
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigations of click-derived 1,2,3-triazoles as keystone ligands for complexation with transition metals: a review†
Tayebeh Hosseinnejad
 *,
Fatemeh Ebrahimpour-Malmir
and
Bahareh Fattahi
*,
Fatemeh Ebrahimpour-Malmir
and
Bahareh Fattahi
Department of Chemistry, Faculty of Physics & Chemistry, Alzahra University, Vanak, Tehran, Iran. E-mail: tayebeh.hosseinnejad@alzahra.ac.ir; Fax: +98-21-8804-1344; Tel: +98-9124775800
First published on 29th March 2018
Abstract
In recent years, metal complexes of organo 1,2,3-triazole click-derived ligands have attracted significant attention as catalysts in many chemical transformations and also as biological and pharmaceutical active agents. Regarding the important applications of these metal–organo 1,2,3-triazole-based complexes, in this review, we focused on the recently reported investigations of the structural, electronic, and spectroscopic aspects of the complexation process in transition metal complexes of 1,2,3-triazole-based click ligands. In line with this, the coordination properties of these triazole-based click ligands with transition metals were studied via several quantum chemistry calculations. Moreover, considering the complexation process, we have presented comparative discussions between the computational results and the available experimental data.
 Tayebeh Hosseinnejad | Tayebeh Hosseinnejad was born in 1979 in Tehran, Iran. She received her BSc, MSc, and PhD degrees from the University of Tehran in 2001, 2003, and 2007, respectively. She completed her doctoral thesis under the supervision of Prof. Hassan Behnejad in the field of nonequilibrium statistical thermodynamics. She currently works as an assistant professor in Alzahra University, Tehran, Iran. Her research interests focus on computational organic and organometallic chemistry and statistical thermodynamics. She has published more than 31 papers in reputed journals and has been recently serving as the guest editor of the thematic issue inCurrent Organic Chemistry. |
 Fatemeh Ebrahimpour-Malmir | Fatemeh Ebrahimpour-Malamir was born in Tehran, Iran in 1990. She received her B.Sc. degree in Applied Chemistry from Sharif University of Technology, Tehran, Iran (2014) and her M.Sc. degree in Organic Chemistry at Alzahra University, Tehran, Iran (2016) under the supervision of Prof. Majid M. Heravi and the advision of Dr Tayebeh Hosseinnejad. Her researches are concentrated on the catalyzed click reactions from the combined experimental and computational viewpoints. |
 Bahareh Fattahi | Bahareh Fattahi was born in Tehran, Iran in 1988. She received her B.Sc. in Applied Chemistry from Islamic Azad University, Tehran, Iran (2010) and her M.Sc. degree in Organic Chemistry at Alzahra University, Tehran, Iran (2016) under supervision of Professor Majid M. Heravi. She has also trained and investigated in the field of computational chemistry by the supervision of Dr Tayebeh Hosseinnejad. Her researches are focused on heterocyclic chemistry, catalysis, click reactions and computational chemistry. |
1. Introduction
1,3-Dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to afford a five-membered ring. Mechanistic study and the synthetic application of this important reaction were investigated initially via Rolf Huisgen's study reported in the 1960s.1,2Although this reaction is often referred to as a 1,3-dipolar cycloaddition reaction, it is worth noting that the specific reaction is actually between an organic azide and an alkyne to construct a common and useful heterocyclic system of 1,2,3-triazoles, as initially realized by Rolf Huisgen1,2 (Scheme 1). It was the American chemist K. Barry Sharpless who referred to this cycloaddition as “the cream of the crop” of click chemistry. Click chemistry reactions are usually high yielding and wide in scope and also generate only by-products that can be removed easily with no chromatography required. Click reactions are regioselective and stereospecific, simple to conduct and can be also performed in easily removable or benign solvents.3
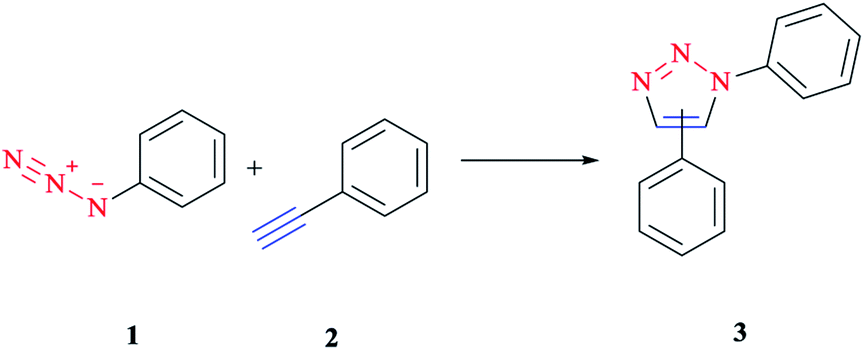 | ||
| Scheme 1 Synthesis of a mixture of 1,4- and 1,5-triazoles via an azide–alkyne cycloaddition reaction. | ||
An outstanding variant of the Huisgen 1,3-dipolar cycloaddition is the copper(I)-catalyzed cycloaddition click reaction, in which organic azides and terminal alkynes are reacted to afford 1,4-regioisomers of 1,2,3-triazoles as the only products under mild reaction conditions and in a much shorter reaction time.
After the discovery of the regioselective copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, so-called click reaction,4,5 the synthesis of click ligands (the archetypal example of click chemistry) attracted significant attention due to its reliability, simplicity, and mild reaction conditions.
It should be mentioned that the mild-functional-group-tolerant click synthesis of 1,2,3-triazole species led to their widespread applications as building blocks throughout synthetic chemistry. The application of the CuAAC click reaction in the synthesis of 1,2,3-triazoles as a stable linkage between chemical or biological components and a coordinating agent with metal ions and also their potential in catalytic systems were assessed, subsequently, increasing their impact far beyond the initial purpose of click chemistry.6–10
As a matter of fact, part of the special interest and focus on this type of reaction is due to the significant and diverse biological activities shown by 1,2,3-triazoles.11–15 These compounds can act as rigid linking units and can mimic the atom placement and electronic properties of a peptide bond, but with no hydrolytic cleavage effect. It is important to note that 1,2,3-triazoles have the following structural and electronic features, which have a significant high impact on their biological pharmaceutical applications. The dipole moment of triazoles is much stronger than that of an amide bond, which leads to increasing the hydrogen bond donor and acceptor properties of triazoles, subsequently enhancing the peptide bond mimicry.15,16
Due to the aforementioned potential of 1,2,3-triazoles to mimic the peptide bond role, several biological activities have been screened for 1,2,3-triazoles, including anti-HIV activity,17 selective β3 adrenergic receptor inhibition,18 anti-bacterial activity,19 and potent anti-histamine activity.20 It is important to note that 1,2,3-triazole click compounds have anticancer potency, which can be subsequently increased upon complexation with transition metal ions.21–23
On the other hand, based on the solid-phase click chemistry, switching from the solution-phase to solid-phase chemistry provides a playground for the application of triazoles to act as linkers to attach other functionalities to solid-phase resins24 as well as being able to functionalize metal surfaces for modification of their properties25 and to enable prompt adhesion.26
Recently, the promise of metal complexes of organo 1,2,3-triazole-based ligands has been demonstrated in the catalysis of some chemical transformations,27,28 such as hydroformylation and intramolecular hydroamination.29–36
It was shown that the catalytic efficiency of these complexes essentially depends on the appropriate electron-donating ability of atoms in 1,2,3-triazole moieties. Precisely speaking, the 1,2,3-triazole unit in the click ligand has an N-donor coordinating role to play through two types of nitrogen atoms: N(2) and N(3).37 From a mechanistic viewpoint, such a set of donor groups may stabilize intermediate species via coordination and increase reactivity and can also reduce the energy of transition states by partial dissociation to consequently improve the catalytic efficiency of the complex.
To the best of our knowledge, a few research studies have reported on the synthesis of 1,2,3-triazole organo-based ligands and the applications of their metal complexes as catalysts.38–42
In addition to their catalysis applications, some transition metal complexes of 1,2,3-triazole-based ligands have attracted interest for their luminescent properties and consequent applications as radiopharmaceutical agents in bioimaging techniques.43–45
Envisaging the importance of such complexes and their several applications, the structural, electronic, and spectroscopic features of these metal complexes with 1,2,3-triazole-based ligands have been widely investigated from experimental and computational viewpoints.
In line with this, several spectroscopic techniques have been used to assess the structural and electronic properties of these complexes. On the other hand, the veracity of computational methods was ascertained by comparison of the calculated results with experimental observations. However, the calculated geometrical parameters of these complexes could be compared with the available X-ray crystal structure and also the frequency calculations can be attributed to FT-Raman spectra. It should be mentioned that Raman spectroscopy can probe the nature of electronic transitions and also provide an insight into the structural character of the donor and acceptor MOs.46 Clearly speaking, the resonance effect enhances the Raman spectra bands corresponding to the structural changes associated with the photoexcitation.47–52 So, the pattern of band enhancements can be combined and interpreted with a computational analysis of the vibration normal modes.
On the other hand, the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energy gap of a complex can be correlated to its chemical reactivity and this information can be applied to interpret the catalytic role of these transition metal complexes of 1,2,3-triazole-based ligands.
In this route, the structural and electronic aspects of coordination in a series of mono and multinuclear transition metal complexes containing nitrogen-donating triazole ligands were investigated computationally through structural optimization calculations via density functional theory (DFT)53,54 and ligand field analysis.55 Moreover, the conformational fluxionality of flexible triazole-based click ligands in coordination with metal ion centers was assessed in the formation of trans or anti–trans conformers of six-membered metallacycles, and the isomerization mechanism was also studied to obtain the activation and entropy of the reactions via DFT computations.56,57
Moreover, the structural aspects of complexation between 1,2,3-triazole-based organochalcogen ligands and metal ions were studied via DFT computations and crystallographical methods and showed a disposition of donor atoms around the metal center with several coordination modes through the triazole skeleton.38–41
The functionalization effect on the structural and photophysical properties of the complexation of triazole-based ligands with transition metal ions have been studied and reviewed.58–62
In this respect, the relation between the energy adsorption band of the electronic spectra and metal-to-ligand charge transfer (MLCT) for all the complexes were modeled via DFT approaches.61,63–65
The coordination and electronic features of functionalized triazaphosphole click ligands and their substitution pattern were studied in comparison with triazole-based click ligands using quantum chemical computations, and the results were verified by crystallographical data.66
We recently investigated the synthesis and characterization of various heterogeneous nanocatalysts and their applications in various organic reactions, especially in the regioselective synthesis of 1,2,3-triazoles via a click reaction.67–75 We also recently focused on combining experimental and computational studies to provide a deeper insight into this important reaction. We reviewed the mechanistical aspects of the 1,3-dipolar Huisgen cycloaddition reactions and the regioselective behavior of metal-catalyzed click reactions using computational chemistry.5,76
Armed with these experiences, herein, we show that mild one-pot click methods can be used to readily and rapidly synthesize a family of functionalized multi-dentate 1,2,3-triazole ligand scaffolds, containing electrochemically, photochemically, and biologically active functional groups in a reliable yield. Overall, in this review, we present the experimental and computational features of the synthesis and properties of functionalized click ligands and their complexes with transition metal cations. The structural, photochemical, and biological properties of the complexes will be discussed via the spectroscopic and computational analysis.
2. 1,2,3-Triazole-containing chelators: click synthetic vision
Click ligands are generally prepared using the functional-group-tolerant Cu(I)-catalyzed 1,3-dipolar cycloaddition of organic azides with terminal alkynes (CuAAC reaction),10,77,78 which leads to the rapid generation of a wide range of functionalized ligands in association with the mild reaction conditions.The 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine 4 and [(4-R-1H-1,2,3-triazol-1-yl)methyl]pyridine 5 (Scheme 2) click ligands have emerged as readily modified bipy and phen surrogates.8,79,80
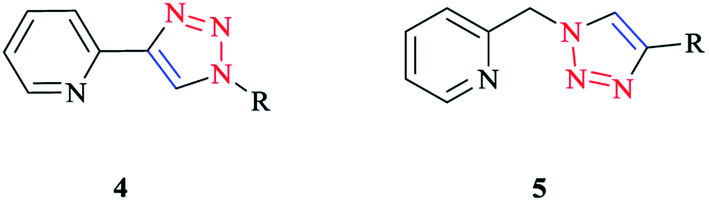 | ||
| Scheme 2 Bidentate N∧N chelators: 2-(1-R-1H-1,2,3-triazol-4-yl)pyridines (4) and [(4-R-1H-1,2,3-triazol-1-yl)methyl]pyridines (5). | ||
These bidentate click ligands form rhenium(I) complexes with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal:ligand ratio. The complexes of these ligands have wide applications in electronic81,82 and luminescent29,83–89 materials, catalysts,90–92 and in metallo-pharmaceuticals.93–95
:1 metal:ligand ratio. The complexes of these ligands have wide applications in electronic81,82 and luminescent29,83–89 materials, catalysts,90–92 and in metallo-pharmaceuticals.93–95
Moreover, with the goal of the development of novel rhenium(I) complexes as luminescent and pharmaceutical agents, rhenium(I) complexes from mono-functionalized 2-pyridyl-1,2,3-triazole derivatives (pyta) were synthesized as alternative ligands to 2,2′-bipyridines.63 Nevertheless, only a few of them possessed a tethering group for further coupling to a biomolecule.63,96
In short, the synthesis of novel pyta derivative grafted onto a bioactive pharmacophore was reported by considering the advantages of trendy click chemistry. Moreover, the corresponding rhenium complexes were synthesized and characterized in the solid and solution states.63,97
In another research, a family of electronically tuned fac-Re(CO)3Cl·2-(1-R-1H-1,2,3-triazol-4-yl)pyridine 6 complexes (Scheme 3) was generated using efficient click reactions.
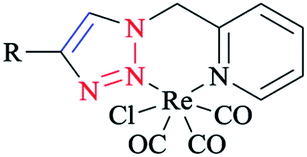 | ||
| Scheme 3 fac-Re(CO)3Cl·2-(1-R-1H-1,2,3-triazol-4-yl)pyridine (6) complexes. | ||
These safe one-pot CuAAC click methodologies produced a series of electronically modified 2-pyridyl-1,2,3-triazole ligand architectures, which were readily converted into the corresponding family of fac-[Re(CO)3Cl] complexes.
Elemental analysis demonstrated that the isolated powders were pure and showed that the bidentate ligands generated rhenium(I) complexes in a 1:1 metal/ligand ratio.64
The coordination chemistry of analogous donor-functionalized triazoles has been widely investigated over the past decades,8,59,79,80,82,94,98,99 whereas the chelating behavior of donor-functionalized 3H-1,2,3,4-triaza-phosphole derivatives is still practically uncharted.100–102 Clearly speaking, the click reaction between donor-functionalized azides and phospha-alkynes enables access to polydentate ligands, so that we can adjust the stereoelectronic properties based on the additional phosphorus heteroatom and the nature of the substituents. However, few investigations have been performed on the influence of an additional heteroatom on the properties of triazole derivatives. So far, only one example of such a species was assessed and characterized crystallographically, namely a tridentate ligand, tripodaltris (triazaphosphole), coordinated toward a Pt(0) center via P dentate, as reported by Jones et al.103 In the next research studies, the click reaction between 2-(azidomethyl)pyridine104 7 with tBu–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P105 and TMS–CP106 (trimethylsilyl is denoted as TMS) was reported according to the standard literature processes. Furthermore, the reaction between equimolar amounts of the azide and tert-butylphospha-ethyne was carried out at room temperature in toluene via a click methodology. Interestingly, it has been also reported that trimethylsilylphospha-ethyne reacts straightforwardly with 2-(azidomethyl)pyridine 7 and proceeds to give the TMS-substituted triazaphosphole derivatives (Scheme 4).66
P105 and TMS–CP106 (trimethylsilyl is denoted as TMS) was reported according to the standard literature processes. Furthermore, the reaction between equimolar amounts of the azide and tert-butylphospha-ethyne was carried out at room temperature in toluene via a click methodology. Interestingly, it has been also reported that trimethylsilylphospha-ethyne reacts straightforwardly with 2-(azidomethyl)pyridine 7 and proceeds to give the TMS-substituted triazaphosphole derivatives (Scheme 4).66
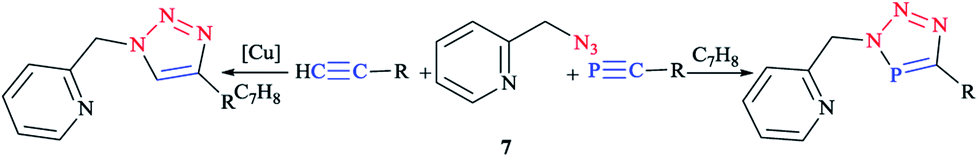 | ||
| Scheme 4 Syntheses of 2-pyridylmethyl-functionalized triazoles (left) and 2-pyridylmethyl-functionalized triazaphospholes (right). | ||
Click-derived3,107 ligands have been introduced as analogs of polypyridyl ligands, in particular of terpyridine (tpy),96,108–111 and successfully used to produce tridentate cyclometalating polypyridyl ligands, namely 1,3-dipyridylbenzene. In this respect, aryl azides react with diethynylbenzene building blocks, in good yields using a standard click procedure.112–114 With the purpose of blocking alternatives, methyl groups were placed at strategic positions when possible and reasonable.115 Therefore, o-xylene was selected as the central ring and mesityl species were selected for the clicked-on functionalities (Scheme 5). It should be mentioned that mesityl was kept as the reference moiety in all the other experiments to discuss the important influences of substituents directly attached to the cyclometalating phenyl ring or the opposing ligand (Scheme 6).
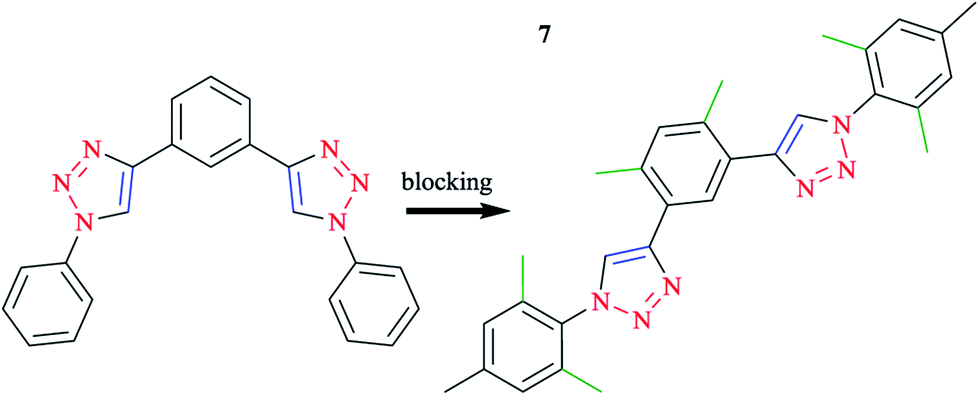 | ||
| Scheme 5 Optimization process, alternative coordination modes (left), and desired coordination mode (right). | ||
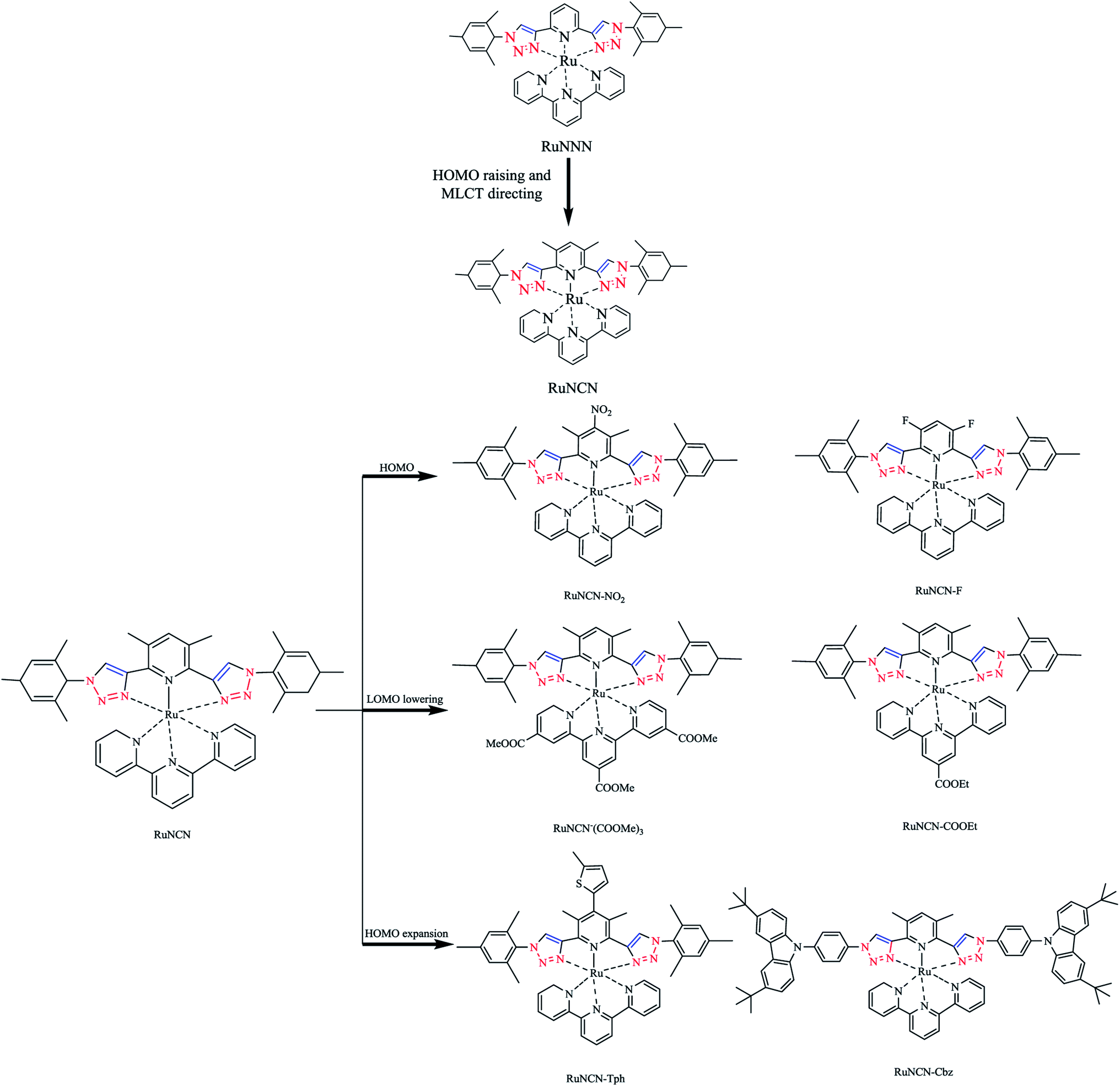 | ||
| Scheme 6 Synthesis procedures of ruthenium(II) complexes. | ||
In this context, a new and systematic series of click-derived tridentate cyclometalated Ru(II) complexes was considered to assess their potential for dye-sensitized solar-cell applications.85 These combined theoretical photophysical studies as well as electrochemical experiments led to a consistent and emergent explanatory picture of the new dyes.116
In another research, the synthesis of 2,6-bis(1,2,3-triazol-4-yl)pyridine 8 ligands were performed via the Cu(I)-catalyzed cycloaddition reaction of hydroxyl methylazide followed by a base-induced cleavage of formaldehyde. It could also be synthesized via a thermal azide–alkyne cycloaddition with azidomethyl pivalate and an internal alkyne. In this azide–alkyne cycloaddition, a mixture of adducts was produced and after cleavage and reprotonation, and free NH–triazole was obtained.7
As illustrated in Scheme 7, the corresponding ruthenium(II) complexes were synthesized in good yields using [RuII(tpy)(MeCN)3](PF6)2 (tpy = 2,2′:6′,2′′-terpyridine) or [RuII(tcmtpy)(MeCN)3](PF6)2 (tcmtpy = 4,4′,4′′-tricarboxymethyl-2,2′:6′,2′′-terpyridine) as precursors.116 The solubilities of the charge-neutral complexes were measured and it was shown that the alkyl chains increases the solubility, allowing the investigation of the photophysical and electrochemical properties (vide infra).117
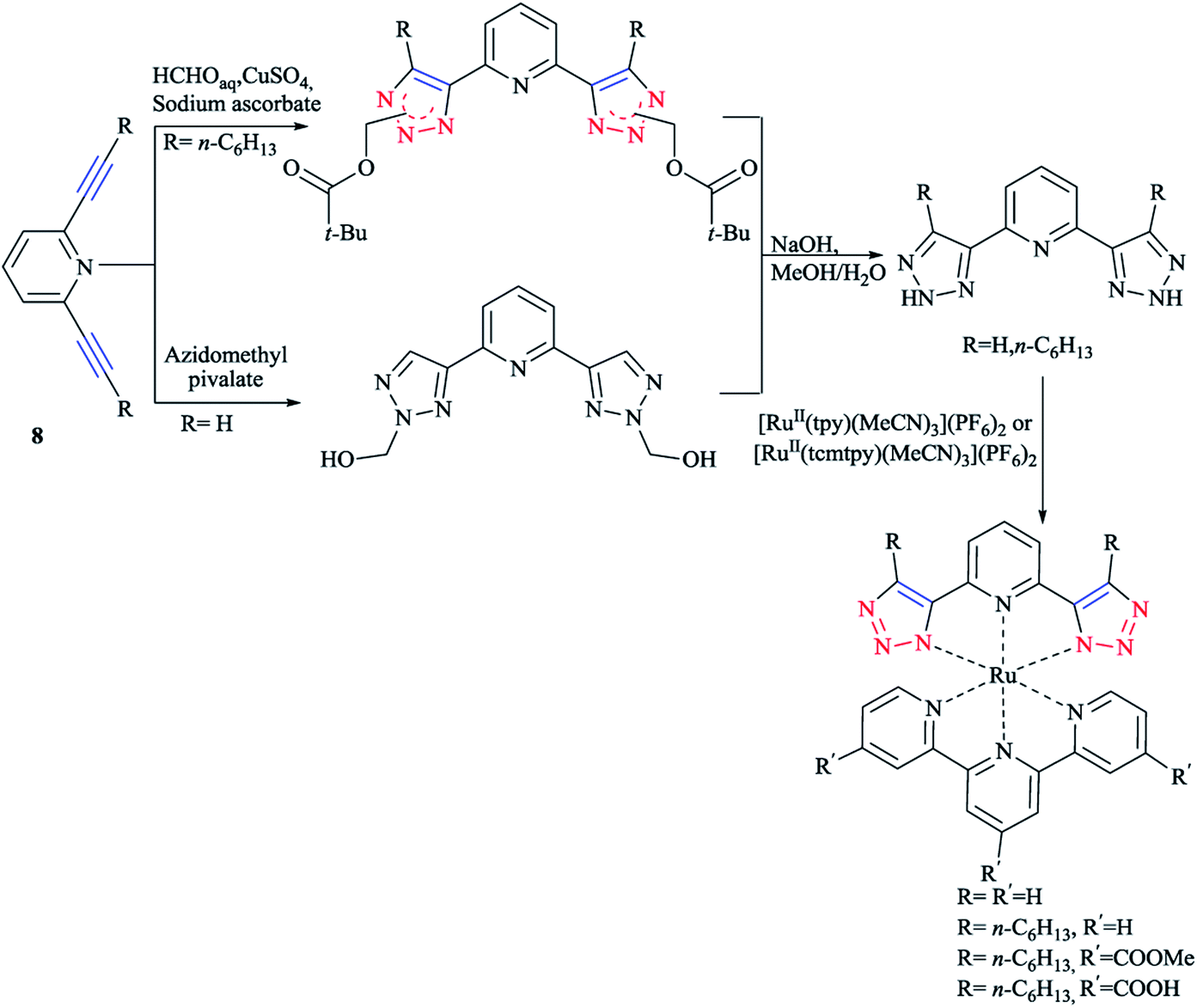 | ||
| Scheme 7 Synthesis of bis(tridentate)ruthenium(II) complexes. | ||
The synthesis and biological activity of Fe, Ru, Co, Pt, and Pd complexes of 2-pyridyl-1,2,3-triazole (Rpytri) 10 bidentate and 2,6-pyridylbis(1,2,3-triazole) (Rtripy) 12 tridentate click ligands (Scheme 8) were investigated. It is important to mention that Rpytri and Rtripy click ligands are commonly considered as analogs of the 2,2′-bipyridine (bipy) 9 and 2,2′:6′,2′′-terpyridine (terpy) 11 ligand systems (Scheme 8).8,79
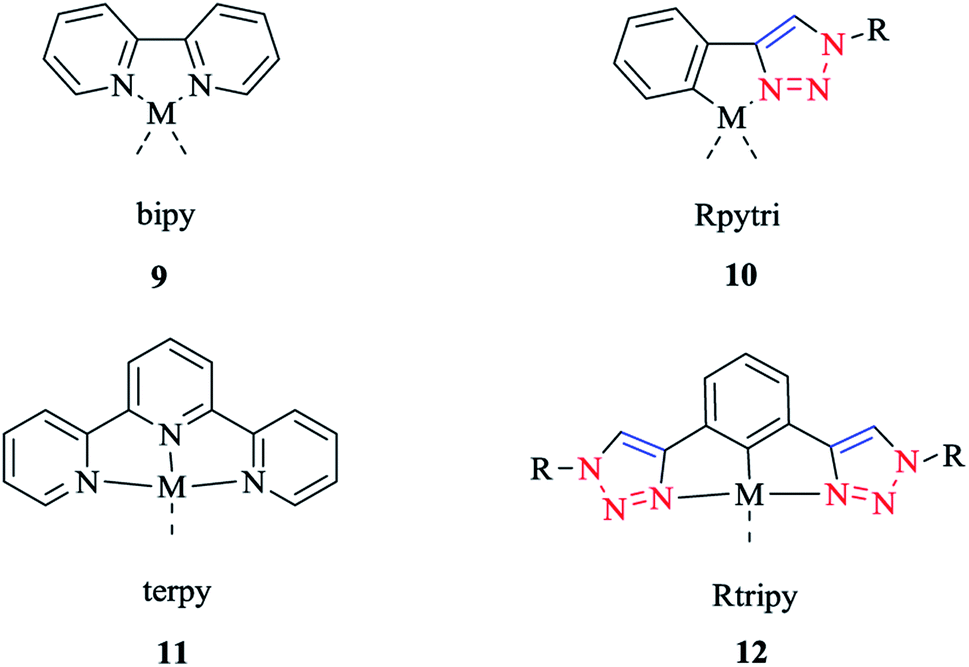 | ||
| Scheme 8 Structures of the 2,2′-bipyridine (bipy) (9) metal complex, 2-pyridyl-1,2,3-triazole (Rpytri) (10) analog, 2,2′:6′,2′′-terpyridine (terpy) (11) metal complex, and 2,6-pyridyl-bis(1,2,3-triazole) (Rtripy) (12) analog. | ||
In this vein, the CuAAC click reaction was exploited to synthesize a series of iron(II),118 ruthenium(II),119 and cobalt(III)120 bis-2-pyridyl-1,2,3-triazole (bis-Rpytri) ligands (Scheme 9). It was demonstrated that iron(II) bis-Rpytri complexes have no biological activity and are rapidly decomposed under biological conditions, while the more inert ruthenium(II) and cobalt(III) bis-Rpytri complexes showed biological stability. The same authors also produced a family of mono- and di-fac-rhenium(I) tricarbonyl complexes using the Rpytri and bis-Rpytri ligands with aliphatic and aromatic substituents and linkers (Schemes 9e and f). These showed modest biological activity, attributed to the low charge on the rhenium centers as well as their molecular shape.121
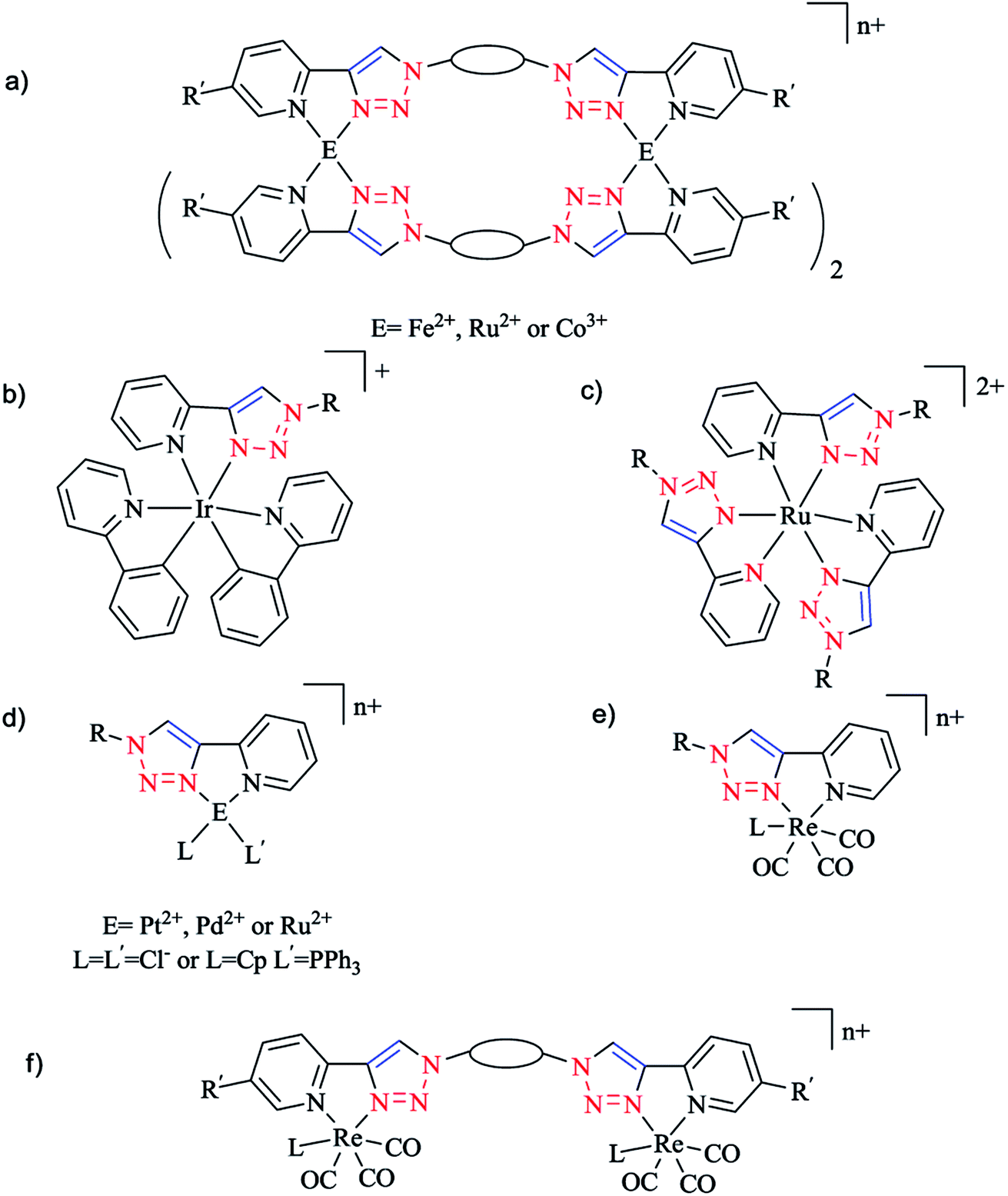 | ||
| Scheme 9 Structures of biologically active click Rpytri complexes. | ||
Moreover, Henderson and coworkers synthesized complexes of iridium(II) with Rpytri ligands including aromatic substituents and assessed their biological potency (Scheme 9b).23
In another work, Aldrich-Wright and coworkers generated a set of six heteroleptic platinum(II) complexes bearing chiral diaminocyclohexane and either aliphatic or aromatic Rpytri ligands (Scheme 9d).122
Pyridyl-substituted [Pd(Rpytri)Cl2] complexes were also synthesized by Sangtrirutnugul and coworkers. The regular Rpytri complex demonstrated a significant catalytic activity in the Suzuki reaction under aerobic conditions without catalyst degradation, indicating it was more resilient than the inverse Rpytri complex (Scheme 10).22,123
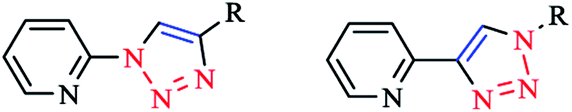 | ||
| Scheme 10 Structures of inverse-Rpytri (left) and regular-Rpytri (right) isomers. | ||
According to the procedure illustrated in Scheme 11, complexes 15 and 16 were synthesized as yellow solids by reacting [(MeCN)2PdCl2] with 13 and 14, respectively, in a 1:1 molar ratio, in THF solution at room temperature, leading to 90% and 85% yields for complexes 15 and 16, respectively. The two air- and moisture-insensitive complexes 15 and 16 were moderately soluble in CHCl3, CH2Cl2, and CH3OH, but had good solubility in CH3CN and DMSO. 13 and 14 and their Pd complexes 15 and 16 were characterized with 1H, 13C{1H} and 77Se{1H} NMR, HR-MS, and IR spectra, and the results were found to be in good confirmation with those reported earlier.37,124
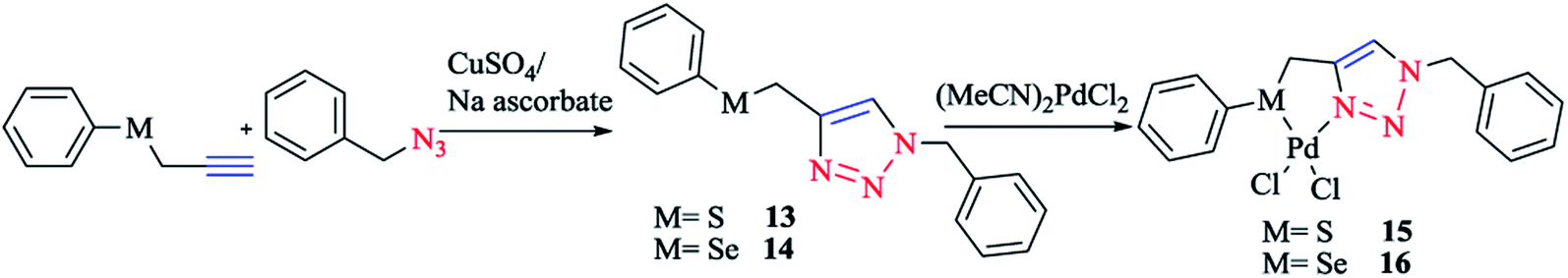 | ||
| Scheme 11 Synthesis of palladium(II) complexes based on the 1,2,3-triazole organosulfur/selenium ligand. | ||
In another work, [Pd(Rpytri)2]2+ complexes were synthesized from the ligands in a head-to-tail arrangement avoiding steric clashes and leading to favorable interligand hydrogen bonding.56,125 Furthermore, related platinum Rpytri compounds are known, including [Pt-(Rpytri)2Cl2] complexes,82,125–129 heteroleptic [Pt(Rpytri)2(L)2]2+ complexes, where L = phenylpyridine130,131 or diaminocyclohexane,122 and bis-pyridyl-triazolato complexes.132 Two bis-2-pyridyl-1,2,3-triazole ligands (L) with methylene spacers were also synthesized, which had the shortest possible “regular” pyridyl-triazole linkage, with the fewest possible degrees of freedom of any “hinged” bis-pyridyl-1,2,3-triazole. Then, theses ligands were used to produce bis-pyridyl-triazole [Pt(L)2]4+ complexes, which were assembled exclusively in a head-to-tail conformation. These Pt(II) metalloligands had vacant peripheral binding sites that could then be employed in the generation of heterometallic complexes. Mixed Pd(II)/Pt(II) macrocycles were synthesized in a concentration-dependent manner, while a mixed metal Cu(I)/Pt(II) was prepared independent of the concentration (Scheme 12).
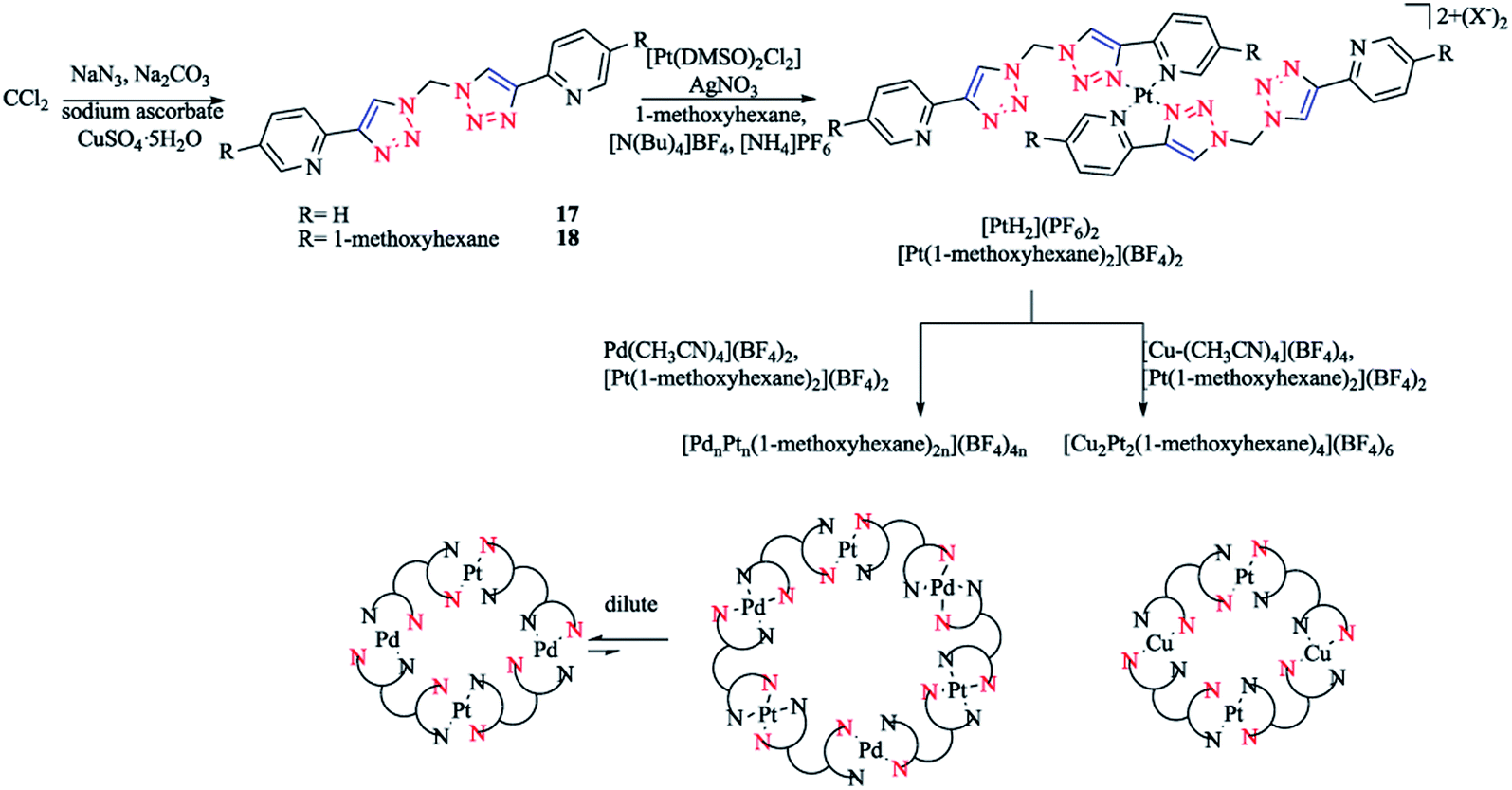 | ||
| Scheme 12 Synthesis of heterometallic [MnPtn(L)2n]x+ macrocycles based on dichloromethane-derivative bis-2-pyridyl-1,2,3-triazole ligands. | ||
While a wide range of bispyridyltriazole ligands133–135 are known, the previously unreported methylene-linked ligands were targeted, to reduce the number of degrees of freedom in the ligand architecture.136–138
The 1,1-bis(2-pyridyl-1,2,3-triazol-4-yl)methane 17 ligand and 1,1-bis(4-(5-(hexyloxy)pyridin-2-yl)-1H-1,2,3-triazol-1-yl)-methane 18 ligand were synthesized through a CuAAC click reaction from sodium azide and dichloromethane(DCM) in a microwave in dimethyl formamide (DMF) up to 110 °C in the presence of carbonate, leading to the explosive diazide in situ, which then reacts with 2-ethynylpyridine or 2-(2-(trimethylsilyl)ethynyl)-5-(hexyloxy)pyridine.139
In the following (Scheme 13), we display the synthesis of a click ligand through a CuAAC one-pot reaction between phenylacetylene 2 and 2-picolylazide 19 and also the coordination of a click ligand to PtII, PdII, CuII, RuII, and AgI using cis-[PtCl2(DMSO)2], [Pd(CH3CN)4](BF4)2, CuCl2, [RuCl(μ-Cl)(η6-p-cymene)]2, and AgNO3. In the case of [Pd(CH3CN)4](BF4)2 and CuCl2, bis-bidentate complexes were obtained, respectively. Synthesis of these two compounds was repeated with a ligand–metal precursor molar ratio of 2:1. Other stable products crystallized out from the reaction mixtures and were then isolated by filtration in a 60–82% yield.99
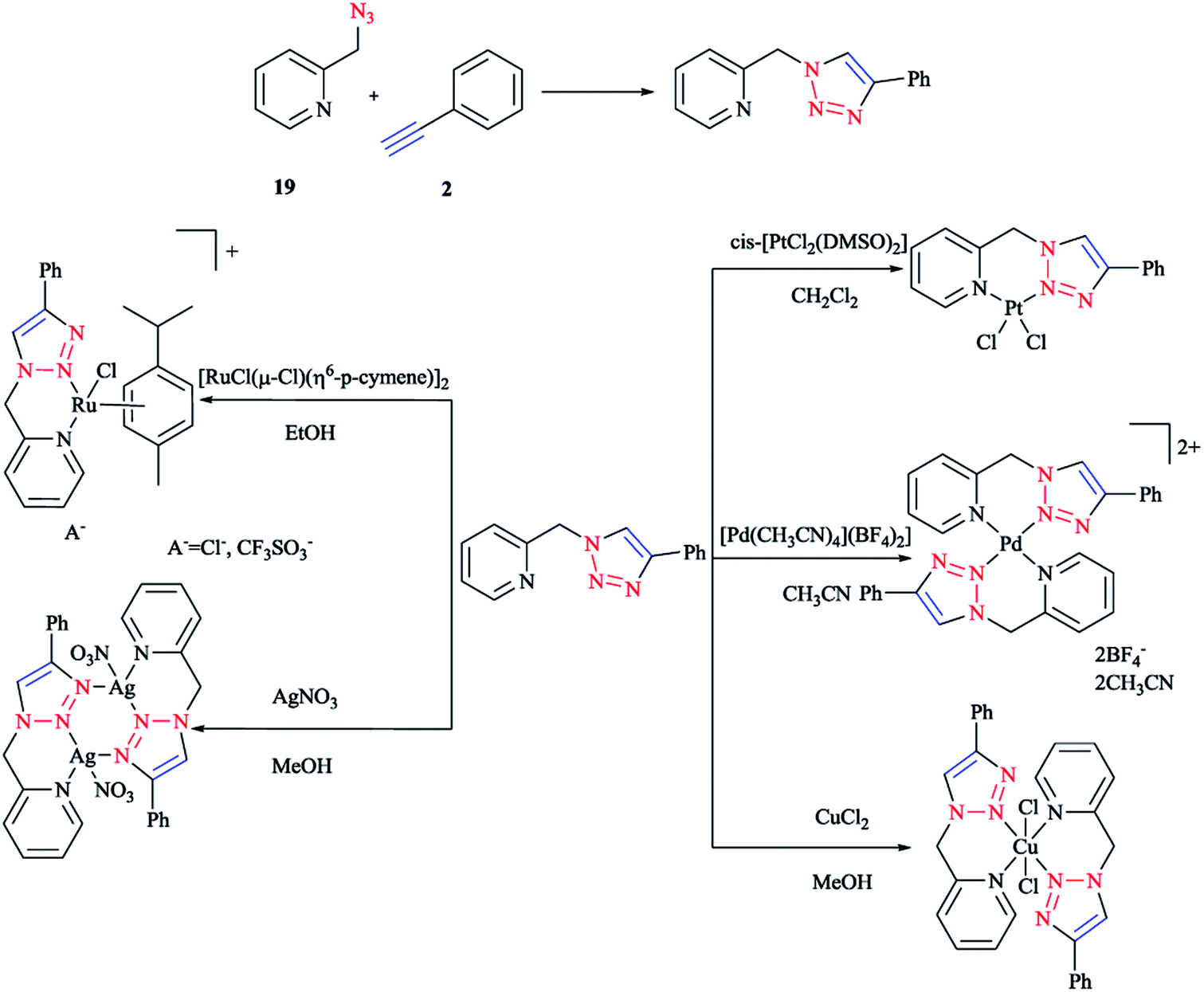 | ||
| Scheme 13 Syntheses of 1-(2-picolyl)-4-phenyl-1H-1,2,3-triazole ligand and some of the complexes investigated. | ||
The synthesis of cyclometalating click ligands was investigated in a highly efficient one-pot consecutive process. In this regard, the acetylene derivative was reacted with the organic azide.140,141 It is important to note that there was no necessity for any further copper(I) catalyst after the formation of the aryl azide for the click reaction. So, 1-decyl-4-phenyl-1H-[1,2,3]triazole 20 and 4-decyl-1-phenyl-1H-[1,2,3] triazole 21 were produced as the potential ligands for the formation of iridium(III) complexes in high yields (Scheme 14).114
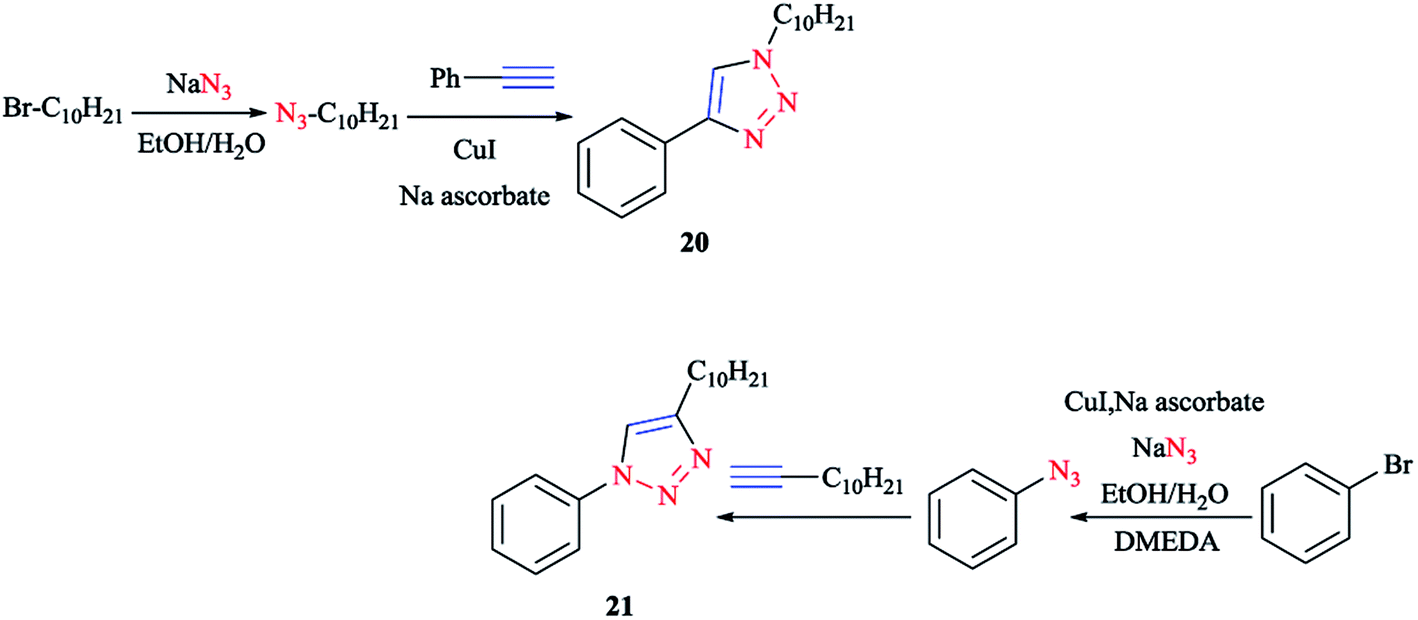 | ||
| Scheme 14 Syntheses of 1- and 4-decyl-4-phenyl-1H-[1,2,3]triazole ligands. | ||
Metallo-macrocycles can be assembled by arranging bis-1,2,3-triazol-5-ylidene 22 carbene ligands in conjunction with linear Ag(I) or Au(I) ions. Crudden and coworkers produced a Ag(I) macrocycle from a bis-1,3(1-aryl-1,2,3-triazol-4-yl)benzene ligand (Scheme 15).142 An equilibrium mixture of mononuclear [AgL]+ and macrocyclic [Ag2L2]+ complexes were synthesized by Frutos and Toree by applying a 1,4-xylyl linked steroid substituted bis-1,2,3-triazol-5-ylidene ligand. In order to expand the range of Au(I) 1,2,3-triazol-5-ylidene catalysts, Crowley and coworkers synthesized a di-Au(I) macrocycle with a 1,3-phenyl linked bis-1,2,3-triazol-5-ylidene ligand.143
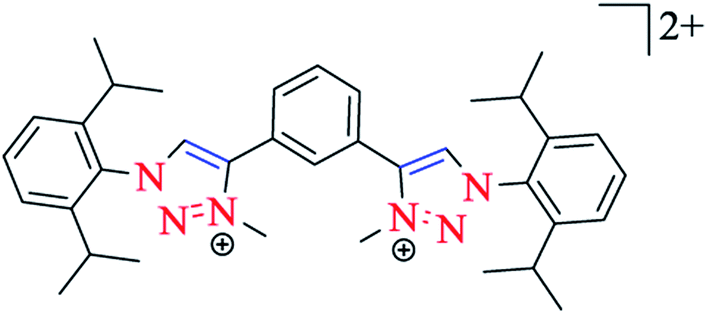 | ||
| Scheme 15 Structure of the bis-monodentate 1,2,3-triazole-5-ylidene (22) ligand. | ||
Furthermore, White and Beer produced four bis-monodentate ligands with a 1,3-(5-tert-butyl)phenyl core.144 The triazole rings bound via N1 or C4 atoms and C4-connected ligands gave cluster-like compounds consisting of four Ag(I) cations and four ligands, while three triazole nitrogens were coordinated to each Ag(I) center and the fourth coordination bond for each metal ion was to water.
Building on their previous Ag(I) metallocycle work, Crowley and coworkers exploited the functional group tolerance of the CuAAC “click” reaction to synthesize a structurally diverse family of bis-triazole ligands. Reaction of a suitable dialkyne with two equivalents of azide gave a range of bis-monodentate ligands with the two triazole groups separated by a linker (either propyl, 1,3-phenyl or 1,4-phenyl units), and substituted with a wide array of benzyl, aryl, and alkyl groups.21,145,146
3. Computational study of electronic energy properties in transition metal complexes of triazole-based ligands
The coordination of Ni(II) with the tripodal ligands tpta (tris[(1-phenyl-1H-1,2,3-triazol-4-yl)-methyl]amine) 23, tbta (tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-amine) 24, and tdta (tris[(1-(2,6-diisopropyl-phenyl)-1H-1,2,3-triazol-4-yl)methyl]amine) 25, and the bidentate ligand pyta (1-(2,6-diisopropylphenyl)-4-(2-pyridyl)-1,2,3-triazole) 26 led to the [Ni-(tpta)2](BF4)2 23′, [Ni(tbta)2](BF4)2 24′, [Ni(tdta)2](BF4)2 25′, and [Ni(pyta)3](BF4)2 26′ complexes (Scheme 16).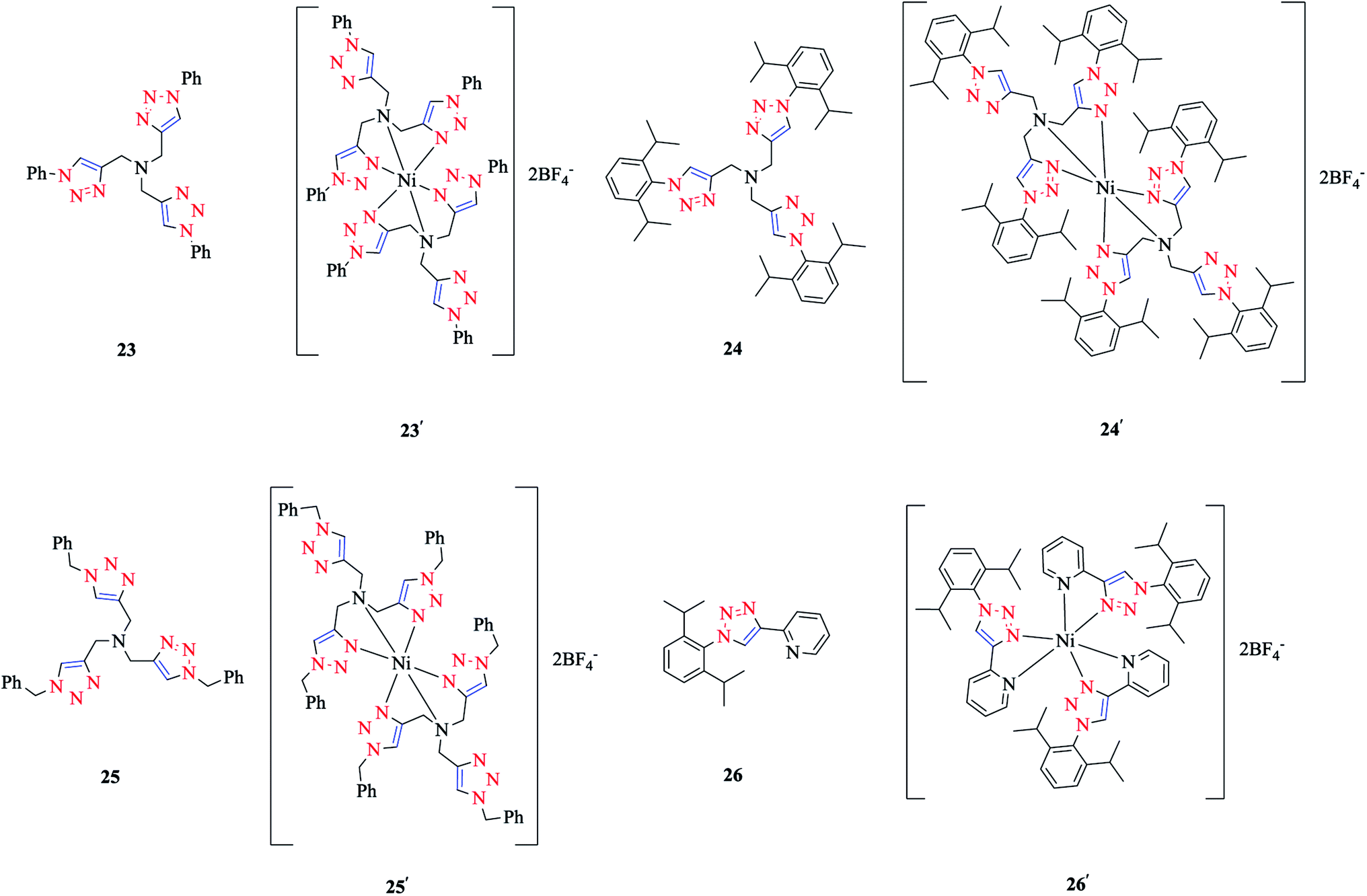 | ||
| Scheme 16 tpta, tbta, tdta, and pyta ligands and their corresponding Ni(II) complexes. | ||
In order to obtain a better insight into their absorption spectra, quantum chemistry calculations at the NEVPT2 level of theory147–154 were performed. Based on the NEVPT2 calculated values, the electronic transitions in the absorption spectra of complexes 23′, 24′, and 26′ were assigned (while corresponding calculations for complex 25′ could not be done due to its large size). The energies of the spin-allowed d–d transitions of complexes 23′, 24′, and 26′ were calculated comparatively based on their X-ray and DFT-geometry-optimized structures.
The experimental absorption spectrum of 23′ in CH3CN was compared with the NEVPT2 calculated values. All the three main bands were adjusted well with three Gaussian distributions, indicate the three-times-three possible transitions, with the sum of these distributions in reliable agreement with the experimental spectrum.
The absorption spectra of 23′, 24′, and 26′, which were calculated at the SA-CASSCF/NEVPT2 level of theory in acetonitrile included 24 excited states (9 triplets and 15 singlets, which with the triplet ground state resulted in a total of 45 microstates of d8). All the triplet states of the absorption spectrum could be attributed to the spin-allowed transitions, while the spin-forbidden transitions were not elucidated.
It should be noted that the excited singlet states can be ascertained by second-order perturbation theory (PT2) and make a minor contribution. In particular, when comparing SA-CASSCF155 with NEVPT2 (ref. 156) approaches, since the dynamic electron correlation is added by the NEVPT2 method, the calculated values of the excited triplet states increase.
Consequently, the zero-field splitting (zfs) parameters D and E are decreased by almost 50%, while zfs qualitatively is inversely proportional to the excited state energies, which can be ascribed as the covalency of the system. The final D values calculated by quasi degenerate perturbation theory (QDPT)157 were 2.94 and 3.79 cm−1 for 23′ and 24′ complexes, respectively, which were in reliable agreement with the values obtained by high-frequency and high-field electron paramagnetic resonance HFEPR.158
Comparison of QDPT with PT2 approaches revealed that QDPT yielded more accurate results due to the fact that QDPT considers all contributions to the zfs, but this approach does not allow discussing the single contribution of excited states to the total zfs in comparison with PT2.
For complex 26′, the angular overlap model (AOM) parameters159 were derived via a least-squares fit to the V matrices (with a rather small standard deviation) by using the exact structural data of complex 26′ obtained from the X-ray and DFT-optimized structure.
A comparison of the calculated results for the two ligands revealed that Ni–N bonding was dominated by Ni–Nta and Ni–Npy σ-antibonding interactions. On the other hand, the parameters epyπs = −131 cm−1 and etaπs = −276 cm−1 (attributed to the out-of-plane π-bonding) had very small and negative values.
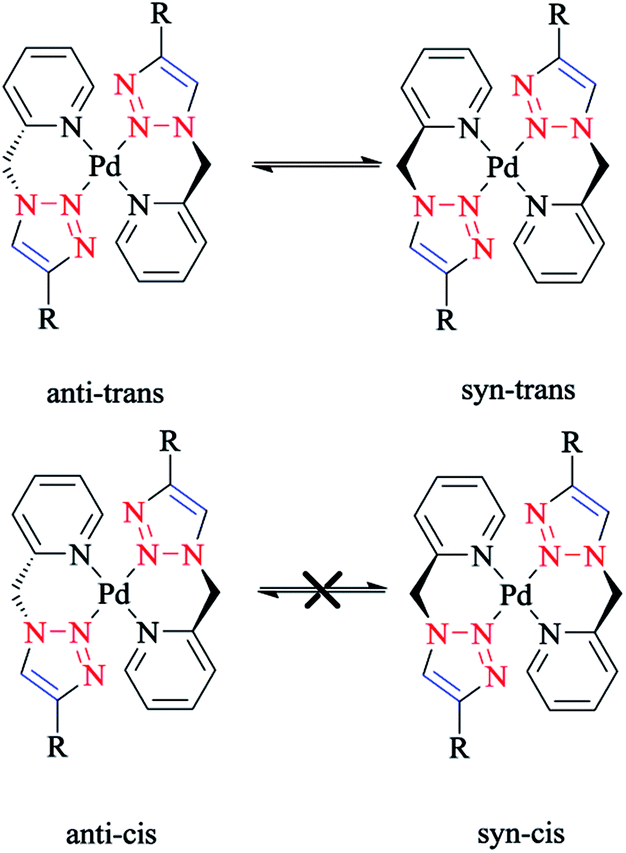
In another research study, the interconversion between two boat conformers in a palladium(II) complex of the flexible click chelator (Pd{(py)2CHNHR}Cl2)160 was assessed computationally. The rate constants for the interconversion between the two conformers were calculated. The calculated values for the activation enthalpy and activation entropy (from the Eyring plot) were 13.0 ± 0.4 kcal mol−1 and 2.7 ± 1.6 cal mol−1 K−1, respectively, which were in reliable agreement with other literature results for boat-to-boat inversions in some similar palladocycles.161 It should be noted that the low activation entropy can be attributed to a nondissociative isomerization mechanism.
The species involved in equilibrium, including syn- and anti-conformers of the trans isomer, namely 27 and 28, respectively, and the structure with an anti–cis arrangement 29 (Scheme 17) were investigated via DFT calculations. The calculated geometrical data clearly indicated that the change in the square-planar core was not considerable when going from the anti (27) to the syn (29) conformer; except the Pd–N2 bond, which was slightly elongated to 0.014 Å.
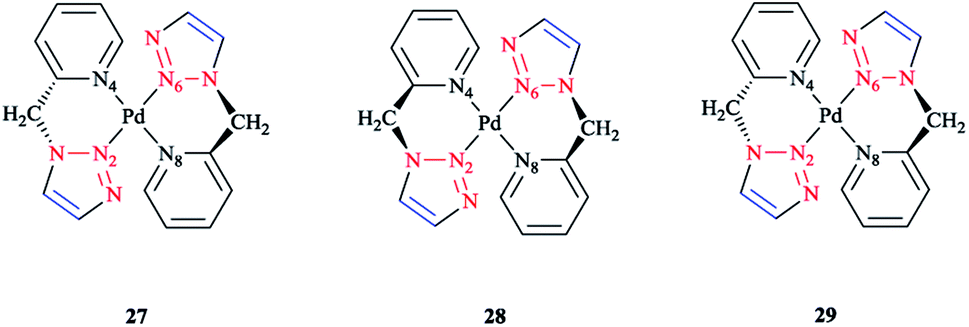 | ||
| Scheme 17 Schematic structures of anti–trans-[Pd(L)2]2+, syn–trans-[Pd(L)2]2+, and anti–cis-[Pd(L)2]2+ isomers based on the 4-phenyl-1-(2-picolyl)-1,2,3-triazole ligand (L). | ||
Considering the calculated values of the electronic energy as well as the gas phase free energy, isomer 28 was found to be more stable than 27 by about 1 kcal mol−1, thus predicting the equilibrium was shifted in the direction of 28.
Moreover, the calculated value of steric repulsion, DEster, (which was defined as the sum of the Pauli repulsion and electrostatic interaction via the Ziegler–Rauk decomposition framework162,163) was smaller between ligands in 28 (DEster = −48.68 kcal mol−1) than in 27 (DEster = −50.36 kcal mol−1), while the calculated value of the orbital interaction (DEoi) was maintained at about 463 kcal mol−1. This feature could be due to the slightly shorter N4–N6 distance in 27 (3.016 Å) than in 28 (3.062 Å).
To assess the kinetics of the isomerization procedure theoretically, we focused on the determination of the transition state (TS) connecting 27 and 28 via a one-step mechanism, as illustrated in Fig. 1. It was shown that the calculated activation barrier values and the entropy contributions were in a good agreement with the experimental data.57
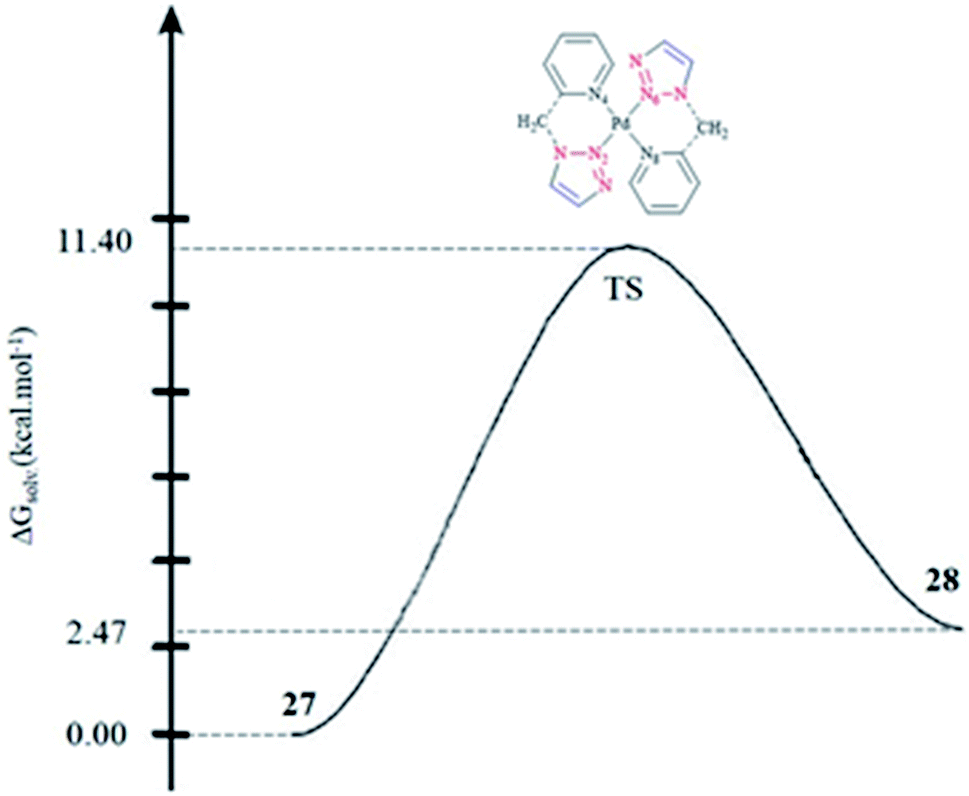 | ||
| Fig. 1 One-step mechanism for the isomerization of 27 into 28. | ||
It was shown that in the following Pd complexes, the calculated electron densities at Se, Pd, and N(3) of the triazole ring were higher in complex 16 than in complex 15, mainly due to the strong metal–ligand interaction in complex 16 and its subsequent higher stability and lower reactivity relative to complex 15.37,56,126 Moreover, in these complexes, there was a good agreement between the experimentally observed and calculated bond distances and angles. However, some of calculated Pd–M bond distances (M = S/Se) deviated from the corresponding observed ones.124
In another research study, the geometric and electronic structures of ruthenium complexes with bidentate ligands, including bis pyridyl and bis triazole ligands (denoted as bpy and btz ligands, respectively), were investigated computationally (Scheme 18). In the first step, the optimized geometry of the complexes were obtained and then the localization of the dz2-like and dx2−y2-like antibonding dσ* orbitals were assessed to recognize the participating Ru–N bonds. In the next step, a pair of stimulated starting geometries was designed by elongation of the main participating Ru–N bonds and were then optimized with a spin multiplicity of 3.
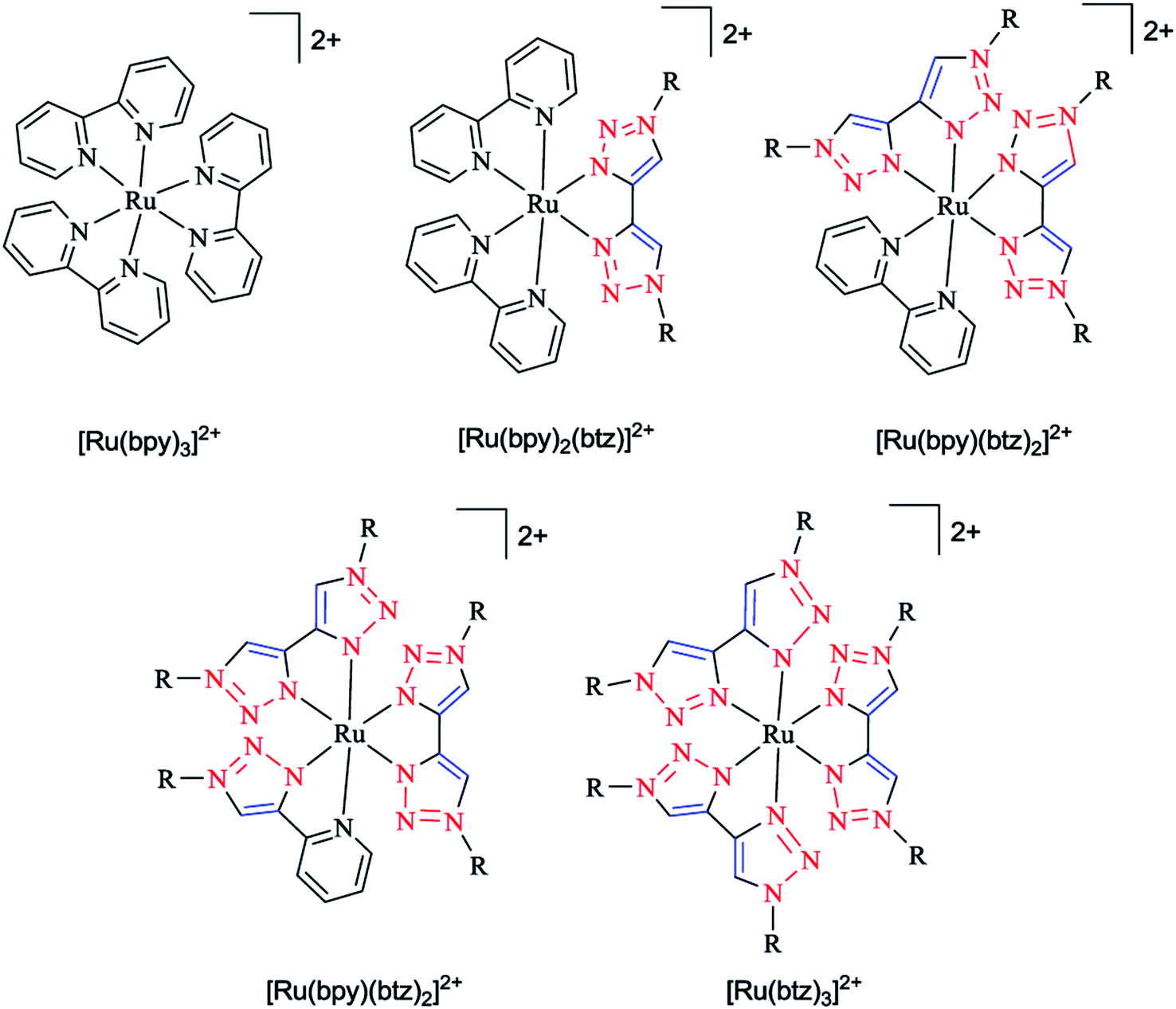 | ||
| Scheme 18 Structures of Ru(II) tris(bidentate) complexes (in experimental studies R = benzyl, to limit the computational expense in the calculations. R is simplified to methyl). | ||
It was shown that the Ru–N bond length increases due to the population of dz2-like dσ* orbitals. Moreover, the dx2−y2-like dσ* orbital population leads to the distortion of all three ligands. It was also demonstrated that a metal–ligand orbital overlap could be deducted based on the elongation of both the Ru–N bonds of one chelate ligand participating in the dσ* orbital, showing the stabilizing electronic effect.164
4. Structural properties and frontier molecular orbital analysis in the complexation reactions
4.1 Ni(II) and Zn(II) complexes
The coordination behaviors of the complexes of Ni(II) with tripodal and bidentate ligands were investigated using X-ray diffraction studies and computational methods, specifically [Ni(tbta)2](BF4)2 24′ as a tripodal ligand and [Ni(pyta)3](BF4)2 26′ as a bidentate ligand. In the [Ni(tbta)2](BF4)2 24′ complex, the Ni(II) center was coordinated by four triazole (Nta) and two trans amine (Nam) nitrogen donors, in a distorted-octahedral space, while in [Ni(pyta)3](BF4)2 26′, Ni(II) was coordinated by pyridine (Npy) and triazole (Nta) N-donors in a meridional arrangement.As could be deduced from the crystal structure of [Ni(tbta)2](BF4)2 24′, the nickel center was coordinated by each tbta ligand via the nitrogen atoms of two 1,2,3-triazole rings and the central amine nitrogen donors, while the third triazole ring of the ligand remained uncoordinated. The three nitrogen donors of triazole rings are located at the equatorial coordination position around the nickel center. It should be mentioned that the amine nitrogen atoms are poorer donors than the triazole nitrogen atoms, and this property leads to the larger Ni–Nam distances in comparison with the Ni–Nta distances. Furthermore, it was shown that the average values of the axial–equatorial angle, denoted by α, and distortion angle, denoted by δα, were significantly distorted from 90°. In the next step, the structural properties of the complexes were reproduced by using DFT geometry optimization in conjunction with a polarized continuum model (COSMO),165 which showed a good agreement with the X-ray results.55
Petitjean and coworkers investigated systematically the synthesis and structural properties of Fe(II), Ni(II), and Zn(II) ions with 1,2,3-triazol-4-yl pyridine chelators. They presented the spacer and substitution pattern for the complexation and assessed the various factors affecting the self-assembly procedure and also the stereochemical features of these architectures.135
In another work, it was reported that the reaction of bis-(2-pyridyl-1,2,3-triazole) ligands including 1,3-propyl, 1,4-xylyl, or 2,6-dimethyl naphthalene spacers with Fe(II) or Ni(II) ions led to the production of M2L3 cylinders. It should be mentioned that by using several functional groups, a large series of bis-(2-pyridyl-1,2,3-triazole) ligands were synthesized via the CuAAC procedure and were then studied in the reaction with Fe(II), Ni(II), and Zn(II) ions.135,166
In the case of Zn(II) complexes, the synthesis and structure of the [2 + 2] macrocycle of Zn(II) ions with the bis-(4-(2-pyridylthiomethyl)triazolyl) ligand including a pentyl linker were assessed. The zinc(II) ions were coordinated to two N3 triazole nitrogen atoms and two chloride anions in a tetrahedral coordination geometry. It was also demonstrated that the ethyl- and butyl-linked ligands formed [(ZnCl2)2L] dinuclear complexes so that each metal ion was coordinated to both a triazole and chelating pyridine.21,167
4.2 Pd(II) and Pt(II) complexes
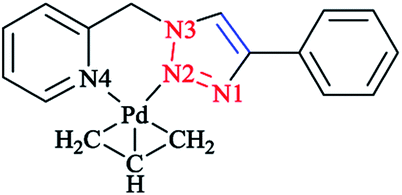
The calculated optimized geometry for the [Pd(η3-C3H5)(1-(2-(methylthio)ethyl)-4-phenyl-1H-1,2,3-triazole)]+ (Scheme 19) complex 30 was assessed computationally. The computations were performed at the M06 level of theory, including with the C-PCM solvation model and in the presence of acetone as a solvent, and the results highlighted the binding geometry adopted by the ligand.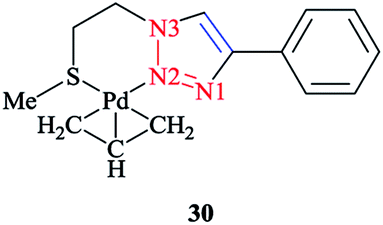 | ||
| Scheme 19 Structure of the [Pd(η3-C3H5)(1-(2-(methylthio)ethyl)-4-phenyl-1H-1,2,3-triazole)]+ complex (30). | ||
Then, a comparison was made between the calculated results and the single-crystal data for the tetrafluoroborate salt of the cationic complex, which demonstrated a reliable confirmation. Moreover, based on the X-ray data, it could be seen that palladium was coordinated to N2 and the sulfur atoms and consequently formed a six-membered ring with the metal center.
The phenyl substituent and the triazole were coplanar, while the angle between N2, Pd, and S was around 95°. Another important structural feature of the complex was that the C-allyl-terminal trans to S and trans to triazole-N2 deviated from planarity specified by Pd, S, and N2, by 0.180 Å and 0.095 Å, respectively. It is important to mention that there was a similar coordination behavior between the triazole species in [Pd(η3-C3H5)(1-(2-(methylthio)ethyl)-4-phenyl-1H-1,2,3-triazole)]+ 30 with the pyridine–triazole Pd(II) allyl complex (Scheme 20).91,168 Strictly speaking, coordination of the S-donor moiety of (1-(2-(methylthio)ethyl)-4-phenyl-1H-1,2,3-triazole) to the [Pd(η3-C3H5)]+ can be evaluated with other neutral bidentate ligands consisting of thioether groups and can generate six-membered rings with the metal center.169–173 This coordination feature was assessed computationally in a number of complexes through adopting starting geometries where the coordination of the triazole fragment occurred through the N1 nitrogen atom. The calculated results demonstrated that, in all cases, the obtained optimized structure was similar to the X-ray structural data, where a Pd–N2 bond was clearly formed.174
 | ||
| Scheme 20 Structure of the [Pd(η3-C3H5)(2-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-pyridine)]+ complex. | ||
In another research study, experimental and theoretical DFT studies on the structural properties of [Pd(L)2](BF4)CH3CN (L = 4-phenyl-1-(2-picolyl)-1,2,3-triazole) as a bischelate complex showed that the central square-planar palladium(II) cation was coordinated by two L substrates in trans position, each through pyridine and triazole N2 nitrogen atoms, generating two six-membered metallacycles. More precisely, anti–trans-[Pd(L)2]2+ and syn–trans-[Pd(L)2]2+ were produced as two boat-like conformations, in which the picolyl methylene carbons were anti or syn, respectively, depending on the palladium coordination plane, and could be easily converted to each other in solution (Scheme 21). It should be mentioned that [Pd(L)2](BF4)CH3CN was synthesized from 4-phenyl-1-(2-picolyl)-1,2,3-triazole (L) and [Pd(CH3CN)4](BF4)2 in CH3CN.99
 | ||
| Scheme 21 The solid-state structures under investigation by their molecular drawing (R = Ph). | ||
Frontier molecular orbital analysis was also performed for palladium(II) complexes of sulfur–selenium 1,2,3-triazole-based ligands using DFT calculations to rationalize the nature of bonding within these complexes and their reactivity. In both the complexes, the HOMOs were mainly located over Pd and Cl atoms and included the interaction of a d orbital of Pd(II) and a p orbital of chlorine atoms. More precisely, in the HOMO-1 of both complexes' metal d orbitals, p orbitals of a chalcogen donor atom, chlorine atoms, and one of the nitrogen of the triazole ring overlap with each other. Since the high reactivity of a complex may be partially related to a low value for the HOMO–LUMO energy gap, among the two complexes, complex 15, which had the lower HOMO–LUMO energy gap, was supposed to be a better catalyst than 16, as was observed experimentally.124
In another research study, the chelation properties of some mononuclear bis-bidentate triazole-based complexes (as illustrated in Scheme 22) were reported via X-ray crystallographical analysis. The X-ray structures of Pt and Pd complexes showed a square-planar coordination environment of the central Pt and Pd. The coordination sphere around the platinum atom consisted of two nitrogen atoms of the ligand and two chloride ions in the cis position, while in the palladium mononuclear bis-bidentate complex, the palladium ion is trans-chelated by two ligands. There was a close agreement between the bond distances in the coordination sphere of platinum in its complex with those found in the X-ray crystal structure of other square-planar complexes of Pt(II).175 In the crystal geometry of the palladium complex, it was shown that the diagonal bonds are nonequivalent, so that N2–Pd is shorter than N4–Pd. In a copper complex, the metal ion is in a distorted-octahedral coordination environment with two additional chloride ions involved, and the ligands are located in a trans configuration comparable to that of the palladium complex. This displacement of two axial N2 atoms from the basal plane formed by two pyridine N4 atoms and two chloride ions leads to a pyramidal distortion of the octahedral geometry. In another reported ionic complex of [Cu(L1)2(CH3CN)-(ClO4)](ClO4),176 the copper cation displays the square-planar geometry of four nitrogen atoms with CH3CN and ClO4− ligands at the axial positions.
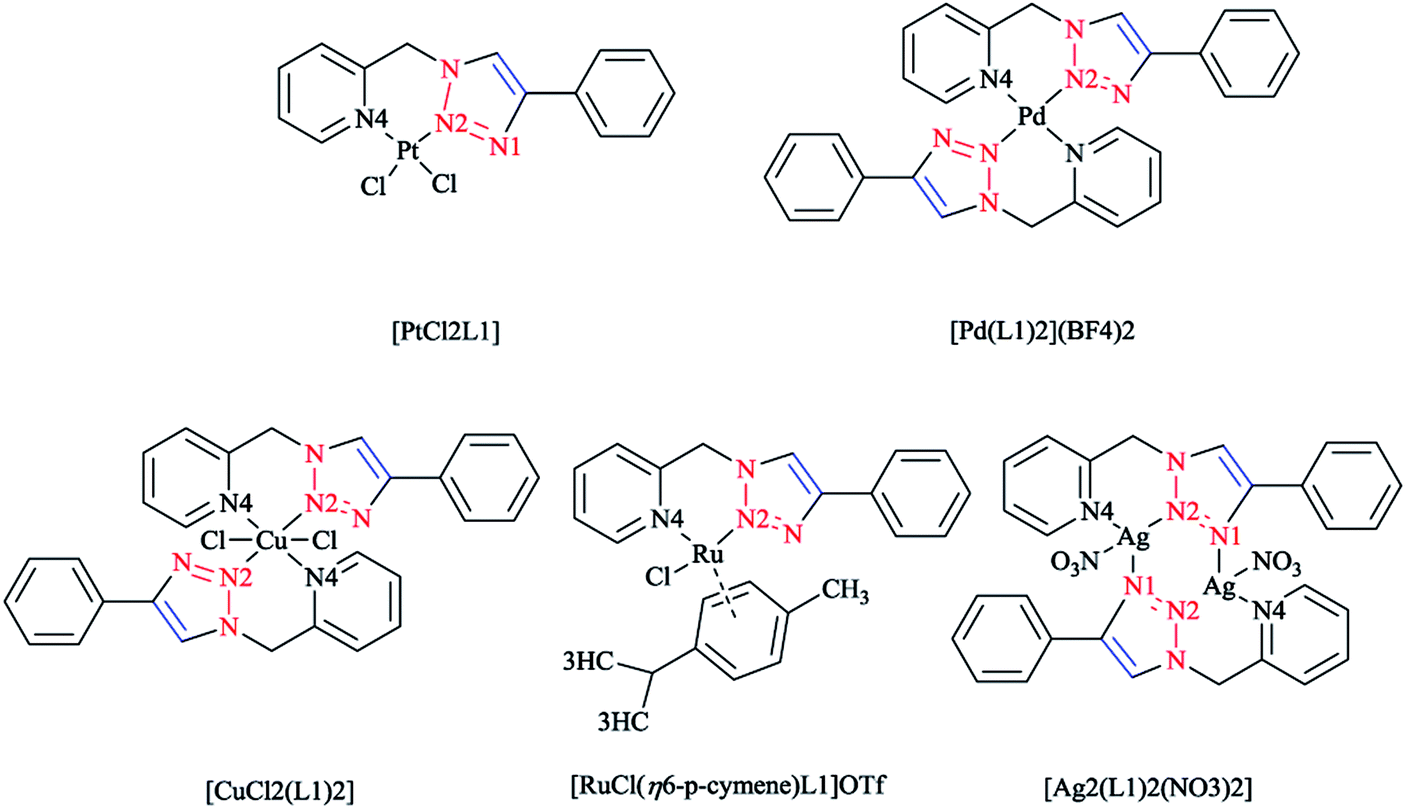 | ||
| Scheme 22 Structures of [PtCl2L1], [Pd(L1)2](BF4)2, [CuCl2(L1)2], [RuCl(η6-p-cymene)L1]OTf and [Ag2(L1)2(NO3)2] complexes based on 1-(2-picolyl)-4-phenyl-1H-1,2,3-triazole ligand (L1). | ||
In the crystal structure of the ruthenium complex, a three-legged piano-stool geometry with the metal center coordinated by p-cymene in an η6 fashion, a chloride ion, and ligand L1 was observed. The distances between Ru(II) and the nitrogen atoms of L1 were not equivalent, similarly to Pt and Pd complexes.
The X-ray geometry of the Ag complex showed two silver centers bridged by two L1 molecules. Both metal ions in the structure were coordinated by two nitrogen atoms from one L1 ligand (N4 from the pyridine and N2 from the triazole part) and another triazole N1 atom from the second L1 ligand in the complex. This showed a distorted tetrahedral coordination consisting of the oxygen atom of a nitrate anion coordinated to both silver atoms in a monodentate mode. Alternatively, in the crystal structure of the Ag complex, each silver atom can be displaced from the plane of the coordinating nitrogen atoms and then coordination of the silver atoms can also be assumed to involve a distorted trigonal planar. In the aromatic L1, it was observed that the angle between the plane of the pyridine ring and the plane of the triazole ring was 87.7° and a distortion was found when ligand was bound to the metal center. Coordination and structural assessments of traditional tetrahedral complexes of Ag(I) and Cu(I) with 1,2,3-triazol-4-yl-pyridine click ligands revealed that these complexes can be used as appropriate motifs for the self-assembly of supramolecular architectures.177
For computational investigation of this series of complexes, in accordance with our elucidations, the complexes with central atoms of copper, palladium, and platinum, [MLn(NH3)2]2+ (M = Cu, Pd, Pt), were considered with a coordination number of 4, while silver complexes [AgLnNH3]+ were modeled with a coordination number 1. It is important to notice that the expected X-ray square-planar (M = Pd, Pt) and trigonal-planar (M = Ag) geometries were obtained with a singlet ground state via DFT computations. The natural bond orbital (NBO) analysis affirmed the donation of the in-plane lone pairs of the appropriate nitrogen atoms of the ligand and two ammonia to the empty s and d orbitals of the metal, which decreased the charge localized on the metal. On the other hand, since in [AgLnNH3]+ the d orbitals are filled, electron donation occurs only to the 5s orbital of the metal, which leads to a weaker metal–ligand interaction. It should be mentioned that these calculated results are in agreement with the observed smallest stabilization energy for [AgLnNH3]+ complexes.
Frontier molecular orbital analysis demonstrated that the dz2 orbital of the metal (dz2 is σ interacting in the D4h symmetry) is combined with the high-lying orbitals of the triazole ring that can be observed in this series of complexes. It was also deduced that due to these interactions, the metal d electrons delocalize more efficiently to the π system of the ligand and so increase the aromaticity and also the stability of the complexes with the ligand. This stabilization effect was confirmed by the second-order perturbation theory analysis of the Kohn–Sham matrix.99
The geometrical and energetic properties of [Pt(Bnpytri)2]2+ 31 complexes (where Bnpytri = 2-(1-benzyl-1H-1,2,3-triazol-4-yl)pyridine and its schematic structure are illustrated in Scheme 23), and the corresponding palladium(II) analogs were assessed computationally through DFT calculations at the B3LYP/LanL2DZ/6-31G(d) level of theory and using dimethyl sulfoxide (DMSO) as the solvent. The calculated results of [Pt(Bnpytri)2]2+ 26 complexes revealed that the head-to-tail configuration was significantly favored from a thermodynamic point of view, as was obtained similarly for the corresponding palladium(II) analog.56,125 Under special conditions that allow the formation of the thermodynamic product, the head-to-tail configuration would be the dominant form for the Pt(II) complexes.
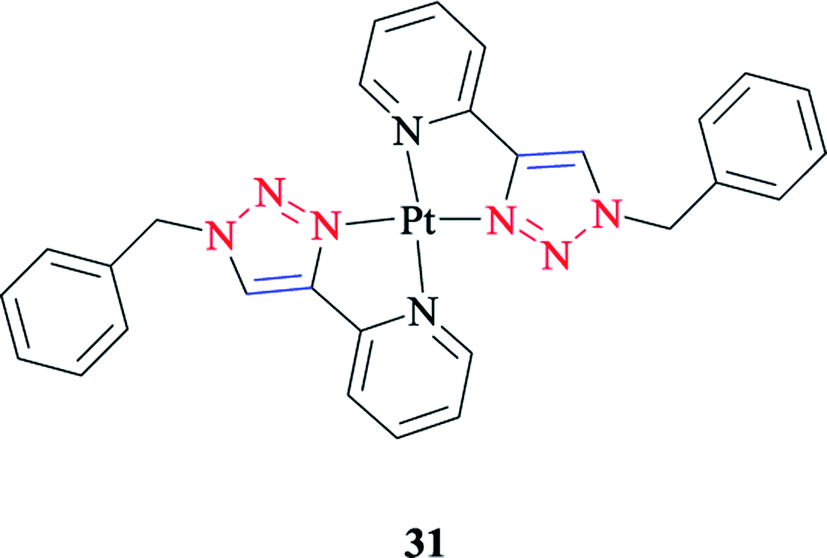 | ||
| Scheme 23 Structure of the [Pt(Bnpytri)2]2+(31) complex. | ||
Moreover, for tetramer complexes, [Pd2Pt2(L)4]8+ and also [Cu2Pt2(L)4]6+ (Scheme 12), DFT geometry optimization was performed at the B3LYP/LanL2DZ/6-31G(d) level of theory in the presence of nitromethane as the solvent. The [Pd2Pt2(L)4]8+ tetramer complex showed the expected box-like geometry, while optimization for the hexamer (with hexyloxy chains) gave the expected geometry. It was also extracted that applying a labile metal with a tetrahedral geometry might have less scope for the production of larger macrocycles, in addition to closing the Pt(II) centers and stabilizing the tetramer through platinum–platinum and π-stacking interactions.
In the case of di-magnetic Cu(I) and Ag(I) [M(Rpytri)2]+complexes, the calculated results showed that there was a tendency to adopt a head-to-tail square-planar geometry with bispyridyl-triazoles,129,133 contrary to the primary preference of Ag(I) for a tetrahedral geometry over a square-planar geometry.178
The crystal structure and DFT calculated structural data for [Cu2Pt2(L)4]6+ complexes demonstrated their tetrahedral geometry,129,177,179 but only using the asymmetric ligands with orthogonal binding sites in combination with cis-protecting directional bonding strategies at the Pt(II) centers.139,180,181
A family of bis-(2-pyridyl-1,2,3-triazole) ligands 32 (Scheme 24) were synthesized by employing in situ azide formation conditions and then by methylene-bridging of the bis-(2-pyridyl-1,2,3-triazole) ligands 32 to produce larger heterometallic macrocycles.
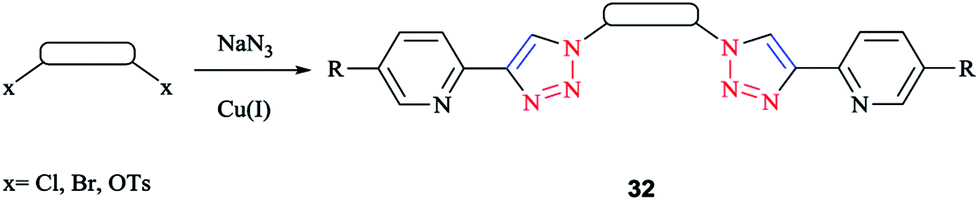 | ||
| Scheme 24 Formation of the bis-bidentate pyridyl-triazole ligand (32). | ||
[Pt(L)2]2+ metallo-ligands showed a square-planar structure with 2-pyridyl-1,2,3-triazole 33 units adopting a head-to-tail arrangement with vacant peripheral binding sites.
In this vein, tetracopper(I) macrocycles were prepared using the bis-(2-pyridyl-1,2,3-triazole)-1,2-xylene ligand so that each pair of copper(I) ions was bridged by two iodides.
Furthermore, [Cu2L2]4+ macrocycles were generated from Cu(II) ions and bis-2-pyridyl-1,2,3-triazole ligands via embedding a m-xylene (1,3-) or 2,7-dimethylnaphthalene spacer unit21,182 (Scheme 25).
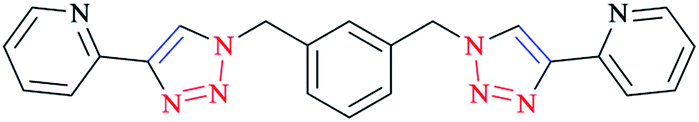 | ||
| Scheme 25 Structure of the bis-2-pyridyl-1,2,3-triazole ligand (33). | ||
4.3 Re(I) complexes
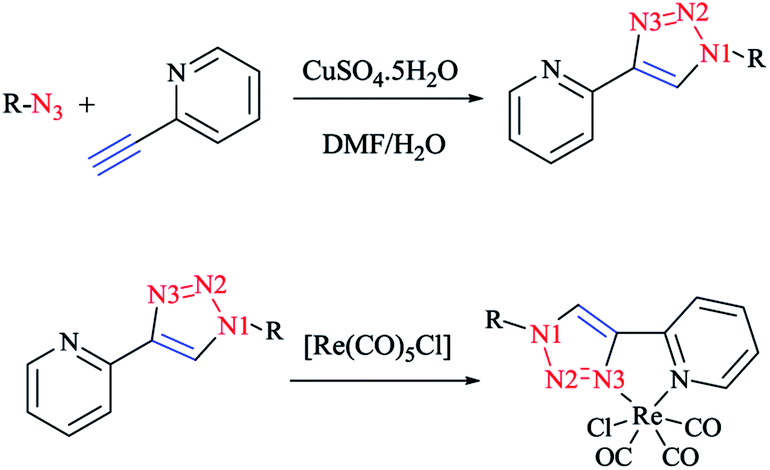
In Scheme 26, the production of {Re(CO)3Cl} bidentate 2-pyridyl-1,2,3-triazole coordination compounds was illustrated via the reaction of the cycloadducts with the commercially available [Re(CO)5Cl].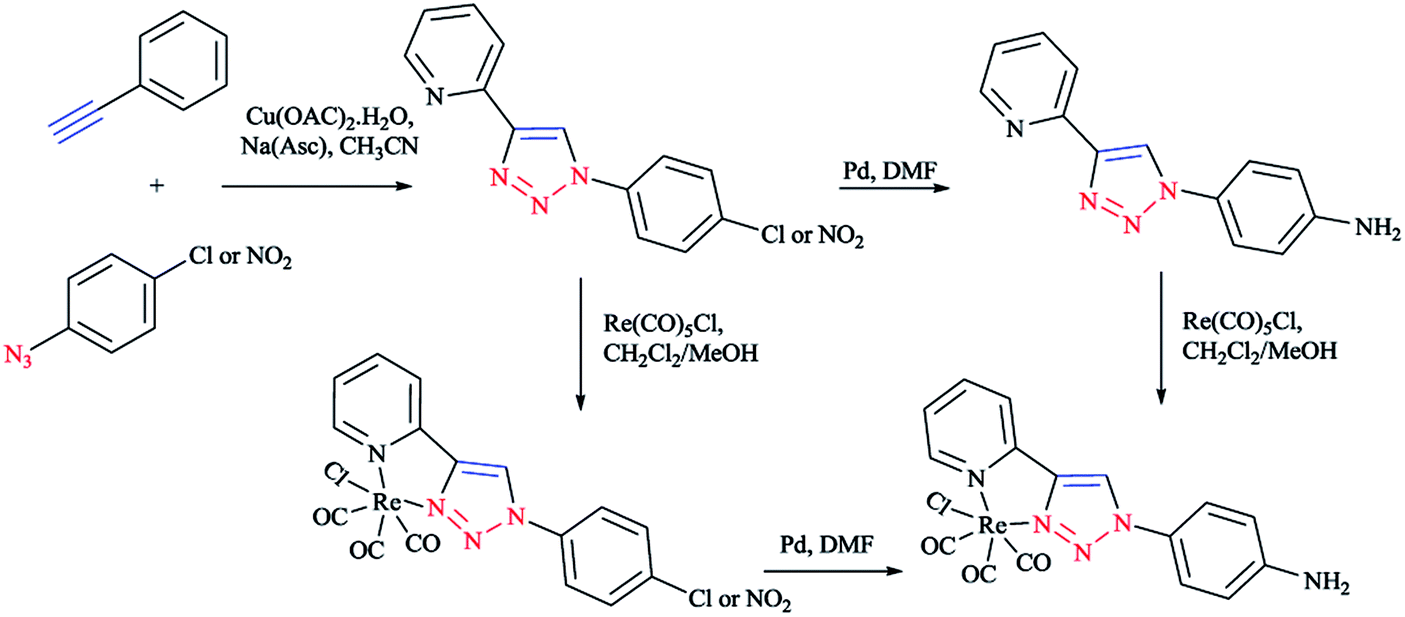 | ||
| Scheme 26 Production of bidentate 2-pyridyl-1,2,3-triazole ruthenium complexes. | ||
X-ray diffraction analysis was also performed to identify the structural properties of the aforementioned rhenium complexes and the results compared with the DFT-optimized geometries (via C-PCM computations in methanol solution). In the optimized structures of the complexes, the rhenium ion attains the usual distorted-octahedral coordination. It should be noted that the obtained bond lengths and angles were in the usual range of tricarbonyl rhenium complexes based on bidentate 2-pyridyl-1,2,3-triazole derivatives59 (Scheme 27).
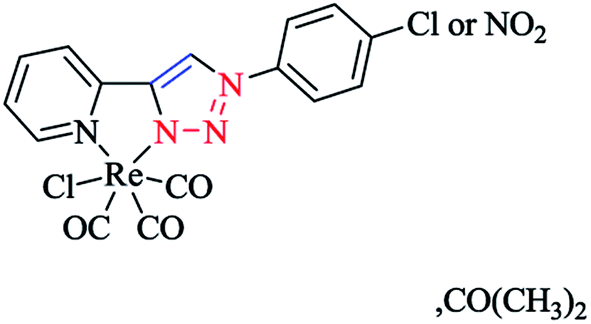 | ||
| Scheme 27 Structure of the [(C13H9N5O2)ReCO3Cl (or NO2)]·CH3COCH3 complex. | ||
Policar and coworkers deeply investigated the complexes of Re(I) centers coordinated with a series of functionalized 1,2,3-triazole ligands in an octahedral geometry as noninvasive imaging probes. In this vein, a series of pyridyl-1,2,3-triazole and quinolinyl-1,2,3-triazole ligands bearing various substituted alkyl side chains were designed and synthesized using click procedures. Furthermore, their photophysical properties were characterized and it was shown that the luminescent properties are enhanced in complexes bearing long alkyl chains.44
In some rhenium complexes, the inverse click ligands are generally coordinated to the rhenium(I) center in a bidentate fashion through the pyridyl and triazole nitrogen atoms. In this context, the Re(I) complexes of the [(4-R-1H-1,2,3-triazol-1-yl)methyl] pyridine ligand series (Scheme 28) were investigated structurally through X-ray crystallography, which showed that all of them were crystallized in the triclinic space group.
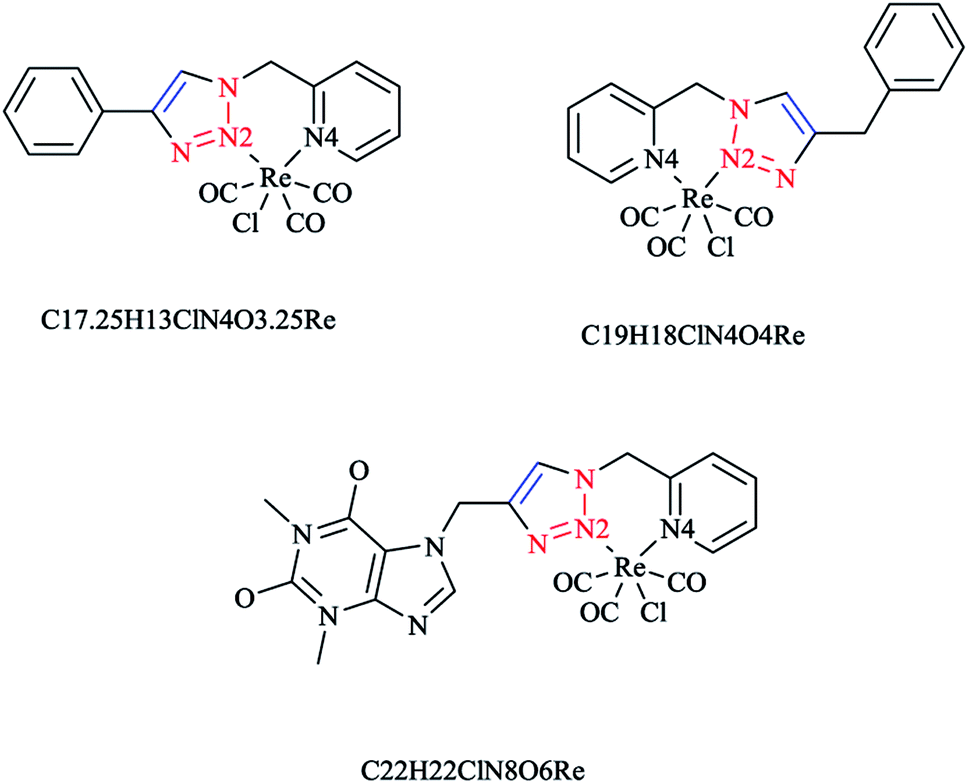 | ||
| Scheme 28 Structures of the C17.25H13ClN4O3.25Re, C19H18ClN4O4Re, and C22H22ClN8O6Re complexes. | ||
As expected, the rhenium metal center forms a distorted-octahedral coordination environment and is coordinated to a chloride and three carbonyl ligands in a facial array. Moreover, due to the presence of a methylene spacer between the triazole and pyridyl groups, a six-membered chelate ring is generated and consequently, the bite angles of the ligands vary little, ranging from 81.98(8) to 83.5(3)°. Chelation of the ligand to the metal center leads to the formation of six-membered rings in boat conformations, with the fold angles between the pyridyl and triazole rings ranging from 111.09 to 114.22°. It has been shown that the triazole ring substituent effect is not considerable on the metric properties of rhenium complexes and so it can be deduced that side-arm substitution leads to little electronic tuning of the ligand. Furthermore, in these [(4-R-1H-1,2,3-triazol-1-yl)methyl]pyridine 5 set of ligands, it was found that the Npy–Re bond length value (2.198(5)–2.218(9) Å) is bigger than the Ntrz–Re bonds (2.155(2)–2.180(7) Å) that occur in other metal complexes of this ligand set. Further, it was assessed experimentally and computationally56,81,126 that metal complexes of the related regular 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine 4 ligands were more stable than those with the [(4-R-1H-1,2,3-triazol-1-yl)methyl]pyridine 5 inverse click ligands. Clearly speaking, regardless of whether the triazole is coordinated to the Re center, the obtained values of the Re–N2trz and Re–N2trz–N4py bond lengths and angles in the entire series of regular five-membered rhenium click complexes63,64,97,183,184 (Schemes 27 and 29) were very near to those of inverse six-membered rhenium click complexes, and this feature has been also studied and confirmed in the Pd(II) complexes of these ligands.56,95
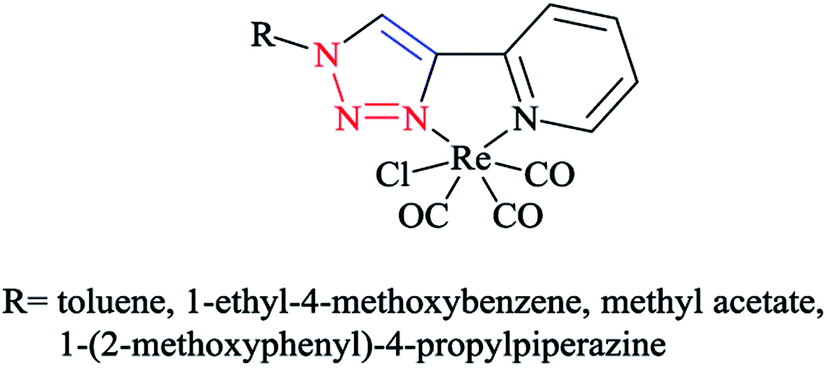 | ||
| Scheme 29 Structure of the [(N–N)Re(CO)3Cl] regular five-membered rhenium click complexes. | ||
It was observed in the crystal structure of these complexes that double-stranded supramolecular polymers are formed via the many C–H⋯Cl hydrogen bonds and CH- π or face-to-face π-stacking interactions.185–188 It was also observed that the chloride of one complex bonds to the adjacent complex through the acidic methylene CH (Cl⋯H = 2.638(2) Å, Cl⋯C = 3.569(7) Å), leading to the formation of a single-stranded linear polymer. A face-to-face π-stacking interaction (centroid–centroid distance of 3.736 Å) occurs between the phenyl group and 1,2,3-triazole units on adjacent complexes to produce the double-stranded structure. In addition, CH–π interactions between the benzyl substituent methylene hydrogens and the phenyl unit of the adjacent complex were found with CH-centroid distances of 3.032 and 3.122 Å. More interestingly, in the caffeine-substituted complex, the individual complexes are folded into a U-shaped conformation and two complexes interact through C–H⋯O hydrogen bonds between a carbonyl of the caffeine moiety and an acidic CH of the pyridyl ring, which consequently leads to a supramolecular cage that is filled with two acetone molecules as the solvent. These face-to-face π-stacking interactions result in the formation of a slipped stacked polymer with a centroid–centroid distance of 3.381 Å between the caffeine groups.
The structures of 4-n-propyl- and 4-phenyl-substituted fac-Re(CO)3 complexes coordinated with the tridentate click ligand (2,6-bis(4-substituted-1,2,3-triazol-1-ylmethyl)pyridine) (Scheme 30) were also investigated via X-ray crystallography and DFT methods. Based on the X-ray and DFT computational results, it was shown that both complexes had the expected facial coordination geometry about distorted-octahedral rhenium(I) centers. More concisely, the Re(I) cations were coordinated to three carbonyl donors and to the pyridyl and triazoyl nitrogen atoms of the ligands.58
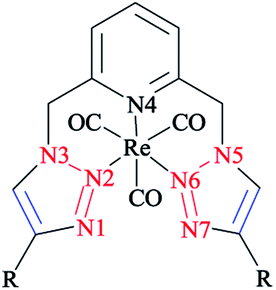 | ||
| Scheme 30 Structure of the 2,6-bis(4-substituted-1,2,3-triazol-1-yl-methyl)pyridine complexes (R = Ph, C3H7). | ||
In another research study, the complex of rhenium(I) with functionalized 2-pyridyl-1,2,3-triazole derivatives (pyta)-based ligands was crystallized by slow evaporation in dichloromethane/methanol solution and then investigated using the X-ray diffraction method.
A comparative study between the structural features of rhenium(I) complexes of the related pyta derivatives82,133 exhibited an anti-arrangement in them adopted by the N(1) and N(2) atoms of the triazole and pyridine rings, respectively. In the 1,2,3-triazole ring, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond length of the 1,2,3-triazole ring was decreased and became shorter than the adjacent N–C and N–N bonds. Moreover, a network of intra-layer hydrogen bonds between the ligands and lattice waters was observed that led to the crystal cohesion. Consequently, the lattice water molecule forms O–H⋯N bonds with one nitrogen of the piperazinyl moiety (d(O–H⋯N(5)) = 3.013 Å, O–H–N(5) = 173.08°) (Scheme 31). It was observed that the ethylene bridge leads to a large distance between the (2-methoxyphenyl)piperazine group with the chelating part of the molecule. So, the components near the coordination sphere do not interact through coordination, hydrogen-bonding interaction, or by folding of the pendant arm toward the metal center, which was previously reported in other flexible spacers by Santos' group.189 The structural results show that the interaction of the metal center with the bioactive fragments is not considerable and so this suggested a suitable affinity for the serotonin receptors.63
N bond length of the 1,2,3-triazole ring was decreased and became shorter than the adjacent N–C and N–N bonds. Moreover, a network of intra-layer hydrogen bonds between the ligands and lattice waters was observed that led to the crystal cohesion. Consequently, the lattice water molecule forms O–H⋯N bonds with one nitrogen of the piperazinyl moiety (d(O–H⋯N(5)) = 3.013 Å, O–H–N(5) = 173.08°) (Scheme 31). It was observed that the ethylene bridge leads to a large distance between the (2-methoxyphenyl)piperazine group with the chelating part of the molecule. So, the components near the coordination sphere do not interact through coordination, hydrogen-bonding interaction, or by folding of the pendant arm toward the metal center, which was previously reported in other flexible spacers by Santos' group.189 The structural results show that the interaction of the metal center with the bioactive fragments is not considerable and so this suggested a suitable affinity for the serotonin receptors.63
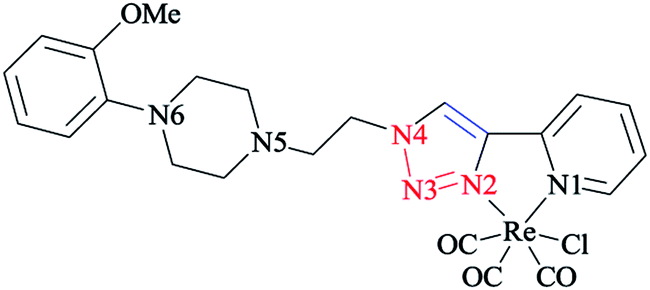 | ||
| Scheme 31 Structure of the 4-piperazinyl-2-pyridyl-1,2,3-triazole rhenium(I) complex. | ||
Time-dependent DFT (TD-DFT) calculations190 on Re(I) complexes of readily functionalized bidentate pyridyl-1,2,3-triazole click ligands (Scheme 32) demonstrated the MLCT nature of the HOMO → LUMO transitions (the blue-shift of Re → bpy MLCT, which lies at 633 nm).
 | ||
| Scheme 32 Syntheses of [Re(CO)3Cl] with bidentate pyridyl-1,2,3-triazole click ligands and [(pytri-R)Re(CO)3Cl] complexes. | ||
It was also found that the emission from Re → pytri-R MLCT (where R = benzyl) is blue-shifted at 538 nm in CH3CN. It should be mentioned that the electronic role of pytri-R in communication between the differing emitting centers has also been studied systematically.88 It was reported that Ir(ppy)2 34 and Ir(phprz)2 35 (where phprz is 1-phenylpyrazole) (Scheme 33) fragments could bind to pytri-R and were then connected via the (N1) sp2 triazole nitrogen to produce dimer and trimer structures. Furthermore, it was shown that the modulation of the linkers through the N1 sp2 triazole did not affect the photophysical properties of the individual Ir units.
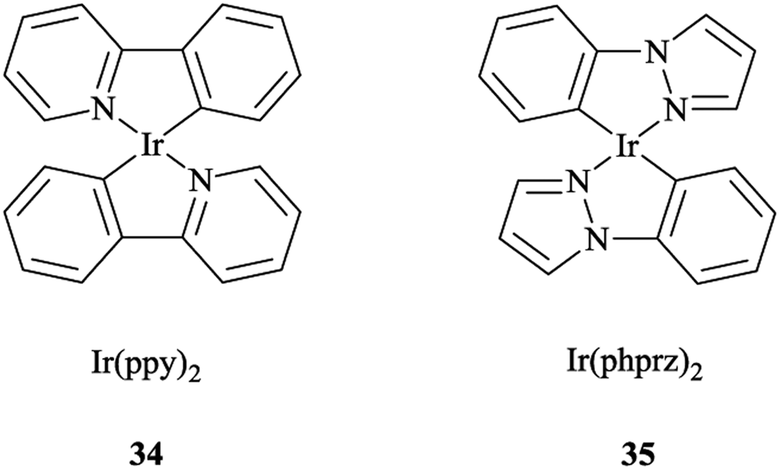 | ||
| Scheme 33 Structures of the Ir(ppy)2 (34) and Ir(phprz)2 (35) complexes. | ||
The coordination behavior of ruthenium ion with (pytri-R)(bpy(CO2H)2)(CNS)2 as a pytri-R based ligand was investigated via DFT analysis85 (Scheme 34).
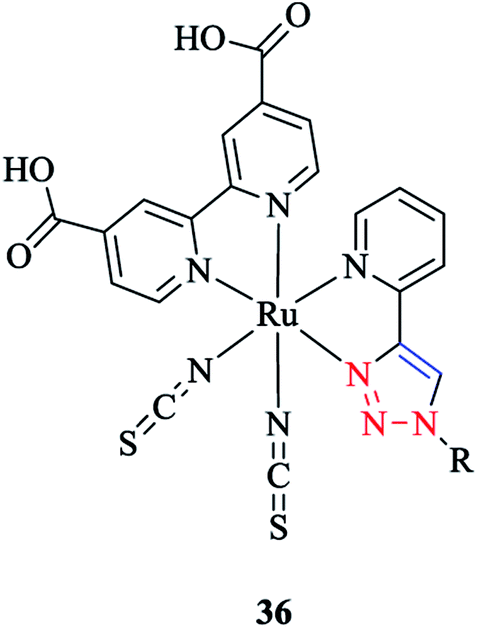 | ||
| Scheme 34 Structure of a ruthenium complex with the (pytri-R)(bpy(CO2H)2)(CNS)2 ligand. | ||
It should be mentioned that these complexes are widely used in dye-sensitized solar cells. DFT computations showed that the LUMO and LUMO+1 of [Ru(pytri-R)(bpy(CO2H)2)(CNS)2] 36 were based on the bpy ligand. Moreover, a systematic study of the substituent effects on the ruthenium complexes with substituted pytri-R ligands was performed by Happ et al.87 In this respect, the pytri-R ligand was substituted with ethynylphenyl groups at either the 5-pyridyl or N1 triazole positions, and it was shown that substitution of the 5-pyridyl position had a significant effect on the emission properties and increased the emission wavelength from 602 nm with the 1-ethynyl-4-methoxybenzene substituent to 674 nm with the 1-ethynyl-4-nitrobenzene substituent. Interestingly, the [Ru(pytri-R)(bpy(CO2H)2)(CNS)2] 36 complex substituted by 1-ethynyl-4-nitrobenzene at N1 triazole nitrogen showed the emission wavelength at 610 nm. This behavior revealed that the linkage of the 1-ethynyl-4-nitrobenzene substituent with the triazole insulated the electron-withdrawing power of this substituent from the Ru center.
In another research study, the properties of [Ru(TAP)2(pytri-R)]2+ 37 complexes with a DNA active moiety (where TAP = 1,4,5,8 tetraazaphenanthracene) were also assessed (Scheme 35).191
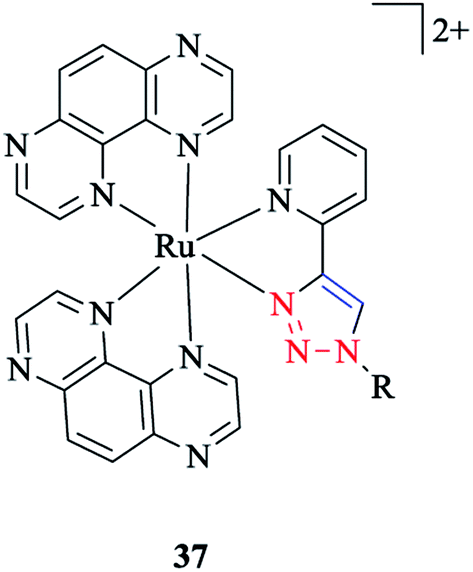 | ||
| Scheme 35 Structure of a ruthenium complex with the (TAP)2(pytri-R) ligand. | ||
It was demonstrated that the d and p orbitals of ruthenium are stabilized at 50 mV relative to L = 1,10-phenanthroline, which could be attributed to a poorer overlap between the dπ and π* (pytri-R) molecular orbitals. Moreover, it was investigated and shown that alkaline and selected first row transition metals have no considerable effect on varying levels of emission intensity and no tuning of the emission frequency.192 On the other hand, the N1 substituent does not shift the emission spectrum for [(pytri-R)Re(CO)3Cl] complexes. For instance, by using R = (CH2)2CO2H and (2-methoxyphenyl) piperazine as substituents in the [(pytri-R)Re(CO)3Cl] complex, the emission maxima were see to lie at 526 nm and 522 nm in DMF/MeOH solution, respectively.63,183 DFT calculations on the molecular orbital properties of these systems demonstrated a modest contribution from the N1 substituent in the LUMOs.
Moreover, a series of electronically tuned fac-Re(CO)3Cl·2-(1-R-1H-1,2,3-triazol-4-yl)pyridine complexes were prepared (Scheme 36). In this systematic study, the substituent effect on the electronic structure of a series of fac-[(pytri-R)Re(CO)3Cl] complexes was examined. The substituents were chosen so that they could couple conjugatively with the triazole and could insulate it via a CH2 group from such communication. The nature of the lowest energy transition resonance was studied via Raman spectroscopy in conjunction with DFT calculations. The lowest excited state properties were also established using time-resolved emission spectroscopy. Then, the calculated values of a number of bond lengths and angles were compared with X-ray crystallographical data, and it was seen that there was a reliable agreement with the experimental data.
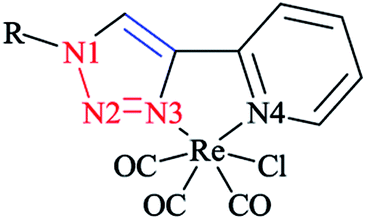 | ||
| Scheme 36 Structure of the Re(CO)3Cl complex series with 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine. | ||
A comparison of the calculated Raman wavenumbers and intensity values with the corresponding experimental data showed a mean absolute deviation (MAD) of <10 cm−1 for bands between 400 and 1650 cm−1 that had more than 20% of the maximum band intensity.
The electronic absorption behavior of all the complex series was investigated. The lowest energy transition at approximately 330 nm was not changed by substitution, regardless of whether was unconjugated or conjugated with the triazole ring.
The band at 330 nm could be assigned to a metal–ligand charge transfer (MLCT)97 as was confirmed by the TD-DFT data. The higher energy MLCT transitions (at 290 nm) and additional transitions associated with the ligand (at 275 nm) were obtained via TD-DFT computations. It is important to note that DFT methods systematically overestimate the conjugative effects and so, the effect of the conjugated group was predicted computationally to be stronger than the experimental values by up to 0.4 eV.193,194
Moreover, it could be observed that conjugated and non-conjugated ligands affect the nature of the MLCT band at 330 nm. Precisely speaking, the acceptor orbitals are localized solely on the triazole species for the unconjugated complexes, while in the case of conjugated complexes, they are spread over the R substituent in addition to the triazole moiety.
This different behavior can be attributed to the overweighing of the conjugation interactions associated with DFT methods, which can be examined through measuring the resonance Raman spectroscopy.
It was shown that different substitution on the triazole ring resulted in different Raman spectra; for instance NO2-substituted complexes showed a distinguished band at 1349 cm−1 attributed to NO2 functionality.195,196
A significant aspect was observed in the resonance Raman spectra, which was the identical spectra for the complexes with similar pytri species (bands at 1285, 1571, 1587, and 1625 cm−1). It should be stated that DFT normal mode calculations also produced these bands and showed that they are based on the pytri moiety. Small differences in the Raman spectra of the conjugated systems were observed. It should be noticed that the Raman spectra showed pytri modes, which were also observed for non-conjugated systems. Two bands were observed due to the stretching of the R-triazole moiety and an enhancement of the NO2 group195,196 was present, which was in agreement with the acceptor molecular orbital nature of a nitro functional group.
From the computational viewpoints, the DFT calculated values revealed that the LUMO was nitrobenzene based, and also the TD-DFT computations for the lowest energy transitions demonstrated no or only a minor LUMO character. The calculated results were in confirmation with the resonance Raman data, which indicated an enhancement of nitro vibration, reflecting the role of the nitro group on the electronic transition.64
In order to compare the stereoelectronic properties of triazoles with triazaphospholes, Re(I) complexes containing these chelating ligands were assessed (Scheme 37). The coordination behavior of the Re(I) cation with the least nucleophilic nitrogen atom N3 in complexes 38–41 is depicted in (Scheme 38).
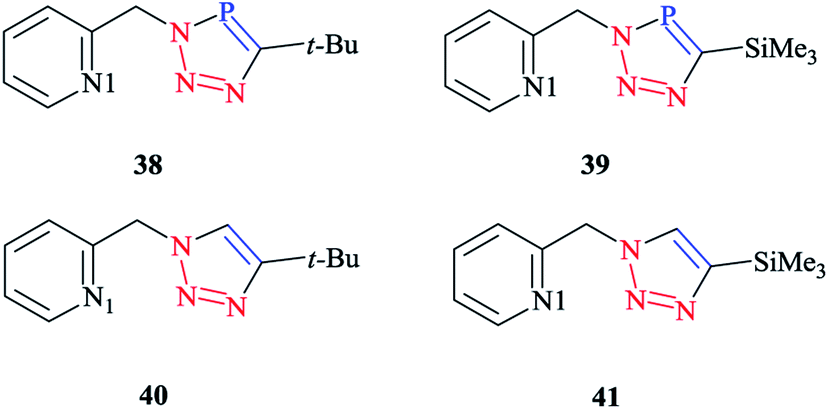 | ||
| Scheme 37 Structures of click-derived chelating phosphorus- and nitrogen-containing heterocycles. | ||
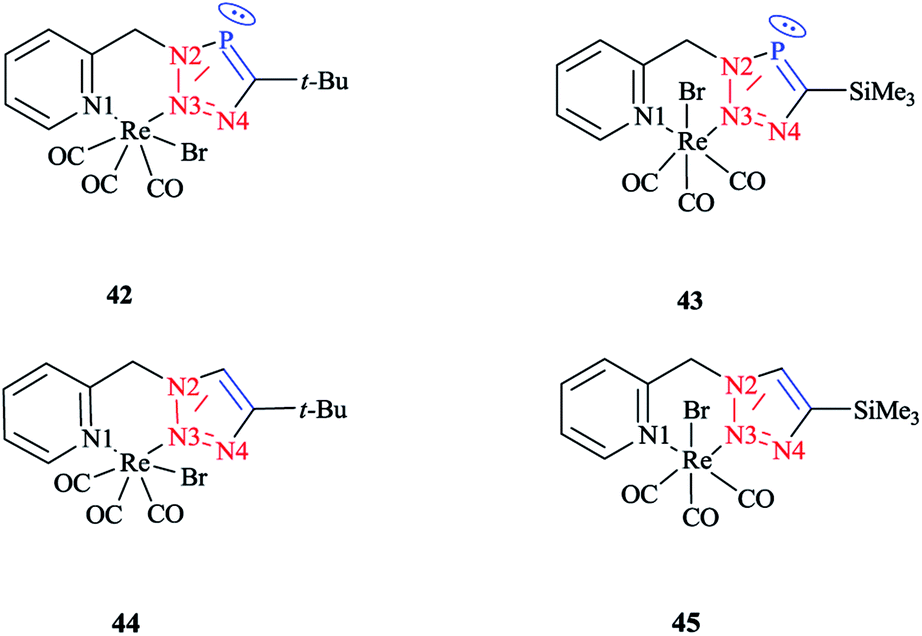 | ||
| Scheme 38 Syntheses of Re(I) complexes 35–38. | ||
The crystallographic properties for two complexes of [L2Re(CO)3Br] were comparatively characterized. The crystal structure revealed that the Re(I) cation is coordinated to chelating ligands via the N(3) atom of the triazaphosphole species, rather than via the phosphorus atom. Since, coordination through the N(3) atom of the triazaphosphole species has not been observed for triazaphospholes before, it can be claimed that this behavior was enforced by the chelating effect of the ligand.
As was observed in the crystal geometry of compounds 42 and 43, the nitrogen atoms N(3) of the triazole units were coordinated to the Re(I) centers in the facial geometry of complexes 44 and 45, [(40)Re(CO)3Br] and [(41)Re(CO)3Br]. Moreover, we can claim that replacing the phosphorus atom in 42 and 43 by an isoelectronic C–H group in 44 and 45 had no considerable effect on the bite angle N(1)–Re(I)–N(3).
DFT calculations at the B3LYP level of theory54 were performed on compound 42 (R = t-Bu) to understand the electronic structure of the triazaphospholes. A large coefficient of π symmetry at the phosphorus atom was obtained on the LUMO of low-coordinate phosphorus heterocycle compounds, which showed the π-accepting nature of these systems. In HOMO-1, a large value of the coefficient of σ symmetry was obtained at the nitrogen atoms N4 and N3 of the heterocycle ring and HOMO-7 clearly indicated the π-donor properties of triazaphospholes.
It is important to mention that transition metal cation centers coordinate preferentially to the nitrogen atom N3 in triazoles, but in the ligands of 38 and 39 this nitrogen atom cannot coordinate via σ bonds due to the chelating effects. Furthermore, in the HOMO-4 orbital, similar large coefficients of σ symmetry at both the phosphorus atom and nitrogen atom N3 appeared. More importantly, the coordination of a transition metal center with the pyridyl nitrogen and the nitrogen N3 or with the pyridyl nitrogen and the phosphorus via a σ coordination mode led to a small energetic difference, and so both coordination modes could be possible.66
4.4 Rh(III) and Ir(III) complexes
Some 1,2,3-triazole-based organochalcogen ligands and their Rh(III)/Ir(III) complexes (Schemes 39 and 40) were synthesized and characterized using elemental analysis, multinuclear NMR, IR, X-ray crystallography, and mass spectral data.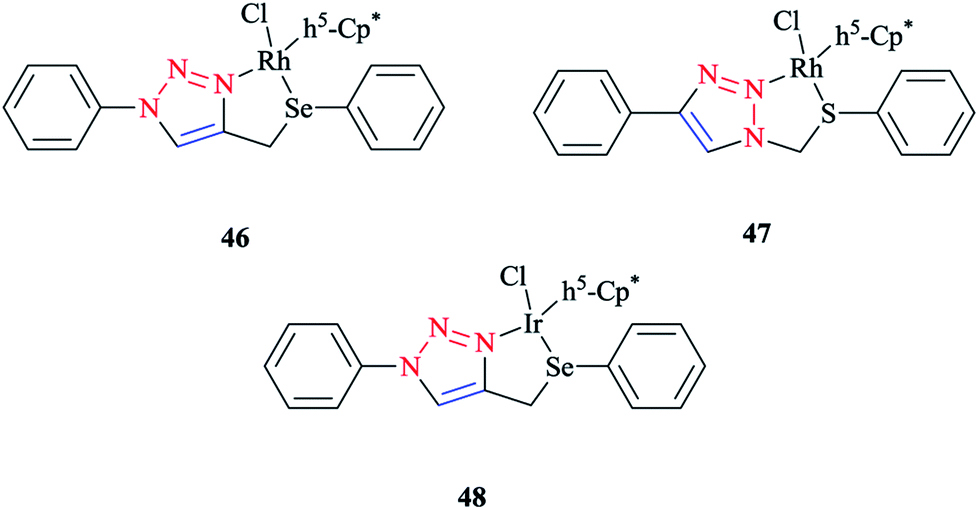 | ||
| Scheme 39 Structures of Rh(III)/Ir(III) complexes with 1,2,3-triazole-based organochalcogen ligands. | ||
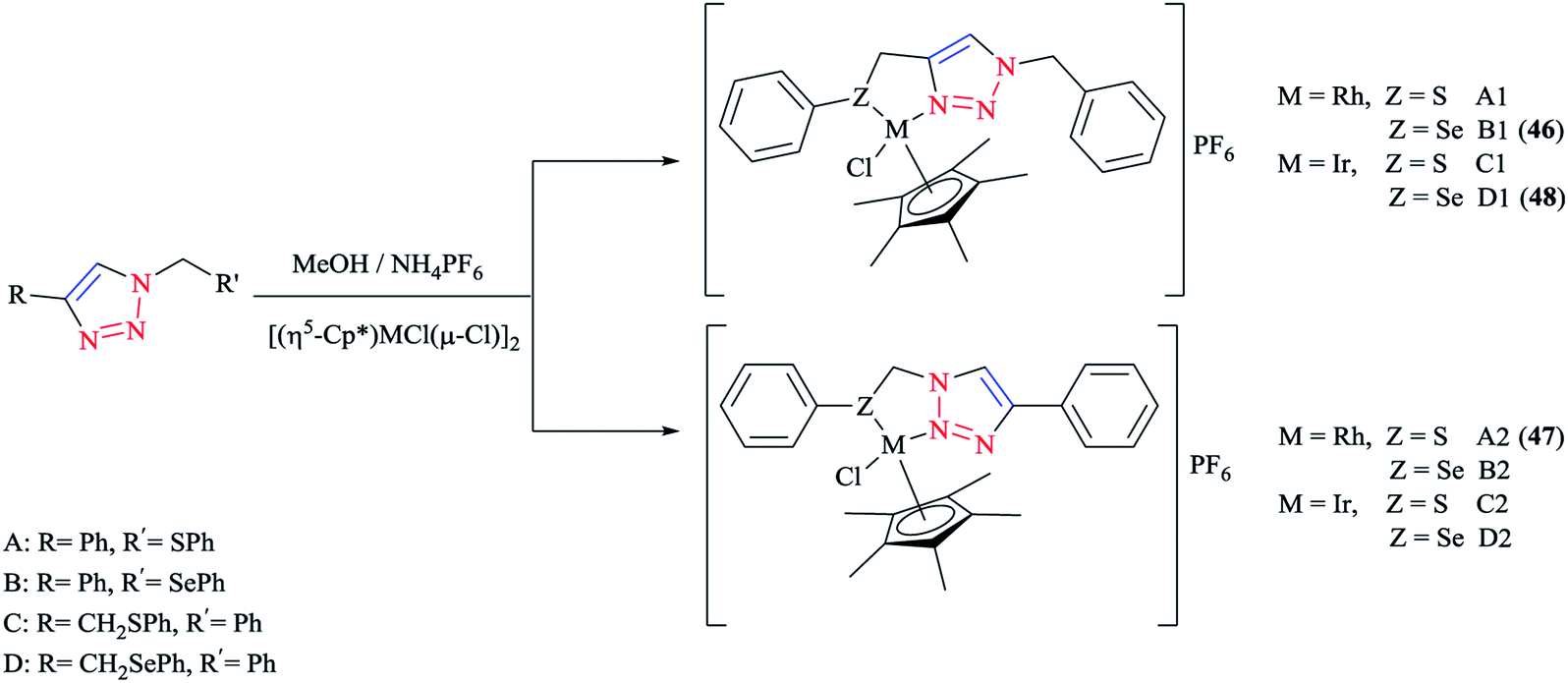 | ||
| Scheme 40 Syntheses of A–B ligands and A1–D1 and A2–D2 complexes. | ||
To synthesize the complexes, the chloro bridge cleavage of [(η5-Cp*)RhCl(μ-Cl)]2/[(η5-Cp*)-IrCl(μ-Cl)]2 occurred, followed by the reaction with 1-benzyl-4-((phenyl-thio/phenylseleno)methyl)-1H-1,2,3-triazole (A/B) or 4-phenyl-1-(phenylthio/phenylseleno)methyl-1H-1,2,3-triazole (C/D) at room temperature. Consequently, rhodium(III) (A1, B1, A2, and B2) and iridium(III) (C1, D1, C2, and D2) half-sandwich complexes were generated and the process was facilitated by anion exchange with NH4PF6.
The nitrogen of triazole five-membered ring is coordinated to the metal center in all of the complexes. Almost similar X-ray molecular structures were obtained for all the rhodium and iridium complexes, which demonstrated an overall three-legged piano-stool conformation. More clearly, a pseudo-octahedral half-sandwich “piano-stool” disposition of donor atoms is formed around the Rh/Ir metal center. The centroid of the η5-Cp* ring occupies nearly the center of a triangular face of an octahedron. The coordination sphere is occupied with a chlorine donor atom and nitrogen and chalcogen atoms, which form a chelate ring with the metal center.
In comparison with complex 46, the Rh–Se bond length of the half-sandwich complex of Rh(III) with the ligand 2-(phenylselenomethyl)pyridine was 2.487 Å (ref. 197) and for the complex [η5-Cp*RhCl{η2-(SePPh2)2N}] was 2.5266 Å,198 which were larger than those obtained for the half-sandwich complex of Rh(III) with the ligand 1,2-dicarba-closo-dodeca-borane-1,2-dichalcogen.199
In complex 47, the Rh–S bond distance was 2.432 Å and was also greater than the corresponding value for the complexes [η5-Cp*RhCl(1,1′-(1,2-ethanediyl)bis(3-methylimidazole-2-thione))]Cl at 2.3967 Å (ref. 200) and [η5-Cp*RhCl{2-(phenylthiomethyl)pyridine}]PF6 at 2.383 Å (ref. 197) and [η5-Cp*RhCl{η2-S,P-Ph2P(S)NHPPh2}]BF4 at 2.404 Å.201 Furthermore, in complex 47, the Rh–N bond distance was 2.091 Å, which was in reliable agreement with those of [η5-Cp*Rh(2,6-(mesityl)2C6H3S)(bpy)][B(3,5-(CF3)2C6H3)4].202 The obtained bond length values for Rh–C(Cp* centroid) in complexes 46 and 47 were in the normal range, i.e., 1.784–1.786 Å.201
In the case of complex 48, the Ir–Se bond distance value was 2.4822 Å, which was longer than the values obtained for the complex [η5-Cp*Ir-{Se2C2(CO2Me)2}] (2.3494 and 2.3520 Å)203and near to the distances reported for [(η5-Cp*)Ir((2-arylselenomethyl)pyridine)Cl][PF6] at 2.453 Å,197 [η5-Cp*IrCl(μ-SeCOC6H5)(κ2-SeCOC6H4)Ir(η5-Cp*)] at 2.445–2.495 Å,204 and [η5-Cp*Ir(μ3-Se)2{PtTol-(PPh3)}2] at 2.416–2.422 Å.205 Moreover, the Ir–N bond distance was 2.094 Å, which was very close to the Ir–N bond length in [(η5-Cp*)Ir(2-arylthiopyridyl) pyridineCl][PF6] at 2.099 Å (ref. 197) and shorter than those of Ir complexes of the pyridine-triazole ligand at 2.137 Å (ref. 29) and 2-(1-benzyl-1H-1,2,3-triazol-4-yl)pyridine at 2.151–2.159 Å.206 The Ir–C (η5-Cp*centroid) bond length in complex 48 was 1.791 Å, which was consistent with the Ir–C bond distance in [(η5-Cp*)Ir((2 arylselenomethyl)pyridine)Cl][PF6] at 1.781 Å and shorter than the reported value for the complex [(η5-Cp*)Ir(phpy)Cl] at 1.863 Å.207
More interestingly, in complexes 46–48, C–H⋯F secondary interactions and consequent chain-like structures have been found.
Theoretical analysis of the frontier molecular orbitals and charge distribution of complexes A1–D1 and A2–D2 were carried out through DFT approaches. Computational results on N(1)-coordinated complexes showed that the HOMOs were essentially located over the metal center and the π orbital of the Cp* ring. Clearly speaking, the HOMO orbitals consist of d orbital of the metal interacting with the π orbital of the Cp* ring and p orbitals of N(1) in the triazole ring and chalcogen atom, while in the HOMOs of N(2)-coordinated complexes, the π orbital of 4-phenyl-1,2,3-triazole interacts with the metal d orbital and π orbital of the Cp* ring and the p orbital of the chlorine atom. The HOMO–LUMO energy gap was also investigated to interpret the chemical reactivity of the complexes. The calculated results indicated that the HOMO–LUMO energy gaps of complexes A1, B1, A2, and B2 were smaller than those of C1, D1, C2, and D2.
It should be stated that the obtained result was in confirmation with the higher reactivity of Rh complexes in comparison to Ir complexes. Moreover, it can be predicted that among the complexes A1, B1, A2, and B2, complexes A2 and B2 could be expected to be more catalytically active than A1 and B1 complexes, and within the Ir complexes, the reactivity of complex D2 should be more than the other three Ir complexes. It should be mentioned that the obtained DFT results were in reliable agreement with the elucidations in terms of the catalytic activities of the various complexes.
From the electronic viewpoint, the coordination strength can be assumed to be a significant parameter in the stability of a complex. In complexes A1–D1, the calculated values of electron density at the coordinated N(1) of the triazole ring were quite high and so these complexes are more stable electronically, while for the complexes where N(2) of the triazole ring is involved in the coordination, the calculated electron density on the N(2) atom and consequently the stability of the complex was reduced. In conclusion, the electronic stability of complexes involving N(1) in coordination was greater than in those involving N(2) and so their reactivity is less.37,56,126,208 It should be noted that the crystallographical structural data for complexes 46, 47, and 48 were in good agreement with the calculated values.
It is important to note that all DFT computations were performed using the B3LYP functional. In the case of Ir and Rh, effective core potential (LANL2DZ) calculations209–212 were applied for core electrons, while for other elements, the 6-311G** basis set were used.
The initial geometry coordinates were directly specified as a single-crystal X-ray structure. In order to establish the stationary points as the minima, frequency calculations were carried out at the end of the geometry optimization process and the molecular orbital properties were calculated with respect to the optimized coordinates using the same level of theory and the same basis set.213
The frontier molecular orbital analysis of iridium(III) complexes with phenyl-1H-[1,2,3]triazoles showed that for all the complexes, the HOMO corresponded to the 5dxy-orbital, while the LUMO was a π*-orbital. In the case of [Ir(19)2(bpy)]+ 49, a π-orbital of the cyclometalating ligand was intercalated within the occupied 5d-orbitals. Interestingly, this antibonding orbital was located on the ancillary ligand in the case of [Ir(19)2(bpy)]+ 49 and [Ir(19)2(pic)] 50, but was on the cyclometalating ligand in the case of [Ir(19)2(acac)] 51 (schematic structures of the iridium(III) complexes are displayed in Scheme 41).
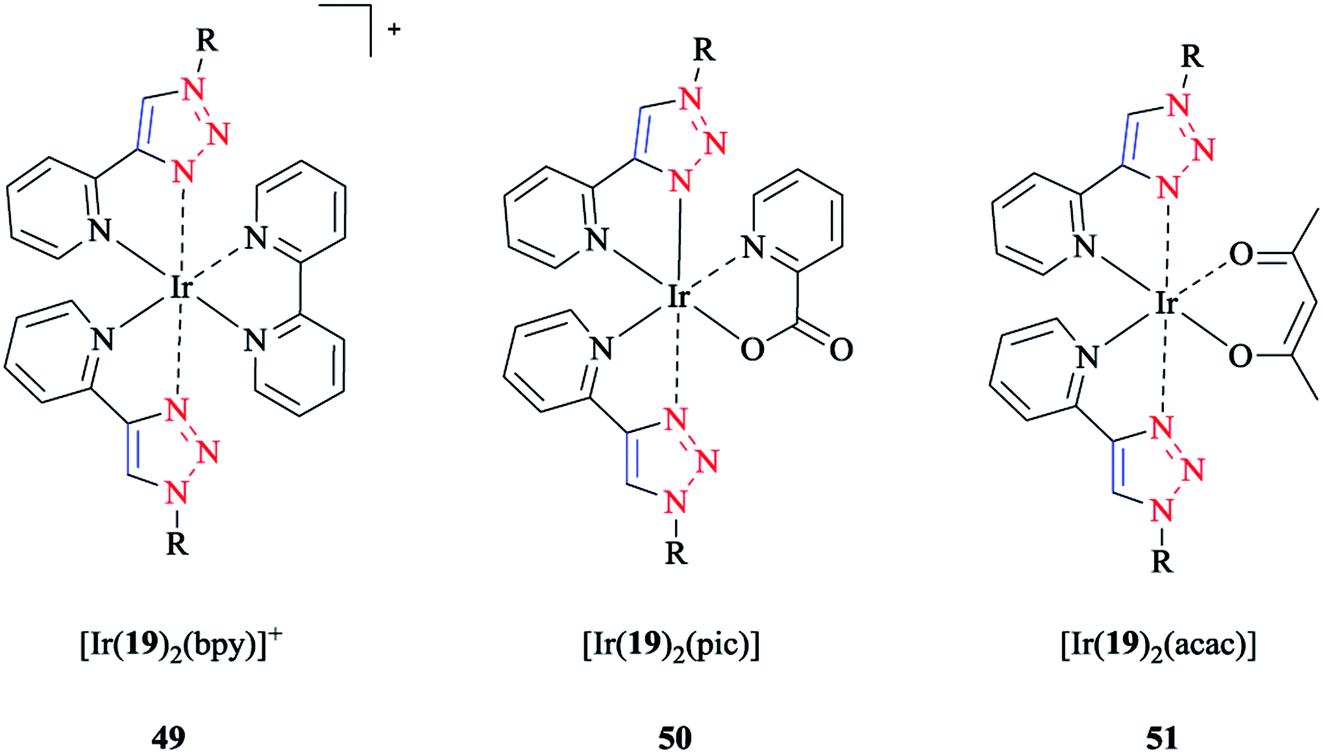 | ||
| Scheme 41 Structures of the [Ir(19)2(bpy)]+ (49), [Ir(19)2(pic)] (50), and [Ir(19)2(acac)] (51) complexes based on 1-decyl-4-phenyl-1H-[1,2,3]triazole ligand (19). | ||
Since the strong ligand field effect of the phenyl anion ligand increased the gap between the t2g and eg orbitals, the eg orbitals of Ir(III) were strongly destabilized in these complexes. The calculations demonstrated that the substituents on the cyclometalating ligands as well as on the ancillary ligand destabilized the HOMO and stabilized the LUMO, and so decreased the HOMO–LUMO energy gap.
The calculated values of the LUMO levels and the theoretical band gaps Eg showed particularly an overestimation compared to the corresponding experimental data. It should be mentioned that the relative change in the complex series was in agreement with the LUMO energies and the HOMO–LUMO energy gaps (Eacacg > Epicg > Ebpyg).
The calculated results for [Ir(19)2(acac)] 51 could be used to evaluate the influence of the cyclometalating ligand on the HOMO–LUMO gap. Apparently, 1,2,3-triazole ligands increase the energy gap in comparison with the ppy ligand-based complex, which can be attributed to the stabilization of HOMO and destabilization of LUMO, and so can be caused by the increased π-acceptor ability of the 1,2,3-triazole moiety. Moreover, back-bonding effects for receiving electron density in the π*-orbitals of the ligand-centered π-system stabilized the occupied t2g orbitals of the iridium complexes. On the other hand, it also destabilized the unoccupied π*-orbitals of the ligands including the LUMO level.
Computational frontier orbitals assessment on the effect of the ancillary ligand (bpy, acac, pic) led to the following results: first, a larger band gap was obtained for the neutral compounds in comparison to the cationic [Ir(19)2(bpy)]+ 49 complex and Ir(III) complexes. More precisely, the HOMO energies were stabilized in the cationic compound in comparison to in the neutral complexes. However, the LUMO was considerably more stabilized than the HOMO, and so the HOMO–LUMO energy gap was reduced. Second, it was shown that the major differences between the neutral compounds (i.e., [Ir(19)2(pic)] 50 and [Ir(19)2(acac)] 51) were mainly due to the LUMO energies rather than the HOMO energies through a comparison between the neutral compounds (i.e., [Ir(19)2(pic)] 50 and [Ir(19)2(acac)] 51). As mentioned earlier, both LUMOs showed a π*-character, but were located on the cyclometalating and on the ancillary ligand, respectively. For the In [Ir(19)2(pic)] 50 complex, the LUMO  on the ancillary ligand was stabilized with respect to the π*and orbital in [Ir(19)2(acac)] 51, which had higher energy values than the π*-orbital of the cyclometalating ligands.114
on the ancillary ligand was stabilized with respect to the π*and orbital in [Ir(19)2(acac)] 51, which had higher energy values than the π*-orbital of the cyclometalating ligands.114
4.5 Ru(II) complexes
Next, we report the crystallographical assessments of some ruthenium complexes of click-generated 1,2,3-triazole-based organosulfur–selenium ligands (Schemes 42 and 43). | ||
| Scheme 42 Structures of Ru(II) complexes with 1,2,3-triazole-based organosulfur/selenium ligands. | ||
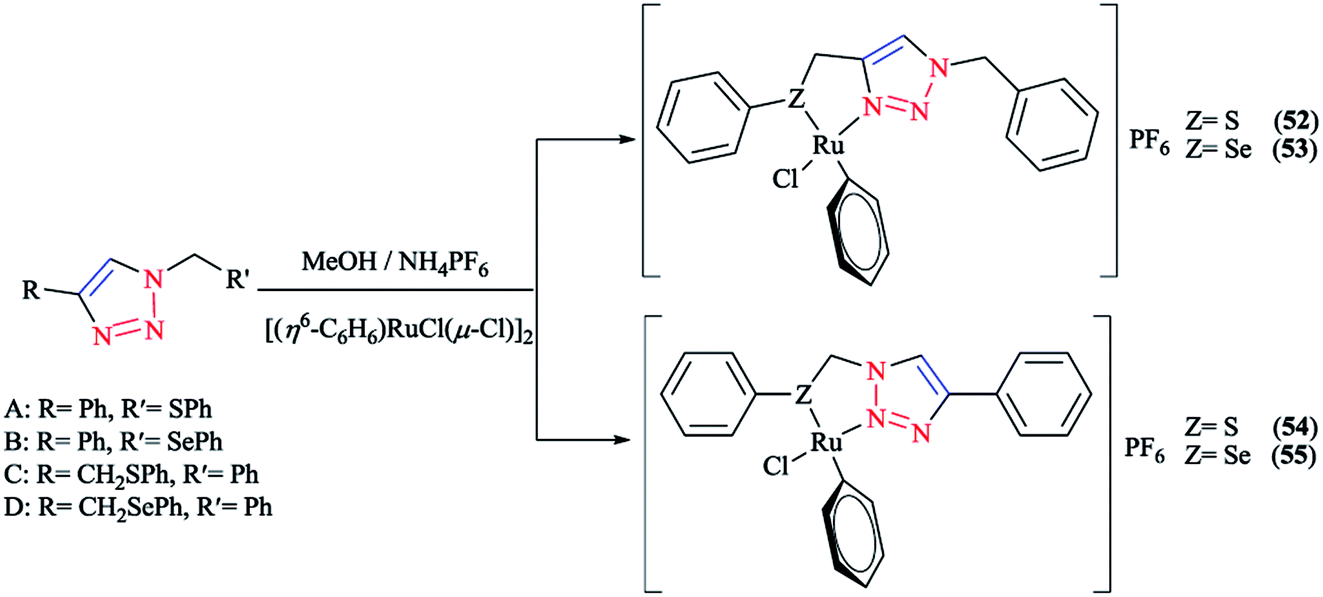 | ||
| Scheme 43 Syntheses of A–D ligands and complexes 52–55. | ||
A pseudo-octahedral half-sandwich “piano-stool” disposition of donor atoms occurs around the Ru metal center in complexes 52–55. More precisely, one face of an octahedron in all four complexes was occupied with a benzene ring and a chelate ring is formed with nitrogen and chalcogen atoms around the metal center, while a chlorine donor atom completes the coordination sphere. Overall, a three-legged piano-stool conformation was observed in the X-ray crystal structure.
The Ru–S bond lengths were obtained as 2.3847 Å in complex 52 and 2.3893 Å in complex 54, which are consistent with each other.
On the other hand, the Ru–Se bond distance in complex 53 was obtained as 2.497 Å, which was longer than that of complex 55 at 2.4859 Å, but was similar with the values for [(η6-C6H6)RuCl(N-{2-(phenylseleno)ethyl}pyrrolidine)]+ at 2.480 Å and for [(η6-C6H6)Ru(2-MeSC6H4CHNCH2CH2SeC6H5)]2+ at 2.4848 Å. However, the Ru–Se bond length for [RuCp(CO)(CCPh)(μ-Se)ZrCp2] at 2.494 Å was in confirmation with that of complex 53. In complexes 52–55, the Ru–N bond lengths varied in a narrow range (2.072–2.101 Å) and were similar with the reported Ru–N bond distance for [Ru(2,6-bis(1-benzyl-1,2,3-triazol-4-yl)pyridine)2](PF6)2 at 2.050 Å (ref. 214) and [(η6-C6H6)Ru(2-MeSC6H4CHN–CH2CH2SC6H5)]2+ at 2.073 Å. In addition, the Ru–C bond lengths of 2.154–2.22 Å and the C–Ru–C bond angles of complexes 52–55 were in the normal range.215–217 The PF6-groups were involved in C–H⋯F secondary interactions in all the complexes 52–55.
From the computational viewpoint, the cases of Ru–Cl, Ru–N, and Ru–C (centroid) showed an excellent agreement between the DFT calculated and experimental bond distances values. Furthermore, the X-ray values of the bond angles were very close to the calculated ones.
In the next step, DFT calculations were performed to assess qualitatively the optimized geometries, frontier orbitals, and charge distribution of complexes 52–55. In complexes 52 and 53, the HOMOs are positioned primarily over the Ru and Cl atoms and include the orbital of Ru(II) interacting with a π orbital of the η6-benzene ring, a p orbital of the triazole nitrogen, and a chalcogen atom. In complexes 54 and 55, the HOMOs constitute a π orbital of 4-phenyl-1,2,3-triazole and a d orbital of the metal. However, the d orbital of Ru(II) interacts with the π orbital of the η6-benzene ring, the p orbital of triazole nitrogen, and a chalcogen donor atom in HOMO-1.
DFT calculations indicated that the HOMO–LUMO energy gaps in complexes 54 and 55 were lower than those of 52 and 53, which corresponds to the higher reactivity of complexes 54 and 55.
It is important to note that in complexes 54 and 55, there is N(2) coordinating moiety, which can be attributed to the low energy gaps. On the other hand, the calculated electron densities of the coordinated N(1) triazole ring in complexes 52 and 53 were higher than the calculated value for N(2) in complexes 54 and 55. This electronic feature can be interpreted as due to a strong coordination between N(1) of triazole and Ru in complexes 52 and 53, which leads to greater electronic stability in comparison with 54 and 55.37
In another work, the crystal structure of Ru(II) complexes with NNN, NCN, NCN–NO2, NCN–F, and NCN–Tph ligands were determined via the X-ray diffraction method and some considerable changes were observed attributed to the packing effects, such as a strong distortion within RuNCN–F.
In the RuNCN–Tph complex 60, the thiophene-phenyl torsion angle was obtained as 30.78°, which confirmed the calculated results. This feature led to the partial extension of conjugation into the thiophene ring. It was shown that when a dative Ru–N bond of the poly-pyridyl-type complex RuNNN was replaced with a covalent organometallic Ru–C1 bond in RuNCN complex 57, the bond distance was reduced from 2.02 to 1.98 Å. This behavior could be mainly attributed to s donation and additional p donation as well as by electrostatic interactions with the anionic, aromatic carbon donor. Moreover, the triazole N–Ru bond length was increased due to the decrease in the orbital overlap by the small bite angle, while, a trans influence occurs and the opposed Ru–N bond distance is increased to 2.01 Å based on the considerable electron-donation ability of the carbanion. More interestingly, the outer pyridine N–Ru bonds were shortened, which could be ascribed to the p back donation from the electron-rich Ru(II) metal center (Schemes 44 and 45).
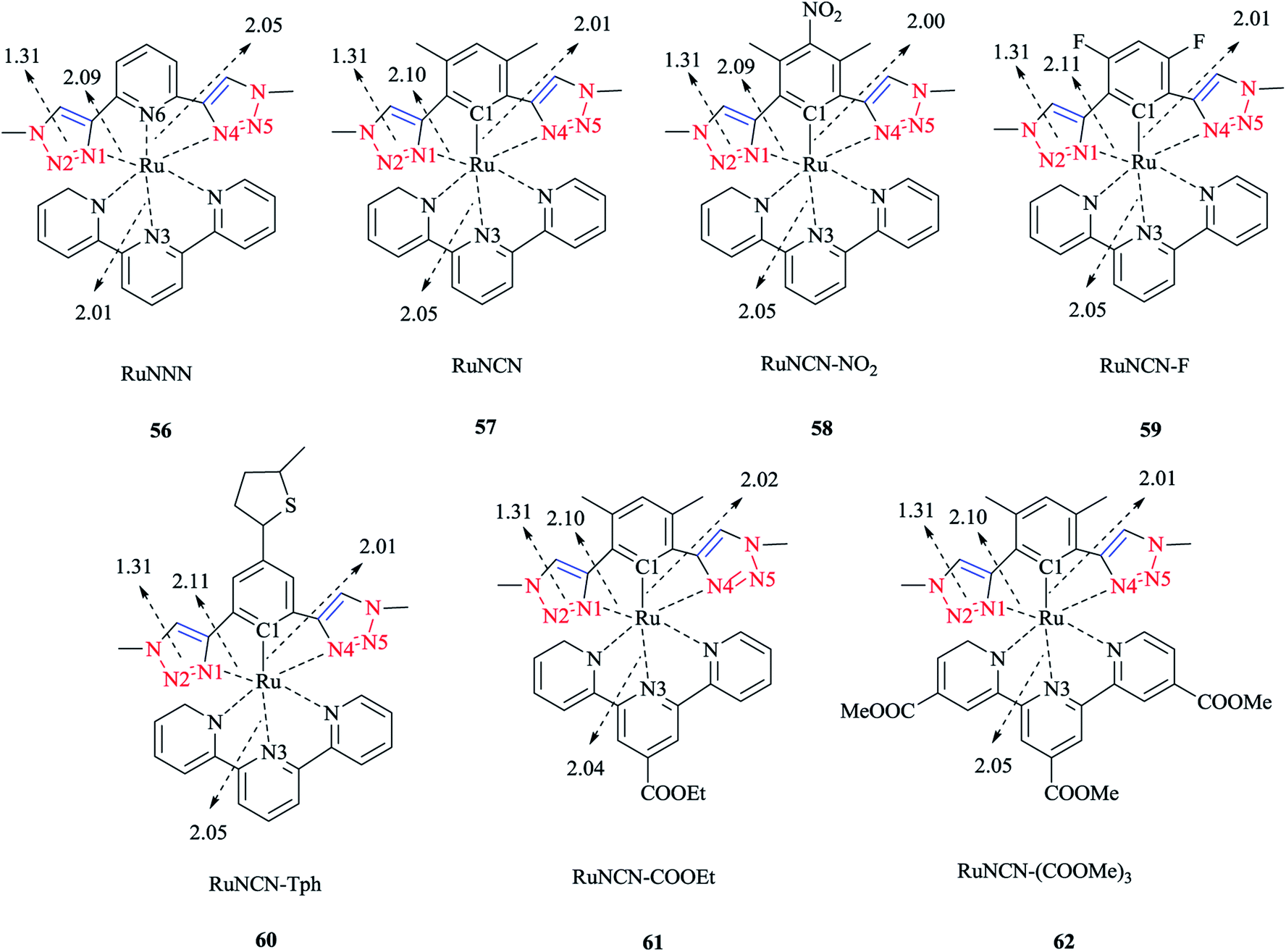 | ||
| Scheme 44 Structures of Ru(II) complexes with some selected calculated bond distances (Å). | ||
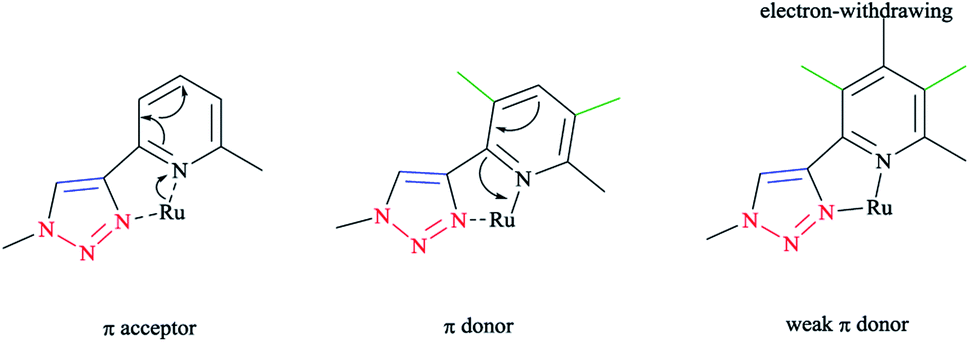 | ||
| Scheme 45 Schematic of the electronic features of the cyclometalation and an electron-withdrawing group. | ||
Further, the bond length of the N–N double bond in the triazole ring was increased due to the increasing p back donation into the π* orbitals. By the substitution of NO2 as an electron-withdrawing group, it was indicated that the cyclometalation process was affected less than by installing a CN-withdrawing moiety in the RuNCN complex 57.
Another important structural feature is the dihedral angle with the central phenyl ring (as illustrated in Scheme 44), which has a 51.88 X-ray determined value and 51.68 calculated value and can lead to a strong decrease in the π-donation ability of the cyclometalating carbanion in the para position, whereby consequently, the Ru–C1 bond is elongated to 2.00 Å.
In the case of the RuNCN–F complex 59, it was found that substitution of the fluorine atom as a strong s-accepting group with a moderate p-donation character, in the meta position, has a not considerable effect on the carbanion and so results in a very short Ru–C1 bond of 1.98 Å. Moreover, it should be mentioned that a decrease in the bond length of the triazole N2–N3 double bond arises from the fluoro and nitro substituent effect, which causes a reduction of the p back electron-donation ability of the carbanion. Strictly speaking, the fluorine substituent essentially affects the σ donation and indirectly reduces the energy of the p system as an inductive effect. This occurs while the NO2 group polarizes the p system additionally and so lowers its p-donation character via a mesomeric effect. It was also demonstrated for the RuNCN–F complex 59 that the HOMO is stabilized and located on Ru(II) metal center and also covers the fluoro-substituted cyclometalating phenyl ring. This occurs while an additional LUMO stabilization occurs in RuNCN–NO2 complex 58 since the LUMO is located on the opposed tpy ligand and so this can be related to the effect of the aromatic p system and d orbitals of Ru(II).116
In another report, the electronic properties of ruthenium(II) complexes with 2,6-bis(1,2,3-triazol-4-yl)pyridine 7 ligands were assessed using DFT calculations. The calculations showed that the highest occupied molecular orbital (HOMO) of the complexes included a metal d orbital and π orbitals located on the triazolate rings, which were in confirmation with the electron-rich π system of the anionic ring.218–220 Based on the electron repulsion between the anionic ligand and the metal center, the HOMO is strongly destabilized in comparison to polypyridyl complexes such as [Ru(tpy)2](PF6)2 63 (Scheme 46). For complexes with terpyridine ligands, the lowest unoccupied molecular orbital (LUMO) is mainly composed of π* orbitals of the ligand. Considering the electron-withdrawing groups, the HOMO and, in particular, the LUMO are stabilized due to the electron-withdrawing effect, leading to a significantly smaller HOMO–LUMO gap. A similar effect on the frontier orbitals energies was observed using –COOH anchoring groups. In contrast, for a deprotonated complex, the HOMO and LUMO of the complex are destabilized and the HOMO–LUMO gap slightly increased, which is attributed to the electron-donating effect of the –COO-groups.
 | ||
| Scheme 46 Structure of [Ru(tpy)2](PF6)2 complex. | ||
There was good confirmation between the calculated electronic excitations and the experimental UV-vis absorption spectrum. The lowest energy absorption was a pure HOMO–LUMO transition, which could be assigned to a metal-to-ligand charge transfer (MLCT) with some ligand-to-ligand charge-transfer (LLCT) character.117
5. Conclusions
In summary, 1,2,3-triazole-based ligands synthesized via CuAAC click reaction are introduced as diverse biological and pharmaceutical compounds and also their complexes with transition metals were widely applied as efficient catalysts in chemical reactions and reported here. The coordination properties of Ni, Pd, Pt, Re, Rh, Ru, and Ir transition metal complexes with a series of triazole-based click ligands were investigated via computational chemistry approaches by the calculation of their structural properties, electronic energies, and by frontier molecular orbital analysis. More clearly, in the aforementioned metal complexes of triazoles- and triazaphospholes-based ligands, their X-ray crystal structure, FT-IR and NMR spectroscopic features, metal-to-ligand charge transfer, trans and cis chelation modes, and emission properties were computationally assessed via comparison with the experimental data, which showed reliable agreement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Alzahra University Research Council. Tayebeh Hosseinnejad is also thankful to Iran National Science Foundation (INSF) for partial financial support.Notes and references
- R. Huisgen, Angew. Chem., 1963, 75, 604–637 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- M. M Heravi, M. Tamimi, H. Yahyavi and T. Hosseinnejad, Curr. Org. Chem., 2016, 20, 1591–1647 CrossRef.
- J. Lauko, P. H. Kouwer and A. E. Rowan, J. Heterocycl. Chem., 2017, 54, 1677–1699 CrossRef CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675–696 RSC.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- A. H. Kategaonkar, P. V. Shinde, A. H. Kategaonkar, S. K. Pasale, B. B. Shingate and M. S. Shingare, Eur. J. Med. Chem., 2010, 45, 3142–3146 CrossRef CAS PubMed.
- S. Demirci, S. Basoglu, A. Bozdereci and N. Demirbas, Med. Chem. Res., 2013, 22, 4930–4945 CrossRef CAS.
- W. S. Horne, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 9372–9376 CrossRef CAS PubMed.
- W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed.
- M. H. Palmer, R. H. Findlay and A. J. Gaskell, J. Chem. Soc., Perkin Trans. 2, 1974, 420–428 RSC.
- R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. D. Clercq, C.-F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS PubMed.
- L. L. Brockunier, E. R. Parmee, H. O. Ok, M. R. Candelore, M. A. Cascieri, L. F. Colwell, L. Deng, W. P. Feeney, M. J. Forrest and G. J. Hom, Bioorg. Med. Chem. Lett., 2000, 10, 2111–2114 CrossRef CAS PubMed.
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester and D. K. Hutchinson, J. Med. Chem., 2000, 43, 953–970 CrossRef CAS PubMed.
- D. R. Buckle and C. J. Rockell, J. Chem. Soc., Perkin Trans. 1, 1982, 627–630 RSC.
- R. A. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402–2414 RSC.
- Q. V. van Hilst, N. R. Lagesse, D. Preston and J. D. Crowley, Dalton Trans., 2018, 47, 997–1002 RSC.
- S. C. Hockey, G. J. Barbante, P. S. Francis, J. M. Altimari, P. Yoganantharajah, Y. Gibert and L. C. Henderson, Eur. J. Med. Chem., 2016, 109, 305–313 CrossRef CAS.
- S. Löber, P. Rodriguez-Loaiza and P. Gmeiner, Org. Lett., 2003, 5, 1753–1755 CrossRef PubMed.
- J. P. Collman, N. K. Devaraj and C. E. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS PubMed.
- D. D. Díaz, S. Punna, P. Holzer, A. K. Mcpherson, K. B. Sharpless, V. V. Fokin and M. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef.
- A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Dalton Trans., 2013, 42, 5200–5223 RSC.
- F. Saleem, G. K. Rao, P. Singh and A. K. Singh, Organometallics, 2013, 32, 387–395 CrossRef CAS.
- M. Felici, P. Contreras-Carballada, Y. Vida, J. M. Smits, R. J. Nolte, L. De Cola, R. M. Williams and M. C. Feiters, Chem.–Eur. J., 2009, 15, 13124–13134 CrossRef CAS PubMed.
- H. Yang, L. Li, L. Wan, Z. Zhou and S. Yang, Inorg. Chem. Commun., 2010, 13, 1387–1390 CrossRef CAS.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed.
- M. Felici, P. Contreras-Carballada, J. Smits and R. Nolte, Molecules, 2010, 15, 2039 CrossRef CAS PubMed.
- C. Hua, K. Q. Vuong, M. Bhadbhade and B. A. Messerle, Organometallics, 2012, 31, 1790–1800 CrossRef CAS.
- N. V. Dubrovina, L. Domke, I. A. Shuklov, A. Spannenberg, R. Franke, A. Villinger and A. Börner, Tetrahedron, 2013, 69, 8809–8817 CrossRef CAS.
- D. C. Goldstein, J. R. Peterson, Y. Y. Cheng, R. G. Clady, T. W. Schmidt and P. Thordarson, Molecules, 2013, 18, 8959–8975 CrossRef CAS PubMed.
- K. Q. Vuong, M. G. Timerbulatova, M. B. Peterson, M. Bhadbhade and B. A. Messerle, Dalton Trans., 2013, 42, 14298–14308 RSC.
- F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2013, 32, 3595–3603 CrossRef CAS.
- M. Schuster, M. Botoshansky and M. Gandelman, Angew. Chem., Int. Ed., 2008, 47, 4555–4558 CrossRef PubMed.
- H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, Chem.–Eur. J., 2008, 14, 6173–6183 CrossRef CAS PubMed.
- E. M. Schuster, G. Nisnevich, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 5025–5031 CrossRef CAS.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 7001–7005 CrossRef CAS.
- H. V. Ching, X. Wang, M. He, N. Perujo Holland, R. Guillot, C. Slim, S. Griveau, H. C. Bertrand, C. Policar and F. Bedioui, Inorg. Chem., 2017, 56, 2966–2976 CrossRef CAS PubMed.
- S. Hostachy, C. Policar and N. Delsuc, Coord. Chem. Rev., 2017, 351, 172–188 CrossRef CAS.
- H. C. Bertrand, S. Clède, R. Guillot, F. Lambert and C. Policar, Inorg. Chem., 2014, 53, 6204–6223 CrossRef CAS PubMed.
- A. Dazzi and C. Policar, in Biointerface characterization by advanced IR spectroscopy, Elsevier, 2011, pp. 245–278 Search PubMed.
- M. G. Fraser, A. G. Blackman, G. I. Irwin, C. P. Easton and K. C. Gordon, Inorg. Chem., 2010, 49, 5180–5189 CrossRef CAS PubMed.
- S. L. Howell, K. C. Gordon and J. J. McGarvey, J. Phys. Chem. A, 2005, 109, 2948–2956 CrossRef CAS PubMed.
- S. L. Howell, B. J. Matthewson, M. I. Polson, A. K. Burrell and K. C. Gordon, Inorg. Chem., 2004, 43, 2876–2887 CrossRef CAS PubMed.
- B. J. Matthewson, A. Flood, M. I. J. Polson, C. Armstrong, D. L. Phillips and K. C. Gordon, Bull. Chem. Soc. Jpn., 2002, 75, 933–942 CrossRef CAS.
- A. Y. Hirakawa and M. Tsuboi, Science, 1975, 188, 359–361 CAS.
- R. J. Clark and T. J. Dines, Angew. Chem., Int. Ed. Engl., 1986, 25, 131–158 CrossRef.
- A. Flood, R. B. Girling, K. C. Gordon, R. E. Hester, J. N. Moore and M. I. Polson, J. Raman Spectrosc., 2002, 33, 434–442 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- D. Schweinfurth, J. Krzystek, I. Schapiro, S. Demeshko, J. Klein, J. Telser, A. Ozarowski, C.-Y. Su, F. Meyer and M. Atanasov, Inorg. Chem., 2013, 52, 6880–6892 CrossRef CAS PubMed.
- K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334–6346 CrossRef CAS PubMed.
- B. Pinter, A. Demšar, D. Urankar, F. De Proft and J. Košmrlj, Polyhedron, 2011, 30, 2368–2373 CrossRef CAS.
- C. B. Anderson, A. B. Elliott, J. E. Lewis, C. J. McAdam, K. C. Gordon and J. D. Crowley, Dalton Trans., 2012, 41, 14625–14632 RSC.
- M. Wolff, L. Munoz, A. François, C. Carrayon, A. Seridi, N. Saffon, C. Picard, B. Machura and E. Benoist, Dalton Trans., 2013, 42, 7019–7031 RSC.
- D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 553–561 CrossRef CAS.
- P. A. Scattergood, A. Sinopoli and P. I. Elliott, Coord. Chem. Rev., 2017, 350, 136–154 CrossRef CAS.
- P. A. Scattergood and P. I. Elliott, Dalton Trans., 2017, 46, 16343–16356 RSC.
- A. Seridi, M. Wolff, A. Boulay, N. Saffon, Y. Coulais, C. Picard, B. Machura and E. Benoist, Inorg. Chem. Commun., 2011, 14, 238–242 CrossRef CAS.
- T. Y. Kim, A. B. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391–1398 CrossRef CAS.
- J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC.
- J. A. Sklorz, S. Hoof, M. G. Sommer, F. Weißer, M. Weber, J. Wiecko, B. Sarkar and C. Müller, Organometallics, 2014, 33, 511–516 CrossRef CAS.
- F. Ebrahimpour-Malamir, T. Hosseinnejad, R. Mirsafaei and M. M. Heravi, Appl. Organomet. Chem., 2017 DOI:10.1002/aoc.3913.
- T. Baie Lashaki, H. A. Oskooie, T. Hosseinnejad and M. M. Heravi, J. Coord. Chem., 2017, 1–20 Search PubMed.
- T. Hossiennejad, M. Daraie, M. M. Heravi and N. N. Tajoddin, J. Inorg. Organomet. Polym. Mater., 2017, 1–10 Search PubMed.
- R. Mirsafaei, M. M. Heravi, T. Hosseinnejad and S. Ahmadi, Appl. Organomet. Chem., 2016, 10, 823–830 CrossRef.
- R. Mirsafaei, M. M. Heravi, S. Ahmadi and T. Hosseinnejad, Chem. Pap., 2016, 70, 418–429 CAS.
- T. Hosseinnejad and M. Dinyari, Comput. Theor. Chem., 2015, 1071, 53–60 CrossRef CAS.
- R. Mirsafaei, M. M. Heravi, S. Ahmadi, M. H. Moslemin and T. Hosseinnejad, J. Mol. Catal. A: Chem., 2015, 402, 100–108 CrossRef CAS.
- T. Hosseinnejad, M. M. Heravi and R. Firouzi, J. Mol. Model., 2013, 19, 951–961 CrossRef CAS PubMed.
- M. M. Heravi, T. Hosseinnejad and N. Nazari, Can. J. Chem., 2017, 95, 530–536 CrossRef CAS.
- T. Hosseinnejad, B. Fattahi and M. M. Heravi, J. Mol. Model., 2015, 21, 264 CrossRef PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7–17 CAS.
- J. D. Crowley and D. A. McMorran, in Click Triazoles, Springer, 2012, pp. 31–83 Search PubMed.
- D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nachr. Chem., 2011, 59, 937–941 CrossRef CAS.
- P. M. Guha, H. Phan, J. S. Kinyon, W. S. Brotherton, K. Sreenath, J. T. Simmons, Z. Wang, R. J. Clark, N. S. Dalal and M. Shatruk, Inorg. Chem., 2012, 51, 3465–3477 CrossRef CAS PubMed.
- D. Schweinfurth, R. Pattacini, S. Strobel and B. Sarkar, Dalton Trans., 2009, 9291–9297 RSC.
- M. Felici, P. Contreras-Carballada, J. M. Smits, R. J. Nolte, R. M. Williams, L. De Cola and M. C. Feiters, Molecules, 2010, 15, 2039–2059 CrossRef CAS PubMed.
- S. Zanarini, M. Felici, G. Valenti, M. Marcaccio, L. Prodi, S. Bonacchi, P. Contreras-Carballada, R. M. Williams, M. C. Feiters and R. J. Nolte, Chem.–Eur. J., 2011, 17, 4640–4647 CrossRef CAS PubMed.
- I. Stengel, A. Mishra, N. Pootrakulchote, S.-J. Moon, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, J. Mater. Chem., 2011, 21, 3726–3734 RSC.
- S. Liu, P. Müller, M. K. Takase and T. M. Swager, Inorg. Chem., 2011, 50, 7598–7609 CrossRef CAS PubMed.
- B. Happ, D. Escudero, M. D. Hager, C. Friebe, A. Winter, H. Görls, E. Altuntas, L. González and U. S. Schubert, J. Org. Chem., 2010, 75, 4025–4038 CrossRef CAS PubMed.
- M. Juríček, M. Felici, P. Contreras-Carballada, J. Lauko, S. R. Bou, P. H. Kouwer, A. M. Brouwer and A. E. Rowan, J. Mater. Chem., 2011, 21, 2104–2111 RSC.
- M. Juríček, P. H. Kouwer and A. E. Rowan, Chem. Commun., 2011, 47, 8740–8749 RSC.
- G. Zhang, Y. Wang, X. Wen, C. Ding and Y. Li, Chem. Commun., 2012, 48, 2979–2981 RSC.
- E. Amadio, M. Bertoldini, A. Scrivanti, G. Chessa, V. Beghetto, U. Matteoli, R. Bertani and A. Dolmella, Inorg. Chim. Acta, 2011, 370, 388–393 CrossRef CAS.
- A. D'Amora, L. Fanfoni, D. Cozzula, N. Guidolin, E. Zangrando, F. Felluga, S. Gladiali, F. Benedetti and B. Milani, Organometallics, 2010, 29, 4472–4485 CrossRef.
- I. Bratsos, D. Urankar, E. Zangrando, P. Genova-Kalou, J. Košmrlj, E. Alessio and I. Turel, Dalton Trans., 2011, 40, 5188–5199 RSC.
- D. Urankar, A. Pevec, I. Turel and J. Košmrlj, Cryst. Growth Des., 2010, 10, 4920–4927 CAS.
- C. B. Anderson, A. B. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Organometallics, 2013, 32, 788–797 CrossRef CAS.
- B. Happ, C. Friebe, A. Winter, M. D. Hager, R. Hoogenboom and U. S. Schubert, Chem.–Asian J., 2009, 4, 154–163 CrossRef CAS PubMed.
- M. Obata, A. Kitamura, A. Mori, C. Kameyama, J. A. Czaplewska, R. Tanaka, I. Kinoshita, T. Kusumoto, H. Hashimoto and M. Harada, Dalton Trans., 2008, 3292–3300 RSC.
- D. Schweinfurth, C.-Y. Su, S.-C. Wei, P. Braunstein and B. Sarkar, Dalton Trans., 2012, 41, 12984–12990 RSC.
- D. Urankar, B. Pinter, A. Pevec, F. De Proft, I. Turel and J. Košmrlj, Inorg. Chem., 2010, 49, 4820–4829 CrossRef CAS PubMed.
- S. L. Choong, A. Nafady, A. Stasch, A. M. Bond and C. Jones, Dalton Trans., 2013, 42, 7775–7780 RSC.
- S. V. Chapyshev, Synlett, 2009, 1–8 CAS.
- S. Chapyshev, U. Bergsträßer and M. Regitz, Chem. Heterocycl. Compd., 1996, 32, 59–64 CrossRef.
- S. L. Choong, C. Jones and A. Stasch, Dalton Trans., 2010, 39, 5774–5776 RSC.
- C. Pardin, I. Roy, W. D. Lubell and J. W. Keillor, Chem. Biol. Drug Des., 2008, 72, 189–196 CAS.
- G. Becker, G. Gresser and W. Uhl, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1981, 36, 16–19 Search PubMed.
- J. G. Cordaro, D. Stein, H. Rüegger and H. Grützmacher, Angew. Chem., Int. Ed., 2006, 45, 6159–6162 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem.–Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- M. Ostermeier, M. A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Chem.–Eur. J., 2010, 16, 10202–10213 CrossRef CAS PubMed.
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, U. Köhn, E. Altuntas, A. Baumgaertel, M. D. Hager and A. Winter, Chem.–Eur. J., 2011, 17, 5494–5498 CrossRef CAS PubMed.
- M. Beley, J. P. Collin, R. Louis, B. Metz and J. P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8521–8522 CrossRef CAS.
- J. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. González and U. S. Schubert, Organometallics, 2009, 28, 5478–5488 CrossRef CAS.
- A. J. Wilkinson, H. Puschmann, J. A. Howard, C. E. Foster and J. G. Williams, Inorg. Chem., 2006, 45, 8685–8699 CrossRef CAS PubMed.
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp and B. Dietzek, Chem.–Eur. J., 2012, 18, 4010–4025 CrossRef CAS PubMed.
- S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, J. Kübel, B. Dietzek, C. P. Berlinguette and U. S. Schubert, Inorg. Chem., 2014, 53, 1637–1645 CrossRef CAS PubMed.
- S. K. Vellas, J. E. Lewis, M. Shankar, A. Sagatova, J. D. Tyndall, B. C. Monk, C. M. Fitchett, L. R. Hanton and J. D. Crowley, Molecules, 2013, 18, 6383–6407 CrossRef CAS PubMed.
- S. V. Kumar, W. K. Lo, H. J. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1–6 CrossRef CAS.
- R. A. Vasdev, D. Preston, S. Ø. Scottwell, H. J. Brooks, J. D. Crowley and M. P. Schramm, Molecules, 2016, 21, 1548 CrossRef PubMed.
- S. V. Kumar, W. K. Lo, H. J. Brooks, L. R. Hanton and J. D. Crowley, Aust. J. Chem., 2016, 69, 489–498 CrossRef CAS.
- B. J. Pages, J. Sakoff, J. Gilbert, Y. Zhang, F. Li, D. Preston, J. D. Crowley and J. R. Aldrich-Wright, J. Inorg. Biochem., 2016, 165, 92–99 CrossRef CAS PubMed.
- S. Jindabot, K. Teerachanan, P. Thongkam, S. Kiatisevi, T. Khamnaen, P. Phiriyawirut, S. Charoenchaidet, T. Sooksimuang, P. Kongsaeree and P. Sangtrirutnugul, J. Organomet. Chem., 2014, 750, 35–40 CrossRef CAS.
- F. Saleem, G. Rao, A. Kumar, S. Kumar, M. P. Singh and A. K. Singh, RSC Adv., 2014, 4, 56102–56111 RSC.
- W. K. Lo, G. S. Huff, J. R. Cubanski, A. D. Kennedy, C. J. McAdam, D. A. McMorran, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2015, 54, 1572–1587 CrossRef CAS PubMed.
- K. J. Kilpin and J. D. Crowley, Polyhedron, 2010, 29, 3111–3117 CrossRef CAS.
- D. Schweinfurth, S. Strobel and B. Sarkar, Inorg. Chim. Acta, 2011, 374, 253–260 CrossRef CAS.
- S. Yano, H. Ohi, M. Ashizaki, M. Obata, Y. Mikata, R. Tanaka, T. Nishioka, I. Kinoshita, Y. Sugai and I. Okura, Chem. Biodiversity, 2012, 9, 1903–1915 CAS.
- E. P. McCarney, C. S. Hawes, S. Blasco and T. Gunnlaugsson, Dalton Trans., 2016, 45, 10209–10221 RSC.
- M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952–2955 CrossRef CAS PubMed.
- D. Sooksawat, S. J. Pike, A. M. Slawin and P. J. Lusby, Chem. Commun., 2013, 49, 11077–11079 RSC.
- M. Prabhath, J. Romanova, R. J. Curry, S. R. P. Silva and P. D. Jarowski, Angew. Chem., Int. Ed., 2015, 54, 7949–7953 CrossRef CAS PubMed.
- J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC.
- J. D. Crowley, P. H. Bandeen and L. R. Hanton, Polyhedron, 2010, 29, 70–83 CrossRef CAS.
- N. Wu, C. F. Melan, K. A. Stevenson, O. Fleischel, H. Guo, F. Habib, R. J. Holmberg, M. Murugesu, N. J. Mosey and H. Nierengarten, Dalton Trans., 2015, 44, 14991–15005 RSC.
- C. Ducani, A. Leczkowska, N. J. Hodges and M. J. Hannon, Angew. Chem., Int. Ed., 2010, 49, 8942–8945 CrossRef CAS PubMed.
- Y. Parajó, J. Malina, I. Meistermann, G. J. Clarkson, M. Pascu, A. Rodger, M. J. Hannon and P. Lincoln, Dalton Trans., 2009, 4868–4874 RSC.
- S. Phongtongpasuk, S. Paulus, J. Schnabl, R. K. Sigel, B. Spingler, M. J. Hannon and E. Freisinger, Angew. Chem., Int. Ed., 2013, 52, 11513–11516 CrossRef CAS.
- D. Preston, R. A. Tucker, A. L. Garden and J. D. Crowley, Inorg. Chem., 2016, 55, 8928–8934 CrossRef CAS PubMed.
- Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697–6700 CrossRef CAS PubMed.
- J. Andersen, U. Madsen, F. Björkling and X. Liang, Synlett, 2005, 2209–2213 CAS.
- E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2011, 31, 456–461 CrossRef.
- K. J. Kilpin, U. S. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC.
- N. G. White and P. D. Beer, Supramol. Chem., 2012, 24, 473–480 CrossRef CAS.
- S. Ø. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117–12124 RSC.
- J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035–4037 RSC.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC.
- M. Atanasov, P. Comba, S. Helmle, D. Müller and F. Neese, Inorg. Chem., 2012, 51, 12324–12335 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- C. Angeli, S. Borini, M. Cestari and R. Cimiraglia, J. Chem. Phys., 2004, 121, 4043–4049 CrossRef CAS PubMed.
- C. Angeli, B. Bories, A. Cavallini and R. Cimiraglia, J. Chem. Phys., 2006, 124, 054108 CrossRef PubMed.
- B. O. Roos, Advances in Chemical Physics: Ab Initio Methods in Quantum Chemistry Part 2, John Wiley & Sons, 2007, pp. 399–445 Search PubMed.
- S. Guo, M. A. Watson, W. Hu, Q. Sun and G. K.-L. Chan, J. Chem. Theory Comput., 2016, 12, 1583–1591 CrossRef CAS PubMed.
- H. Nakano, R. Uchiyama and K. Hirao, J. Comput. Chem., 2002, 23, 1166–1175 CrossRef CAS PubMed.
- J. Telser, J. Krzystek and A. Ozarowski, J. Biol. Inorg Chem., 2014, 19, 297–318 CrossRef CAS.
- P. E. Hoggard, Angular overlap model parameters, Springer, 2004, pp. 37–57 Search PubMed.
- J. J. Wilson, J. Fedoce Lopes and S. J. Lippard, Inorg. Chem., 2010, 49, 5303–5315 CrossRef CAS PubMed.
- V. A. Polyakov and A. D. Ryabov, J. Chem. Soc., Dalton Trans., 1986, 589–593 RSC.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS.
- I. M. Dixon, J.-L. Heully, F. Alary and P. I. Elliott, Phys. Chem. Chem. Phys., 2017, 19, 27765–27778 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- K. A. Stevenson, C. F. Melan, O. Fleischel, R. Wang and A. Petitjean, Cryst. Growth Des., 2012, 12, 5169–5173 CAS.
- L. Jiang, Z. Wang, S.-Q. Bai and T. A. Hor, CrystEngComm, 2013, 15, 10451–10458 RSC.
- E. Amadio, A. Scrivanti, G. Chessa, U. Matteoli, V. Beghetto, M. Bertoldini, M. Rancan, A. Dolmella, A. Venzo and R. Bertani, J. Organomet. Chem., 2012, 716, 193–200 CrossRef CAS.
- J. Herrmann, P. S. Pregosin, R. Salzmann and A. Albinati, Organometallics, 1995, 14, 3311–3318 CrossRef CAS.
- P. Barbaro, A. Currao, J. Herrmann, R. Nesper, P. S. Pregosin and R. Salzmann, Organometallics, 1996, 15, 1879–1888 CrossRef CAS.
- M. Gómez, S. Jansat, G. Muller, M. A. Maestro and J. Mahía, Organometallics, 2002, 21, 1077–1087 CrossRef.
- A. Ros, D. Monge, M. Alcarazo, E. Álvarez, J. M. Lassaletta and R. Fernández, Organometallics, 2006, 25, 6039–6046 CrossRef CAS.
- S. J. Roseblade, A. Ros, D. Monge, M. Alcarazo, E. Álvarez, J. M. Lassaletta and R. Fernández, Organometallics, 2007, 26, 2570–2578 CrossRef CAS.
- E. Amadio, A. Scrivanti, M. Bortoluzzi, M. Bertoldini, V. Beghetto, U. Matteoli and G. Chessa, Inorg. Chim. Acta, 2013, 405, 188–195 CrossRef CAS.
- A. Maisonial, P. Serafin, M. Traïkia, E. Debiton, V. Thery, D. J. Aitken, P. Lemoine, B. Viossat and A. Gautier, Eur. J. Inorg. Chem., 2008, 298–305 CrossRef CAS.
- W. S. Brotherton, H. A. Michaels, J. T. Simmons, R. J. Clark, N. S. Dalal and L. Zhu, Org. Lett., 2009, 11, 4954–4957 CrossRef CAS PubMed.
- O. Fleischel, N. Wu and A. Petitjean, Chem. Commun., 2010, 46, 8454–8456 RSC.
- A. G. Young and L. R. Hanton, Coord. Chem. Rev., 2008, 252, 1346–1386 CrossRef CAS.
- P. Thongkam, S. Jindabot, S. Prabpai, P. Kongsaeree, T. Wititsuwannakul, P. Surawatanawong and P. Sangtrirutnugul, RSC Adv., 2015, 5, 55847–55855 RSC.
- C. Schouwey, M. Papmeyer, R. Scopelliti and K. Severin, Dalton Trans., 2015, 44, 2252–2258 RSC.
- M. L. Saha, Z. Zhou and P. J. Stang, Chem.–Asian J., 2016, 11, 2662–2666 CrossRef CAS PubMed.
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Dalton Trans., 2013, 42, 14064–14067 RSC.
- A. Boulay, A. Seridi, C. Zedde, S. Ladeira, C. Picard, L. Maron and E. Benoist, Eur. J. Inorg. Chem., 2010, 5058–5062 CrossRef CAS.
- S. Clède, F. Lambert, C. Sandt, Z. Gueroui, M. Réfrégiers, M.-A. Plamont, P. Dumas, A. Vessières and C. Policar, Chem. Commun., 2012, 48, 7729–7731 RSC.
- L. Wei, S. R. Banerjee, M. K. Levadala, J. Babich and J. Zubieta, Inorg. Chim. Acta, 2004, 357, 1499–1516 CrossRef CAS.
- T. Tabeya, M. Abe, A. Mitani, K. Tsuge and Y. Sasaki, J. Nucl. Radiochem. Sci., 2005, 6, 157–159 CrossRef CAS.
- R. Díaz, A. Francois and B. Loeb, Polyhedron, 2011, 30, 697–701 CrossRef.
- S. Wanniarachchi, B. J. Liddle, J. Toussaint, S. V. Lindeman, B. Bennett and J. R. Gardinier, Dalton Trans., 2010, 39, 3167–3169 RSC.
- E. Palma, J. D. Correia, Â. Domingos, I. Santos, R. Alberto and H. Spies, J. Organomet. Chem., 2004, 689, 4811–4819 CrossRef CAS.
- E. Runge and E. K. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- A. Mattiuzzi, I. Jabin, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2011, 40, 7395–7402 RSC.
- D. Schweinfurth, K. I. Hardcastle and U. H. Bunz, Chem. Commun., 2008, 2203–2205 RSC.
- T. M. Clarke, K. C. Gordon, D. L. Officer, S. B. Hall, G. E. Collis and A. K. Burrell, J. Phys. Chem. A, 2003, 107, 11505–11516 CrossRef CAS.
- T. M. Clarke, K. C. Gordon, D. L. Officer and D. K. Grant, J. Phys. Chem. A, 2005, 109, 1961–1973 CrossRef CAS PubMed.
- M. R. Waterland and K. C. Gordon, J. Raman Spectrosc., 2000, 31, 243–253 CrossRef CAS.
- M. R. Waterland, K. C. Gordon, J. J. McGarvey and P. M. Jayaweera, J. Chem. Soc., Dalton Trans., 1998, 609–616 RSC.
- O. Prakash, P. Singh, G. Mukherjee and A. K. Singh, Organometallics, 2012, 31, 3379–3388 CrossRef CAS.
- M. Valderrama, R. Contreras, M. P. Lamata, F. Viguri, D. Carmona, F. J. Lahoz, S. Elipe and L. A. Oro, J. Organomet. Chem., 2000, 607, 3–11 CrossRef CAS.
- M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Organometallics, 2000, 19, 4289–4294 CrossRef CAS.
- W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612–5620 RSC.
- E. Simón-Manso, M. Valderrama, P. Gantzel and C. P. Kubiak, J. Organomet. Chem., 2002, 651, 90–97 CrossRef.
- M. Sakamoto, Y. Ohki and K. Tatsumi, Organometallics, 2010, 29, 1761–1770 CrossRef CAS.
- S. Nagao, H. Seino, T. Okada, Y. Mizobe and M. Hidai, J. Chem. Soc., Dalton Trans., 2000, 3546–3553 RSC.
- L. B. Kumbhare, A. P. Wadawale, V. K. Jain, M. Sieger and W. Kaim, Inorg. Chem. Commun., 2010, 13, 475–478 CrossRef CAS.
- T. Nakagawa, H. Seino and Y. Mizobe, Organometallics, 2010, 29, 2254–2259 CrossRef CAS.
- R. Lalrempuia, H. Müller-Bunz and M. Albrecht, Angew. Chem., Int. Ed., 2011, 50, 9969–9972 CrossRef CAS PubMed.
- Z. Liu, L. Salassa, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Inorg. Chem., 2011, 50, 5777–5783 CrossRef CAS PubMed.
- A. Bastero, D. Font and M. A. Pericàs, J. Org. Chem., 2007, 72, 2460–2468 CrossRef CAS PubMed.
- T. H. Dunning Jr and P. J. Hay, in Methods of electronic structure theory, Springer, 1977, pp. 1–27 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2014, 33, 2341–2351 CrossRef CAS.
- C. Zhang, X. Shen, R. Sakai, M. Gottschaldt, U. S. Schubert, S. Hirohara, M. Tanihara, S. Yano, M. Obata and N. Xiao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 746–753 CrossRef CAS.
- S. J. Ahmed, M. I. Hyder, S. E. Kabir, M. A. Miah, A. J. Deeming and E. Nordlander, J. Organomet. Chem., 2006, 691, 309–322 CrossRef CAS.
- A. K. Singh, M. Kadarkaraisamy, M. Mishra, J. Sooriyakumar, J. Drake, M. Hursthouse, M. Light and J. P. Jasinski, Inorg. Chim. Acta, 2001, 320, 133–140 CrossRef CAS.
- H. Mishra and R. Mukherjee, J. Organomet. Chem., 2006, 691, 3545–3555 CrossRef CAS.
- C.-W. Hsu, S.-T. Ho, K.-L. Wu, Y. Chi, S.-H. Liu and P.-T. Chou, Energy Environ. Sci., 2012, 5, 7549–7554 CAS.
- K.-L. Wu, H.-C. Hsu, K. Chen, Y. Chi, M.-W. Chung, W.-H. Liu and P.-T. Chou, Chem. Commun., 2010, 46, 5124–5126 RSC.
- P. G. Bomben, K. C. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS.
Footnote |
| † Dedicated to the unique essence of a great teacher, Professor Majid M. Heravi, who is the realization of simorgh in the form of a scientist. |
| This journal is © The Royal Society of Chemistry 2018 |