produced by the dissolution reactions of the metal oxides shown in eqn (1), evaporates at 175 °C and does not coordinate the metals dissolved in the ionic liquids. The removal of water from the right side of the equation (by evaporation) shifts the reaction equilibrium towards the products. Although four water molecules are required for each dimer [Ln2(bet)8][Tf2N]6˙, the reaction is able to produce only three. The small percentage of dissolved metals can be explained by the presence of other less stable compounds, such as traces of Nd2O3. In general, the coordination sphere of the metal cations cannot be completely saturated in the absence of water, since the coordinating [Hbet] is too large to have eight or nine of these around one Ln(III). As for the [Tf2N]− it is a weak-coordinating ligand and therefore added to a higher extent to confer hydrophobicity to the IL. The metal complexes in the liquid phase are not stable and the dissolution process is very slow.
Leaching processes with ionic liquids at temperatures between 100 °C and the decomposition temperature of the ionic liquid, at atmospheric pressure, have not been explored so far. New insights into the metal leaching mechanism and the metal coordination are provided by replacing water with other coordinating molecules, such as nitrate and chloride salts. This additive should be (1) not (or barely) volatile at the working conditions, (2) a strong ligand, (3) small, to minimise the steric hindrance, and (4) acidic, to be able at the same time to coordinate but also to leach the metal.
Conclusions
This paper provides new insights in the dissolution mechanism of metal oxides by [Hbet][Tf2N] at leaching temperatures higher than typically used in hydrometallurgical leaching processes. The leaching of NdFeB magnets and production scrap with the ionic liquid [Hbet][Tf2N] at 175 °C and at atmospheric pressure has been studied, focusing in particular on metal coordination and the leaching mechanism. It is shown that two factors affect the leaching yields and the selectivity: the roasting step and the absence of water. The neodymium in the NdFeB magnets or production scrap is converted in the very stable NdFeO3 structure during the roasting. This complex oxide dissolve much slower than the pure metal oxides (Fe2O3 or Nd2O3). The roasting conditions are to be optimised to obtain Nd2O3 and Fe2O3 instead than the complex oxide NdFeO3. Water has the crucial role to saturate the coordination sphere of the metals. It has been shown that in absence of water, improved leaching rates can be achieved by the addition to the ionic liquid of small, metal coordinating groups such as CaCl2 and that an efficient leaching system has to contain a dissolving agent and a coordinating agent as well.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results received funding from the European Community's Horizon 2020 Programme ([H2020/2014–2019]) under Grant Agreement no. 674973 (MSCA-ETN DEMETER). This publication reflects only the authors' view, exempting the Community from any liability. Project website: http://etn-demeter.eu/. The authors also want to thank Magneti Ljubljana for providing the magnets and the production scrap samples for the carried out research, Less-Common Metals for the dross samples, Tony Debecker and Kevin Wierinckx for crushing the magnets, Jeroen Jordens for the particle size measurements, Tobias Hertel for the quantitative XRD analysis, Jeroen Sniekers for the SEM and EDS analysis, Mathieu Meerts for the viscosity measurements and Thomas Quanten for the luminescence measurements. TVDH thanks the FWO Flanders (Belgium) for a postdoctoral fellowship.Notes and references
- European Commission, Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs, Study on the Review of the List of Critical Raw Materials: Executive Summary, 2017 Search PubMed.
- O. Gutfleisch, M. A. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821–842 CrossRef CAS PubMed.
- T. E. Graedel, E. M. Harper, N. T. Nassar and B. K. Reck, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6295–6300 CrossRef CAS PubMed.
- E. Balomenos, P. Davris, E. Deady, J. Yang, D. Panias, B. Friedrich, K. Binnemans, G. Seisenbaeva, C. Dittrich, P. Kalvig and I. Paspaliaris, Johnson Matthey Technol. Rev., 2017, 61, 142–153 CrossRef.
- U.S. Department of Energy, Critical Materials Strategy, 2011 Search PubMed.
- UNEP, Metal Recycling. Opportunities, Limits, Infrastructure, International Resource Panel, 2007, vol. 8 Search PubMed.
- F. Habashi, Hydrometallurgy, 2005, 79, 15–22 CrossRef CAS.
- K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan and P. Veyssière, Encycl. Mater. Sci. Technol., 2nd edn, 2010, pp. 1–135 Search PubMed.
- K. Binnemans and P. T. Jones, J. Sust. Metall., 2017, 3, 570–600 CrossRef.
- A. P. Abbott, A. Z. M. Al-Bassam, A. Goddard, R. C. Harris, G. R. T. Jenkin, F. J. Nisbet and M. Wieland, Green Chem., 2017, 19, 2225–2233 RSC.
- A. P. Abbott, R. C. Harris, F. Holyoak, G. Frisch, J. Hartley and G. R. T. Jenkin, Green Chem., 2015, 17, 2172–2179 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 10031–10033 RSC.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 471–481 RSC.
- J. A. Whitehead, G. A. Lawrance and A. McCluskey, Green Chem., 2004, 6, 313 RSC.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 2150–2163 RSC.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- K. Larsson and K. Binnemans, J. Sust. Metall., 2015, 1, 161–167 CrossRef.
- T. Vander Hoogerstraete, B. Onghena and K. Binnemans, J. Phys. Chem. Lett., 2013, 4, 1659–1663 CrossRef PubMed.
- B. Onghena, J. Jacobs, L. Van Meervelt and K. Binnemans, Dalton Trans., 2014, 43, 11566–11578 RSC.
- P. Davris, D. Marinos, E. Balomenos, A. Alexandri, M. Gregou, D. Panias and I. Paspaliaris, Hydrometallurgy, 2018, 175, 20–27 CrossRef CAS.
- P. Davris, E. Balomenos, M. Taxiarchou, D. Panias and I. Paspaliaris, Berg- Huettenmaenn. Monatsh., 2017, 162, 245–251 CrossRef CAS.
- P. Davris, E. Balomenos, D. Panias and I. Paspaliaris, Hydrometallurgy, 2016, 164, 125–135 CrossRef CAS.
- P. Davris, E. Balomenos, D. Panias and I. Paspaliaris, in Rare Earths Industry, ed. I. B. De Lima and W. L. Filho, Elsevier, Boston, 2016, pp. 183–197 Search PubMed.
- N. I. Poulimenou, I. Giannopoulou and D. Panias, Mater. Manuf. Processes, 2015, 30, 1403–1407 CrossRef CAS.
- L. F. Audrieth, A. Long and R. E. Edwards, J. Am. Chem. Soc., 1936, 58, 428–429 CrossRef CAS.
- K. Starke, Can. J. Res., 1950, 28, 225–233 CrossRef.
- B. R. Hollebone, W. A. Morgan and D. R. Wiles, Trans. Metall. Soc. AIME, 1967, 239, 97–99 CAS.
- P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed.
- Z. Zhou, L. Guo, H. Yang, Q. Liu and F. Ye, J. Alloys Compd., 2014, 583, 21–31 CrossRef CAS.
- L. K. Jakobsson, G. Tranell and I. Jung, Metall. Mater. Trans. B, 2016, 48, 60–72 CrossRef.
- W. Sławiński, R. Przeniosło, I. Sosnowska, M. Brunelli and M. Bieringer, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 254, 149–152 CrossRef.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2002, vol. 7 Search PubMed.
- B. Thijs, PhD thesis, KU Leuven, 2007.
- T. Vander Hoogerstraete, B. Blanpain, T. Van Gerven and K. Binnemans, RSC Adv., 2014, 4, 64099–64111 RSC.
- A. Belsky, M. Helderman, V. L. Karen and P. Ulkch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- S. Cotton, Lanthanide and Actinide Chemistry, Chichester, UK, 2006 Search PubMed.
- K. V. Klementev, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 448, 299–301 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- V. A. Streltsov and N. Ishizawa, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1–7 Search PubMed.
- P. Nockemann, B. Thijs, K. Lunstroot, T. N. Parac-Vogt, C. Gorller-Walrand, K. Binnemans, K. Van Hecke, L. Van Meervelt, S. Nikitenko, J. Daniels, C. Hennig and R. Van Deun, Chem.–Eur. J., 2009, 15, 1449–1461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00198g |
| This journal is © The Royal Society of Chemistry 2018 |