
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCipadessains A–K, eleven limonoids from the fruits of Cipadessa cinerascens†
Dong-Mei Sun,
Fa-Liang An,
Shan-Shan Wei,
Yan-Qiu Zhang,
Xiao-Bing Wang ,
Jun Luo* and
Ling-Yi Kong*
,
Jun Luo* and
Ling-Yi Kong*
State Key Laboratory of Natural Medicines, Department of Natural Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, People's Republic of China. E-mail: luojun1981ly@163.com; cpu_lykong@126.com; Fax: +86-25-8327-1405; Tel: +86-25-8327-1405
First published on 14th March 2018
Abstract
Eleven new mexicanolide-type limonoids, cipadessains A–K (1–11), were isolated from the fruits of Cipadessa cinerascens (Pellegr) Hand.-Mazz. Their planar structures were determined based on IR, UV, 1D and 2D NMR spectra and HRESIMS data. The absolute configuration of 1 was elucidated by single-crystal X-ray diffraction using mirror Cu Kα radiation, and that of compounds 2–8 were determined by ECD analysis. Two mexicanolides bearing methoxybutenolide moiety originated from the furan ring 3 and 6, showed significant cytotoxicity against HepG2 cell line with IC50 values of 5.23 ± 0.12, 8.67 ± 1.02 μM, respectively; and NO inhibitory activities in LPS-activated RAW 264.7 macrophages at nontoxic concentration (IC50 5.79 ± 0.18, 6.93 ± 0.89 μM, respectively).
Introduction
Cipadessa cinerascens (Pellegr) Hand.-Mazz is a species of Cipadessa genus and known as ‘Ya Luo Qing’ or ‘Lao Ya Fan’ by the Dai.1 Its roots and leaves have been used in Chinese folk medicine for the treatment of rheumatism, stomachache, dysentery, malaria, scald, and skin itch.1–3 Previous reports demonstrated that terpenoids, flavonoids, steroids, and limonoids are the main bioactive constituents of Cipadessa genus.2,3 Among those compounds, limonoids have become a point of interest in the field of natural products, because of its diverse skeletons (mexicanolides, methyl angolensates, trijugins, and cipadesins) and various biological effects such as cytotoxic, insecticidal, antioxidant, and trypanocidal activities.2–6 Previous studies on Cipadessa genus focused on their leaves, roots, barks and stems.4–15 To our knowledge, there have been few phytochemical investigations on the fruits of Cipadessa species. In continuing search for new bioactive limonoids, 11 new mexicanolide limonoids with various modified furan rings, cipadessains A–K (1–11), were isolated from the fruits of C. cinerascens. All the isolates were evaluated for NO inhibitory activities in LPS activated RAW 264.7 macrophages and cytotoxicity against HepG2 cell line. In this paper, we describe the isolation, identification of 1–11, as well as their bioactivity screening.Results and discussion
Cipadessain A (1), was isolated as white powder, and assigned to a molecular formula of C32H40O9 on the basis of HRESIMS at m/z 591.2560 [M + Na]+ (calcd C32H40O9Na, 591.2565), corresponding with 13 degrees of unsaturation. Its IR absorption bands at 3460 and 1728 cm−1 showed the presence of hydroxyl and carbonyl functionalities. The NMR data (Table 1) revealed the characteristic resonances for β-substituted furan ring [δH 7.93 (H-21), 7.45 (H-23), and 6.50 (H-22), δC 143.4 (C-23), 142.4 (C-21), 120.5 (C-20), 109.7 (C-22)], four tertiary methyls (δH 1.42, 1.11, 0.85, 0.83); a characteristic downfield H-17 signal (δH 5.43); a ketone carbonyl (δC 217.3), three ester carbonyls (δC 175.1, 168.6, 167.2) and two unsaturated bonds (δH 6.90, 5.39; δC 139.6, 137.5, 127.6, 125.6). In the HMBC spectrum (Fig. 2), both angular methyls (C-28, C-29) were attached to C-4 (δC 38.7), were elucidated by the cross peaks from H3-28 (δH 0.83) to C-29 (δC 20.3), C-5 (δC 41.8) and C-3 (δC 77.6), and from H3-29 (δH 0.85) to C-28 (δC 22.9), C-5 (δC 41.8) and C-3 (δC 77.6); a C-6/7 esterificated appendage (δH 3.74; δC 52.3, 175.1) was easily verified by the HMBC cross peaks from H2-6 (δH 2.98, 2.40) to C-5 (δC 41.8), C-7 (δC 175.1) and from OCH3 (δH 3.74) to C-7 (δC 175.1). From cross peaks of H-3 (δH 4.83) to C-5 (δC 41.8), C-30 (δC 125.6), of H-30 (δH 5.39) to C-9 (δC 63.9), C-1 (δC 217.3), and of H3-19 (δH 1.42) to C-1 (δC 217.3), C-5 (δC 41.8), and C-9 (δC 63.9), the characteristic [3, 3, 1] A/B ring system could be elucidated.16 Furthermore, the HMBC correlations from H-17 (δH 5.43) to C-12 (δC 45.4), C-18 (δC 21.4), C-14 (δC 45.4), from H-15b (δH 2.73) to C-13 (δC 37.4), C-16 (δC 168.6), from H-15a (δH 2.85) to C-8 (δC 137.5), from H3-18 (δH 1.11) to C-17 (δC 76.7), C-14 (δC 45.4) indicated the ring C/D in 1.14,16 These aforementioned data strongly suggested 1 was a B,D-seco mexicanolide-type limonoid,7,19 typical constituent of Cipadessa genus.| Position | 1a | 2a | 3a | 4a | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| a 1H (500 MHz) NMR data of compounds.b Signals were overlapped. | ||||||||
| 1 | 217.3 | 216.9 | 217.2 | 216.6 | ||||
| 2 | 3.56, dd (8.8, 7.7) | 48.9 | 3.50, t (9.7) | 48.9 | 3.50b | 49.2 | 3.56, t (10.0) | 48.8 |
| 3 | 4.83, d (9.4) | 77.6 | 4.81, d (9.5) | 77.2 | 4.83, d (9.2) | 77.3 | 4.83, d (9.3) | 77.3 |
| 4 | 38.7 | 38.7 | 38.9 | 38.7 | ||||
| 5 | 3.49, d (10.4) | 41.8 | 3.39, dd (7.9, 3.8) | 41.4 | 3.51b | 40.4 | 3.37, d (9.5) | 41.7 |
| 6 | 2.40, dd (16.8, 10.8) | 32.2 | 2.38, m | 33.0 | 2.36, dd (17.3, 10.4) | 33.1 | 2.39, dd (17.0, 9.5) | 32.8 |
| 2.98, d (16.8) | 2.38, m | 2.48, d (17.3) | 2.23, d (17.0) | |||||
| 7 | 175.1 | 174.1 | 173.3 | 174.5 | ||||
| 8 | 137.5 | 141.2 | 138.0 | 137.5 | ||||
| 9 | 2.19, d (9.8) | 63.9 | 2.72, dd (12.2, 5.5) | 53.0 | 2.20b | 56.5 | 2.25b | 56.4 |
| 10 | 49.7 | 50.1 | 50.6 | 50.0 | ||||
| 11 | 4.60, td (10.4, 4.6) | 65.8 | 1.64b | 20.2 | 1.73b | 21.1 | 1.76, m | 20.5 |
| 2.07, dd (13.0, 4.1) | 2.29, td (13.2, 3.5) | 2.07, m | ||||||
| 12 | 1.39, d (11.8) | 45.4 | 1.33, d (13.5) | 28.6 | 1.43, td (14.0, 3.5) | 34.8 | 1.59, m | 34.2 |
| 1.82, d (11.8) | 1.98, dd (13.5, 4.1) | 1.91, d (14.0) | 1.83, m | |||||
| 13 | 37.4 | 41.3 | 36.8 | 37.3 | ||||
| 14 | 2.24, d (5.8) | 45.4 | 73.4 | 2.22b | 45.4 | 2.24b | 45.4 | |
| 15 | 2.85, dd (18.8, 5.8) | 30.0 | 2.99, d (18.0) | 39.2 | 2.79, m | 29.2 | 2.80, m | 29.5 |
| 2.73, d (18.8) | 2.92, d (18.0) | 2.78, m | 2.79, m | |||||
| 16 | 168.6 | 168.6 | 167.8 | 167.0 | ||||
| 17 | 5.43, s | 76.7 | 5.66, s | 77.4 | 5.54, s | 76.6 | 5.73, s | 78.1 |
| 18 | 1.11, 3H, s | 21.4 | 1.08, 3H, s | 15.9 | 1.00, 3H, s | 22.8 | 1.07, 3H, s | 21.9 |
| 19 | 1.42, 3H, s | 18.7 | 1.16, 3H, s | 15.9 | 1.16, 3H, s | 15.7 | 1.16, 3H, s | 15.8 |
| 20 | 120.5 | 120.1 | 136.1 | 164.1 | ||||
| 21 | 7.93, s | 142.4 | 7.84, s | 142.4 | 168.2 | 4.98, dd (18.0, 2.8) | 72.3 | |
| 22 | 6.50, s | 109.7 | 6.49, s | 110.0 | 7.23, s | 147.8 | 6.21, d (1.08) | 118.7 |
| 23 | 7.45, s | 143.4 | 7.42, s | 143.0 | 5.80, s | 102.4 | 172.9 | |
| 28 | 0.83, 3H, s | 22.9 | 0.78, 3H, s | 22.5 | 0.79, 3H, s | 22.6 | 0.78, 3H, s | 22.5 |
| 29 | 0.85, 3H, s | 20.3 | 0.82, 3H, s | 20.6 | 0.81, 3H, s | 20.9 | 0.82, 3H, s | 20.4 |
| 30 | 5.39, td (7.5, 11.7) | 125.6 | 5.63, d (6.8) | 124.5 | 5.34, d (6.6) | 123.4 | 5.35, d (6.9) | 124.3 |
| 7-OCH3 | 3.74, 3H, s | 52.3 | 3.71, 3H, s | 52.3 | 3.64, 3H, s | 52.1 | 3.69, 3H, s | 52.5 |
| 23- OCH3 | 3.58, 3H, br s | 57.6 | ||||||
| 1′ | 167.2 | 176.7 | 167.3 | 167.1 | ||||
| 2′ | 127.6 | 2.62, m | 33.9 | 127.8 | 127.7 | |||
| 3′ | 6.90, m | 139.6 | 1.15, 3Hb | 19.2 | 6.91, m | 139.3 | 6.88, m | 139.4 |
| 4′ | 1.80, 3H, s | 11.9 | 1.17, 3Hb | 18.6 | 1.84, 3H, s | 12.0 | 1.82, 3H, mb | 12.0 |
| 5′ | 1.68, 3H, d (7.1) | 14.7 | 1.82, 3H, d (7.3) | 14.8 | 1.82, 3H, mb | 14.9 | ||
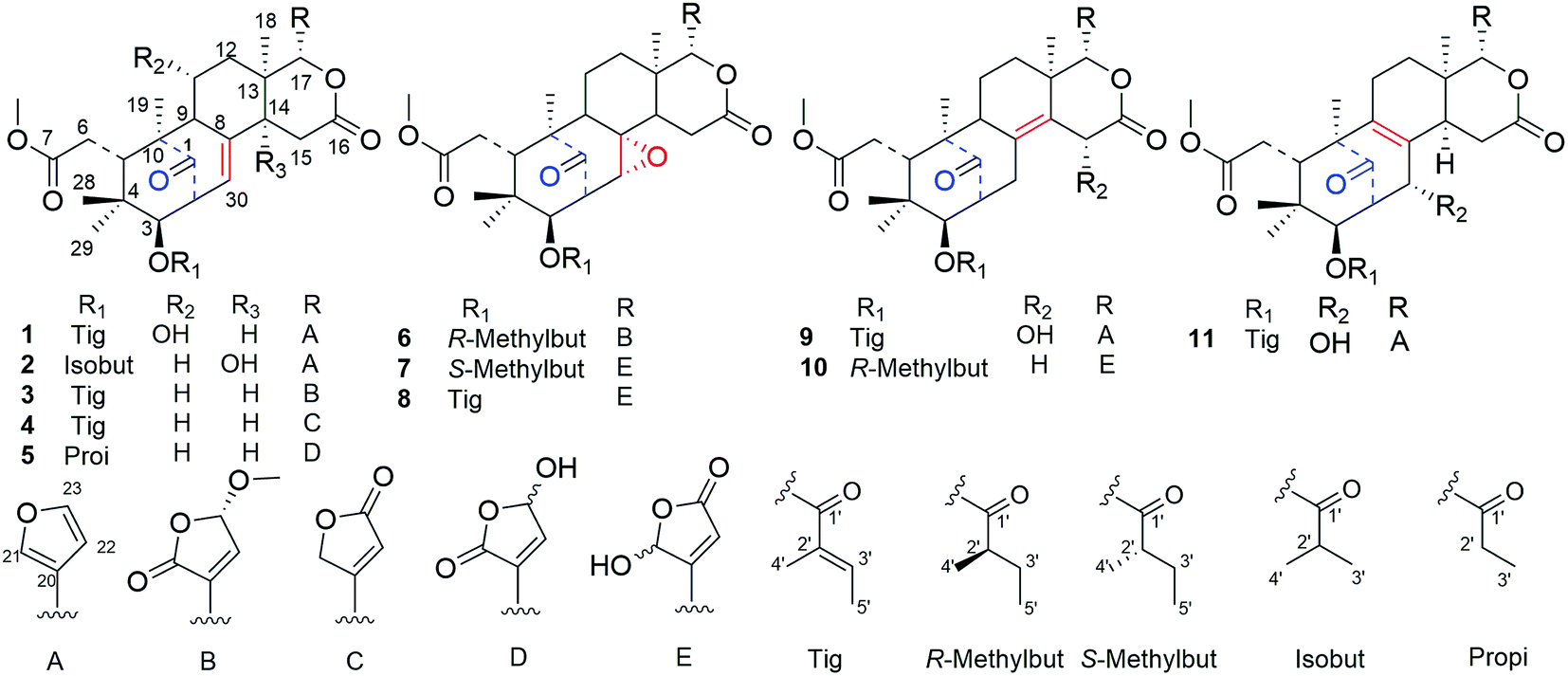 | ||
| Fig. 1 Chemical structures of cipadessains A–K (1–11). | ||
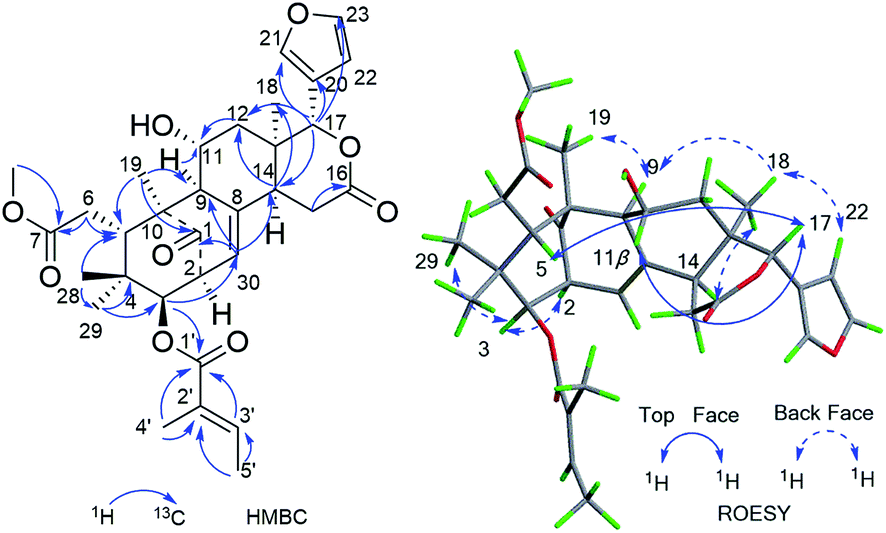 | ||
| Fig. 2 Key HMBC and ROESY correlations of compound 1. | ||
Furthermore, the existence of Δ8,30 double bond was determined by the HMBC correlations from H-30 (δH 5.39) to C-9 (δC 63.9) and C-14 (δC 45.4), and from H-2 (δH 3.56) to C-1 (δC 217.3), C-30 (δC 125.6). The correlation from H-3 (δH 4.83) to C-1′ (δC 167.2) of the tigloxyl moiety [δH 6.90 (H-3′), 1.80 (H-4′), 1.68 (H-5′); δC 167.2 (C-1′), 139.6 (C-3′), 127.6 (C-2′), 14.7 (C-5′), 11.9 (C-4′)] showed the presence of a tigloxyl group at C-3. The NMR data of 1 were similar to those of 6-desoxyswietenine,17 except for the additional of a hydroxyl group at C-11 in 1. This inference was verified by the HSQC correlation of H-11 (δH 4.60) with C-11 (δC 65.8) and HMBC correlations from H-9 (δH 2.19) and H2-12 (δH 1.82, 1.39) to C-11 (δC 65.8). Thus, the planar structure of 1 was determined as shown in Fig. 1.
The ROESY correlations between H-17 (δH 5.43)/H-11 (δH 4.60), H-5 (δH 3.49)/H3-28 (δH 0.83) suggested these protons were the same configuration (Fig. 2). In turn, the ROESY correlations between H-2 (δH 3.56)/H-3 (δH 4.83), H-3 (δH 4.83)/H3-29 (δH 0.85), H-14 (δH 2.24)/H3-19 (δH 1.42) and H-9 (δH 2.19)/H3-19 (δH 1.42) revealed that they were co-facially oriented. Accordingly, the structure of 1 was proposed as shown. Fortunately, compound 1 was recrystallized in the CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture to yield prisms. On the basis of single-crystal X-ray diffraction data (CCDC 1815118), the absolute stereochemistry of 1 was elucidated to be 2S, 3R, 5S, 9S, 10S, 11R, 13S, 14R, 17S (Fig. 3). Finally, the structure of 1, with a Δ8,30 double bond and a 11-hydroxyl group, was established as depicted in Fig. 1.
:1) mixture to yield prisms. On the basis of single-crystal X-ray diffraction data (CCDC 1815118), the absolute stereochemistry of 1 was elucidated to be 2S, 3R, 5S, 9S, 10S, 11R, 13S, 14R, 17S (Fig. 3). Finally, the structure of 1, with a Δ8,30 double bond and a 11-hydroxyl group, was established as depicted in Fig. 1.
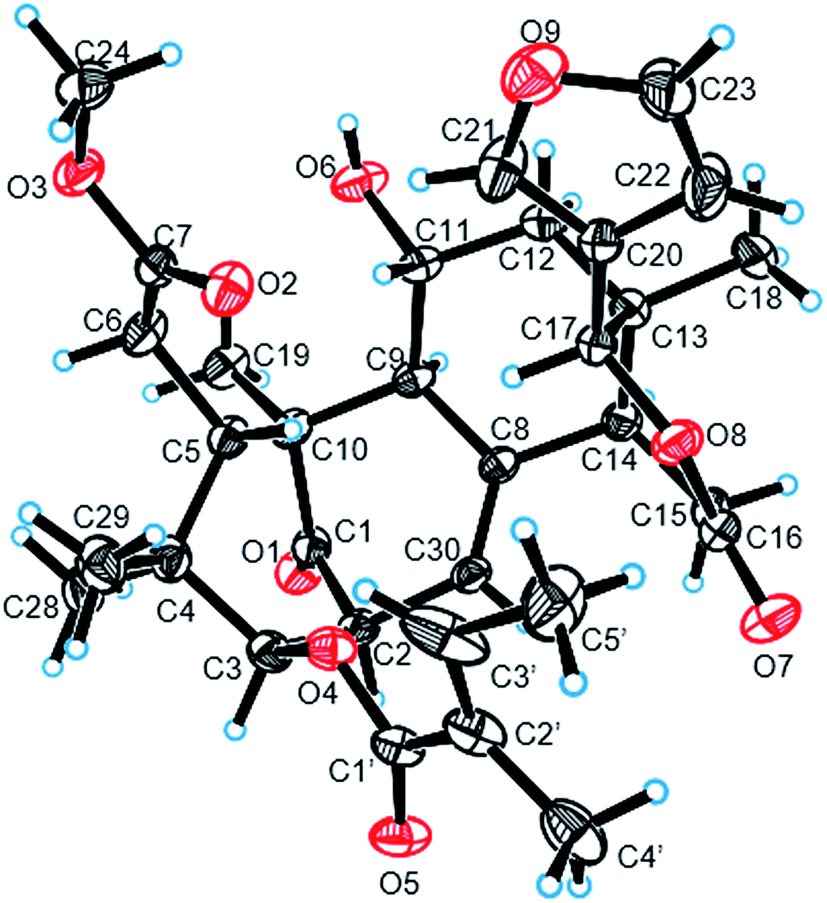 | ||
| Fig. 3 Single-crystal X-ray diffraction (Cu Kα radiation) of compound 1. | ||
Compound 2, was obtained as a colorless needles, which had a sodium adduct ion peak at m/z 579.2568 [M + Na]+ in the HRESIMS spectrum corresponding to a molecular formula of C31H40O9. The NMR spectroscopic data of 2 (Table 1) exhibited closed similarity to those of 1. In comparison to 1, the correlations from H3-3′ (H3-4′) to C-1′ (δC 176.7) and from C-2′ (δC 33.9), H-3 (δH 4.81) to C-1′ (δC 176.7) in the HMBC spectrum indicated the presence of an isobutyryloxyl group at C-3 in 2 instead of a tigloxyl group in 1. An upfield shift at δC 20.2 (Δ − 45.6 ppm) of C-11 and a downfield shift at δC 73.4 (Δ + 28.0 ppm) of C-14 in the 13C NMR spectrum of 2 as compared with the corresponding signals of 1 implied that the hydroxyl group was located at C-14. The deduction was supported by the HMBC correlations of C-14 (δC 73.4) and H2-15 (δH 2.99, 2.92), H-17 (δH 5.66), H-30 (δH 5.63). The α-oriented of OH-14 was determined by comparison of NMR data of 2 with those of two known compounds Khasenegasin D and Khasenegasin E.14 Therefore, the structure of 2 was established as shown (Fig. 1).
Compound 3 was isolated as a white amorphous powder and showed the HRESIMS ion peak at m/z 621.2669 [M + Na]+ (calcd for C33H42O10Na, 621.2670). The spectroscopic data of 3 were similar to those of 6-desoxyswietenine,17 except for the presence of 23-methoxybutenolide ring moiety in 3 instead of the β-substituted furan moiety as found in 6-desoxyswietenine. This was confirmed by the HMBC correlations of OCH3-23 (δH 3.58)/C-23 (δC 102.4); of H-23 (δH 5.80)/C-21 (δC 168.2), C-20 (δC 136.1); of H-22 (δH 7.23)/C-23 (δC 102.4), C-20 (δC 136.1), C-21 (δC 168.2); of H-17 (δC 5.54)/C-21 (δC 168.2), C-20 (δC 136.1). Thus, the planar structure of 3 was identified as shown. The relative configuration of 3 was mainly elucidated by its ROESY data and comparison of its NMR spectroscopic data with those of similar reported compounds.17 The correlations between H-17/H-11b (δH 2.29), H-17/H-15b (δH 2.78), H-17/H-5 (δH 3.51), indicated these groups were on the same side of the structure, and they were assigned as β-configuration. Correlations of H3-19 (δH 1.16)/H-9 (δH 2.20)/H-2 (δH 3.50), H-9/H-3 (δH 4.83)/H3-29 (δH 0.81), H-3/H-14 (δH 2.22) and cross peaks of OCH3-23/CH3-18 revealed that these protons were α-oriented. Hence, the structure of 3 was elucidated as shown in Fig. 4 and named cipadessain C.
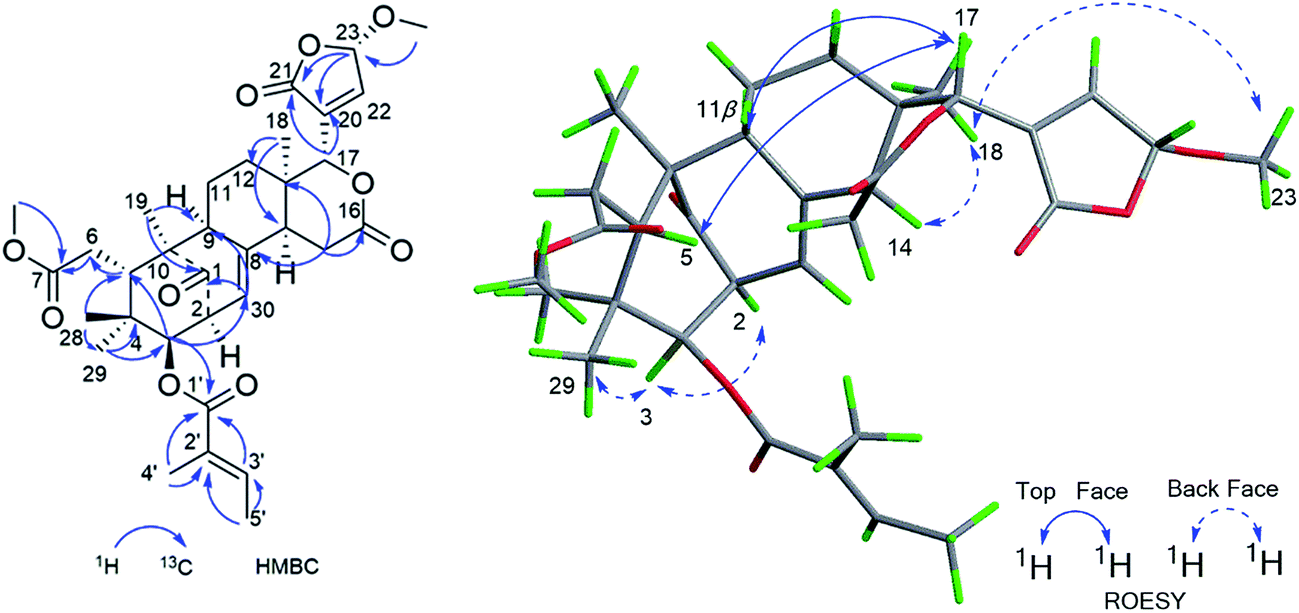 | ||
| Fig. 4 Key HMBC and ROESY correlations of compound 3. | ||
Compound 4, a white, amorphous powder, displayed a molecular formula C32H40O9, on the basis of its HRESIMS ion at m/z 591.2568 ([M + Na]+, C32H40O9Na, calcd 591.2565). The 1H and 13C NMR data (Table 1) of 3 and 4 were almost the same except for signals of the E ring. The appearance of α,β-unsaturated-γ-lactone ring portion linked to C-17 in 4 was determined by the HMBC correlations between H-17 (δH 5.73)/C-20 (δC 164.1), C-21 (δC 72.2), C-22 (δC 118.7) and H2-21 (δH 4.98)/C-20 (δC 164.1), C-22 (δC 118.7), C-23 (δC 172.9). The ROESY correlations were observed between H-17 (δH 5.73)/H-5 (δH 3.37), H-5/H-11b (δH 2.07), H-5/H3-28 (δH 0.78), H3-29 (δH 0.82)/H-3 (δH 4.83), H-3/H-2 (δH 3.56) suggesting the relative configuration of 4 was similar to that of 3. Thus, the structure of 4 was finally determined and represented in Fig. 1.
Compound 5, a white amorphous powder, afforded a molecular formula of C30H38O10 as assigned by the (+)-HRESIMS ion peak at m/z 581.2353 [M + Na]+ (calcd 581.2357) and the 13C-NMR data. The NMR data (Table 2) of 5 resembled those of 3-de(2-methylbutanoyl)-3-propanoylcipadesin18 except for the additional presence of 21-hydroxybutenolide unit attached to C-17 in 5. This was confirmed by the cross peaks from H-17 (δH 5.60) to C-20 (δC 135.1/135.0), C-22 (δC 149.8/149.6) in the HMBC spectrum. The appearance of pairs of proton and carbon resonances in the NMR spectra of 5 indicated the presence of epimers at C-21.19 Thus, based on these data, the planar structure of 5 was established.
| Position | 5a | 6a | 7a | 8a | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| a 1H (500 MHz) NMR data of compounds.b Signals were overlapped. | ||||||||
| 1 | 217.3/217.2 | 214.3 | 213.9 | 214.2 | ||||
| 2 | 3.49, m | 48.8 | 3.57b | 48.9 | 3.58, dd (9.5, 3.2) | 48.9 | 3.66, d (9.3, 2.4) | 49.3/49.1 |
| 3 | 4.75, d (9.2) | 77.4b | 5.09, d (9.2) | 77.1 | 5.09, dd (9.5, 2.4) | 77.1 | 5.03, d (9.3) | 78.1/77.5 |
| 4 | 38.6 | 39.5 | 39.5/39.3 | 39.4/39.2 | ||||
| 5 | 3.39, d (10.0) | 40.7/40.7 | 3.21, t (5.0) | 42.6 | 3.20, d (9.3) | 42.9/42.6 | 3.27, d (9.4) | 43.0/42.6 |
| 6 | 2.43, dd (17.1, 5.8) | 33.1/33.0 | 2.35, d (5.0) | 33.1 | 2.36 | 33.3 | 2.40, d (16.6) | 33.2 |
| 2.37b | 2.35, d (5.0) | 2.33 | 2.39, dd (16.6, 9.7) | |||||
| 7 | 173.9/173.6 | 174.3 | 174.8 | 174.9 | ||||
| 8 | 137.9/137.8 | 60.6 | 60.1 | 60.1 | ||||
| 9 | 2.23b | 56.3/56.2 | 1.90b | 56.1 | 1.94b | 54.9/54.6 | 1.94, m | 54.8/54.4 |
| 10 | 50.5/50.2 | 48.4 | 48.3/48.2 | 48.3/48.2 | ||||
| 11 | 1.73, br s | 20.9 | 1.76, m | 19.5 | 1.78, m | 19.5 | 1.85, m | 19.8/19.6 |
| 2.23b | 1.88, m | 1.93b | 2.01, m | |||||
| 12 | 1.92, d (14.0) | 34.5 | 1.13b | 32.8 | 2.13, d (13.9) | 33.1 | 2.10, m | 33.3 |
| 1.44, m | 2.25, d (16.3) | 1.39, m | 1.41, m | |||||
| 13 | 36.8/36.7 | 36.8 | 36.5/36.4 | 36.3/26.2 | ||||
| 14 | 2.24b | 45.5 | 1.49b | 46.4 | 1.56, m | 46.6/46.5 | 1.56, dd (12.7, 4.0) | 46.5/46.1 |
| 15 | 2.84, m | 29.6 | 3.76, m | 34.7 | 3.50, dd (16.4, 13.7) | 33.6/33.5 | 3.40, dd (16.1, 13.6) | 33.1 |
| 2.84, m | 2.85, dd (15.5, 3.7) | 2.84, dd (16.4, 4.3) | 2.77, dd (16.5, 4.0) | |||||
| 16 | 169.0/169.0 | 171.3 | 170.7 | 170.4 | ||||
| 17 | 5.60, s | 77.2 | 5.10, s | 76.5 | 5.30, br s/5.15, br s | 79.2/78.4 | 5.30, br s/5.12, br s | 78.8 |
| 18 | 1.06/1.04, 3H, s | 23.0 | 1.04, 3H, s | 26.5 | 1.23/1.22, 3H, br s | 16.3/16.2 | 1.20/1.07, 3H, br s | 26.2/24.5 |
| 19 | 1.14, 3H, s | 15.7/15.7 | 1.06, 3H, s | 15.8 | 1.08/1.06, 3H, br s | 26.7/26.2 | 1.08, 3H, br s | 16.4 |
| 20 | 135.1/135.0 | 133.7 | 162.7 | 162.7/160.2 | ||||
| 21 | 168.6 | 169.8 | 6.32, br s/6.14, br s | 98.7/97.7 | 6.22, br s/6.12, br s | 98.8/97.7 | ||
| 22 | 7.35/7.34, br s | 149.8/149.6 | 7.28, s | 148.8 | 6.33, br s/6.22, br s | 122.5/121.8 | 6.30, br s | 122.4/121.3 |
| 23 | 6.18/6.17, br s | 97.1/96.7 | 5.82, br s/5.78, br s | 103.1 | 169.4/169.0 | 169.1 | ||
| 28 | 0.76, 3H, s | 22.5 | 0.81, 3H, s | 21.2 | 0.81, 3H, s | 20.9/20.9 | 0.83, 3H, s | 23.1/23.0 |
| 29 | 0.79, 3H, s | 21.0/20.9 | 0.79, 3H, s | 22.5 | 0.78, 3H, s | 22.7 | 0.82, 3H, s | 21.0 |
| 30 | 5.33, d (6.5) | 123.3/123.3 | 3.31, br s | 63.7 | 3.30, d (2.5) | 63.6/63.5 | 3.20, d (2.2) | 63.7/63.4 |
| 7-OCH3 | 3.66/3.64, 3H, br s | 52.4/52.3 | 3.72, 3H, s | 52.5 | ||||
| 23-OCH3 | 3.62, br s/3.56, br s | 57.8 | 3.72, 3H, s | 52.8/52.6 | 3.74, 3H, s | 52.8/52.7 | ||
| 1′ | 174.3/174.2 | 175.8 | 176.1/175.9 | 167.0 | ||||
| 2′ | 2.39b | 27.2/27.2 | 2.56, m | 41.7 | 2.55, m | 41.5/41.3 | 127.6/127.2 | |
| 3′ | 1.12, d (7.5) | 9.0 | 1.77, m | 26.8 | 1.74, m | 26.5 | 7.03, m | 140.6/140.3 |
| 1.54, m | 1.52, m | |||||||
| 4′ | 1.25, 3H, d (7.0) | 17.5 | 1.24, 3H, d (7.0) | 17.3/17.3 | 1.91, 3H, mb | 12.5 | ||
| 5′ | 0.97, 3H, t (7.4) | 12.0 | 0.97, 3H, t (7.4) | 11.9/11.8 | 1.90, 3H, mb | 14.9 | ||
Compound 6 gave a molecular formula of C33H44O11 from the positive ion peak at m/z 639.2771 [M + Na]+ (calcd for C33H44O11Na, 639.2776). The 1D and 2D NMR data (Table 2) of 6 were similar to those of 2′R-cipadesin A,7 a mexicanolide-type limonoid with a Δ8,30 epoxide ring from Cipadessa fruticosa. The structures 6, featuring 23-methoxybutenolide ring in the C-17 side-chain, was verified by the key HMBC cross peaks of OCH3-23 (δH 3.62/3.56)/C-23 (δC 103.1); H-23 (δH 5.82/5.78)/C-21 (δC 169.8), C-20 (δC 133.7); H-22 (δH 7.28)/C-17 (δC 76.5). In the ROESY spectrum, the cross peak of H-23 (δH 5.82) to H-17 (δH 5.10) confirmed the β-configuration of H-23 in 6. The detailed analysis of the ROESY correlations based on the general rule,7 in which the cross peaks of H3-4′ (δH 1.25) to H-15b (δH 2.85) and H-30 (δH 3.31), of H-2′ (δH 2.56) to H-30 (δH 3.31), of H3-5′ (δH 0.97) and H3-29 (δH 0.79), revealed the remaining relative configurations of 6 (Fig. 5) were identical to those of 2′R-cipadesin A.
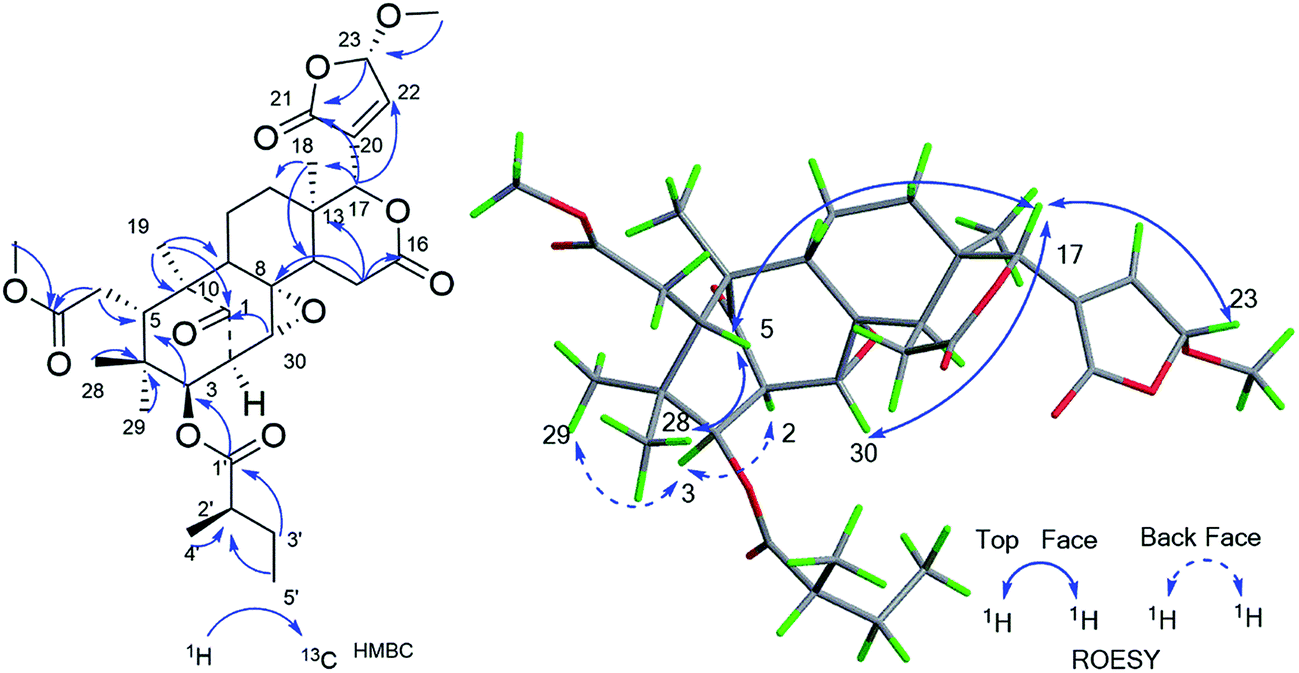 | ||
| Fig. 5 Key HMBC and ROESY correlations of compound 6. | ||
Compound 7, a white amorphous powder, exhibited the molecular formula of C32H42O11 based on the pseudo-molecular ion peak at m/z 625.2620 [M + Na]+ (calcd for C32H42O11Na, 625.2619). The 1H and 13C-NMR data of 7 were closely comparable to those of 6 for rings A–D (Table 2), while the 23-methoxybutenolide group was absent, and concomitantly a γ-hydroxylbutenolide group. This difference was supposed by the broad singlet signals of H-21 at δH 6.32/6.14 and H-22 at δH 6.34/6.22 and by carbon signals at δC 162.7/160.4 (C-20), 98.7/97.7 (C-21), 122.5/121.8 (C-22), 169.4/169.0 (C-23). The α-configuration of epoxide ring at C-8/C-30 was supposed from the coupling constant value of H-30 (J = 2.5 Hz).20 The appearance of pairs of most proton and carbon resonances in the NMR spectra (Table 2) of 7 identified the presence of C-21 epimers. Thus, compound 7 was elucidated as a mexicanolide-type limonoid bearing γ-hydroxybutenolide group. According to the general ruler,7 the ROESY correlations between H3-5′ (δH 0.97) and H-30 (δH 3.30), between H-2′ (δH 2.55) and H-15α (δH 3.50), between H3-4′ (δH 1.24) and H3-29 (δH 0.78) confirmed its relative configurations were as the same as that of cipadesin A.
The molecular formula of 8 was determined to be C32H40O11 from the HRESIMS peak at m/z 623.2464 (calcd for C32H40O11Na, 623.2463), 2 mass units less than 7. The NMR data of 8 (Table 2) exhibited overall similarity to those of 7, but revealed the presence of a tigloxyl moiety instead of a 2-methylbutyryl moiety attached to C-3, this was proved by the HMBC correlations of H3-4′ (δH 1.91) to C-1′ (δC 167.0), C-3′ (δC 140.6/140.2), of H3-5′ (δH 1.90) to C-2′ (δC 127.6/127.2), of H-3 (δH 5.03) to C-1′ (δC 167.0). Therefore, the planar structure of 8 was assigned as shown. The α-configuration of epoxide ring at C8/30 was determined by the coupling constant value of H-30 (J = 2.3 Hz).20 The relative configuration of the tigloxyl moiety at C-3 was β-oriented according to the observed ROESY correlations between H-3 and H-2, and the coupling constant value of H-3 (J = 9.4 Hz).21 The appearance of pairs of most proton and carbon resonances in the NMR spectra (Table 2) of 8 suggested the presence of C-21 epimers. Its relative configurations were as the same as that of 7 by ROESY experiment. Therefore, the structure of 8 was elucidated as shown in Fig. 1.
The similar electronic circular dichroism spectra of 1–8 (see S1 in ESI†) indicated that the basic skeletons of these compounds possessed absolute configurations as Fig. 1.
Compound 9 was isolated as a white powder. Its molecular formula, C32H40O9 was deduced from the HRESIMS data (m/z 567.2597 [M − H]−, calcd for C32H39O9, 567.2600). Analysis of its NMR spectra indicated that 9 was an analogue of proceranolide.22 A proton signal at δH 4.97 was correlated with a carbon signal at δC 65.4 in HSQC spectrum and the HMBC correlation from the hydroxyl group (δH 3.18 br s) to C-15 (δC 65.4) indicated that the hydroxyl group was located at C-15. Furthermore, a tigloxyl moiety was located at C-3′, which was confirmed by the HMBC correlation from H-3 (δH 4.89) to C-1′ (δC 167.2). In the ROESY spectrum, the H-15 (δH 4.97) was correlated with H-17 (δH 5.54), which suggested the H-15 was β-oriented. The other configurations of 9 were similar to that of proceranolide by ROESY spectroscopic analysis.
Compound 10, was isolated as a white powder and its molecular formula C32H42O10 was determined by the molecular ion peak at m/z 609.2675 (calcd for C32H42O10Na, 609.2670) in the HRESIMS. The 1D and 2D NMR data indicated that 10 was also a mexicanolide-type limonoid bearing a γ-hydroxylbutenolide unit as 8. A comparison of their NMR data (Table 3) revealed the main differences being the presence of the Δ8,14 double bond and the absence of the Δ8,30 epoxide ring. The conclusion was confirmed from the HMBC correlations of H-15b (δH 3.46) to C-8 (δC 129.2/128.9), of H-30b (δH 2.12) to C-14 (δC 130.8/130.5), of H-2 (δH 3.19) to C-8 (δC 129.2/128.9). Moreover, the tigolxyl group was replaced by the R-methylbutyryl at C-3 in 10, which can be proved from the H-3 (δH 4.97) to C-1′ (δC 176.3). The appearance of pairs of proton and carbon resonances in the NMR spectra of 10 indicated that the presence of epimers at C-21. Therefore, the structure of 10 was shown in Fig. 1. The relative structure of 10 was displayed by the ROESY experiment and general ruler.7
| Position | 9a | 10b | 11a | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a 1H (500 MHz) NMR data of compounds.b 1H (600 MHz) NMR data of compound.c Signals were overlapped. | ||||||
| 1 | 217.7 | 217.5/217.3 | 213.4 | |||
| 2 | 3.26, m | 48.4 | 3.19, m | 48.1 | 3.19, dd (7.9, 1.9) | 57.0 |
| 3 | 4.89, m | 79.3 | 4.97, d (9.8) | 78.0/77.9 | 5.17, d (7.9) | 77.1 |
| 4 | 38.8 | 38.4/38.2 | 38.4 | |||
| 5 | 3.37, dd (11.0, 1.5) | 40.7 | 3.11, d (10.7) | 41.4/41.2 | 2.83, t (6.0) | 48.4 |
| 6 | 2.42, d (16.5, 11.0) | 33.4 | 2.45, m | 33.9 | 2.29, d (6.0) | 32.4 |
| 2.37, d (16.5) | 2.36, m | |||||
| 7 | 174.7 | 176.3/175.7 | 174.1 | |||
| 8 | 135.7 | 129.2/128.9 | 130.7 | |||
| 9 | 2.11, d (6.6) | 51.9 | 2.05c | 51.3 | 142.1 | |
| 10 | 53.6 | 53.1/52.8 | 50.9 | |||
| 11 | 1.75, m | 18.7 | 1.79, m | 18.7/18.6 | 2.03, m | 21.6 |
| 1.89, m | 1.94, m | 2.38, m | ||||
| 12 | 1.00 | 28.6 | 1.99, m | 28.6 | 1.58c | 29.1 |
| 1.78, m | 1.27, m | 1.51c | ||||
| 13 | 1.02, m | 38.8 | 39.1 | 35.8 | ||
| 14 | 135.0 | 130.8/130.5 | 2.45c | 38.2 | ||
| 15 | 4.97, s | 65.4 | 3.77, m | 32.9 | 2.80, dd (14.0, 3.0) | 31.4 |
| 3.46, d (21.0) | 2.41, d (14.0) | |||||
| 16 | 174.5 | 168.7/168.1 | 172.7 | |||
| 17 | 5.54, s | 81.9 | 5.62, br s/5.35, br s | 79.7 | 4.91, s | 81.3 |
| 18 | 1.02, 3H, s | 16.9 | 1.08, 3H, s | 17.4 | 0.74, 3H, s | 20.4 |
| 19 | 1.18, 3H, s | 16.7 | 1.17, 3H, s | 16.9 | 1.08, 3H, s | 17.2 |
| 20 | 120.6 | 162.8 | 120.8 | |||
| 21 | 7.62, s | 142.0 | 6.10, s | 98.9/98.0 | 7.43, s | 140.5 |
| 22 | 6.51, s | 110.0 | 6.30, s | 122.8/122.1 | 6.40, s | 109.8 |
| 23 | 7.42, s | 143.0 | 169.6/159.4 | 7.43, s | 143.4 | |
| 28 | 0.76, 3H, s | 20.4 | 0.68, 3H, s | 23.5 | 1.14, 3H, s | 26.5 |
| 29 | 0.83, 3H, s | 23.9 | 0.81, 3H, s | 20.6 | 0.89, 3H, s | 25.9c |
| 30 | 2.99,dd, (15.9, 1.9) | 34.6 | 2.72, d (15.0) | 33.1 | 4.54, br s | 71.4 |
| 2.30, dd, (15.9, 6.6) | 2.12, d (6.0) | |||||
| 7-OCH3 | 3.73, 3H, s | 52.6 | 3.71, 3H, s | 52.8/52.2 | 3.69, 3H, s | 52.2 |
| 15-OH | 3.18, br s | |||||
| 1′ | 167.2 | 176.3 | 167.0 | |||
| 2′ | 129.4 | 2.40, m | 41.3 | 128.5 | ||
| 3′ | 6.90, m | 138.6 | 1.73, dt (20.0, 7.0) | 27.3 | 6.92, m | 139.1 |
| 1.51, dt (14.0, 7.0) | ||||||
| 4′ | 1.87, 3H, m | 12.5 | 1.21, 3H, m | 16.2 | 1.89, 3H, s | 12.5 |
| 5′ | 1.79, 3H, d (7.0) | 14.6 | 0.91, 3H, dd (7.1, 5.5) | 11.3 | 1.86, 3H, d (7.1) | 14.9 |
Compound 11, was isolated as white and amorphous powder, shown as a positive HRESIMS ion peak at m/z 591.2562 ([M + Na]+, calcd for C32H40O9Na 591.2565). The 1H and 13C-NMR data of 11 (Table 3) suggested that its structure was closely related to that of khasengasin N with Δ8,9 double bond.14 The only difference between 11 and khasengasin N was that a hydroxyl group linked to C-3 as found in khasengasin N was replaced by a tigloxyl group [δH 1.89 (s), δH 1.86 (d, J = 7.1 Hz) and 6.92 (m)] in 11. Further, it was evidenced by HMBC correlation of H-3 (δH 5.17) to C-1′ (δC 167.0) of the tigloxyl group. The relative configuration of 11 was assigned by the ROESY experiment, in which key cross-peaks between H-17/H-5, H-5/H-30 indicated H-30 to be β-oriented (Fig. 6). Therefore, the structure of 11 was demonstrated as shown in Fig. 1.
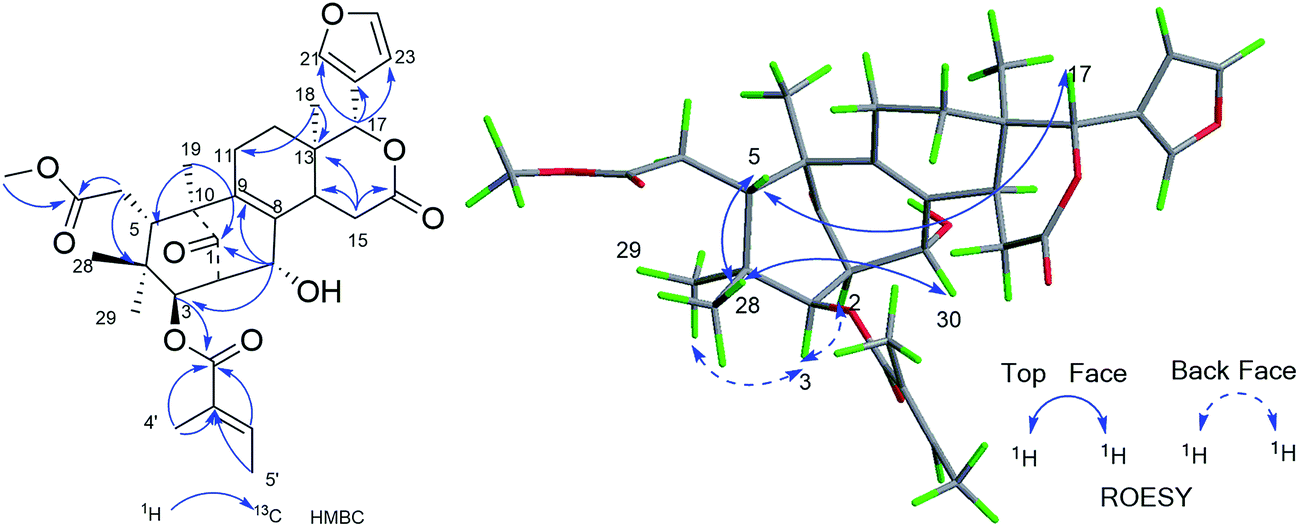 | ||
| Fig. 6 Key HMBC and ROESY correlations of compound 11. | ||
All the isolates were evaluated for their cytotoxicity against HepG2 cancer cell line with doxorubicin as the positive control. As shown in Table 4, compounds 3 and 6 showed significant cytotoxic activities. These isolates were further tested for their inhibitory effects on NO production of LPS-activated RAW 264.7 macrophages with N-monomethyl-L-arginine as the positive control (IC50 41.88 ± 0.91 μM). The results showed that compounds 3 and 6 exhibited significant NO inhibitory activities with IC50 values of 5.79 ± 0.18 μM, 6.93 ± 0.89 μM, respectively. On the contrary, those mexicanolides bearing complete furan moiety or hydroxybutenolide furan moiety were inactive (1, 2, 5, 8–11). According to the continuing study,14–16 the modified furan moiety may play important role on the bioactivity of these limonoids.
| Compounds | NO inhibition RAW 264.7 macrophages | Cytotoxicity (HepG2) |
|---|---|---|
| a Values were expressed as the means ± SD based on three independent experiments.b Compounds 1,2,5,8–11 were inactive.c Positive controls. | ||
| 3 | 5.79 ± 0.18 | 5.23 ± 0.12 |
| 4 | 23.90 ± 2.1 | >50 |
| 6 | 6.93 ± 0.89 | 8.67 ± 1.02 |
| 7 | 20.54 ± 0.63 | >50 |
| L-NMMAc | 41.88 ± 0.91 | — |
| Doxorubicinc | — | 1.04 ± 0.37 |
Conclusions
In summary, eleven new mexicanolide-type limonoids, cipadessains A–K (1–11) were isolated from the fruits of Cipadessa cinerascens. Their structures were elucidated on the NMR data and HRESIMS. The absolute configuration of 1 was determined by X-ray diffraction. In bioactivity screening, two mexicanolides (3 and 6) bearing methoxybutenolide moiety originated from the furan ring showed significant cytotoxicity activities against HepG2 cell line and NO inhibitory activities in LPS-activated RAW 264.7 macrophages, which were more excellent than those limonoids with complete furan moiety or hydroxybutenolide furan moiety. This finding adds the complexity and diversity of mexicanolide-type limonoids in Cipadessa cinerascens. It's worthy of paying more attention to the cytotoxicity activities against HepG2 cell line of the limonoids with modified furan rings from the Cipadessa genus.Experimental
General experimental procedures
UV and IR were recorded on a Shimadzu UV-2450 spectrophotometer (Shimadzu, Tokyo, Japan) and Bruker Tensor 27 spectrometer (Bruker, Karlsruhe, Germany), optical rotations were measured on a JASCO P-1020 polarimeter (Jasco, Tokyo, Japan). ECD spectra were obtained on a JASCO J-810 spectropolarimeter (Jasco, Tokyo, Japan) respectively. 1D and 2D NMR spectra were measured by a Bruker AVIII-600 and AVIII-500 NMR instrument at 600 and 500 MHz (1H), 150 MHz (13C) and 125 MHz (13C) in CDCl3. HRESI mass spectra were conducted on an Agilent 6520B UPLC-Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). MCI gel (Mitsubishi Chemical Corp., Tokyo, Japan), MPLC (Beijing H&E Co., Ltd., Beijing, China), and RP-C18 (40–63 μm, FuJi), silica gel (Qingdao Haiyang Chemical Co., Ltd.) were used as column chromatography. Preparative HPLC was performed on a Shimadzu LC-6AD instrument with a SPD-10A detector using a shim-pack reversed-phase C18 column (20 × 200 mm, 10 μm). Analytical HPLC was carried out on an Agilent 1260 Series instrument with a DAD detector using a shim-pack VP-ODS column (250 × 4.6 mm, 5 μm). All solvents used were analytical grade (Jiangsu Hanbon Science and Technology Co., Ltd.). The mouse macrophage cell line RAW264.7 and human hepatocellular carcinoma (HepG2) cell were obtained from the Cell Bank of the Shanghai Institute of Cell Biology and Biochemistry, Chinese Academy of Sciences (Shanghai, China). All cells were cultured in Dulbecco's modified Eagle medium (DMEM, GIBCO Invitrogen Corp., Carlsbad, CA, USA) with 10% FBS, penicillin (100 U ml−1), and streptomycin (100 μg ml−1) in a humidified atmosphere with 5% CO2 at 37 °C.Plant material
The air-dried fruits of Cipadessa cinerascens (Pellegr) Hand.-Mazz (Meliaceae) were collected from Xishuangbanna, Yunnan province, People's Republic of China, in July 2013, and authenticated by Professor Mian Zhang, Research Department of Pharmacognosy, China Pharmaceutical University. A voucher specimen (no. 2015-CCHM) has been deposited in the Department of Natural Medicinal Chemistry, China Pharmaceutical University.Extraction and isolation
The dried and powdered fruits of Cipadessa cinerascens (5.0 kg) were extracted by refluxing with 95% ethanol (25 L × 4), for 4 h, 4 h, 4 h, 3 h. The ethanol extract was concentrated under reduced pressure to afford the crude extract (637 g), it was suspended in H2O (1.5 L) and partitioned sequentially with petroleum ether (PE), CH2Cl2, EtOAc. The oily PE and CH2Cl2 part were subjected to a silica gel column gradient eluted with CH2Cl2/MeOH (150:1 to 0:100) to afford collect six fractions A–F by TLC analysis. Fraction C (120 g) was run on a D101-macroporous absorption resin, eluted with EtOH/H2O (30%, 50%, 70%, and 90%, v/v) to give six sub-fractions (C1–C6) monitored by HPLC. Fr. C3 (12.8 g) was chromatographed on an ODS-C18 column eluted with MeOH/H2O (30:70 to 100:0) to obtain fourteen sub-fractions (C3A–C3N). Fraction C3A was subjected to an ODS-C18 column eluted with MeOH/H2O (40:60, v/v) to obtain ten sub-fractions (C3A1–C3A10). C3A5 was purified by semi-preparative HPLC yielding 5 (2.2 mg). Fraction C3F was applied to a silica gel column (CH2Cl2/MeOH; 100:1 to 0:100) to give five sub-fractions (C3F1–C3F5). Fraction C3F2 (102 mg) was separated by the semi-preparative HPLC (MeOH/H2O; 65:35, v/v, 10 mL min−1) to yield 3 (5.2 mg). Using the same purification procedures, fraction C3E yielded 4 (2.3 mg), 7 (2.1 mg) and 10 (8.6 mg). Fraction C3I was subjected to a Sephadex LH-20 gel, using CH2Cl2/MeOH (1:1) as the eluent, to obtain three sub-fractions, C3I1–C3I3. C3I2 was separated by the semi-preparative HPLC (MeOH/H2O; 65:35, v/v, 10 ml min−1) to obtain 8 (1.8 mg). Fraction C3K (2.01 g) was subjected to MPLC to give six sub-fractions (C3K1–C3K6). Fraction C3K1 was repeatedly chromatographed over semi-preparative HPLC (MeCN/H2O; 50:50, v/v, 10 ml min−1) to afford 1 (2.2 mg), 2 (2.0 mg), 9 (4.5 mg), 11 (4.3 mg) in turn. Fraction C3K3 (350 mg) was subjected to silica gel column (CH2Cl2/MeOH; 100:1 to 0:100), further purified by the semi-preparative HPLC (MeCN/H2O; 55:45) to obtain 6 (1.8 mg).
:CH2Cl2 = 1:1); [α]25D −47.6 (c 0.10, MeOH); UV (MeOH) λmax (logε) 229 (3.58) nm; ECD (MeOH, Δε) 200 (−14.074), 239 (+1.894), 294 (−4.559) nm; IR (KBr) vmax 3451, 2922, 1640, 1384 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 591.2560 [M + Na]+ (cacld for C32H40O9Na, 579.2565).:CH2Cl2 = 1:1); [α]25D −119.3 (c 0.13, MeOH); UV (MeOH) λmax (logε) 227 (3.58) nm; ECD (MeOH, Δε) 200 (−11.079), 212 (+4.854), 293 (−2.852) nm; IR (KBr) vmax 3452, 1730, 1640, 1384 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 579.2568 [M + Na]+ (cacld for C31H40O9Na, 579.2565).ε) 210 (3.91) nm; ECD (MeOH, Δε) 200 (−43.442), 229 (+5.615), 294 (−5.617) nm; IR (KBr) vmax 3453, 1727, 1647, 1437, 1383, 1263 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 621.2669 [M + Na]+ (calcd for C33H42O10Na, 621.2670).ε) 234 (3.47) nm; ECD (MeOH, Δε) 200 (−13.199), 255 (+0.468), 294 (−3.856) nm; IR (KBr) vmax 3455, 1726, 1643, 1455 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 591.2568 [M + Na]+ (calcd for C32H40O9Na, 591.2565).ε) 208 (3.61) nm; ECD (MeOH, Δε) 200 (−22.845), 224 (+7.992), 297 (−5.732) nm; IR (KBr) vmax 3455, 1726, 1643, 1455 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 581.2353 [M + Na]+ (calcd for C31H38O8Na, 581.2357).ε) 219 (3.64) nm; ECD (MeOH, Δε) 200 (−4.342), 252 (+1.559), 296 (−3.587) nm; IR (KBr) vmax 3451, 1639, 1384, 617 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 639.2771 [M + Na]+ (calcd for C33H44O11Na, 639.2771).ε) 210 (3.86) nm; ECD (MeOH, Δε) 209 (−11.148) 246 (+4.267), 294 (−6.172) nm; IR (KBr) vmax 3460, 2969, 1768, 1635, 1460 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 625.2620 [M + Na]+ (calcd for C32H42O11Na, 625.2619).ε) 230 (3.35) nm; ECD (MeOH, Δε) 200 (−16.272), 224 (+7.992), 297 (−5.732); IR (KBr) vmax 3451, 2969, 1768, 1641, 1457 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 623.2463 [M + Na]+ (calcd for C32H40O11Na, 623.2464).ε) 213 (3.80) nm; ECD (MeOH, Δε) 200 (−20.403), 222 (+15.345) nm; IR (KBr) vmax 3400 1725, 1645, 1460 cm−1; 1H and 13C NMR data, see Table 3; HRESIMS m/z 567.2597 [M − H]− (cacld for C32H39O9Na, 567.2600).ε) 228 (4.14) nm; ECD (MeOH, Δε) 200 (−5.196), 208 (+2.013), 225 (−5.931), 258 (+8.547), 296 (−11.285) nm; IR (KBr) vmax 3445, 1730, 1643, 1142 cm−1; 1H and 13C NMR data, see Table 3; HRESIMS m/z 609.2675 [M + Na]+ (calcd for C32H42O10Na, 609.2670).ε) 229 (3.48) nm; ECD (MeOH, Δε) 200 (+12.576), 294 (−3.937) nm; IR (KBr) vmax 3460, 2931, 1728, 1648 cm−1; 1H and 13C NMR, see Table 3; HRESIMS m/z 591.2565 [M + Na]+ (cacld for C32H40O9Na, 579.2565).:1). Crystal data were obtained on a Bruker Smart-1000 CCD with a graphite monochromator with Cu Kα radiation (λ = 1.54184 Å) at 290(2) K. The structure was solved by direct methods using the SHELXS-97 (ref. 23) and expanded with difference Fourier techniques, refined with the SHELXL-97.24163 reflections measured (8.072° ≤ 2Θ ≤ 142.798°), 5564 unique (Rint = 0.0242, Rsigma = 0.0160) which were used in all calculations. The final R1 was 0.0420 (I > 2σ (I)) and wR2 was 0.1182 (all data). Flack parameter = −0.03 (5).Anti-inflammatory activities
All new compounds were evaluated for their inhibitory effects on NO production in lipopolysaccharide-activated RAW 264.7 macrophages as described in the literature.25 L-NMMA were used as positive control. All experiments were conducted for three independent replicates.In vitro cytotoxicity assay
All new compounds were evaluated for their cytotoxicities against HepG2 by MTT assay.26 Cells were plated in 96-well culture plates (5 × 103 cells per well). After incubation overnight, the cells were treated with different concentrations of each compound for 48 h. DMSO (0.1%) was used as a vehicle. MTT (5 mg mL−1) was dissolved in PBS and filter sterilized, then 20 μL of the prepared solution was added to each well and cells were incubated until a purple precipitate was visible. The formed formazan crystals were dissolved in DMSO (150 μL per well) by constant shaking for 10 min. The absorbance was measured on an ELISA reader (SpectraMax Plus = 384, Molecular Devices, Sunnyvale, CA) at a test wavelength of 570 nm and a reference wavelength of 630 nm. After treatment, cell viability was detected and IC50 values were calculated by the Reed and Muench method.26 All experiments were performed as three independent replicates.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (31470416), the Outstanding Youth Fund of the Basic Research Program of Jiangsu Province (BK20160077), the PhD Programs Foundation of Ministry of Education of China (20120096130002), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R63), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- S. K. Chen, B. Y. Chen and H. Li in Zhongguo Zhiwu Zhi, Science Press, BeiJing, 1997, vol. 43, p. 58 Search PubMed.
- S. G. Liao, H. D. Chen and J. M. Yue, Chem. Rev., 2009, 109, 1092–1140 CrossRef CAS PubMed.
- Q. G. Tan and X. D. Luo, Chem. Rev., 2011, 111, 7437–7522 CrossRef CAS PubMed.
- A. K. R. Bandi and D. U. Lee, Chem. Biodiversity, 2012, 9, 1403–1421 CAS.
- B. Siva, B. Poominma, A. Venkanna, K. Rajendra Prasad, B. Sridhar, V. Lakshma Nayak, S. Ramakrishna and K. Suresh Babu, Phytochemistry, 2014, 98, 174–182 CrossRef CAS PubMed.
- X. Fang, Y. T. Di, C. S. Li, Z. L. Geng, Z. Zhang, Y. Zhang, S. Y. Yang and X. J. Hao, J. Nat. Prod., 2009, 72, 714–718 CrossRef CAS PubMed.
- L. S. Gan, X. N. Wang, Y. Wu and J. M. Yue, J. Nat. Prod., 2007, 70, 1344–1347 CrossRef CAS PubMed.
- Y. T. Di, P. H. He, H. Y. Liu, P. Yi, Z. Zhang, Y. L. Ren, J. S. Wang, F. W. Yang, X. Fang, S. l. Li, H. J. Zhu and X. J. Hao, J. Nat. Prod., 2007, 70, 1352–1355 CrossRef CAS PubMed.
- L. G. Lin, C. P. Tang, C. Q. Ke, Y. Zhang and Y. Ye, J. Nat. Prod., 2008, 71, 628–632 CrossRef CAS PubMed.
- X. Fang, Q. Zhang, C. J. Tan, S. Z. Mu, Y. Lü, Y. B. Lu, Q. T. Zheng, Y. T. Di and X. J. Hao, Tetrahedron, 2009, 65, 7408–7414 CrossRef CAS.
- H. J. Yu, Q. F. Liu, L. Sheng, G. C. Wang and J. M. Yue, Org. Lett., 2016, 18, 444–447 CrossRef PubMed.
- X. Fang, Y. T. Di, G. W. Hu, S. L. Li and X. J. Hao, Biochem. Syst. Ecol., 2009, 37, 528–530 CrossRef CAS.
- J. Ning, Y. T. Di, X. Fang, H. P. He, Y. Y. Wang, Y. Li, S. L. Li and X. J. Hao, J. Nat. Prod., 2010, 73, 1327–1331 CrossRef CAS PubMed.
- H. Li, Y. Li, X. B. Wang, T. Pang, L. Y. Zhang, J. Luo and L. Y. Kong, RSC Adv., 2015, 5, 40465–40474 RSC.
- X. M. Tian, H. Li, F. An, R. J. Li, M. M. Zhou, M. H. Yang, L. Y. Kong and J. Luo, Planta Med., 2017, 83, 341–350 CAS.
- Y. Li, Q. Lu, J. Luo, J. S. Wang, X. B. Wang, M. D. Zhu and L. Y. Kong, Chem. Pharm. Bull., 2015, 63, 305–310 CrossRef PubMed.
- T. R. Govindachari, B. Banumathy, G. Gopalakrishnan and G. Suresh, Fitoterapia, 1991, 70, 106–108 CrossRef.
- X. Y. Wang, C. M. Yuan, G. H. Tang, T. Zou, F. Guo, J. H. Liao, H. Y. Zhang, G. Y. Zuo, G. X. Rao, Q. Zhao, X. J. Hao and H. P. He, J. Asian Nat. Prod. Res., 2014, 16, 795–799 CrossRef CAS PubMed.
- M. Y. Li, X. B. Yang, J. Y. Pan, G. Feng, Q. Xiao, J. Sinkkonen, T. Satyanandamurty and J. Wu, J. Nat. Prod., 2009, 72, 2110–2114 CrossRef CAS PubMed.
- A. C. Leite, J. B. Fernandes, M. Fatima das and G. F. da Silva, Z. Naturforsch. B, 2005, 60, 351–355 CAS.
- K. L. Mikolajczak, D. Weisleder, L. Parkanyi and J. Cladry, J. Nat. Prod., 1988, 51, 606–610 CrossRef CAS.
- B. L. Sondengam, C. S. Kamga and J. D. Connolly, Phytochemistry, 1980, 19, 2488 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Resolution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- Y. S. Li, M. Ishibashi, M. Satake, X. G. Chen, Y. Oshima and Y. Ohizumi, J. Nat. Prod., 2003, 66, 696–698 CrossRef CAS PubMed.
- L. J. Reed and H. A. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D and 2D NMR spectra of compounds 1–11 and ECD spectrum of 1–8 are available as supporting information. CCDC 1815118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra00728d |
| This journal is © The Royal Society of Chemistry 2018 |