
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic alcoholysis of alkaline extracted lignin for the production of aromatic esters over SO42−/ZrO2-ATP
Zhen Wu ab,
Jun Zhanga,
Qingqing Pana,
Xun Lia,
Yu Zhanga and
Fei Wang*a
ab,
Jun Zhanga,
Qingqing Pana,
Xun Lia,
Yu Zhanga and
Fei Wang*a
aCollege of Chemical Engineering, Nanjing Forestry University, Jiangsu Provincial Key Lab for Chemistry and Utilization of Agro-forest Biomass, Jiangsu Key Lab of Biomass-Based Green Fuels and Chemicals, Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing 210037, China. E-mail: hgwf@njfu.edu.cn
bJiangsu Key Laboratory for Biomass-based Energy and Enzyme Technology, School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huaian 223300, China
First published on 3rd April 2018
Abstract
An efficient process for the depolymerization of alkaline extracted lignin (AEL) using attapulgite (ATP)-supported solid catalysts in ethanol was developed in this work. Different ATP-supported catalysts were prepared and used to catalyze the depolymerization of the lignin AEL. The results demonstrated that the addition of ATP-supported catalysts was favorable for controlling the distribution of valuable depolymerization products. The optimal solid catalyst SO42−/ZrO2-ATP (Cat 2) exhibited high catalytic activity and selectivity, which showed a 78.6% conversion of AEL and a 29.4% selectivity to ethyl ferulate (ethyl 4-hydroxy-3-methoxycinnamate) with a catalyst/AEL ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 200 °C for 120 min. The catalyst could be reused and its catalytic activity did not obviously decreased after 6 successive runs. Particularly, a plausible mechanism involving esterification, hydrogenation, and dehydration for the production of aromatic esters from AEL depolymerization over SO42−/ZrO2-ATP in ethanol was also proposed.
:1 at 200 °C for 120 min. The catalyst could be reused and its catalytic activity did not obviously decreased after 6 successive runs. Particularly, a plausible mechanism involving esterification, hydrogenation, and dehydration for the production of aromatic esters from AEL depolymerization over SO42−/ZrO2-ATP in ethanol was also proposed.
1. Introduction
Lignin is a highly cross-linked macromolecule which consists of various oxygen and carbon bridges between alkylated methoxyl-phenol rings. Based on its phenolic functionalities and properties, lignin offers possibilities for high value-added renewable products applications.1 Lignin has been gradually generated as a byproduct in bioethanol production and the pulp/paper industry. However, this readily available lignin is a rich source of aromatic monomers and hence, if depolymerized efficiently, it can produce value-added chemicals and fuels.2 In fact, it has been estimated that less than 2% of lignin byproducts from the paper industry are used for specialty products.3 Therefore, developing efficient technologies for the high value utilization of lignin is crucially important and imperative.4As a natural amorphous three-dimensional polymer consisting mainly of methoxylated phenylpropane units,2,5 lignin is linked to other main components, such as cellulose and hemicellulose, in a variety of ways, including hydrogen, ester, and covalent bonds.6 Depolymerization of lignin is the key step towards making lignin cost-effective as a raw material,7 but its highly random and branched three-dimensional polyphenolic structure leads to its resistance to depolymerization.8
Various catalytic depolymerization methods have been used to depolymerize lignin into monomeric compounds and reduction depolymerization with the existence of hydrogen or a hydrogen donner, e.g. methanol, ethanol and i-propanol, is considered as a splendid process. Chen et al.9 use mesoporous SBA-15 catalysts to depolymerize hydrolyzed lignin and eliminate char in ethanol and the results demonstrate that both catalyst and ethanol play important roles in suppressing the repolymerization reaction. Yan et al.10 have reported the depolymerization of kraft lignin in supercritical ethanol on a MoC1−x/Cu–MgAlOz composite catalyst and a yield of aromatic compounds of 575 mg g−1 lignin is achieved at 330 °C. Hidajat et al.11 report the base-catalyzed depolymerization of lignin in supercritical methanol and they find that the most abundant monomers produced are methoxylated benzene and toluene species because of the unique O-alkylation ability of supercritical methanol. Kim et al.12 illustrate that lignin can be selectively degraded to alkylated phenols efficiently in supercritical t-BuOH as the Mw of lignins decrease after a supercritical t-BuOH treatment, which demonstrating the effectiveness of this method for lignin depolymerization.
Heterogeneous catalysts are currently used extensively in lignin depolymerization, which are particularly suited for large-scale industrial operations due to their many advantages over homogeneous catalysts. The products can be easily separated to allow recovery and reuse of solid catalysts.13 There are many literatures about heterogeneous catalysts, including metal sulfide,14 metal phosphide,15 metal carbide,16,17 and metal nitride,18,19 etc. Although these materials have showed good performance in catalysis, the precursors acting as nitrogen and/or carbon sources are expensive. Moreover, the preparation processes of the precursors are complicated.
Attapulgite (ATP) is a mesoporous clay phyllosilicate mineral, which has an ideal formula (Mg,Al)4Si8O20(OH)2(H2O)4·4H2O and Mg/Al ratio ≤ 1.20 There are significant ATP reserves in various countries such as China, the US, and Spain.21 The high surface area, porosity, thermal resistance, and fibrous morphology of ATP provide much potential for diverse applications such as catalyst supports, nanocomposites, and environmental absorbents.22 Miao et al.23 report that rhodium nanoparticles can be supported on ATP via an ionic liquid (1,1,3,3-tetramethylguanidine lactate, TMG+L−) and the resulting catalyst is active and stable for hydrogenation of cyclohexene. More recently, a new type of hybrid material has been fabricated from the hybridization of metal–organic framework HKUST-1 and ATP whose surface areas are even higher than that of pure HKUST-1. Such hybrid materials exhibit good catalytic activity in the ring-opening reaction of styrene oxide.21 Zirconia, with high catalytic activity, good thermal and chemical stabilities, is commonly used as catalyst supporting material. Meanwhile, coke which may cover active sites is hardly formed on ZrO2.24 Zirconium oxide treated with sulfuric acid exhibits a strong acidity.25
To depolymerize lignin into high-valued monomeric compounds, a novel method was herein developed in this work, in which the advantages of solid catalysts and clay materials are combined to develop a new type of low-cost catalyst with high catalytic activity and selectivity for lignin depolymerization. The objective of the present study is to explore process parameters for the effective depolymerization of lignin over ATP-supported catalysts in alcohols. For this purpose, the effects of lignin source, solvent, composition of catalysts, and reaction conditions were examined in detail. Furthermore, we also performed studies toward the stability of the catalysts prepared. In the proposed strategy, we designed the reaction routes of directional conversion to aromatic esters from lignin over ATP-supported catalysts and the reaction mechanism was also discussed.
2. Materials and methods
2.1 Chemicals and materials
The lignocellulosic materials harvested in the northern part of China were cut into 3–5 mm in length, screened (1 mm × 1 mm sieve), washed with deionized water, and air-dried. The ATP mineral was friendly provided by Xuyi Oubaite Clay Material Co. Ltd. (Huaian, China). The ethanol and sulphuric acid were purchased from Nanjing chemical reagent Co., LTD. The metal precursors (H32Mo7N6O28, Co(NO3)2·6H2O, H6O41SiW12, ZrOCl2·8H2O) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China) and used without further purification.2.2 Preparation of alkaline extracted lignin
Alkaline extracted lignin (AEL) was obtained from mild alkali cooking of as-prepared lignocellulosic material. In brief, a 10 g material was added to 2.5% sodium hydroxide solution at a liquid/solid ratio of 10:1 (v/w) in a three-necked flask and then heated to 105 °C for 2 h with stirring at 200 rpm. The mixture was filtrated at a high temperature and washed with hot distilled water. The filtrate was neutralized to pH 3.5 with 20% sulfuric acid at 60 °C for 30 min. The precipitate was separated by centrifugation and then dried at 50 °C for 72 h in a vacuum oven, sieved to particle size smaller than 40 mesh (<0.425 mm) prior to the depolymerization experiments.
2.3 Catalyst preparation and characterization
The ATP mineral was firstly treated with 0.5 wt% HNO3 to activate its surface to get an activated ATP aqueous dispersion, whose solid content could be controlled in the range of 15–25%. The suspension was stirred at room temperature for 8 h, filtered and washed to neutral, dried at 105 °C for 12 h, and sieved to particle size smaller than 100 mesh (<0.154 mm).The preparation procedure of ATP-supported Co–Mo catalyst, for example, could be described as follows: 20 g of ATP was added into 60 mL deionized water in a round-bottom flask and stirred at 45 °C. 1.85 g of H32Mo7N6O28 and 4.93 g of Co(NO3)2·6H2O were added into the above ATP/deionized water suspension, adjusted to pH 9–10 with ammonia, stirred at 85 °C for 6 h, filtered immediately, dried at 105 °C for 12 h and then calcined at 500 °C for 3 h, sieved to particle size smaller than 100 mesh, and denoted as Co–Mo/ATP. The procedure for preparing ATP-supported silicotungstic acid catalyst was same to that of Co–Mo/ATP, but deionized water was substituted by 50% (v/v) ethanol aqueous solution and calcined at 200 °C for 5 h, sieved to particle size smaller than 100 mesh, and denoted as STA/ATP.
The procedure for preparation of SO42−/ZrO2-ATP was as follows: 50 g of ATP was added into 200 mL deionized water in a round-bottom flask, 10 g of ZrOCl2·8H2O was added into the suspension and stirred at 45 °C for 2 h, adjusted to pH 9–10 with ammonia, aged at room temperature for 24 h, filtered and washed to neutral, dried at 105 °C for 12 h and ground, then it was impregnated in 2 mol L−1 H2SO4 for 4 h, filtered and dried at 105 °C for 12 h, then calcined at 500 °C for 3 h, and sieved to particle size smaller than 100 mesh. Thereafter, SO42−/ZrO2-ATP (Cat 1) was obtained. SO42−/ZrO2-ATP (Cat 2) and SO42−/ZrO2-ATP (Cat 3) were prepared through the same procedure with different ZrOCl2·8H2O mass of 25 g and 50 g, respectively. The procedure for preparing SO42−/ZrO2 and ZrO2-ATP were also same to that of SO42−/ZrO2-ATP (Cat 1), but no ATP was impregnated for SO42−/ZrO2 or without loading of SO42− for ZrO2-ATP. All prepared catalysts were stored in a vacuum dryer before use in any reaction.
X-ray photoelectron spectra (XPS), Brunauer–Emmett–Teller (BET), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), and transmission electron microscopy (TEM) were used to detect and characterize the structural characteristics and properties of the as-prepared catalysts. XPS were recorded with a Kratos Axis Ultra DLD spectrometer, Al Kα radiation as the excitation source, and the X-ray power supply was run at 15 kV and 10 mA. Nitrogen gas adsorption desorption isotherms were measured at 77 K using a Quantachrome Autosorb iQ2 instrument. The specific surface area was estimated by the multipoint BET method using the adsorption data. SEM and TEM images were recorded on a JSM-7600F and JEM-1400 instrument, respectively. FTIR spectrum was obtained on a Nicolet 380 FTIR spectrometer using KBr discs in the range of 4000–400 cm−1.
2.4 Depolymerization of the lignin
The depolymerization reactions were carried out in a 50 mL micro autoclave equipped with a temperature controller and a pressure sensor. For a typical experiment, 0.5 g of AEL, 0.25 g of catalyst, and 15 mL of ethanol were loaded into the autoclave, purged with high-purity nitrogen for three times, then heated to the desired temperature and kept for the desired reaction time. After the reaction, the autoclave was cooled down to room temperature by ice-bath. After releasing the gas, the reaction mixture was filtrated. The solid residue was washed with ethanol and water, dried at 105 °C for 12 h and then weighed and the weight loss rate was calculated as conversion of AEL. The filtrate was collected for gas chromatography-mass spectrometry (GC-MS) analysis.2.5 Products analysis
The organic phase containing the products was injected neatly to a GC-MS system (Agilent GC7890A/MSD5975C) for product qualitative analysis. GC-MS was equipped with EI source, DB-5 capillary column (30 m × 0.25 mm × 0.25 μm) and NIST05 MS data base. An inlet temperature of 290 °C, a He gas flow rate of 20 mL min−1 and a flow split-ratio of 10:1 were used for the GC analysis. A heating schedule for the column was pre-set as: remained at 50 °C for 5 min, increased to 280 °C at a rate of 5 °C min−1 and then remained for 5 min. The solvent delay was set as 2 min for MS detector. Each experiment was conducted at least three times under the same conditions, and their average values were reported.
3. Results and discussion
3.1 Catalyst characterization
In order to illustrate the characteristics of the ATP-supported catalysts, a series of tests were carried out. The as-prepared catalysts were characterized by XPS, and the survey spectra of all ATPs were shown in Fig. 1. It can be seen from ATP curve that the predominant peaks at 712 eV, 532 eV, 285 eV, 103 eV, 75 eV, and 57 eV are attributed to Fe, O, C, Si, Al, and Mg species. XPS analysis showed that the Fe, O, C, Si, Al, Mg, and N mass contents on the surface of ATP were 6.48%, 44.84%, 22.87%, 18.32%, 5.16%, 2.06%, and 0.27%, respectively. As shown in Fig. 1, all the target elements were successfully loaded onto the surface of ATP (see the curves of SO42−/ZrO2-ATP, ZrO2-ATP, STA/ATP and Co–Mo/ATP in Fig. 1).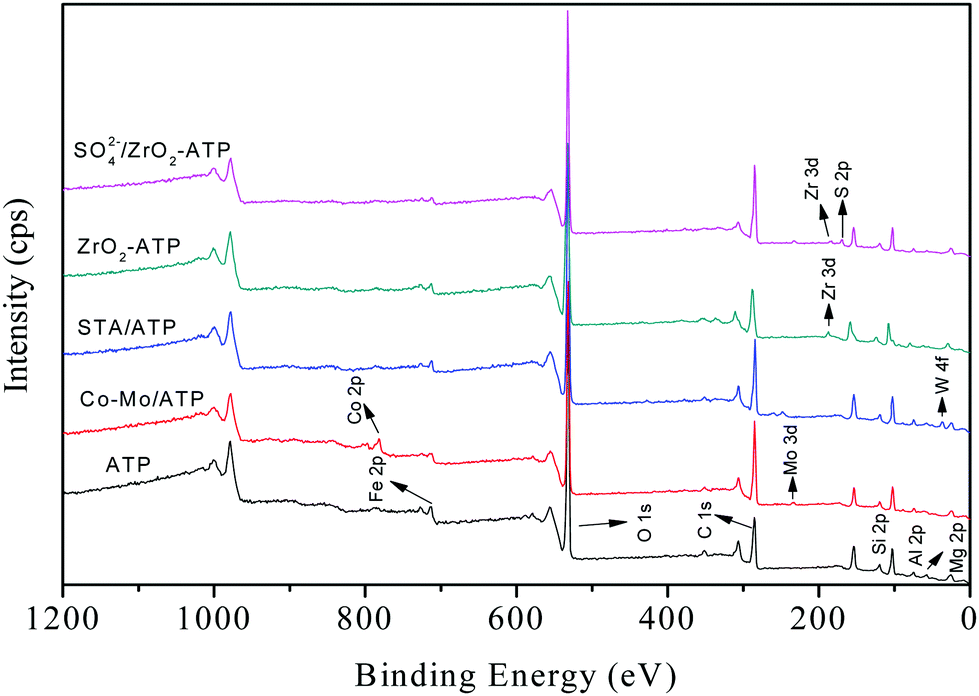 | ||
| Fig. 1 XPS full-scan spectra of ATP and ATP-supported catalysts. | ||
The specific surface area, pore size, and pore volume of the ATP-supported catalysts are listed in Table 1. As shown, the specific surface area decreased obviously after being modified by Co and Mo or STA. One can speculate that in the co-precipitation process of catalyst preparation, the metal oxides dispersed on the surface of the catalyst leading to the decrease of the specific surface area and a part of metal oxides entered into the pore leading to the decrease of the pore volume. Among all of the as-prepared catalysts, ZrO2-ATP was the only one not to present a decline in specific surface area compared to ATP. This can be attributed to the approximate specific surface area and pore size of ZrO2.26,27 However, it is worth noting that the specific surface area and pore volume of ZrO2-ATP decrease dramatically after SO42− loading. The possible reasons may be that fibrous bundles are disaggregated initially, and then the octahedral cations (Mg2+ and Al3+) are dissolved.28
| Catalyst | SBET (m2 g−1) | Pore size (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| ATP | 221.583 | 1.178 | 0.252 |
| Co–Mo/ATP | 208.320 | 2.419 | 0.249 |
| STA/ATP | 171.800 | 1.176 | 0.227 |
| ZrO2-ATP | 228.559 | 1.410 | 0.296 |
| SO42−/ZrO2 | 198.268 | 3.627 | 0.174 |
| SO42−/ZrO2-ATP (Cat 1) | 109.084 | 2.419 | 0.172 |
| SO42−/ZrO2-ATP (Cat 2) | 50.902 | 4.543 | 0.096 |
| SO42−/ZrO2-ATP (Cat 3) | 80.075 | 3.169 | 0.077 |
3.2 Catalytic activity
Inspired by previous studies,15,29,30 ATP-supported catalysts were prepared to catalyze the depolymerization of AEL. As a hydrogen donor, alcohols used in lignin depolymerization can suppress the condensation of the phenolic intermediates.10 In this paper, the catalytic activities of the ATP-supported catalysts were tested at 225 °C in ethanol.The main products obtained from AEL of corncob in the absent/presence of ATP-supported catalysts were verified to be compounds 1–18 as shown in Table 2. The monomeric compounds produced mainly consisted of phenol and its derivatives, including phenols (compounds 1–8), aldehydes (compound 9), esters (compounds 10–15), ethers (compounds 16 and 17), and acids (compound 18). As expected, catalysts showed a large effect on the distribution of depolymerization products. In the absence of catalyst, only 34.6% AEL conversion was obtained (Fig. 2) and the products were complicated and fragmented (Fig. 3(a)). When ATP-supported catalysts were added, AEL conversions were improved to varying degrees and the products distribution changed apparently. A high conversion of AEL (82.3%) was found for the addition of ATP, but the products were still fragmented. Relatively high content of ethyl hydroferulate (ethyl 3-(4-hydroxy-3-methoxyphenyl) propionate, compound 11) was obtained in the presence of Co–Mo/ATP, however, conversion of AEL was increased slightly. The addition of STA/ATP or ZrO2-ATP made the products more dispersed. However, it was worth noting that the contents of esters (compounds 10–15) increased significantly after SO42− loading onto ZrO2. The esterification reaction between ethanol and decomposition products of phenolic oligomers or monomers may be the main contributor to the increase in esters. In addition, previous studies demonstrate that SO42−/ZrO2 shows strong acidity and catalytic activity for lignin depolymerization. For instance, CuO/SO42−/ZrO2 has been explored for the hydrogenolysis of soda lignin by Zhang et al.,31 who report that the main effect of SO42−/ZrO2 is cleaving the β-O-4 linkages, and CuO is cracking the methoxy groups. Moreover, SO42−/ZrO2 with higher acidity is favorable for dehydration and esterification reactions.32
| No. | Products & relative content (area, %) | Catalyst | ||||||
|---|---|---|---|---|---|---|---|---|
| No catalyst | ATP | Co–Mo/ATP | STA/ATP | ZrO2-ATP | SO42−/ZrO2 | SO42−/ZrO2-ATP (Cat 1) | ||
| a Reaction conditions: catalyst (0.25 g), AEL (0.5 g), C2H5OH (15 mL), 225 °C, 300 rpm, 120 min. | ||||||||
| 1 | 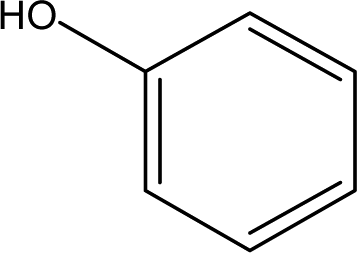 |
0.44 | 3.90 | 1.02 | 1.22 | 1.66 | 0.69 | 0.61 |
| 2 | 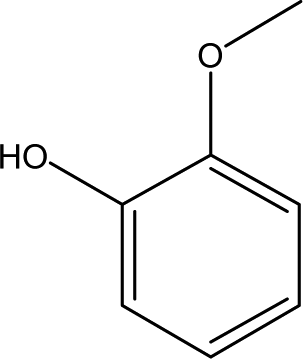 |
1.39 | 12.24 | 4.01 | 4.29 | 2.38 | 0.96 | 2.28 |
| 3 | 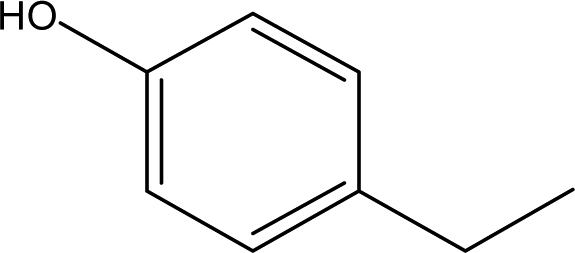 |
2.81 | 3.73 | 5.24 | 5.46 | 6.51 | 5.07 | 6.36 |
| 4 | 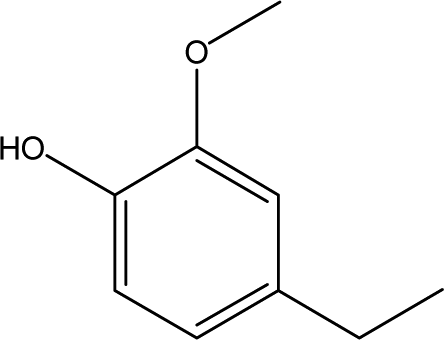 |
10.43 | 14.53 | 5.46 | 5.63 | 5.98 | 4.04 | 5.42 |
| 5 | 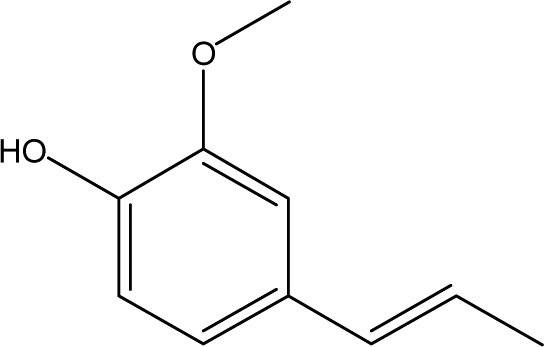 |
5.31 | 0.76 | 1.23 | 0.87 | 1.98 | 2.37 | 0.65 |
| 6 | 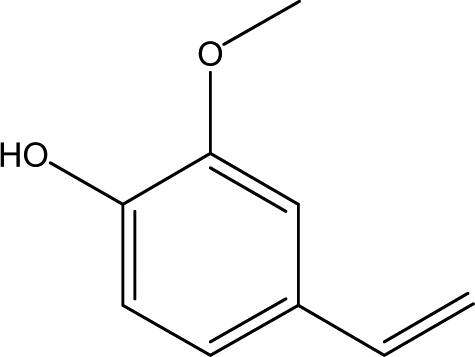 |
13.23 | 1.08 | 2.77 | 3.46 | 2.83 | 1.15 | 2.08 |
| 7 | 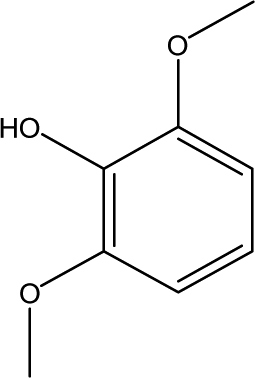 |
3.09 | 7.53 | 3.54 | 3.10 | 3.22 | 2.16 | 1.99 |
| 8 | 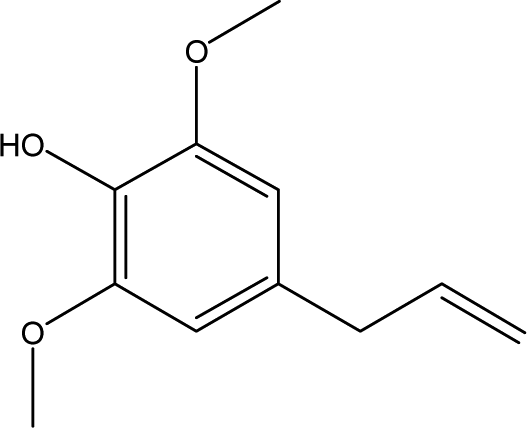 |
2.16 | 0.71 | 0.29 | 1.37 | 1.44 | 0.84 | 0.77 |
| 9 | 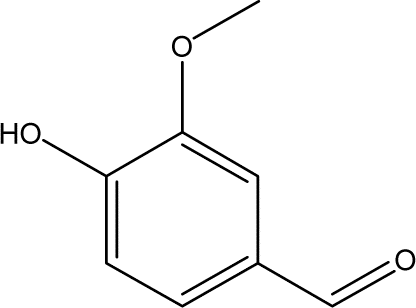 |
3.25 | 1.22 | 0.27 | 0.79 | 0.44 | 0.39 | 0.62 |
| 10 | 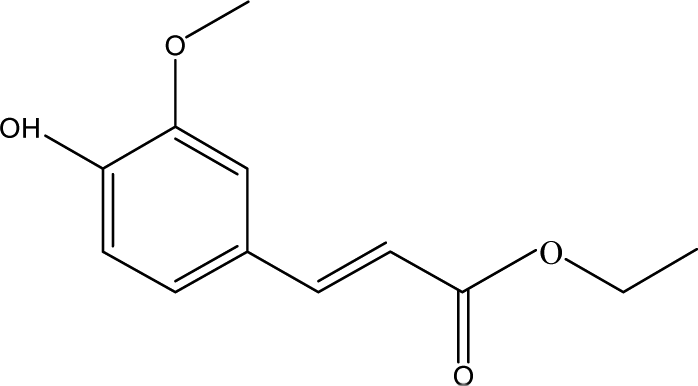 |
4.49 | 1.66 | 7.06 | 0.98 | 6.72 | 13.76 | 22.51 |
| 11 | 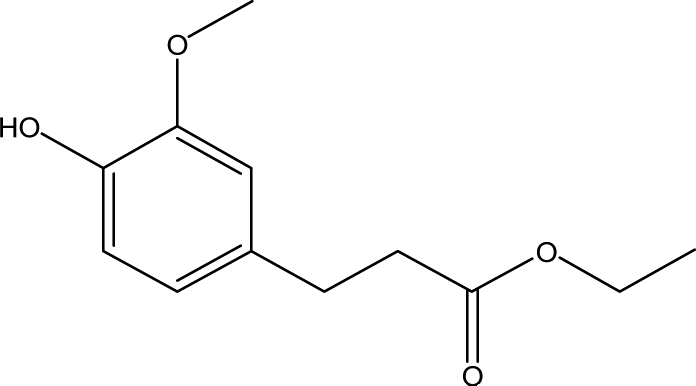 |
1.32 | 7.74 | 13.57 | 3.47 | 4.64 | 8.22 | 4.12 |
| 12 | 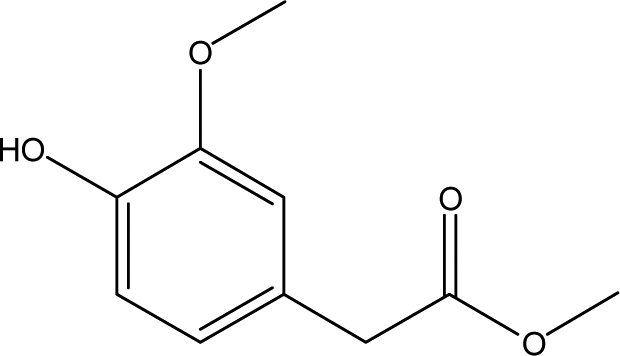 |
— | 3.07 | 1.53 | 3.26 | 2.28 | 7.24 | 9.79 |
| 13 | 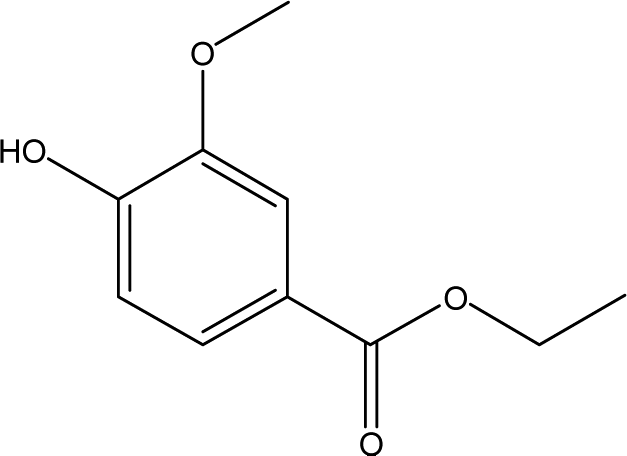 |
1.94 | 0.56 | 1.86 | 0.39 | 0.47 | 1.08 | 1.49 |
| 14 | 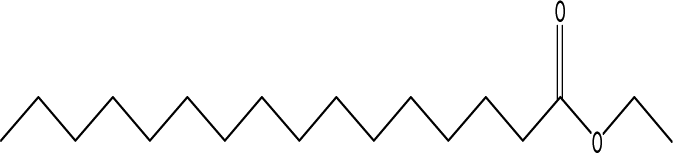 |
2.84 | 3.84 | 5.80 | 6.05 | 3.17 | 7.08 | 4.57 |
| 15 | 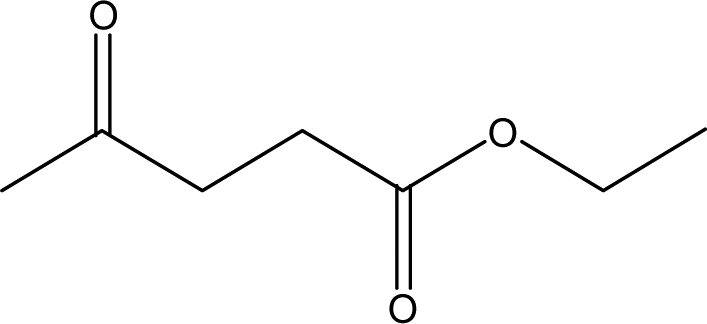 |
— | — | — | — | 1.24 | 1.66 | 2.17 |
| 16 | 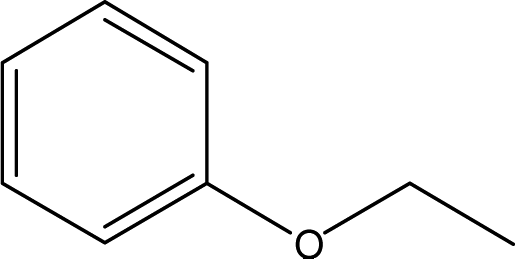 |
— | — | — | 0.18 | — | 0.03 | — |
| 17 | 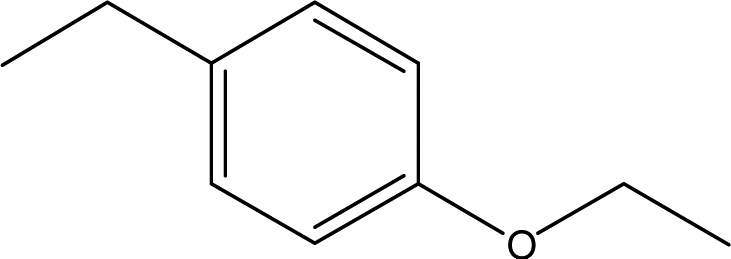 |
— | — | — | 1.70 | — | 1.55 | 0.16 |
| 18 | 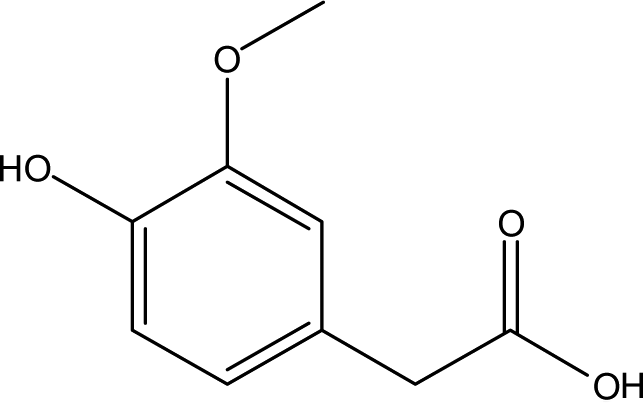 |
— | — | — | 1.95 | 1.02 | — | 1.17 |
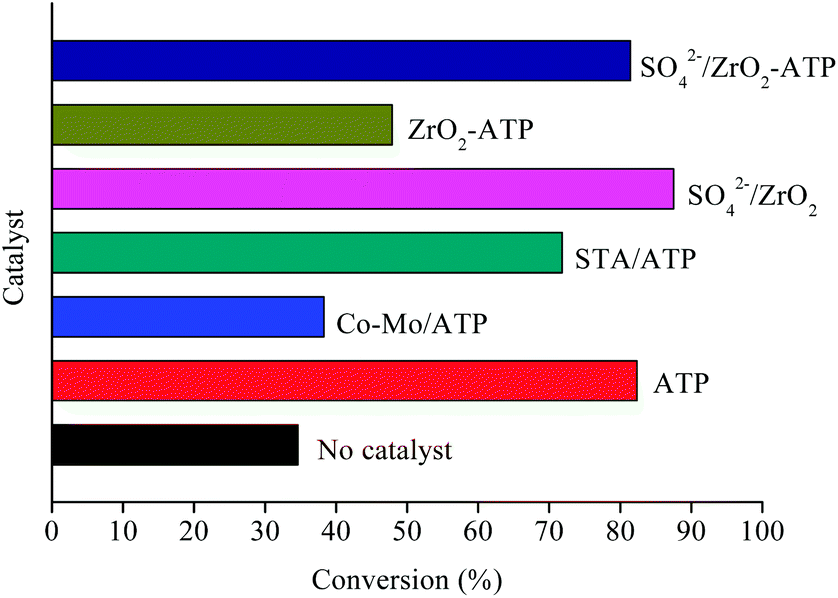 | ||
| Fig. 2 Conversion of AEL catalyzed by ATP-supported catalysts. | ||
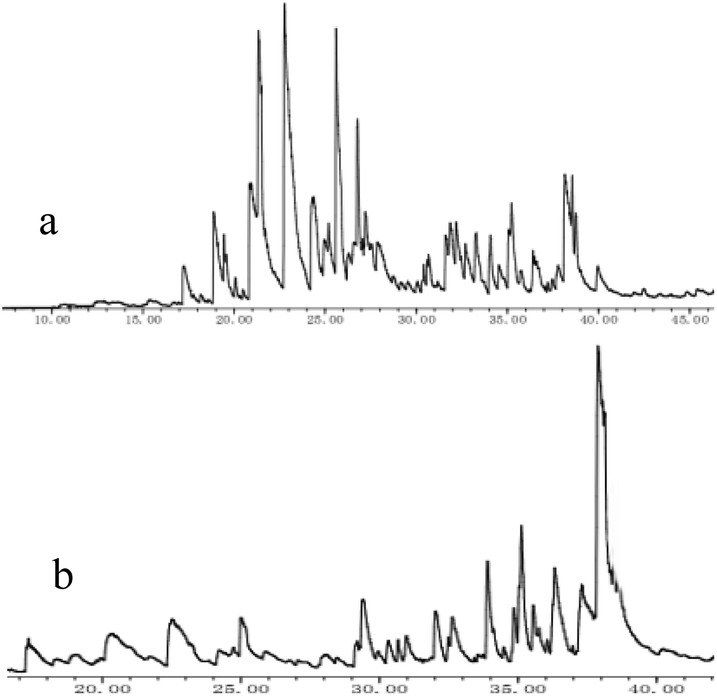 | ||
| Fig. 3 Total ion chromatogram (TIC) of AEL depolymerization products over no catalyst (a) and SO42−/ZrO2-ATP (b). | ||
When catalyst SO42−/ZrO2-ATP (Cat 1) was added to the above-mentioned reaction system, a 81.4% conversion of AEL was achieved and the products distribution changed apparently (Fig. 3(b)). The contents of aromatic esters increased steeply and a 22.5% selectivity was obtained for ethyl ferulate (ethyl 4-hydroxy-3-methoxycinnamate, compound 10, peak at 38.103′), which is a noteworthy compound, not only because of its flavor characteristics but also its antioxidant and functional properties and is currently used as a sun screening agent in commercial sunscreen blends.33,34 Much larger amounts of aromatic esters were produced by the addition of SO42−/ZrO2-ATP, suggesting that the depolymerization reaction of AEL appeared directional conversion over SO42−/ZrO2-ATP. This can be attributed to the enhancement of porosity and the excellent properties for the selective hydrogenation of the acidified ATP-supported catalyst.35,36 Among all ATP-supported catalysts prepared, SO42−/ZrO2-ATP with high catalytic activity and selectivity was selected for further investigations.
To get more insight into the catalyst SO42−/ZrO2-ATP (Cat 1), FTIR spectrum was recorded (Fig. 4). The peak at 3615 cm−1 is assigned to stretching vibration of Al–OH and (Al–Fe)–OH, which is the character of ATP. The peak at 3411 cm−1 is attributed to hydroxyl stretching vibration of coordinated water of ATP. The peak located at 1638 cm−1 can be assigned to an OH2 bending vibration. The Si–O–Si stretching band appears at 1020 cm−1.28,37 The absorbance peaks at 1155 cm−1 and 1078 cm−1, representative of the symmetric stretching vibration of O–S–O,38 is also observed in the spectrum, verifying SO42− has been successfully grafted onto ATP to form SO42−/ZrO2-ATP. The changes of morphology and structure of ATP after modification were also examined by SEM and TEM. It is obvious from Fig. 5 that ATP is well-crystallized and an individual crystalline rod is about 500 nm in length and 50 nm in diameter. Although the surface of the fibrous bundles was partially covered and disaggregated, significant changes in morphology and structure of ATP were not observed and large enough surface area and pore size were maintained after modification, providing much potential for the adequate contact and adsorption of reactants during lignin depolymerization.
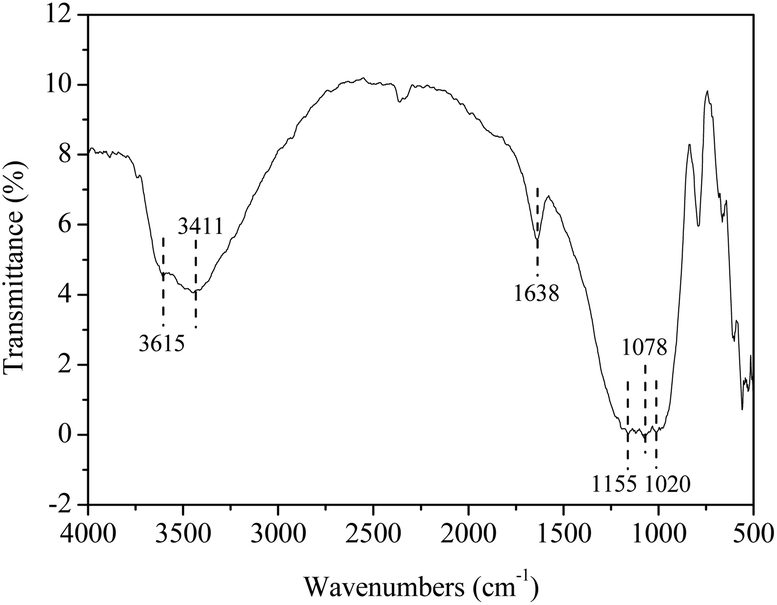 | ||
| Fig. 4 FTIR spectrum of catalyst SO42−/ZrO2-ATP. | ||
 | ||
| Fig. 5 SEM (a) and TEM (b) images of the original (a1, b1) and modified (a2, b2) ATP. | ||
3.3 Effect of lignin source
The structure and properties of different varieties of lignin vary with extraction method, plant type, environmental factors, and even genotype.39 Thus, the structural characteristics of the six kinds of AEL used in this study (corncob lignin: CCL, corn straw lignin: CSL, rice straw lignin: RSL, wheat straw lignin: WSL, poplar lignin: PL, and bamboo lignin: BL) were comparatively analysed before investigating the distributions of aromatic esters in lignin alcoholysis products.It was clearly observed from Fig. 6 that all the lignin samples exhibited the expected functional groups: hydrogen bonded O–H stretching vibration at 3421 cm−1, C–H (CH3, CH2) stretching vibration at 2933 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (conjugated ketone, carbonyl, and ester groups) at 1686 cm−1, aromatic skeletal vibrations at 1600, 1510 and 1421 cm−1, C–H deformation vibration at 1463 cm−1 (connected to benzene ring or in O–CH3), vibration at 1328 cm−1 from syringyl rings, CO stretching vibration at 1243 cm−1 from guaiacyl rings. Overall, the CCL, CSL, RSL, WSL, and BL had more strong absorption peaks at 1157, 1123, and 836 cm−1, those are the typical signal of herbaceous lignin (consist of GSH units).
O stretching vibration (conjugated ketone, carbonyl, and ester groups) at 1686 cm−1, aromatic skeletal vibrations at 1600, 1510 and 1421 cm−1, C–H deformation vibration at 1463 cm−1 (connected to benzene ring or in O–CH3), vibration at 1328 cm−1 from syringyl rings, CO stretching vibration at 1243 cm−1 from guaiacyl rings. Overall, the CCL, CSL, RSL, WSL, and BL had more strong absorption peaks at 1157, 1123, and 836 cm−1, those are the typical signal of herbaceous lignin (consist of GSH units).
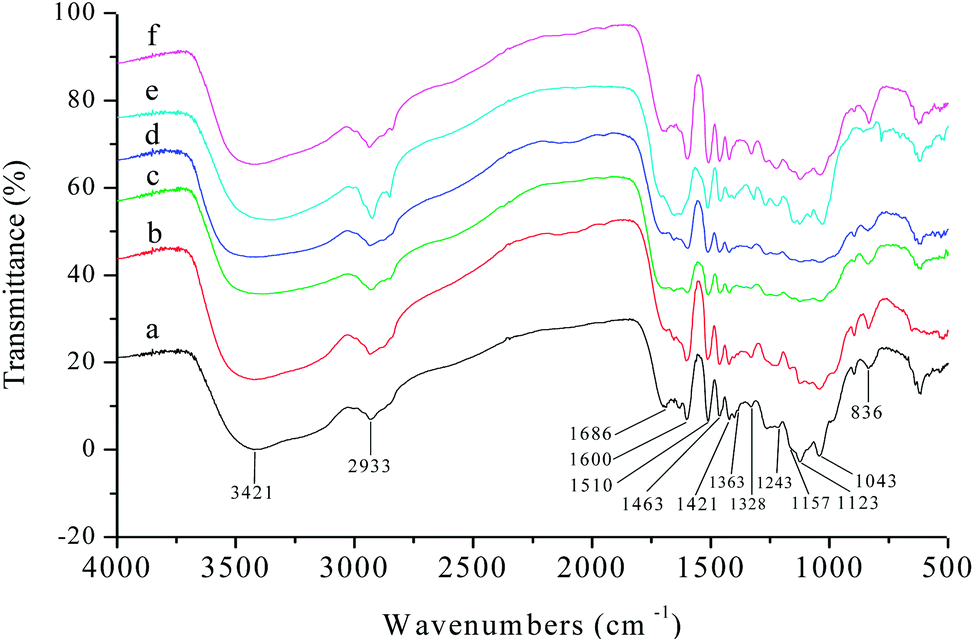 | ||
| Fig. 6 FTIR spectra of lignin fractions: (a) CCL; (b) CSL; (c) RSL; (d) WSL; (e) PL; (f) BL. | ||
The effect of lignin source on AEL depolymerization was investigated and the results in the comparison of the conversion and selectivity of the six different lignin sources were shown in Fig. 7. A lower conversion of lignin and negligible amount of ethyl ferulate (EF) were obtained from the poplar lignin, indicating that the poplar lignin has more recalcitrant properties. Peng et al.40 report that a minor amount of H units is observed in poplar lignin sample, which is connected to G and S units by multiple ester linkages, suggesting a more obstinate and stable structure compared to herbaceous lignin that consist of GSH units. Meanwhile, ferulic acid is commonly found in commelinid plants, mostly in the trans-isomeric form, and esterified with the specific polysaccharides.41 Hence, the lower content of ferulic acid in poplar lignin may be the main contributor to the low selectivity to ethyl ferulate. Among all AELs prepared, CCL exhibited superior selectivity to ethyl ferulate as compared to other herbaceous lignins was selected for the following study.
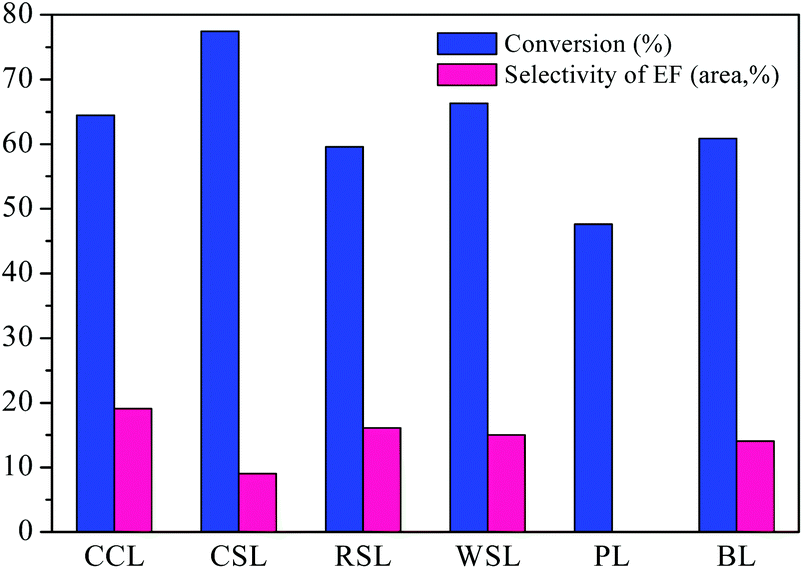 | ||
| Fig. 7 Different lignin depolymerization catalyzed by SO42−/ZrO2-ATP (Cat 1). Reaction conditions: AEL (0.5 g), catalyst (0.25 g), C2H5OH (15 mL), 225 °C, 300 rpm, 60 min. | ||
3.4 Effects of solvent and composition of catalysts
The effects of solvent and composition of catalysts on AEL of corncob depolymerization were examined at 225 °C for a reaction time of 120 min using ATP-supported catalysts SO42−/ZrO2-ATP with different ZrO2-to-ATP mass ratios in methanol, ethanol, and i-propanol, respectively. Fig. 8 shows the conversion (a) and selectivity (b) of the lignin depolymerization over different composition of catalysts in low mass molecule alcohols. As expected, the conversion and selectivity were increased significantly by the use of SO42−/ZrO2-ATP in all alcohols. In addition, different alcohol showed the different alcoholysis ability. In the case of ethanol, conversions of lignin reached 81.4%, 72.5%, and 75.9%, respectively, which were higher than those in methanol or i-propanol. The enhanced lignin depolymerization can be attributed to the excellent in situ hydrogen donating ability of ethanol which is essential for lignin depolymerization.42 Cat 2 exhibited superior catalytic selectivity to aromatic esters as compared to the Cat 1 and Cat 3 catalysts, which could be related to the physicochemical properties of the catalysts. Specific surface area and pore size might be the dominant reason. Cat 2 with the smallest specific surface area and the biggest pore size (shown in Table 1) was propitious to the catalytic alcoholysis reaction.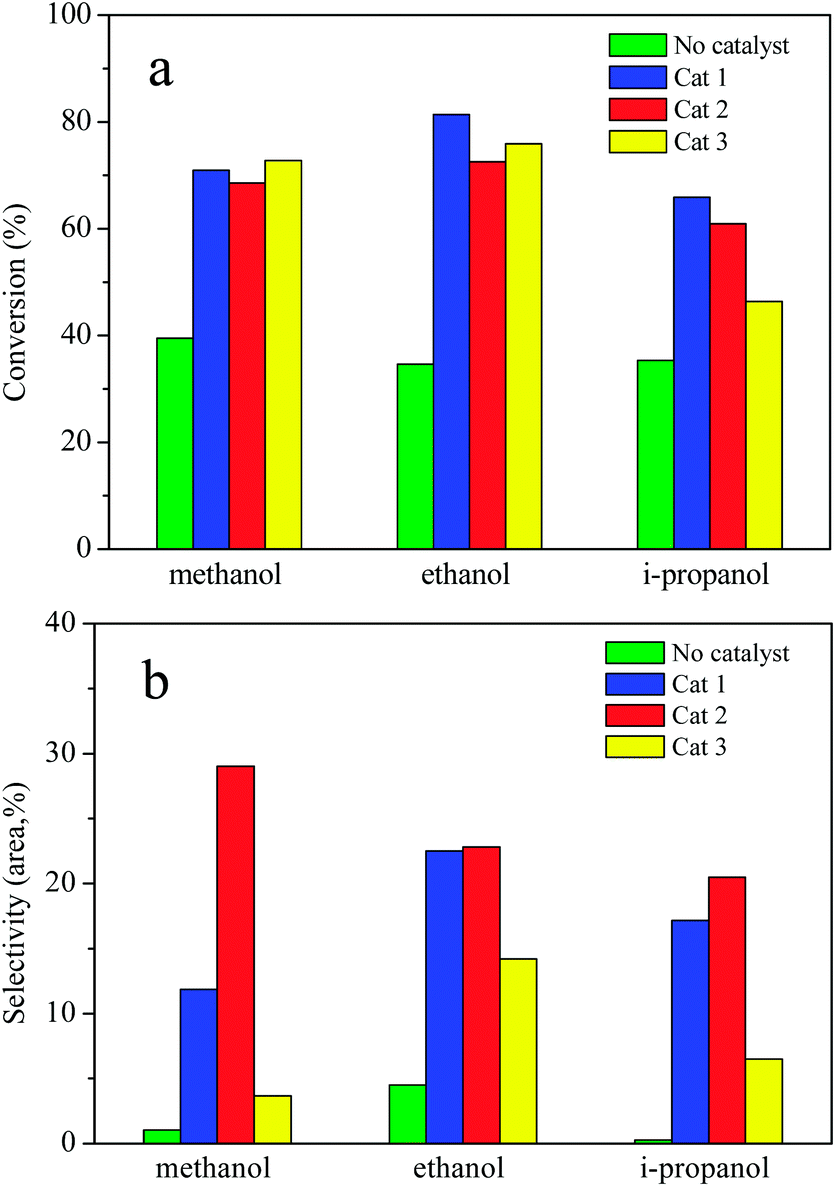 | ||
| Fig. 8 AEL depolymerization in different alcohol over ATP-supported catalysts. Reaction conditions: AEL (0.5 g), catalyst (0.25 g), alcohol (15 mL), 225 °C, 300 rpm, 120 min. | ||
3.5 Effects of reaction temperature, reaction time, and catalyst loading
The effects of reaction temperature, reaction time, and catalyst loading on the conversion of AEL of corncob into ethyl ferulate were studied. The investigation was conducted in ethanol over Cat 2 and the results were given in Fig. 9. As can be seen, the reaction temperature, reaction time, and catalyst loading have a significant influence on content of ethyl ferulate and lignin conversion. In the case of reaction temperature (Fig. 9(a)), the content of ethyl ferulate and conversion of AEL firstly increased and then decreased progressively with the increase of temperature, and reached highest at 200 °C (30.5%) and 225 °C (81.4%), respectively. As be seen from Fig. 9(b), the change in AEL conversion with increasing the reaction time could be fitted by a quadratic function, with a vertex of 81.4% at 120 min. It was worth noting that the content of ethyl ferulate changed slightly after 60 min, which indicated that ethyl ferulate produced was more stable to reaction time comparing to reaction temperature. As shown in Fig. 9(c), the content of ethyl ferulate increased substantially from 11.8% to 28.7% as the catalyst/AEL ratio increased from 0.1/1 to 1/1, meanwhile, the conversion of AEL increased substantially from 42.3% to 82.6%, correspondingly. Overall, the dramatic decline of AEL conversion with the increase of temperature, time or catalyst loading was most likely due to the repolymerization of decomposition products or carbonization of carbonaceous materials. Hence, a reaction temperature of 200 °C, a reaction time of 120 min, and a catalyst/AEL ratio of 1:1 were chosen as the optimal conditions for the conversion of AEL into ethyl ferulate.
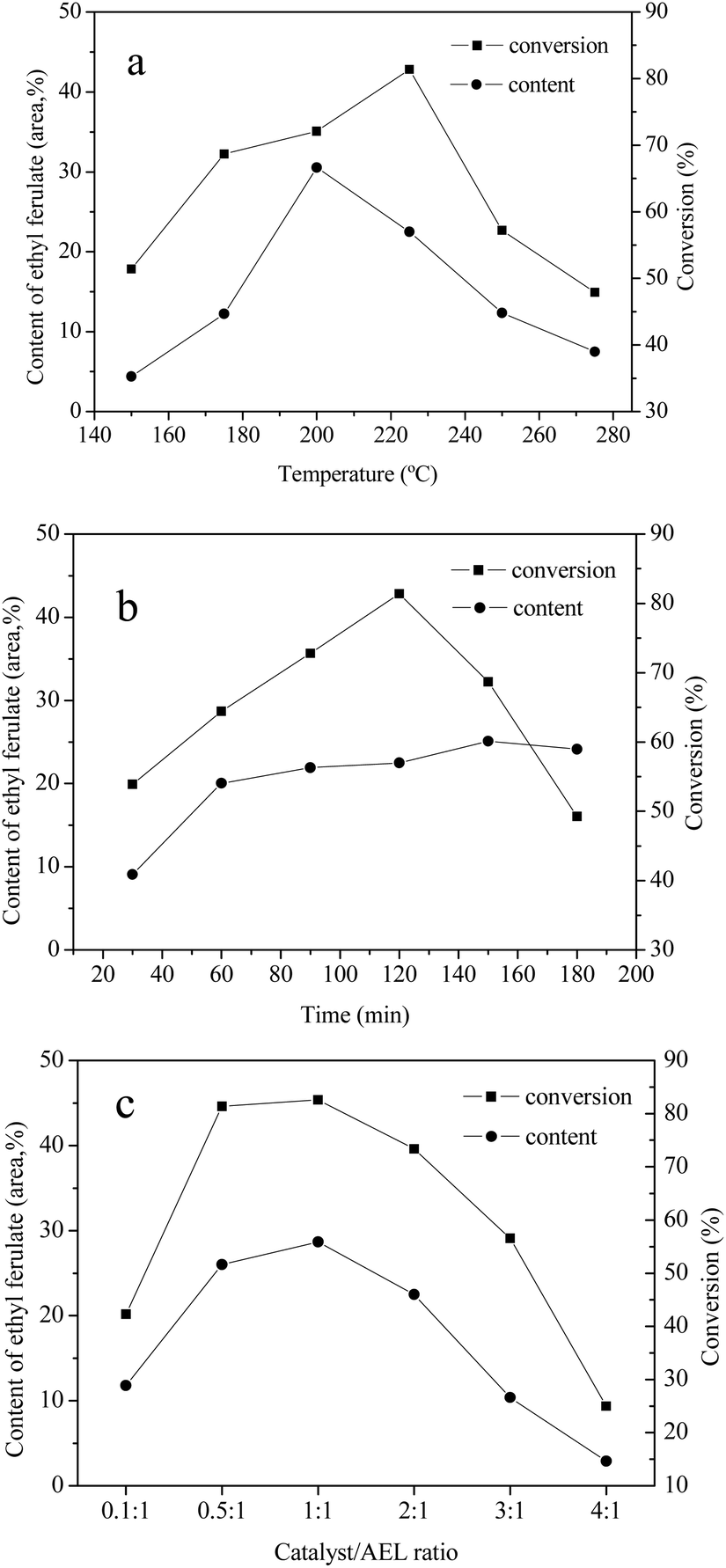 | ||
| Fig. 9 Effects of reaction temperature (a1), reaction time (b2), and catalyst loading (c3) on AEL depolymerization. 1Reaction conditions: catalyst (0.25 g), AEL (0.5 g), C2H5OH (15 mL), 300 rpm, 120 min. 2Reaction conditions: catalyst (0.25 g), AEL (0.5 g), C2H5OH (15 mL), 225 °C, 300 rpm. 3Reaction conditions: AEL (0.5 g), C2H5OH (15 mL), 225 °C, 300 rpm, 120 min. | ||
3.6 Catalyst stability
The reusability or recyclability of the catalyst is an important parameter for the evaluation of heterogeneous catalyst. In this paper, repetitive use of the catalyst SO42−/ZrO2-ATP (Cat 2) was performed to test its stability. The stability was investigated by the depolymerization of AEL and evaluated by conversions of AEL and relative contents of main esters (compounds 10–15, area, %) in decomposition products. The details of the reusability test were as follows: 0.5 g of catalyst, 0.5 g of AEL, and 15 mL of ethanol were loaded into the reactor, 200 °C for 120 min. After the depolymerization reaction, the resulting products were carefully poured out and filtered for the reuse of the catalyst in the reactor, weighed and calculated the weight loss as conversion. Fresh AEL was then added and the process was repeated under the same conditions. This procedure was repeated 8 times. The results of the recycle experiments are shown in Fig. 10. It can be seen that SO42−/ZrO2-ATP kept a good catalytic activity for the conversion of AEL and a 78.6% conversion of AEL and a 29.4% selectivity of ethyl ferulate were achieved after 1 run under the optimal conditions of 200 °C, 120 min, and catalyst/AEL ratio of 1:1. Conversion of 65.3% was still achieved after 6 runs. Meanwhile, the total relative contents of main esters decreased from 49.5% to 27.6% and 18.3% after 6 and 8 runs, respectively, which indicated that the catalyst could be reused at least 6 times. We also noticed that the distribution of esters experienced great changes after the repetitive use of the catalyst. Nearly all the contents of main esters decreased significantly and some even disappeared, which could be attributed to the deterioration of catalytic capacity of the used catalyst.
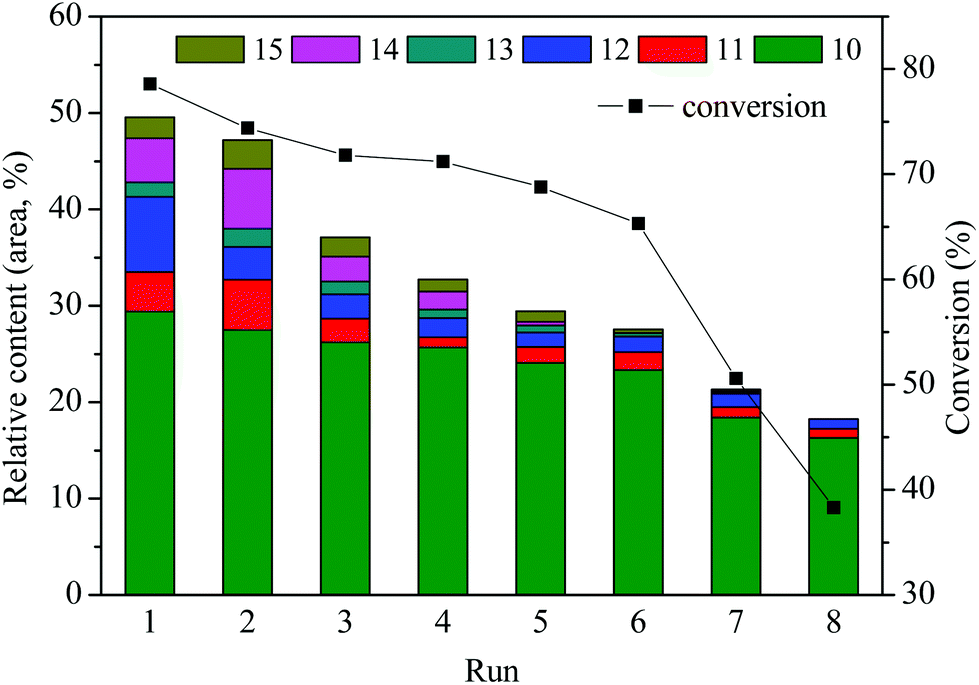 | ||
| Fig. 10 Repeated use of catalyst SO42−/ZrO2-ATP in the depolymerization of AEL. | ||
3.7 Mechanism analysis
Based on the major products observed in Table 2, a β-O-4 model compound, 3-(4-hydroxy-3-methoxyphenyl)-3-hydroxy-2-(2-methoxyphenoxy) propionic acid, was depolymerized in ethanol under the reaction conditions. As a result, compounds 2, 3, 4, 10, 11, 12, and 18 in Table 2 were detected in the model depolymerization products, indicating that the model could explain the basic structure of AEL well. Based on the above analysis, the authors proposed some possible reaction mechanism of depolymerization of AEL catalyzed by SO42−/ZrO2-ATP in ethanol and the possible pathways are summarized in Fig. 11. Given that the β-O-4 bond cleavage (cleavage 3) happened initially at relatively low temperature under acid conditions because of its low dissociation energy.43,44 Aldehydes and phenols (guaiacol, 4-ethylphenol, and 4-ethyl-2-methoxyphenol) were generated by β-O-4 bond cleavages and hydrogenation reaction that probably initiated by hydrogen radicals produced by ethanol. Acids and ketones might be produced by hydrogenation and dehydrogenation reaction. Being the most abundant products, aromatic esters, including ethyl 4-hydroxy-3-methoxycinnamate (compound 10), ethyl 3-(4-hydroxy-3-methoxyphenyl) propionate (compound 11), and methyl 4-hydroxy-3-methoxyphenylacetate (compound 12) (marked with red), could be produced by β-O-4 bond cleavages and reactions of esterification, hydrogenation, and dehydration over SO42−/ZrO2-ATP in ethanol, among which esterification reaction between ethanol and lignin-derived intermediates, such as ferulic acid and phenylacetic acid formed during lignin depolymerization was the main contributor. In another way, aromatic esters might be generated by the transesterification reaction between ethanol and covalent linkages which connect lignin with cellulose or hemicellulose in the form of lignin-ether bond-phenolic acid-ester bond-hemicellulose or lignin-ester bond-phenolic acid.45 These results indicated that ethanol served as not only a solvent but also a reactant for the depolymerization of AEL over SO42−/ZrO2-ATP.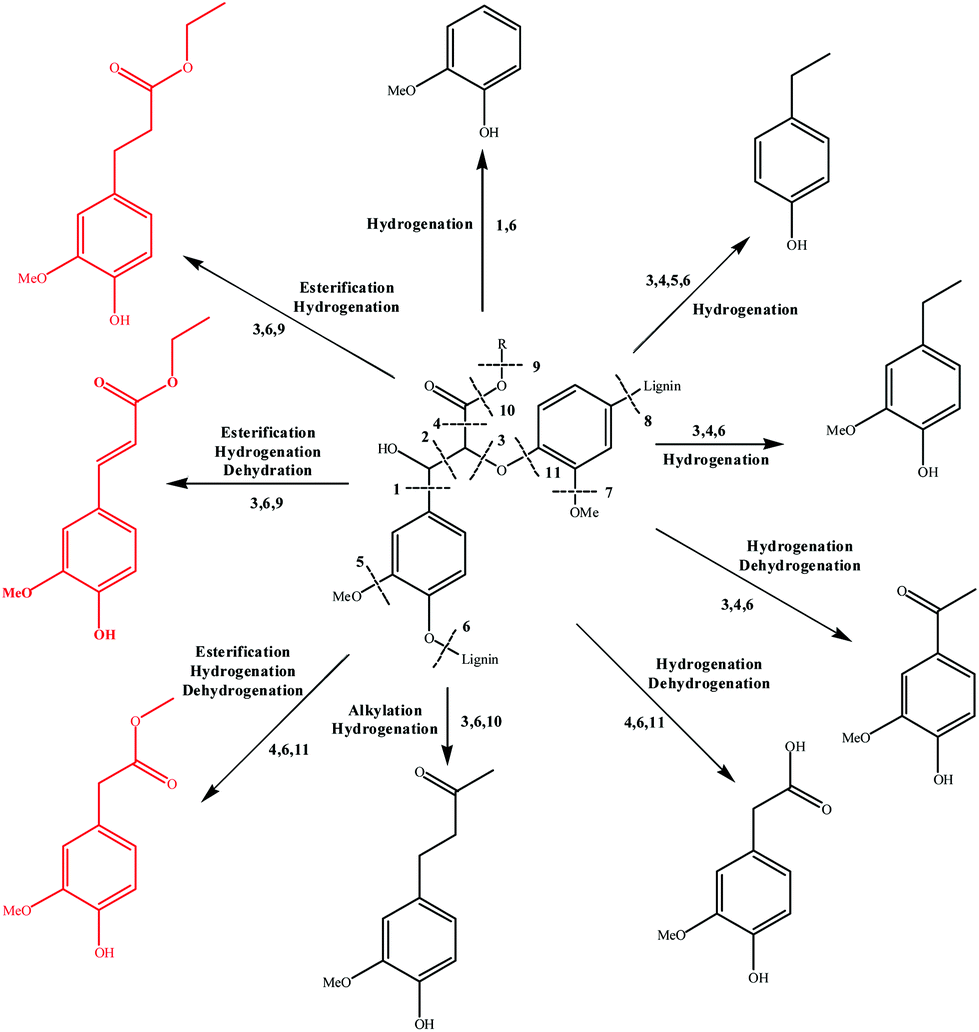 | ||
| Fig. 11 Proposed reaction pathway of AEL depolymerization catalyzed by SO42−/ZrO2-ATP in ethanol. | ||
4. Conclusion
The depolymerization of AEL in alcohols over ATP-supported catalysts was investigated. Among all ATP-supported catalysts prepared, SO42−/ZrO2-ATP with excellent catalytic activity and catalytic selectivity was selected. Herbaceous lignin was found to be more suitable for the preparation of aromatic esters from AEL due to the composition of the lignin fraction and the contents of phenolic acids. Ethanol presented the highest alcoholysis ability was selected as the alcohol solvent. A 78.6% conversion of AEL of corncob and a 29.4% selectivity to ethyl ferulate were achieved under the optimal conditions of 200 °C, 120 min, and catalyst/AEL ratio of 1:1 over Cat 2. The catalyst could be used at least 6 successive runs with a decline from 78.6% to 65.3% in the conversion of AEL and a decline from 49.5% to 27.6% in main esters relative content. Ethanol was found to be not only a solvent but also a reactant for the depolymerization of AEL over SO42−/ZrO2-ATP.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Key Research and Development Plan of China (2016YFD0600801), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), as well as Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP).References
- S. H. Ghaffar and M. Fan, Int. J. Adhes. Adhes., 2014, 48, 92–101 CrossRef CAS.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- J. Y. Kim, J. H. Lee, J. Park, J. K. Kim, D. An, I. K. Song and J. W. Choi, J. Anal. Appl. Pyrolysis, 2015, 114, 273–280 CrossRef CAS.
- Z. Cai, Y. Li, H. He, Q. Zeng, J. Long, L. Wang and X. Li, Ind. Eng. Chem. Res., 2015, 54, 11501–11510 CrossRef CAS.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- S. Y. Park, C. Y. Hong, H. S. Jeong, S. Y. Lee, J. W. Choi and I. G. Choi, J. Anal. Appl. Pyrolysis, 2016, 121, 113–120 CrossRef CAS.
- M. K. Chan, Q. Ye, Z. M. Png, H. N. Zeng, X. Wang and J. Xu, Waste Biomass Valorization, 2016, 1–8 Search PubMed.
- G. Zhu, X. Qiu, Y. Zhao, Y. Qian, Y. Pang and X. Ouyang, Bioresour. Technol., 2016, 218, 718–722 CrossRef CAS PubMed.
- P. Chen, Q. Zhang, R. Shu, Y. Xu, L. Ma and T. Wang, Bioresour. Technol., 2016, 226, 125–131 CrossRef PubMed.
- F. Yan, R. Ma, X. Ma, K. Cui, K. Wu, M. Chen and Y. Li, Appl. Catal., B, 2017, 202, 305–313 CrossRef CAS.
- M. J. Hidajat, A. Riaz, J. Park, R. Insyani, D. Verma and J. Kim, Biochem. Eng. J., 2017, 317, 9–19 CAS.
- J. Y. Kim, J. Park, H. Hwang, J. K. Kim, I. K. Song and J. W. Choi, J. Anal. Appl. Pyrolysis, 2015, 113, 99–106 CrossRef CAS.
- S. Hu, F. Jiang and Y. L. Hsieh, ACS Sustainable Chem. Eng., 2015, 3, 2566–2574 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, Chemcatchem, 2013, 5, 2964–2972 CrossRef CAS.
- Y. Zheng, D. Chen and X. Zhu, J. Anal. Appl. Pyrolysis, 2013, 104, 514–520 CrossRef CAS.
- X. Liu, L. Xu, G. Xu, W. Jia, Y. Ma and Y. Zhang, ACS Catal., 2016, 6, 7611–7620 CrossRef CAS.
- R. Giustetto, J. G. Vitillo, I. Corazzari and F. Turci, J. Phys. Chem. C, 2014, 118, 19322–19327 CAS.
- B. Yuan, X. Q. Yin, X. Q. Liu, X. Y. Li and L. B. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 16457–16464 CAS.
- H. Yang, A. Tang, J. Ouyang, M. Li and S. Mann, J. Phys. Chem. B, 2010, 114, 2390–2398 CrossRef CAS PubMed.
- S. Miao, Z. Liu, Z. Zhang, B. Han, Z. Miao, K. Ding and G. An, J. Phys. Chem. C, 2007, 111, 2185–2190 CAS.
- K. Nagaoka, K. Seshan, J. A. Lercher and K. I. Aika, Catal. Lett., 2000, 70, 109–116 CrossRef CAS.
- L. Grzona, N. Comelli, O. Masini, E. Ponzi and M. Ponzi, React. Kinet. Catal. Lett., 2000, 69, 271–276 CrossRef CAS.
- E. Hernández-Ramírez, J. A. Wang, L. F. Chen, M. A. Valenzuela and A. K. Dalai, Appl. Surf. Sci., 2017, 399, 77–85 CrossRef.
- F. Rahmani, M. Haghighi and B. Mohammadkhani, Microporous Mesoporous Mater., 2017, 242, 34–49 CrossRef CAS.
- R. Dong, Y. Liu, X. Wang and J. Huang, J. Chem. Eng. Data, 2011, 56, 3890–3896 CrossRef CAS.
- M. Ferrari, B. Delmon and P. Grange, Carbon, 2002, 40, 497–511 CrossRef CAS.
- M. V. Bykova, D. Y. Ermakov, V. V. Kaichev, O. A. Bulavchenko, A. A. Saraev, M. Y. Lebedev and V. A. Yakovlev, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS.
- S. Zhang, L. Su, L. Liu and G. Fang, Ind. Crops Prod., 2015, 77, 451–457 CrossRef CAS.
- H. Yan, Y. Yang, D. Tong, X. Xiang and C. Hu, Catal. Commun., 2009, 10, 1558–1563 CrossRef CAS.
- K. Hashizume, T. Ito, T. Ishizuka and N. Takeda, J. Biosci. Bioeng., 2013, 116, 209–213 CrossRef CAS PubMed.
- M. D. Horbury, L. A. Baker, N. D. N. Rodrigues, W. D. Quan and V. G. Stavros, Chem. Phys. Lett., 2017, 673, 62–67 CrossRef CAS.
- P. Pushpaletha and M. Lalithambika, Appl. Clay Sci., 2011, 51, 424–430 CrossRef CAS.
- H. Ma, K. Sun, Y. Li and X. Xu, Catal. Commun., 2009, 10, 1363–1366 CrossRef CAS.
- H. Chen, A. Zhong, J. Wu, J. Zhao and H. Yan, Ind. Eng. Chem. Res., 2012, 51, 14026–14036 CrossRef CAS.
- B. Wang, J. Zhu and H. Ma, J. Hazard. Mater., 2009, 164, 256–264 CrossRef CAS PubMed.
- S. Nanayakkara, A. F. Patti and K. Saito, ACS Sustainable Chem. Eng., 2014, 2, 2159–2164 CrossRef CAS.
- X. P. Peng, S. L. Sun, J. L. Wen, W. L. Yin and R. C. Sun, Fuel, 2014, 134, 485–492 CrossRef CAS.
- N. Kumar and V. Pruthi, Biotechnol. Rep., 2014, 4, 86–93 CrossRef PubMed.
- K. Cui, L. Yang, Z. Ma, F. Yan, K. Wu, Y. Sang, H. Chen and Y. Li, Appl. Catal., B, 2017, 219, 592–602 CrossRef CAS.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- S. Chu, A. V. Subrahmanyam and G. W. Huber, Green Chem., 2013, 15, 125–136 RSC.
- X. Zhang, W. Yang and W. Blasiak, Energy Fuels, 2011, 25, 4786–4795 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |