
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully-automated magnetic stirring-assisted lab-in-syringe dispersive liquid–liquid microextraction for the determination of arsenic species in rice samples
Xiaojun Wang *,
Guoliang Xu,
Peng Chen,
Yueshu Sun,
Xiaoting Yao,
Yan Lv,
Weiwei Guo and
Guozhen Wang
*,
Guoliang Xu,
Peng Chen,
Yueshu Sun,
Xiaoting Yao,
Yan Lv,
Weiwei Guo and
Guozhen Wang
College of Civil Engineering and Architecture, Zhejiang University of Water Resources and Electric Power, 583 Xuelin Street, Hangzhou 310018, Zhejiang Province, People's Republic of China. E-mail: wangxiaoj@zjweu.edu.cn; Fax: +86 571 86926577; Tel: +86 571 86926577
First published on 8th May 2018
Abstract
Fully-automated magnetic stirring-assisted lab-in-syringe dispersive liquid–liquid microextraction (MAS-LIS-DLLME), combined with graphite furnace atomic absorption spectrometry (GFAAS) was developed for the fast and efficient separation and preconcentration of trace levels of inorganic arsenic species in rice samples. This totally automated analytical procedure combines the advantages of lab-in-syringe flow system and dispersive liquid–liquid microextraction (DLLME) aiming at separation of trace arsenite and arsenate species from natural matrix for the first time. With a single syringe pump that is coupled with a multiposition valve, the whole lab-in-syringe microextraction process including cleaning, mixing, microextraction, phase separation, and target analyte collection was implemented in a fully-automated way. Significant factors of the MAS-LIS-DLLME method were sample acidity, concentration of the chelating agent, amounts of ionic liquids (ILs), aspiration speed and matrix interference. Using the present method, the limits of detection (LODs) for As(V) was 0.005 μg L−1. The relative standard deviation (RSDs) for seven replicate measurements of 2.0 μg L−1 of As(V) was 3.7%. The linear dynamic range (LDR) was 0.04–5.0 μg L−1 and the determination coefficients was 0.9990. Under the optimum conditions, the developed totally automated analytical procedure was successfully applied for the trace arsenite and arsenate species studies in natural rice samples and standard reference materials with satisfactory results.
Introduction
Arsenic has been identified as a public health problem because it has serious toxic effects even at low exposure levels and is widespread in the environment.1 Arsenic mainly occurs in two oxidation states, As(III) and As(V), each of which manifests different physicochemical properties and biochemical activity. In fact, it has been shown that As(III) is approximately 25–60 times more toxic than As(V) and, in comparison with organic arsenic forms, As(III) is several hundred times more toxic than organic arsenicals. Therefore, speciation of arsenic as a ubiquitous and potentially toxic element in complex matrix has become an interesting topic in food analysis.In general, several element-specific and sensitive instrumental techniques2–11 have been successfully utilized for direct analysis of total amount of As at trace levels. To obtain information about the toxicities, biochemical, bioavailability and environmental behavior of arsenite and arsenate species, it is necessary to develop sensitive and selective methodologies for the qualitative and quantitative analysis of these individual inorganic arsenic species. However, direct measurement of trace arsenite and arsenate by the above-mentioned sensitive detection technique is limited due to matrix interference and insufficient sensitivity. To obtain information on species, separation and microextraction of arsenite and arsenate species are required prior to the spectrometric determination by an accessible, simple, inexpensive and sensitive non-chromatographic detection technique. In trace and ultratrace analysis, the separation and preconcentration steps are often required. For the speciation of trace arsenite and arsenate species from natural samples, the separation and microextraction methodologies reported in the literature are usually based on ion exchange,12 hydride generation,13 solid phase extraction (SPE)14,15 and liquid–liquid extraction (LLE)16 etc. Although drawbacks such as long extraction time, significant chemical additives, production of secondary wastes and multistage operation limit the use of these separation techniques.17
In order to overcome the shortcomings of these methodologies, a novel mode of liquid phase microextraction (LPME) termed dispersive liquid–liquid microextraction (DLLME) was suggested by Rezaee and co-workers.17 In DLLME procedure, the extractant is dispersed into the sample solution as very fine droplets to form an emulsified solution. Such a strategy enables the mass transfer between the analytes and fine droplets of water-immiscible extractant and significantly enhanced the microextraction recoveries. The extension of the contact surface between the organic extraction solvent and sample solution could effectively reduce sample pretreatment time.18 The advantages of the DLLME technology are short extraction time, low cost, simplicity of operation and high microextraction recoveries. Various studies have been presented by the usage of DLLME method on the separation and microextraction for the elemental speciation analysis at trace levels.19,20
In general, the main shortcomings of the conventional DLLME technology are the necessity of using extractant like chloroform, carbon tetrachloride, octanol and carbondisulfide, isolation of acceptor phase from water phase, loss of extractant containing the target analytes at the phase isolation process and low microextraction recoveries of the target analytes. However, the phase isolation step is considered to be the most time-consuming process in traditional DLLME technology. Some authors have reported that they have spent 15–20 min centrifuging each sample.21,22 To overcome limitations mentioned above, very recently, a novel combined DLLME technology which is termed as lab-in-syringe dispersive liquid–liquid microextraction (LIS-DLLME)23 was proposed and successfully applied for efficient analysis of dye rhodamine B from soft drinks. The so-called lab-in-syringe systems combine the strategies of sequential injection systems24 with flow-batch technologies,25 and the entire process of sample separation and microextraction and measurement can take place inside the integrated system.26 In lab-in-syringe systems, chemical reactions between metal ions and complexing agents, and different operation steps like chelation, preconcentration or separation could perform inside the syringe barrel.27,28 Thus, the lab-in-syringe systems has been successfully used for various interesting designs in fully-automated DLLME for the determination of trace caffeine,26 heavy metals,27,28 and priority phenolic pollutants29 from complex matrix.
In this paper, the applicability of fully-automated magnetic stirring-assisted lab-in-syringe dispersive liquid–liquid microextraction (MAS-LIS-DLLME) using 1-hexyl-3-methylimidazoliumhexafluorophosphate ([C6MIM][PF6]) as extractant, to fast extract trace arsenite and arsenate species in natural rice samples, prior to their analysis by GFAAS was developed. However, to the best of our knowledge, this is the first reported combination of MAS-LIS-DLLME with atomic spectrometric assays for the inorganic arsenic speciation. The significant factors influencing the microextraction recoveries of MAS-LIS-DLLME, such as sample acidity, concentration of the chelating agent, amounts of ionic liquids (ILs), aspiration flow rate and matrix interference, were systematically tested, and the analytical performance of the proposed methodology was evaluated. Furthermore, the developed fully-automated DLLME procedure has been used for the preconcentration and speciation of trace arsenite and arsenate species from rice matrix with satisfactory results.
Experimental
Apparatus
The fully-automated DLLME system (Fig. 1) comprised a rotary 8-port multi-position valve (VICI, Schenkon, Switzerland) coupled to an automatic burette with a 10 mL syringe (Hamilton, Bonaduz, Switzerland) and a three-way solenoid valve (Jiashan Ristron electronic technology Co., Ltd., Zhejiang, China) in the head section. Multiwave 3000 XF100 (Anton Paar, Graz, Austria) microwave digestion system was used for rice digestion. A research Delta 320 pH-meter (Mettler-Toledo, Zurich, Switzerland) were employed for measurement of pH values. The magnetic stirring system was composed by a lab made device and a small magnetic stirrer bar placed inside the syringe, as detailed in previous study.30 A Perkin-Elmer Model AAnalyst 800 atomic absorption spectrometer (Perkin-Elmer, USA) with Zeeman effect background correction was utilized as the detection system. An arsenic hollow cathode lamp operated at 6 mA was utilized as a radiation source. The spectral bandwidth was set at 0.7 nm and the wavelength was set at 193.7 nm resonance line. The detailed temperature program used for graphite atomizer is listed in Table 1. All tubing of the fully-automated DLLME systems was made from polytetrafluoroethylene (PTFE) tubing with 0.79 mm inner diameter.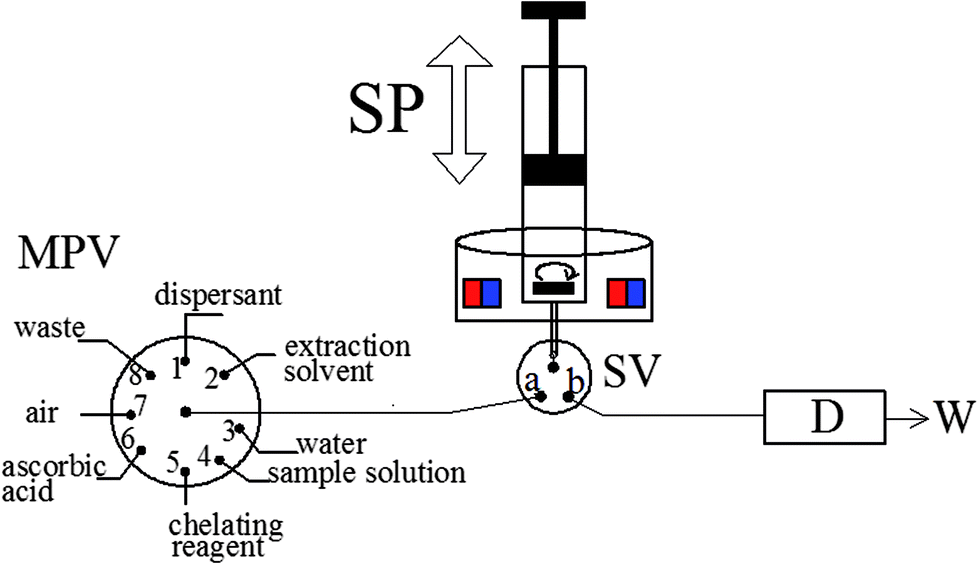 | ||
| Fig. 1 The schematic of the MAS-LIS-DLLME system. D, detection system; SP, syringe pump; SV, three-way solenoid valve; W, waste; MPV, multiposition valve. | ||
| Operation parameters | |
|---|---|
| Drying1 | 110 °C (ramp 1 s, hold 30 s, argon flow rate 250 mL min−1) |
| Drying2 | 130 °C (ramp 15 s, hold 30 s, argon flow rate 250 mL min−1) |
| Pyrolysis1 | 600 °C (ramp 10 s, hold 10 s, argon flow rate 250 mL min−1) |
| Pyrolysis2 | 800 °C (ramp 5 s, hold 10 s, argon flow rate 250 mL min−1) |
| Atomization | 2200 °C (ramp 0 s, hold 3 s, argon flow rate 0 mL min−1) |
| Cleaning | 2400 °C (ramp 1 s, hold 3 s, argon flow rate 250 mL min−1) |
Standard solution and reagents
All the chemicals used in this study were at least of analytical grade, and all solutions were prepared with ultrapure water obtained from a Milli-Q Element water system (Millipore Corporation, USA). Ammonium molybdate heptahydrate was obtained from the Sinopharm Chemical Reagent Corporation by (Shanghai, China). Ionic liquid ([C6MIM][PF6]) was purchased from Shanghai Cheng Jie Chemical Co., Ltd., Shanghai, China. The solution of chelating agent, ammonium molybdate, was prepared daily by dissolving appropriate amount of ammonium molybdate heptahydrate in ultrapure water. The stock standards (1000 mg L−1) of inorganic arsenic species were prepared by dissolving appropriate amounts of Na3AsO3 and As2O5, respectively, (Sigma-Aldrich, St. Louis, MO, USA) in ultrapure water and storing the solutions in a refrigerator at 4 °C. All vessels used for trace analysis were kept in 10% (v/v) nitric acid for at least 24 h and washed three times with ultrapure water prior to use.Extraction procedures
After adjusting the sample pH to 1.5 using diluted nitric acid/ammonia, As(III) and As(V) were separated and enriched using the developed fully-automated DLLME methodology (MAS-LIS-DLLME) systems as follows. Initially, the cleaning of the syringe, magnetic stirring bar and connecting tube is done by the aspiration of 10 mL ultrapure water (MPV in position 3 and SV in position a) followed by discharge to waste (MPV in position 8) for three times. Afterwards, 5 mL of aliquots of the sample or standard solution (MPV in position 4), 0.1 mL of 40 mmol L−1 ammonium molybdate solution (MPV in position 5), 0.1 mL of 10 mmol L−1 ascorbic acid solution (MPV in position 6) and 1 mL of air (at MPV in position 7) were sequentially aspirated into the syringe. The aqueous solution and the mixture of chelating agent and analyte inside the syringe were stirred by 20 s for the complexing reaction development. Then, 500 μL of methanol as the dispersant (MPV in position 1), 45 μL of [C6MIM][PF6] as the extraction solvent (MPV in position 2) and 1 mL of air (at MPV in position 7) were subsequently aspirated into the syringe. Next, the mixture of sample, dispersive solvents and extractant solvents was stirred by 30 s to accelerate the dispersion of [C6MIM][PF6] and to enhance the separation and preconcentration of target analytes. Subsequently, the syringe pump and stirrer are stopped for 60 s enabling phase separation process, and then the extractant phase was sedimented at the bottom of the syringe. After that, the SV moves to position b and the extractant phase is dispensed into the flow cell for the subsequent graphite furnace atomic absorption spectrometric analysis of arsenic.Samples preparation
All the commercially available rice samples of different varieties were acquired from local markets in Hangzhou, China and these rice samples were classified as follows: white rice, brown rice, parboiled rice, glutinous rice and rice flour. The rice samples were washed three times with ultrapure water, and then were dried at 60 °C in air oven. After that, the rice grains were milled by crushing and grinding to 100 mesh in a food processor. The resulting fine powder was stored at 4 °C in a dark environment prior to use.Total arsenic was also measured as As(V) using the developed fully-automated DLLME method. Briefly, portions (0.2000 g) of resulting fine powder of the rice samples were accurately weighed into the PTFE vessel, then 4 mL HNO3 (65%) and 2 mL H2O2 (30%) were added. The rice samples were digested with Multiwave 3000 XF100 (Anton Paar, Graz, Austria) microwave digestion system after the application of the microwave digestion procedure (the first stage: 600 W for 10 min (10 min of ramp); the second stage: 800 W for 10 min (10 min of ramp); the last cooling stage: 0 W for 15 min) and then the resulting solutions were diluted to proper concentrations with ultrapure water.
The inorganic arsenic was also extracted from the rice samples using the developed ultrasound-assisted extraction method and measured as As(V) after oxidation reaction. The ultrasound-assisted extraction of inorganic arsenic from natural samples was proposed in some previous works.31,32 Briefly, 0.5 g of finely powdered rice sample was placed into a 50 mL polyethylene centrifuge tube. A total of 30 mL HNO3 (0.5 mol L−1) was added and the polyethylene centrifuge tube was subjected to sonication for 30 min at 40 °C to extract inorganic arsenic. After the ultrasonic extraction process, the acid extract was separated by centrifugation at 5000 rpm for 5 min. The supernatant was filtered through a 0.45 μm pore size nylon membrane filter and then an aliquot of the supernatant solution was submitted to the developed fully-automated DLLME method for determination of As(V).
Selective oxidation of As(III) to As(V) was performed before the fully-automated DLLME process. The KMnO4 solution was added to the obtaining supernatant above in order to oxidize As(III) to As(V). The mixture was left to stand for 10 min with sonication assistant, and then the resulting solution was subjected to the developed fully-automated DLLME method for the measurement of total inorganic arsenic. In addition, the As(III) concentration was calculated by subtracting the As(V) concentration from the total inorganic As concentration.
Results and discussion
To obtain good sensitivity and precision, the effects of chemical and hydrodynamic parameters, including sample acidity, concentration of the chelating agent, amounts of ionic liquids (ILs) and aspiration flow rate, were thoroughly investigated in the present study. Standard solutions at 2.0 μg L−1 As(III) and As(V) concentration level were applied for the optimization experiments and all the experiments were performed in triplicate.Effect of sample acidity
The effect of the sample acidity on the metal-chelate formation and subsequent extraction into extractant phase was examined between the pH values 0.5–4.5 (Fig. 2). As could be seen, the microextraction recovery of As(V) was increased rapidly with increasing sample solution acidity from 0.5 to 1.5, and kept nearly constant in the pH range of 1.5–2.5, then was decreased with further increasing pH from 2.5 to 4.5. Hence, highly selective separation and preconcentration of As(V) from As(III) can be achieved. For all subsequent experiments, the pH value of 1.5 was chosen to adjust samples and standards before fully-automated DLLME process.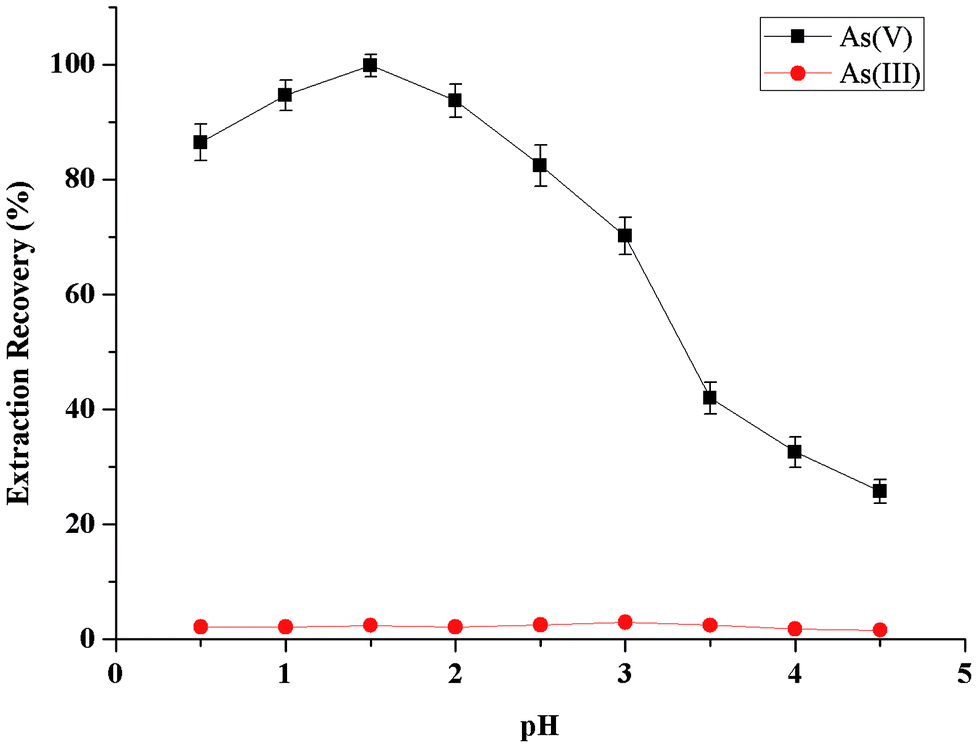 | ||
| Fig. 2 Effect of sample acidity on the extraction recovery of inorganic arsenic obtained from the proposed MAS-LIS-DLLME method. Extraction conditions: sample volume, 5 mL; concentration of inorganic arsenic, 2.0 ng mL−1; amount of ascorbic acid, 100 μL of 10 mmol L−1; extraction solvent volume, [C6MIM][PF6], 45 μL; dispersive solvent volume, methanol, 500 μL; amount of ammonium molybdate, 100 μL of 40 mmol L−1; sample aspiration speed, 2200 step s−1. | ||
Effect of ammonium molybdate concentration
The influence of the concentration of ammonium molybdate as chelating reagent on the sensitivity was studied in the range 20–50 mmol L−1. As can be seen in Fig. 3, the microextraction recovery of As(V) increased by increasing the concentration of ammonium molybdate up to 40 mmol L−1, and then remained nearly constant. On the other hand, no amount of As(V) could be selectively extracted from As(III) in the absence of ammonium molybdate. Thus, a concentration of ammonium molybdate of 40 mmol L−1 was selected to obtain the highest extraction efficiency in the further study.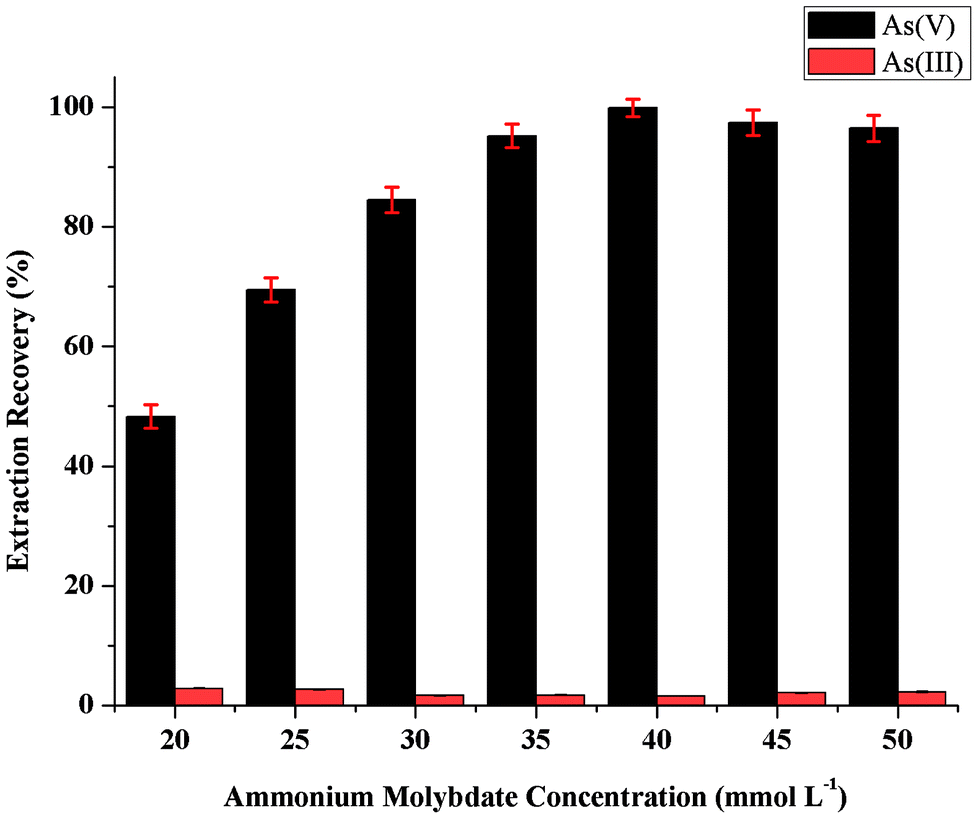 | ||
| Fig. 3 Effect of ammonium molybdate concentration on the extraction recovery of inorganic arsenic obtained from proposed MAS-LIS-DLLME method. Extraction conditions: sample volume, 5 mL; concentration of inorganic arsenic, 2.0 ng mL−1; amount of ascorbic acid, 100 μL of 10 mmol L−1; extraction solvent volume, [C6MIM][PF6], 45 μL; dispersive solvent volume, methanol, 500 μL; volume of ammonium molybdate, 100 μL; sample aspiration speed, 2200 step s−1. | ||
Effect of the amount of extraction solvent
Due to its non-volatile physicochemical properties, [C6MIM][PF6] is a suitable extraction solvent to form biphasic systems. The influence of ionic liquids amount on the microextraction recovery was also studied. For this purpose, 0.5 mL methanol with different amounts of [C6MIM][PF6] (within the range of 20–60 μL) was used for highly selective separation and microextraction of As(V) from As(III). Fig. 4 shows that microextraction recovery of the developed fully-automated lab-in-syringe systems was affected significantly by [C6MIM][PF6] amount; microextraction recovery increased with the amount of ionic liquids with the most As(V) recovered from 45 μL of extraction solvent. An amount of 45 μL [C6MIM][PF6] was marked as the optimum for the developed fully-automated DLLME procedure, while adding more [C6MIM][PF6] have no significant changes in microextraction recovery.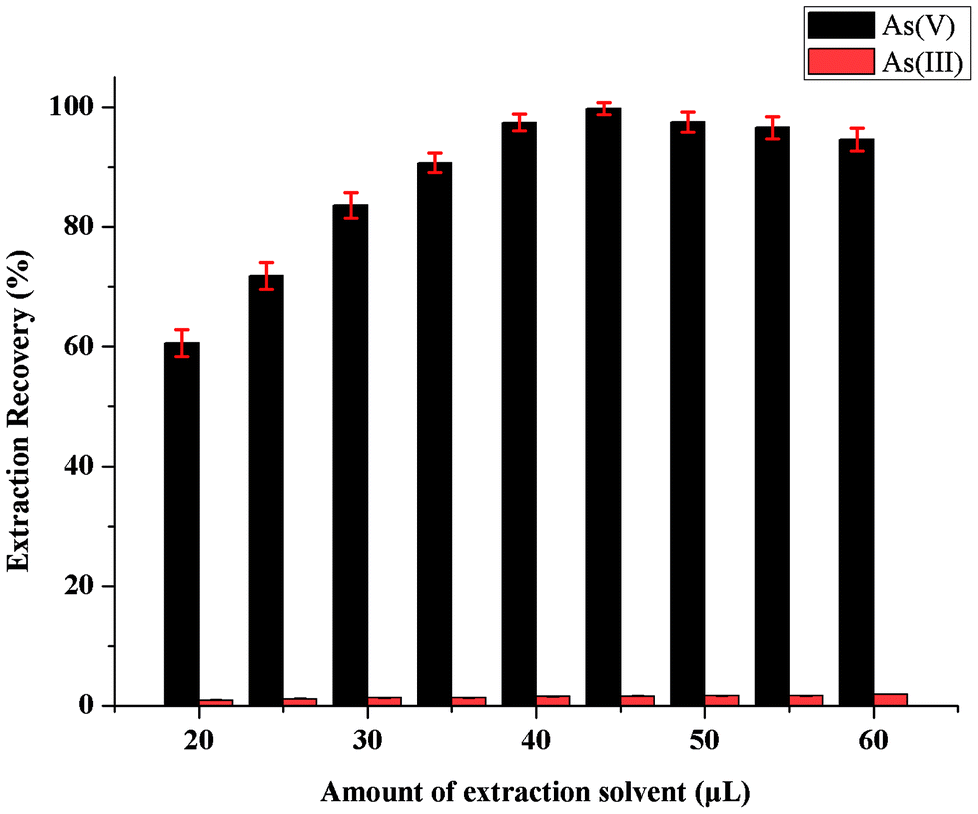 | ||
| Fig. 4 Effect of the extraction solvent ([C6mim][FeCl4]) volume on the extraction recovery of inorganic arsenic obtained from proposed MAS-LIS-DLLME method. Extraction conditions: sample volume, 5 mL; concentration of inorganic arsenic, 2.0 ng mL−1; amount of ascorbic acid, 100 μL of 10 mmol L−1; dispersive solvent volume, methanol, 500 μL; amount of ammonium molybdate, 100 μL of 40 mmol L−1; sample aspiration speed, 2200 step s−1. | ||
Effect of the speed of sample aspiration
The speed of sample aspiration plays a unique role in the separation and preconcentration of As(V) from As(III) in natural samples. In this study, influence of the speed of sample aspiration on microextraction efficiency was investigated by in the range between 1100 and 2400 step s−1 while the other experimental variables were kept constant. The obtained results showed that the microextraction recovery increased as the speed of sample aspiration increased from 1100 to 2200 step s−1. The speed of sample aspiration of 2200 step s−1 was marked as the optimum for the fully-automated lab-in-syringe systems, while faster speed of sample aspiration has no significant changes in microextraction efficiency.Interferences
To test the selectivity and sensitivity of the proposed fully-automated DLLME procedure for the separation and preconcentration of As(V) from As(III) the influence of some potentially interfering anions (SO42−, Cl− and NO3−) and cations (K+, Na+, Ca2+, Mg2+ and Ba2+), which are concomitant and regularly found in complex matrix, was examined. In these experiments, 2.0 ng mL−1 As(V) solution containing the added interferences was processed according to the recommended process under the optimum experimental condition. The tolerance limit was defined as the maximum concentration of interfering ions, in the presence of which the extraction recovery of As(V) could be maintained within the range of 90–110%. As shown in Table 2, alkaline and alkaline earth metal ions such as K+, Na+, Mg2+ and Ba2+, and anions including SO42−, Cl− and NO3− did not influence the As(V) extraction efficiency at the experimental concentrations (extraction recoveries 94.3–106.9%). The tolerance limits of Ni2+, Co2+, Pb2+ and Fe3+ were not as high as other coexisting ions, mainly because those common metal ions can react with chelating agent at working pH. Therefore, the excessive ammonium molybdate as chelating reagent was necessary in this test. Since the concentrations of coexisting ions were generally at trace levels in natural matrix, this developed fully-automated DLLME method may be suitable for the separation and microextraction of As(V) from As(III) in rice matrix.| Interfering ions | Concentration (mg L−1) | Recovery (%) |
|---|---|---|
| Na+ | 4000 | 96.7 |
| K+ | 4000 | 95.2 |
| Ca2+ | 1500 | 101.8 |
| Mg2+ | 1500 | 96.3 |
| Ba2+ | 1500 | 98.6 |
| Co2+ | 5 | 101.3 |
| Fe2+ | 5 | 102.9 |
| Mn2+ | 5 | 99.4 |
| Ni2+ | 5 | 96.9 |
| Zn2+ | 5 | 106.7 |
| Cu2+ | 5 | 98.5 |
| Cd2+ | 5 | 97.1 |
| Pb2+ | 2 | 104.8 |
| Fe3+ | 0.5 | 105.6 |
| Cl− | 4000 | 94.3 |
| SO42− | 4000 | 102.8 |
| NO3− | 4000 | 106.9 |
Analytical performance
Under the optimum experimental conditions, the analytical characteristics of the established fully-automated DLLME method were tested and limits of detection (LOD), relative standard deviation (RSD), and linear dynamic ranges (LDR) are given in Table 3. The precision of this established fully-automated DLLME method was determined by analyzing standard solution at 2.0 ng mL−1 As(V) for seven replicate extraction and measurements and the relative standard deviation (RSD) was 3.7%.34 The limits of detection (LOD), which is defined as CLOD = 3Sd/m (where CLOD, Sd and m are LOD, standard deviation of the blank, and slope of the calibration graph, respectively) according to the IUPAC definition, was 0.005 ng mL−1 for As(V). The calibration graph was linear within the range of 0.04–5.0 ng mL−1 of As(V), with a correlation of coefficient (r) of 0.9990.| Method | Detection | LODa | RSD (%) | LDRb | Sample | Ref. |
|---|---|---|---|---|---|---|
| a Limit of detection (μg L−1).b Linear dynamic range (μg L−1).c Cloud point extraction.d Electrothermal vaporization inductively coupled plasma mass spectrometry.e In situ solvent formation microextraction.f Ionic liquid-based dispersive liquid–liquid microextraction.g Ultrasound assisted emulsification of solidified floating organic drop microextraction.h Solid phase extraction-dispersive liquid–liquid microextraction based on the solidification of floating organic drop.i Hollow fiber-liquid phase microextraction.j Solid phase extraction.k Activated carbon-modified knotted reactor. | ||||||
| MAS-LIS-DLLME | GFAAS | 0.005 | 3.7 | 0.04–5.0 | Rice | This work |
| CPEc | ETAAS | 0.009 | 2.5 | 0.05–10.0 | Rice | 32 |
| DLLME | ETV-ICP-MSd | 0.0025 | 9.7 | 0.01–10 | Environmental water | 33 |
| ISFMEe | ETAAS | 0.006 | 4.78 | 0.02–0.250 | Saline samples | 34 |
| IL-DLLMEf | ETAAS | 0.005 | 4.7 | 0.05–6 | Wine | 35 |
| USAE-SFODMEg | ETAAS | 0.004 | 6.1 | 0.05–2 | Environmental water | 36 |
| SPE-DLLME-SFOh | GFAAS | 0.0025 | 6.8 | 0.01–0.1 | Water | 37 |
| HF-LPMEi | ETAAS | 0.12 | 8 | 1–50 | Fresh waters and human hair | 38 |
| SPEj | ETAAS | 0.02 | 3.5 | 0.03–0.6 | Water | 39 |
| AC-Modified-KRk | ETAAS | 0.004 | 4.3 | 0.01–1.5 | Medicinal herbs and tea infusions | 40 |
A comparison of the developed fully-automated DLLME method with other previously published methods is summarized in Table 3. It clearly shows that the developed fully-automated DLLME method has good sensitivity, selectivity and precision with a low sample consumption in comparison to in situ solvent formation microextraction (ISFME), cloud point extraction (CPE), hollow fiber-liquid phase microextraction (HF-LPME) and solid phase extraction (SPE). In contrast to many previous studies, the LDR of the represented fully-automated DLLME method is acceptable compared with other methodologies. The LOD of the proposed process was comparable to those of other published microextraction methodologies whilst the obtained RSD was better than dispersive liquid–liquid microextraction (DLLME), hollow fiber-liquid phase microextraction (HF-LPME), ultrasound assisted emulsification of solidified floating organic drop microextraction (USAE-SFODME) and solid phase extraction-dispersive liquid–liquid microextraction based on the solidification of floating organic drop (SPE-DLLME-SFO) considered in the comparison.
Analysis of real samples
The proposed fully-automated DLLME method was used for separation and preconcentration of As(V) from As(III) in some natural rice matrix. Moreover, the accuracy of the results was verified by spiking known amount of As(III) and As(V) in different rice samples, including parboiled rice, white rice, brown rice, glutinous rice and rice flour. The obtained results were presented in Table 4. As could been seen, the extraction efficiencies for the spiked samples were in acceptance range (94.2–108.9%), which confirms the reliability of the developed fully-automated DLLME method and its independence from the complex matrix effect.| Samples | Added | Found mean ± S.D.a | Recovery (%) | |||
|---|---|---|---|---|---|---|
| As(III) | As(V) | As(III) | As(V) | As(III) | As(V) | |
| a Standard deviation (n = 3). | ||||||
| Parboiled rice (μg kg−1) | — | — | 37.8 ± 0.81 | 15.9 ± 0.47 | — | — |
| 20.0 | — | 61.2 ± 1.32 | 16.1 ± 0.53 | 105.9 ± 2.29 | 101.3 ± 3.36 | |
| — | 20.0 | 35.6 ± 0.83 | 36.8 ± 0.91 | 94.2 ± 2.21 | 102.5 ± 2.21 | |
| 20.0 | 20.0 | 60.9 ± 1.28 | 38.3 ± 0.95 | 105.4 ± 2.22 | 106.7 ± 2.64 | |
| Brown rice (μg kg−1) | — | — | 53.9 ± 1.21 | 32.6 ± 0.85 | — | — |
| 20.0 | — | 75.6 ± 1.62 | 30.9 ± 0.71 | 102.3 ± 2.19 | 94.8 ± 2.18 | |
| — | 20.0 | 56.8 ± 1.18 | 55.2 ± 1.29 | 105.4 ± 2.19 | 104.9 ± 2.46 | |
| 20.0 | 20.0 | 75.1 ± 1.84 | 50.6 ± 1.15 | 101.6 ± 2.50 | 96.2 ± 2.20 | |
| White rice (μg kg−1) | — | — | 45.3 ± 1.15 | 16.7 ± 0.42 | — | — |
| 20.0 | — | 62.9 ± 1.42 | 17.3 ± 0.39 | 96.3 ± 2.17 | 103.6 ± 2.34 | |
| — | 20.0 | 47.6 ± 1.21 | 35.8 ± 0.99 | 105.1 ± 2.67 | 97.5 ± 2.71 | |
| 20.0 | 20.0 | 64.1 ± 1.37 | 38.5 ± 1.02 | 98.2 ± 2.10 | 104.9 ± 2.78 | |
| Glutinous rice (μg kg−1) | — | — | 30.2 ± 0.75 | 28.9 ± 0.81 | — | — |
| 20.0 | — | 53.6 ± 1.28 | 27.2 ± 0.67 | 106.8 ± 3.52 | 94.1 ± 2.33 | |
| — | 20.0 | 31.9 ± 0.87 | 50.7 ± 1.14 | 105.6 ± 2.87 | 103.7 ± 2.34 | |
| 20.0 | 20.0 | 48.3 ± 1.19 | 48.1 ± 1.06 | 96.2 ± 2.38 | 98.4 ± 2.16 | |
| Rice flour 1 (μg kg−1) | — | — | 18.4 ± 0.56 | 13.7 ± 0.39 | — | — |
| 10.0 | — | 28.1 ± 0.69 | 14.6 ± 0.34 | 98.9 ± 2.43 | 106.6 ± 2.50 | |
| — | 10.0 | 19.2 ± 0.29 | 25.8 ± 0.76 | 104.3 ± 1.60 | 108.9 ± 3.19 | |
| 10.0 | 10.0 | 30.5 ± 0.67 | 24.3 ± 0.53 | 107.4 ± 2.36 | 102.5 ± 2.24 | |
| Rice flour 2 (μg kg−1) | — | — | 16.7 ± 0.41 | 10.8 ± 0.22 | — | — |
| 10.0 | — | 28.6 ± 0.87 | 10.3 ± 0.28 | 107.1 ± 3.25 | 95.4 ± 2.62 | |
| — | 10.0 | 16.4 ± 0.50 | 22.1 ± 0.49 | 98.2 ± 3.02 | 106.3 ± 2.36 | |
| 10.0 | 10.0 | 26.9 ± 0.92 | 20.7 ± 0.34 | 100.7 ± 3.44 | 99.5 ± 1.63 | |
Accuracy of the method
To validate the accuracy of the developed fully-automated DLLME methodology, several standard reference materials (GBW10010 rice, GBW10043 Liaoning rice and GBW10044 Sichuan rice) were detected, and the content of total As are summarized in Table 5. As could be seen, the analyzed values (0.105 ± 0.006, 0.117 ± 0.015 and 0.11 ± 0.02 μg g−1 for three replicate measurements) found by using the developed fully-automated DLLME method for GBW10010 rice, GBW10043 Liaoning rice and GBW10044 Sichuan rice were statistically in good agreement with the certified values of 0.102 ± 0.008, 0.114 ± 0.018 and 0.12 ± 0.03 μg g−1, respectively. As the certified values of standard reference materials were within the 95% confidence interval around the mean of the analyzed values, there is no significant difference between the values. It can be concluded that the developed fully-automated DLLME methodology is sensitive, selective, reliable and free from systematic errors.| Samples | Found | Certified value | t-Test |
|---|---|---|---|
| a Standard deviation (n = 3). | |||
| GBW10010 (Rice) | 0.105 ± 0.006 | 0.102 ± 0.008 | 0.704 |
| GBW10043 (Liaoning rice) | 0.117 ± 0.015 | 0.114 ± 0.018 | 0.866 |
| GBW10044 (Sichuan rice) | 0.11 ± 0.02 | 0.12 ± 0.03 | 0.701 |
Conclusion
In the present work, fully-automated magnetic stirring-assisted lab-in-syringe dispersive liquid–liquid microextraction (MAS-LIS-DLLME) coupled with graphite furnace atomic absorption spectrometry (GFAAS) was successfully employed for highly selective separation and preconcentration of trace arsenite and arsenate species in natural rice samples. Ammonium molybdate and 1-hexyl-3-methylimidazoliumhexafluorophosphate ([C6MIM][PF6]) were chosen as a chelating reagent and extractant in MAS-LIS-DLLME, respectively. The developed fully-automated DLLME methodology (MAS-LIS-DLLME) has some distinct advantages such as sensitive, selective, high extraction capacity, simple operation, low cost and rapid mass transfer. The combination of MAS-LIS-DLLME with GFAAS was shown to be alternative for the quantification of trace arsenite and arsenate species in relatively complicated matrices with good accuracy and reproducibility.Compliance with ethics requirements
This article does not contain any studies with human or animal subjects.Conflicts of interest
Xiaojun Wang, Guoliang Xu, Peng Chen, Yueshu Sun, Xiaoting Yao, Yan Lv, Weiwei Guo, Guozhen Wang declare that they have no conflict of interest.Acknowledgements
This work was supported by the Zhejiang Provincial Soft Science Research Project under Grant number 2018C35027; and Zhejiang Provincial Public Technology Application Research Project under Grant number 2017C33060; and Research Project of Zhejiang Association of Social Sciences under Grant number 2018N20; and Hangzhou Philosophy and Social Science Program under Grant number Z17JC078.References
- T. S. Choong, T. G. Chuah, Y. Robiah, F. G. Koay and I. Azni, Desalination, 2007, 217, 139–166 CrossRef CAS.
- G. M. dos Santos, D. Pozebon, C. Cerveira and D. P. de Moraes, Microchem. J., 2017, 133, 265–271 CrossRef.
- H. İ. Ulusoy, M. Akçay and R. Gürkan, Talanta, 2011, 85, 1585–1591 CrossRef CAS PubMed.
- C. Zeng, Y. Yan, J. Tang, Y. Wu and S. Zhong, Spectrosc. Lett., 2017, 50, 220–226 CrossRef CAS.
- A. F. M. Y. Haider, M. H. Ullah, Z. H. Khan, F. Kabir and K. M. Abedin, Opt. Laser Technol., 2014, 56, 299–303 CrossRef CAS.
- S. Döker, L. Uzun and A. Denizli, Talanta, 2013, 103, 123–129 CrossRef PubMed.
- J. Michon, V. Deluchat, R. Al Shukry, C. Dagot and J. C. Bollinger, Talanta, 2007, 71, 479–485 CrossRef CAS PubMed.
- F. J. Pereira, M. D. Vázquez, L. Debán and A. J. Aller, Talanta, 2016, 152, 211–218 CrossRef CAS PubMed.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Bioanal. Chem., 2015, 407, 919–929 CrossRef CAS PubMed.
- P. Heitland, M. Blohm, C. Breuer, F. Brinkert, E. G. Achilles, I. Pukite and H. D. Köster, J. Trace Elem. Med. Biol., 2017, 41, 36–40 CAS.
- N. Phattamajintatamrong, M. Puanngam and A. Imyim, Curr. Anal. Chem., 2017, 13, 137–143 CrossRef.
- S. Jeong, H. Lee, Y. T. Kim and H. O. Yoon, Microchem. J., 2017, 134, 295–300 CrossRef CAS.
- T. D. Çiftçi and E. Henden, Bull. Environ. Contam. Toxicol., 2016, 97, 272–278 CrossRef PubMed.
- K. Hagiwara, T. Inui, Y. Koike, M. Aizawa and T. Nakamura, Talanta, 2015, 134, 739–744 CrossRef CAS PubMed.
- V. G. Mihucz, L. Bencs, K. Koncz, E. Tatár, T. Weiszburg and G. Záray, Spectrochim. Acta, Part B, 2017, 128, 30–35 CrossRef CAS.
- R. Güell, C. Fontàs, V. Salvadó and E. Anticó, Sep. Purif. Technol., 2010, 72, 319–325 CrossRef.
- M. Rezaee, Y. Assadi, M. R. M. Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- Z. G. Shi and H. K. Lee, Anal. Chem., 2010, 82, 1540–1545 CrossRef CAS PubMed.
- L. Elçi, A. Elçi, T. A. Berg and J. F. Tyson, Int. J. Environ. Anal. Chem., 2013, 93, 1065–1073 CrossRef.
- S. Rabieh, M. Bagheri and B. Planer-Friedrich, Microchim. Acta, 2013, 180, 415–421 CrossRef CAS.
- L. M. Ravelo-Pérez, J. Hernández-Borges, M. Asensio-Ramos and M. Á. Rodríguez-Delgado, J. Chromatogr. A, 2009, 1216, 7336–7345 CrossRef PubMed.
- Q. Zhou, N. Zhao and G. Xie, J. Hazard. Mater., 2011, 189, 48–53 CrossRef CAS PubMed.
- F. Maya, B. Horstkotte, J. M. Estela and V. Cerdà, Anal. Bioanal. Chem., 2012, 404, 909–917 CrossRef CAS PubMed.
- J. Ruzicka and G. D. Marshall, Anal. Chim. Acta, 1990, 237, 329–343 CrossRef CAS.
- P. H. G. D. Diniz, L. F. de Almeida, D. P. Harding and M. C. U. de Araújo, TrAC, Trends Anal. Chem., 2012, 35, 39–49 CrossRef CAS.
- R. M. Frizzarin, F. Maya, J. M. Estela and V. Cerdà, Food Chem., 2016, 212, 759–767 CrossRef CAS PubMed.
- B. Horstkotte, K. Fikarová, D. J. Cocovi-Solberg, H. Sklenářová, P. Solich and M. Miró, Talanta, 2017, 173, 79–87 CrossRef CAS PubMed.
- R. Sánchez, B. Horstkotte, K. Fikarova, H. Sklenářová, S. Maestre, M. Miró and J. L. Todolí, Anal. Chem., 2017, 89, 3787–3794 CrossRef PubMed.
- A. González, J. Avivar and V. Cerdà, Anal. Bioanal. Chem., 2015, 407, 2013–2022 CrossRef PubMed.
- B. Horstkotte, R. Suárez, P. Solich and V. Cerdà, Anal. Chim. Acta, 2013, 788, 52–60 CrossRef CAS PubMed.
- J. F. Paula, R. E. Froes-Silva and V. S. Ciminelli, Microchem. J., 2012, 104, 12–16 CrossRef CAS.
- B. E. dos Santos Costa, N. M. M. Coelho and L. M. Coelho, Food Chem., 2015, 178, 89–95 CrossRef PubMed.
- Y. Liu, M. He, B. Chen and B. Hu, Talanta, 2015, 142, 213–220 CrossRef CAS PubMed.
- B. Majidi and F. Shemirani, Biol. Trace Elem. Res., 2011, 143, 579–590 CrossRef CAS PubMed.
- L. B. Escudero, E. M. Martinis, R. A. Olsina and R. G. Wuilloud, Food Chem., 2013, 138, 484–490 CrossRef CAS PubMed.
- M. Asadollahzadeh, N. Niksirat, H. Tavakoli, A. Hemmati, P. Rahdari, M. Mohammadi and R. Fazaeli, Anal. Methods, 2014, 6, 2973–2981 RSC.
- M. Shamsipur, N. Fattahi, Y. Assadi, M. Sadeghi and K. Sharafi, Talanta, 2014, 130, 26–32 CrossRef CAS PubMed.
- H. Jiang, B. Hu, B. Chen and L. Xia, Anal. Chim. Acta, 2009, 634, 15–21 CrossRef CAS PubMed.
- I. López-García, R. E. Rivas and M. Hernández-Córdoba, Talanta, 2011, 86, 52–57 CrossRef PubMed.
- A. C. Grijalba, E. M. Martinis, G. E. Lascalea and R. G. Wuilloud, Spectrochim. Acta, Part B, 2015, 103, 49–56 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |