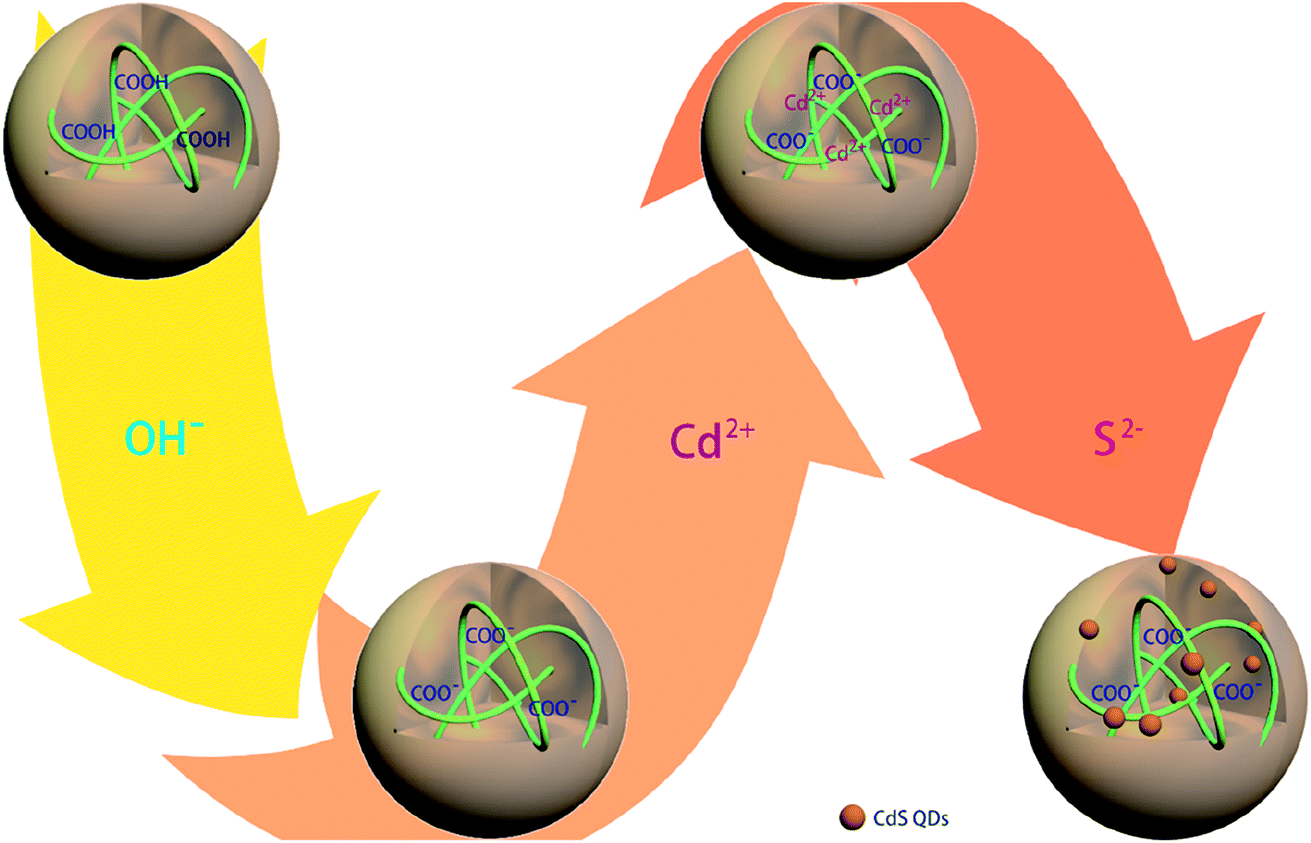DOI:
10.1039/C8RA01855C
(Paper)
RSC Adv., 2018,
8, 16850-16857
Synthesis of poly (N-isopropylacrylamide)-co-(acrylic acid) microgel-entrapped CdS quantum dots and their photocatalytic degradation of an organic dye†
Received
2nd March 2018
, Accepted 29th April 2018
First published on 9th May 2018
Abstract
CdS quantum dots (CdSQDs) were generated inside the network structure of poly (N-isopropylacrylamide)-co-(acrylic acid) (pNIPAm-co-AAc) microgels and their ability to photocatalytically degrade organic dyes was evaluated using rhodamine B (RhB). The microgel-stabilized CdSQDs were generated by first enriching the microgels with Cd2+ followed by their reaction with Na2S. The resultant microgels were characterized, and the CdSQDs were found to be distributed throughout the microgels. We went on to show that the hybrid microgels exhibited photocatalytic properties by exposing them to a solution of RhB followed by exposure to UV irradiation. We found that the hybrid microgels were able to degrade the RhB, while native microgels without CdSQDs present were not capable of the same behavior. Due to the thermo- and pH-responsivity of pNIPAm-co-AAc-based microgels their ability to degrade RhB was also evaluated as a function of environmental temperature and solution pH. We showed that the removal efficiency was highest when the microgels were in their swollen state, which we attribute to more effective mass transfer of the RhB inside the microgels when their porous structure is expanded. Finally, we show that the hybrid microgels can be reused multiple times, although their photocatalytic degradation ability decreases the more they are used, which may be a result of the aggregation and decomposition of the CdSQDs. We conclude that this approach is an effective means of removing RhB from water, which may be modified to photodegrade a variety of other organic compounds.
Introduction
The dumping of industrial waste (e.g., organics and heavy metals) into the environment without treatment has had significant (and often irreversible) negative impacts on vegetation, wildlife and those that live in the region of the release. Due to a lack of government oversight, these issues disproportionately affect those in developing countries. To help mitigate the impacts of the release of these environmental pollutants on the environment, significant research has looked into the development of technologies that can be used to treat environmental contaminants before they are released.1,2 Indeed, many technologies have been introduced to remove contaminants from water streams, although semiconductor photocatalysis has attracted significant attention due to its ability to use sunlight as the energy source for decomposing organic compounds.2,3 Related to this, a variety of materials/agents capable of photocatalytic degradation have been identified, e.g., simple oxides (Bi2O3),4 complex oxides (Bi2WO6),5 and nitrides (C3N4).6 Of interest to this study are CdS quantum dots (CdSQDs) that are known for their unique electronic and optical properties and light-driven production of reactive oxygen species (ROS).7–9 It has been shown that the ROSs can degrade pollutants ultimately producing H2O and CO2.10,11 However, due to their high specific surface area, CdSQDs are not colloidally stable, and readily aggregate and precipitate in solution. Thus, surface modification of CdSQDs has been used to stabilize the particles and prevent their aggregation.12,13
Here, we show that stimuli-responsive polymers can be used as stabilizers for CdSQDs and the resultant materials used to degrade organic compounds upon exposure to light. Since their discovery, stimuli-responsive polymers have been used for a variety of applications mainly due to their ability to “sense” their environment and “react” to it chemically and/or physically.14–16 Of the various stimuli-responsive polymers, poly(N-isopropylacrylamide) (pNIPAm) has received the most attention, and has previously been used to stabilize nanoparticles.17,18 pNIPAm is well known to be thermoresponsive, exhibiting a lower critical solution temperature (LCST) and collapsing/deswelling above 32 °C in water. The deswelling/reswelling process is fully reversible over many cycles. pNIPAm-based networks can also be generated via polymerization in the presence of a crosslinker. Furthermore, colloidally stable pNIPAm-based nano and micro particles (nanogels/microgels, respectively) can also be generated,14 and multiple responsivities can be introduced into the microgels via copolymerization with functional monomers.19,20 Previous studies in our group and by others have shown that pNIPAm-based microgels can serve as pH- and thermosensitive absorbents for the removal of organic dyes from water, e.g., Orange II.21–24 Furthermore, these additional functional monomers have also been used to introduce nanomaterials into their network structure. For instance, pNIPAm microgels have been loaded with semiconductor nanomaterials, e.g., CdSe/CdS nanoparticles, via simple mixing.2 The hybrids can exhibit both the photophysical properties of the nanomaterial and thermoresponsivity. In a previous study, we also showed that pNIPAm-co-acrylic acid (pNIPAm-co-AAc) microgels could be used as a scaffold for the in situ generation of Ag nanoparticles, which we showed could be used as a colorimetric sensor for H2O2.25 Although many have generated semiconductor nanomaterials in pNIPAm-based microgels,26,27 we are not aware of any previous reports of their use as a photocatalyst for degradation of water pollutants.
In this investigation, we show that pNIPAm-based microgels can be used as a scaffold for the generation and stabilization of CdSQDs; we hypothesize that the resultant microgels will be capable of UV-stimulated degradation of the dye, rhodamine B (RhB). Specifically, we propose an in situ synthetic route for the preparation of CdSQDs@pNIPAm-co-AAc hybrid microgels. Cadmium ions were first enriched in the microgels through deprotonation of the pNIPAm-co-AAc microgels. CdSQDs were subsequently generated in the pNIPAm-co-AAc microgels utilizing a previously reported hydrothermal method.28 The hybrid materials were also shown to exhibit UV-stimulated photocatalytic degradation of RhB. This concept can be further modified in the future to allow sunlight-driven degradation of other contaminations or antimicrobial activity.
Experimental section
Materials and methods
N-isopropylacrylamide was purchased from TCI (Portland, OR) and purified by recrystallization from hexanes (ACS reagent grade, purchased from EMD, Gibbstown, OR, USA). N,N-Methylenebisacrylamide (BIS), acrylic acid (AAc), ammonium persulfate (APS), cadmium chloride (CdCl2), sodium hydroxide (NaOH), hydrochloric acid (HCl), sodium sulfide (Na2S) and RhB were purchased from Sigma-Aldrich (Oakville, ON). Microgel samples were lyophilized using a VirTis bench-top K-manifold Freeze Dryer (Stone Ridge, New York, USA). Deionized (DI) water with a resistivity of 18.2 MΩ cm was obtained from a Milli-Q Plus system (Billerica, MA, USA).
Synthesis of pNIPAm-co-AAc microgels
pNIPAm-co-AAc microgels were synthesized via surfactant-free, free radical precipitation polymerization, as previously described.25,28 The monomer, NIPAm (10.54 mmol), and the crosslinker, BIS (0.703 mmol), were fully dissolved in water (99 mL) with stirring in a beaker for 1 h. The mixture was then filtered through a 0.2 μm filter affixed to a 20 mL syringe into a 250 mL, 3-necked round bottom flask. The flask was then equipped with a thermometer, a condenser/N2 inlet/outlet, and a stir bar. The monomer solution was bubbled with N2 gas for ∼1 h while stirring and heating to 70 °C. AAc (2.812 mmol) and APS (0.046 g in 1.0 mL water) was then added to the pre-heated solution, respectively. The reaction continued for 4 h. After cooling down, the turbid solution was filtered through glass wool to remove any large aggregates. The coagulum was rinsed and the collected liquid was diluted to 100 mL. Aliquots of the microgel solution (33 mL) were centrifuged at a speed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 relative centrifugal force (RCF) at 20 °C for 45 min. The microgels were isolated and redispersed to their original volume (∼33 mL) with DI water. This centrifugation/resuspension procedure was repeated 6 times. Finally, all of the centrifuged particles were combined into one tube and diluted to 30 mL with DI water for storage. The microgel yield was estimated to be 80%, which was determined by lyophilizing 1 mL of the 30 mL stock solution and dividing it by the sum of the mass of NIPAm, BIS and AAc that is expected to be in that 1 mL aliquot (i.e., 3.3% of total mass of those components added to the initial reaction solution). The final concentration of the 30 mL stock microgel solution was 40 mg mL−1.
000 relative centrifugal force (RCF) at 20 °C for 45 min. The microgels were isolated and redispersed to their original volume (∼33 mL) with DI water. This centrifugation/resuspension procedure was repeated 6 times. Finally, all of the centrifuged particles were combined into one tube and diluted to 30 mL with DI water for storage. The microgel yield was estimated to be 80%, which was determined by lyophilizing 1 mL of the 30 mL stock solution and dividing it by the sum of the mass of NIPAm, BIS and AAc that is expected to be in that 1 mL aliquot (i.e., 3.3% of total mass of those components added to the initial reaction solution). The final concentration of the 30 mL stock microgel solution was 40 mg mL−1.
Synthesis of CdSQDs@pNIPAm-co-AAc hybrid microgels
CdSQDs@pNIPAm-co-AAc hybrid microgels were prepared according to a previously reported method modified with a final hydrothermal treatment.27,28 Typically, 25 mL of the 40 mg mL−1 microgel stock solution was used and its pH was adjusted to 8.3 by addition of NaOH followed by equilibration for 1 h. The resulting microgel solution was mixed with CdCl2 solution (500 mM, 25 mL) and incubated with gentle shaking overnight to allow for the ion exchange between Na+ and Cd2+. The solution was then washed by centrifugation and resuspension in DI water 6 times to remove any unbound Cd2+. Then, 25 mL of the purified Cd2+-loaded microgel solution and 175 mL of DI water were combined into a three-necked round bottom flask (500 mL) equipped with a thermometer, a condenser/N2 inlet/outlet, and a stir bar. The solution was bubbled with N2 for 1 h and heated to 80 °C followed by the drop-wise addition of 25 mL of a 500 mM Na2S solution. After adding Na2S, the reaction was allowed to proceed for an additional 2 h and the color of the solution changed to yellow. Finally, the cooled solution was washed by centrifugation and resuspension in DI water 6 times to yield the purified CdSQDs@pNIPAm-co-AAc hybrid microgels. The resultant microgel solutions were combined into one tube and diluted to 25 mL with DI water for storage. The concentration of the hybrid microgels was estimated to ∼40 mg mL−1, which was determined by lyophilizing 1 mL of the 25 mL stock hybrid microgel solution.
Photocatalytic degradation of RhB
The photocatalytic activity of CdSQDs@pNIPAm-co-AAc hybrid microgels was assessed by degradation of RhB under a 350 W UV lamp at 10 °C. 15 mL of RhB solution (0.021 mmol L−1, pH 7.0) was pipetted into a Petri dish (diameter: 140 mm) and magnetically stirred (500 rpm). Then, 0.40 g of the hybrid microgels (10 mL) was introduced into the Petri dish. The distance between the Petri dish to the light source was fixed at 5 cm. During the irradiation, sampling (200 μL) was done at 1 h intervals. The RhB degradation efficiencies (eqn (1)) in samples were determined by recording variations of the UV-vis absorption band maximum (553 nm). It follows from the Beer–Lambert law (A = εbc, A: absorbance; ε: molar absorptivity; b: light path length; c: concentration) that concentration and absorbance are directly related, hence degradation efficiencies can be calculated by simply considering variations in absorbance.| |
|
Removal efficiency = 1 − c/c0 = 1 − A/A0
| (1) |
where C0 and C were attributed to the concentration of RhB before and after photocatalysis, respectively, while A0 and A were assigned to the absorbance of RhB at λ553 before and after photocatalysis, respectively.
Characterization
UV-vis absorption spectra of CdSQDs@pNIPAm-co-AAc hybrid microgels were recorded on a Hewlett Packard 8453 UV-vis Spectrophotometer (USA). The size and morphology of CdSQDs@pNIPAm-co-AAc hybrid microgels were determined at 60 to 120 kV accelerating voltage by a Hitachi H-7650 transmission electron microscope (TEM, Japan), and the electron diffraction pattern for the composite microgels was also measured. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos AXIS Ultra spectrometer equipped with a monochromated Al Kα (hν = 1486.6 eV) X-ray source (Kratos Analytical, UK). A Rigaku XRD Ultima IV (Japan) using Cu Kα X-ray source (40 kV, 44 mA) and continuous scan mode was used to collect data, which was examined using JADE software. Samples for Raman spectroscopy were placed onto 2 cm2 glass slides. Measurements were conducted using a Senterra Raman spectrometer (Bruker, USA) equipped with a laser diode (λ = 785 nm) of 10 mW laser power. Mass spectrometry was used to analyze the photodegradation products of RhB using a Voyager Elite MALDI-TOF mass spectrometer (AB Sciex, Canada) equipped with a pulsed nitrogen laser (337 nm, 3 ns pulse).
Results and discussion
The in situ synthetic route for generating CdSQDs@pNIPAm-co-AAc hybrid microgels is shown schematically in Scheme 1. The pNIPAm-co-AAc microgels were composed of COOH groups that could be deprotonated by increasing the solution pH above the pKa for AAc (4.25), which rendered them negatively charged. The negative charges in the pNIPAm-co-AAc microgels could increase the interaction strength of the microgels with the Cd2+ allowing its enrichment in the microgels.27 As detailed in the Experimental section above, the microgels were then washed 6 times, and the Cd2+-loaded pNIPAm-co-AAc microgels subsequently exposed to S2− to form CdSQDs.
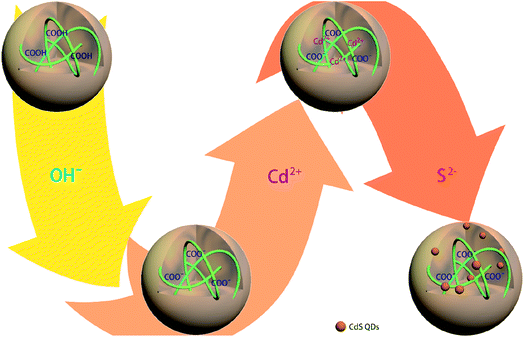 |
| | Scheme 1 Schematic illustration of the synthetic route used for generating the CdSQD@pNIPAm-co-AAc hybrid microgels. | |
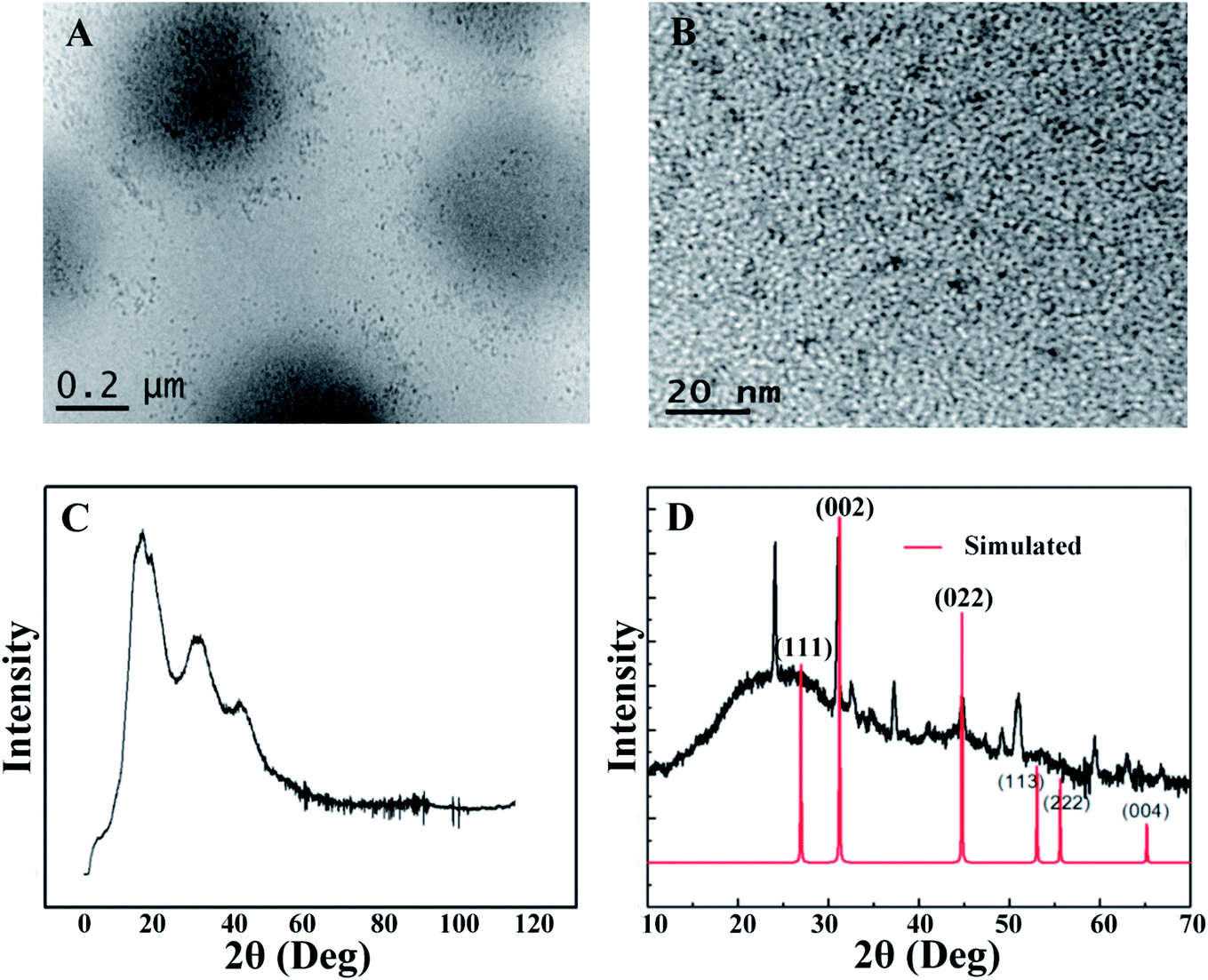
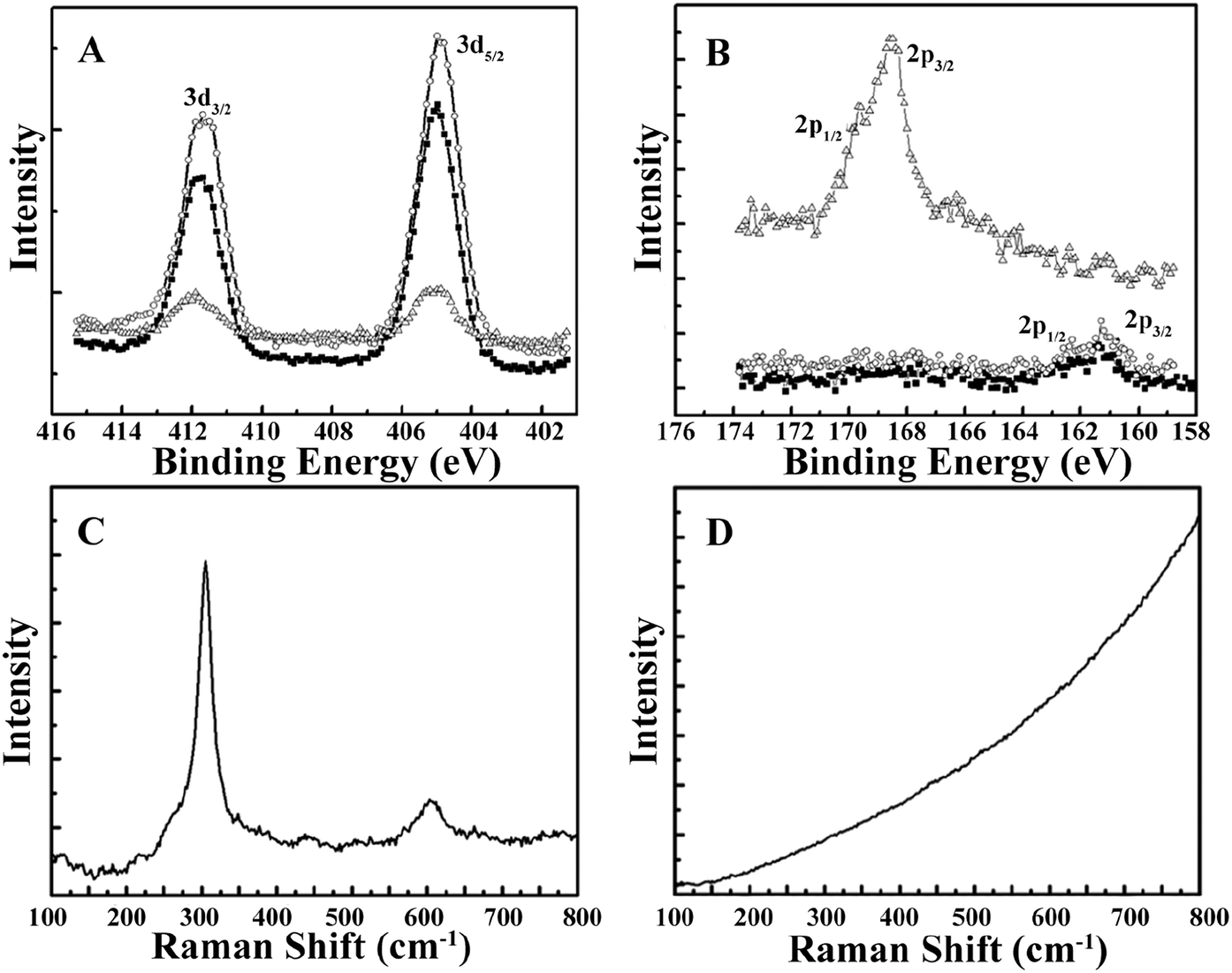
The morphology of the CdSQDs@pNIPAm-co-AAc hybrid microgels was determined using TEM imaging (Fig. 1A and B). As can be seen, the microgel scaffold has a low TEM contrast and a diameter of ∼700 nm. The images also showed that each microgel contained between 100–200 nanoparticles with a diameter of 8.0 ± 2.5 nm (Fig. S1†). Of note, the location of a considerable number of particles are distributed around the edges of the microgels, possibly due to the steric hindrance of the Cd2+ and/or S2− entering the “core” of the microgels. The formation of CdSQDs inside the pNIPAm-co-AAc microgels was subsequently investigated using selected area electron diffraction (SAED). The SAED pattern of the composite microgels (Fig. S2†), revealed the polycrystalline nature of CdSQDs in the microgels, which was further investigated using XRD analysis. As can be seen in Fig. 1C, the native microgels exhibited broad peaks 17°, 34° and 40°, showing amorphous phases from the polymer.14 After the generation of CdSQDs in the pNIPAm-co-AAc microgels, a series of new sharp and intense peaks ranging from 20° to 60°, appeared (Fig. 1D), due to the presence of polycrystalline CdSQDs.7,29 Fig. 2A shows the XPS spectra of CdSQDs incorporated in pNIPAm-co-AAc microgels, exhibiting two peaks at 412.2 eV and 405.9 eV, which are attributed to Cd 3d3/2 and Cd 3d5/2, respectively, indicating the Cd2+ oxidation state.30 Also, the high-resolution XPS spectrum of S 2p exhibited the expected peaks at 162.1 eV (2p1/2) and 161.2 eV (2p3/2) verifying the existence of CdS.30 Raman scattering measurements were also performed to investigate the photophysical properties of nanostructured semiconductor CdS within pNIPAm-co-AAc microgels. Raman peaks of the hybrid microgels in Fig. 2C are located at 305 cm−1 and 607 cm−1, assigned to the characteristic first- and second-order longitudinal optical (LO) phonons of the CdS semiconductor,31 while no such peaks were found in the Raman spectrum of the native microgels (Fig. 2D).
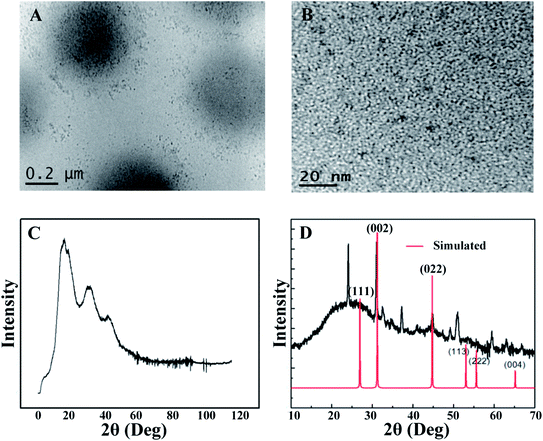 |
| | Fig. 1 (A) and (B) Representative TEM images of the CdSQDs@pNIPAm-co-AAc hybrid microgels and CdSQDs within the microgels, respectively; (C) and (D) X-ray diffraction pattern of pNIPAm-co-AAc microgels and CdSQDs@pNIPAm-co-AAc hybrid microgels, respectively. | |
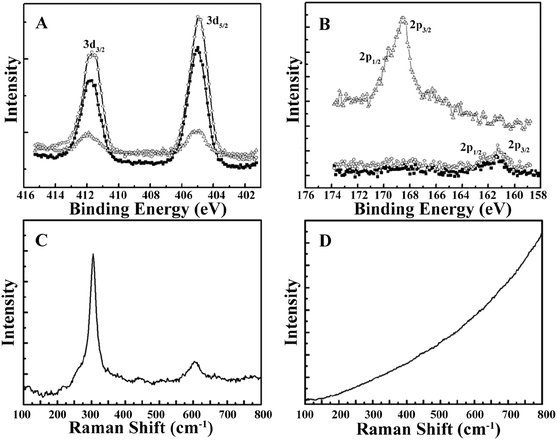 |
| | Fig. 2 XPS spectra with characteristic peaks labeled for (A) cadmium and (B) sulfur (■: CdSQDs@pNIPAm-co-AAc hybrid microgels, ○: mixed solution of the hybrid microgels with RhB, △: supernatant of mixed solution of the hybrid microgels with RhB after photocatalytic degradation). (C) Raman spectrum of the CdSQDs@pNIPAm-co-AAc hybrid microgels via excitation by the 514 nm line of an argon laser, and (D) Raman spectrum of the unmodified microgels via excitation by the 514 nm line of an argon laser. | |
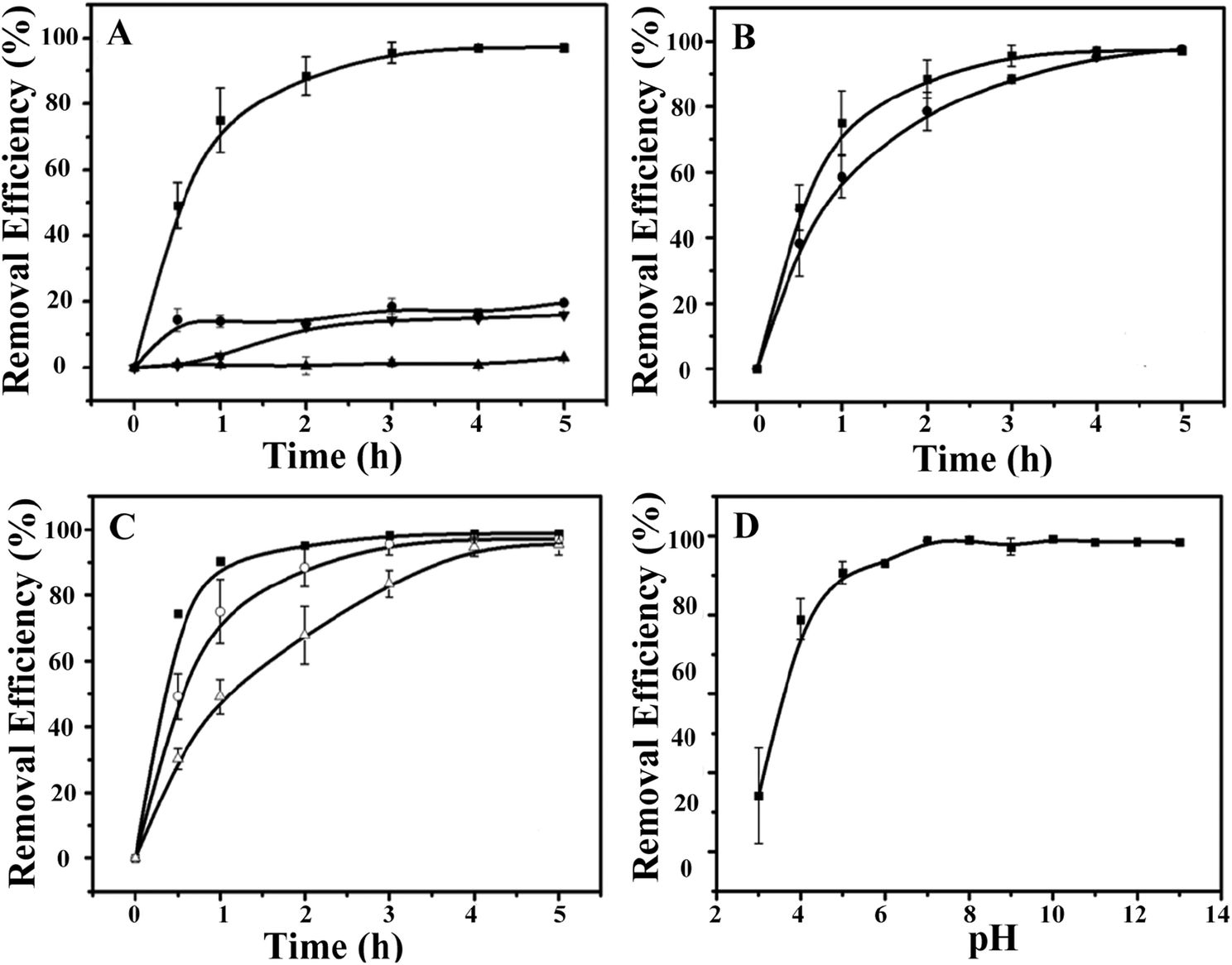
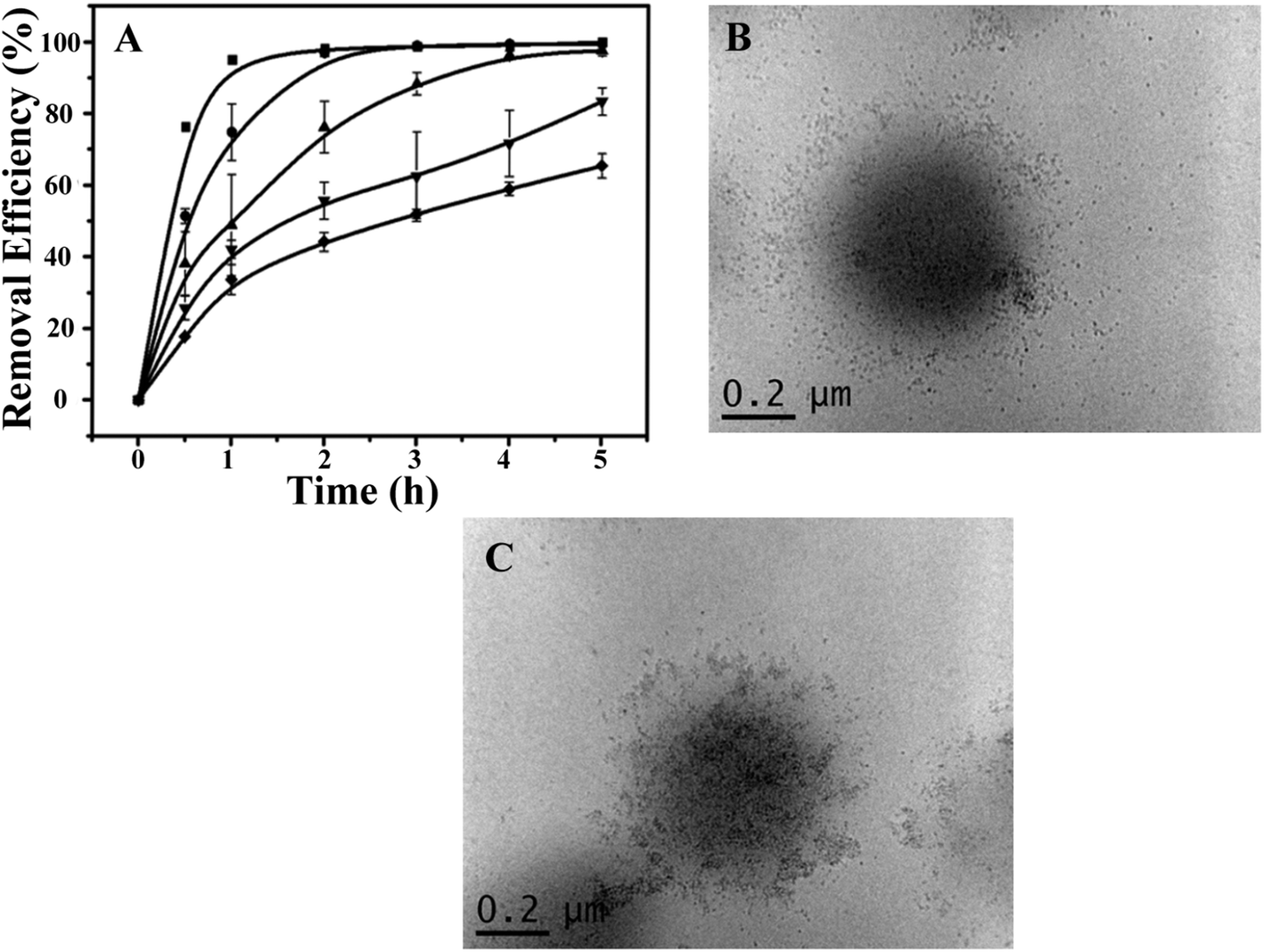
Next, the ability of the CdSQDs@pNIPAm-co-AAc hybrid microgels to photocatalytically degrade RhB was investigated. To do this, the UV-vis absorbance spectra were collected for RhB (λmax = 553 nm) as a function of time. To begin, we verified that UV irradiation of the RhB solution alone yielded minimal change in the UV-vis absorbance maximum for RhB (Fig. 3A). After introduction of the CdSQDs@pNIPAm-co-AAc hybrid microgels and subsequent exposure to same UV irradiation, we showed that all of the RhB could be degraded within 3 h. Of note, RhB could be removed from solution via absorption/adsorption by the microgels themselves (without exposure to UV irradiation),21–24 but the removal efficiency was insignificant relative to the CdSQD-loaded microgels. For example, as can be seen in Fig. 3A, both the native microgels exposed to RhB with UV irradiation and the hybrid microgels exposed to RhB without UV irradiation could only remove ∼20% of RhB from solution within 3 h. Therefore, we conclude that the photocatalytic degradation is the primary mechanism for RhB removal from solution. Previous studies have shown that the following photodegradation mechanism is possible for CdSQDs:32–34
| CdS + light → CdS holes (h+) + CdS electrons (e−) |
| H2O2 + O2−˙→ ˙OH + OH−+ O2 |
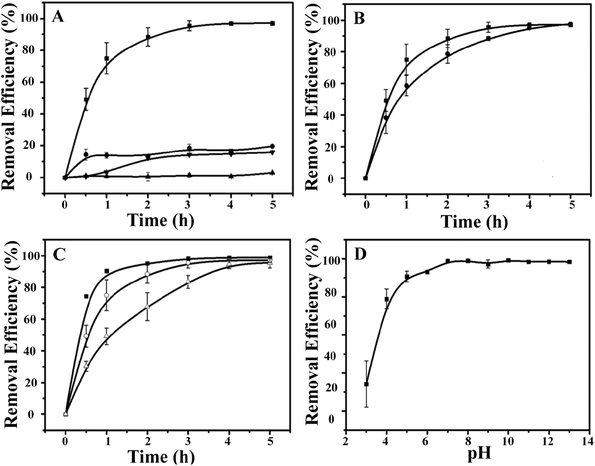 |
| | Fig. 3 (A) The removal efficiency (as a percent) of RhB as a function of time (degradation kinetics) at 10 °C. (■: hybrid microgels + UV, ●: hybrid microgels + dark, ▼: unmodified microgels + UV, ▲: no microgels + UV). (B) The degradation kinetics of RhB at different temperatures (■: 10 °C, ●: 45 °C) with hybrid microgels. (C) Comparison of the degradation kinetics of RhB for different amounts of hybrid microgels (■: 0.6 g, ○: 0.4 g, △: 0.2 g). (D) Working pH range for the hybrid microgels. Data are mean values ± standard deviation of three independent experiments. | |
As indicated above, UV irradiation activates the CdSQDs to generate electron/hole pairs and reactive oxygen species (ROS), including H2O2, O2−˙, and ˙OH, which we think is responsible for the RhB degradation into colorless compounds. To determine what the degradation byproducts were, we performed mass spectrometry (MS). RhB shows a peak at 443.3 m/z in the MS spectrum (Fig. S3A†) that we assigned as cationized RhB (C28H31N2O3+). To get a good determination of the possible degradation byproducts, the UV-triggered degradation was only allowed to continue for 1.5 h and the solution sampled. As can be seen in Fig. S3B,† the resultant byproducts add a series of new peaks in the MS spectrum, mainly at 415.3 m/z, 387.3 m/z and 354.5 m/z, as a result of the loss of CH2CH2 or CO from RhB. The products could thus be attributed to be a mixture of deethylations and carboxylations of RhB, which is in good agreement with previous reports.35–37 Together, a possible RhB degradation process could be thus be depicted as shown in Fig. S4,† according to the aforementioned investigations.
We then investigated the impact of solution temperature, pH, and hybrid microgel concentration on RhB degradation. First, the photocatalytic degradation was investigated at 45 °C (>LCST for pNIPAm) and 10 °C (<LCST for pNIPAm). As can be seen in Fig. 3B, the RhB removal efficiency kinetics were higher at 10 °C than at 45 °C, where the hybrid microgels were swollen. We hypothesize that the swollen microgels allow easier access of RhB to CdSQDs for more efficient photocatalytic degradation. Fig. 3C shows how the hybrid microgel concentration impacts the RhB degradation rate. As can be seen, the RhB removal efficiency kinetics are enhanced as the amount of the hybrid microgels in solution increases (Fig. 3C). Finally, the microgels used here were exposed to high solution pH, where any remaining AAc groups in the microgel will be deprotonated and the microgels will swell that we hypothesize will lead to enhanced RhB degradation kinetics, which we observe in Fig. 3D.
Using the optimized conditions (i.e., 0.6 g of the hybrid microgels, at 10 °C and pH 7.0), we further investigated the recyclability of the photocatalytic hybrid microgels. As can be seen in Fig. 4A, the kinetics of RhB degradation gradually decreased as the number of times they were recycled increased. Specifically, only 60% removal efficiency could be achieved after the microgels were recycled 5 times after 5 h UV exposure (Fig. S5†). Although, complete removal could be achieved by simply extending the irradiation time. The decrease in RhB removal efficiency indicated possible physicochemical changes of the CdSQDs@pNIPAm-co-AAc hybrids microgels, which was investigated by employing TEM imaging for examining the microgel structure and XPS for examining their composition. Fig. 4B and C show representative TEM images of the hybrid microgels after 5 photocatalytic cycles. As can be seen, the CdSQDs appear to aggregate and form irregular clusters with sizes from tens to a hundred nanometers. We hypothesize that the aggregates hamper the mass transfer of the RhB into the microgels, hence limiting the ability of RhB to be photodegraded. Furthermore, due to the ability of S2− to be oxidized to S+6 could lead to the degradation of the CdSQDs. Indeed, the S+6 state was observed in the XPS spectrum (Table S1† and Fig. 2B). This was also supported by the simultaneous appearance of Cd and S in the XPS spectrum (Table S1† and Fig. 2A and B) of the supernatant recovered from the photodegraded RhB solution. Of note, the atomic ratio of Cd to S in the hybrid microgel was 1.01:0.13, while this ratio was changed to 0.10:1.16 in the supernatant of the UV-irradiated mixed solution, indicating much less leakage of Cd from the microgel when the CdSQDs were degraded. This is important as it can prevent the leakage of toxic heavy metals to the environment, while still affording the hybrid microgel with photodegradation properties.
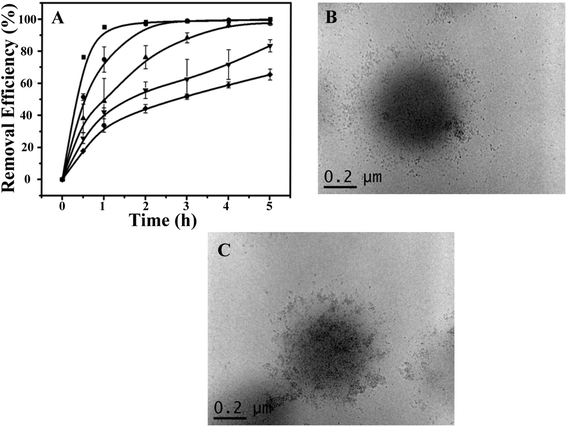 |
| | Fig. 4 (A) Degradation kinetics as a function of the number of times the hybrid microgels were recycled (■: 1st time, ●: 2nd time, ▲: 3rd time,▼: 4th time, ◆: 5th time). Representative TEM images of hybrid microgels (B) before and (C) after their use for photodegradation. Data in (A) are mean values ± standard deviation of three independent experiments. | |
Conclusion
In summary, we reported an approach for generating CdSQDs@pNIPAm-co-AAc hybrid microgels, which were shown to have the ability to photocatalytically degrade the organic dye RhB in an aqueous solution. We fully characterized the synthesized hybrid microgels, and demonstrated that the hybrid microgels have the ability to degrade RhB into products that were characterized via mass spectrometry. We went on to show that the hybrid microgels most efficiently photodegraded the RhB when the microgels were swollen at low temperature and high pH. Finally, we demonstrated that the hybrid microgels could be recycled and reused multiple times, although their activity was diminished as the number of times they were recycled increased. While we attribute the loss of activity to CdSQD degradation, we do not observe a significant leakage of Cd2+ into solution. While this study was only done with RhB as a proof of concept, it is a promising approach for removing other organic contaminants from aqueous solutions in a light triggered manner.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
MJS acknowledges funding from the University of Alberta (the Department of Chemistry and the Faculty of Science), the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), the Alberta Advanced Education & Technology Small Equipment Grants Program (AET/SEGP), Grand Challenges Canada and IC-IMPACTS. XZ and TS acknowledge support from the National Natural Science Foundation of China (Grant No. 21727815), the Beijing Municipal Science and Technology Commission (z131102002813058), Beijing Natural Science Foundation (2184107) and the Fundamental Research Funds for the Central Universities (FRF-TP-15-096A1). MJS, XZ and TS acknowledge the Research Fund for International Young Scientists of the National Natural Science Foundation of China (21750110449).
References
- A. Khatri, M. H. Peerzada, M. Mohsin and M. White, J. Cleaner Prod., 2015, 87, 50–57 CrossRef CAS.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- L. S. Zhang, W. Z. Wang, J. O. Yang, Z. G. Chen, W. Q. Zhang, L. Zhou and S. W. Liu, Appl. Catal., A, 2006, 308, 105–110 CrossRef CAS.
- L. S. Zhang, W. Z. Wang, Z. G. Chen, L. Zhou, H. L. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526–2532 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Zhang, Y. Guo, H. Fang, W. Jia, H. Li, L. Yang and K. Wang, New J. Chem., 2015, 39, 6951–6957 RSC.
- X. Fu, Y. Zhang, P. Cao, H. Ma, P. Liu, L. He, J. Peng, J. Li and M. Zhai, Radiat. Phys. Chem., 2016, 123, 79–86 CrossRef CAS.
- Q. Fan, Y. Huang, C. Zhang, J. Liu, L. Piao, Y. Yu, S. Zuo and B. Li, Catal. Today, 2016, 264, 250–256 CrossRef CAS.
- X. Li, J. Zhu and H. X. Li, Appl. Catal., B, 2012, 123, 174–181 CrossRef.
- H. Zhu, R. Jiang, L. Xiao, Y. Chang, Y. Guan, X. Li and G. Zeng, J. Hazard. Mater., 2009, 169, 933–940 CrossRef CAS PubMed.
- Z. Yang, L. Lu, C. J. Kiely, B. W. Berger and S. McIntosh, Ind. Eng. Chem. Res., 2016, 55, 11235–11244 CrossRef CAS.
- S. Akshya, P. S. Hariharan, V. Vinod Kumar and S. P. Anthony, Spectrochim. Acta, Part A, 2015, 135, 335–341 CrossRef CAS PubMed.
- X. Li and M. J. Serpe, Adv. Funct. Mater., 2016, 26, 3282–3290 CrossRef CAS.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- N. Lu, J. Liu, J. Li, Z. Zhang, Y. Weng, B. Yuan, K. Yang and Y. Ma, J. Mater. Chem. B, 2014, 2, 3791–3798 RSC.
- H. Jia, R. Roa, S. Angioletti-Uberti, K. Henzler, A. Ott, X. Lin, J. Möser, Z. Kochovski, A. Schnegg, J. Dzubiella, M. Ballauff and Y. Lu, J. Mater. Chem. A, 2016, 4, 9677–9684 CAS.
- X. Wu, R. H. Pelton, A. E. Hamielec, D. R. Woods and W. McPhee, Colloid Polym. Sci., 1994, 272, 467–477 CAS.
- T. Hoare and R. Pelton, Macromolecules, 2004, 37, 2544–2550 CrossRef CAS.
- D. Parasuraman and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 4714–4721 CAS.
- D. Parasuraman and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 2732–2737 CAS.
- D. Parasuraman, E. Leung and M. J. Serpe, Colloid Polym. Sci., 2012, 290, 1053–1064 CAS.
- D. Parasuraman, A. K. Sarker and M. J. Serpe, Colloid Polym. Sci., 2013, 291, 1795–1802 CAS.
- D. M. Han, Q. M. Zhang and M. J. Serpe, Nanoscale, 2015, 7, 2784–2789 RSC.
- A. Salcher, M. S. Nikolic, S. Casado, M. Vélez, H. Weller and B. H. Juárez, J. Mater. Chem., 2010, 20, 1367–1374 RSC.
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS PubMed.
- A. M. Holi, Z. Zainal, Z. A. Talib, H. N. Lim, C. C. Yap, S. K. Chang and A. K. Ayal, J. Mater. Sci.: Mater. Electron., 2016, 27, 7353–7360 CrossRef CAS.
- N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, J. Catal., 2013, 303, 60–69 CrossRef CAS.
- J. J. Zhang, W. S. Li, Y. Li, L. Zhong and C. J. Xu, Appl. Catal., B, 2017, 217, 30–36 CrossRef CAS.
- L. Ma, M. Liu, D. Jing and L. Guo, J. Mater. Chem. A, 2015, 3, 5701–5707 CAS.
- H. M. Jia, W. W. He, W. G. Wamer, X. N. Han, B. B. Zhang, S. Zhang, Z. Zheng, Y. Xiang and J. J. Yin, J. Phys. Chem. C, 2014, 118, 21447–21456 CAS.
- Y. Hu, Y. Liu, H. S. Qian, Z. Q. Li and J. F. Chen, Langmuir, 2010, 26, 18570–18575 CrossRef CAS PubMed.
- X. Y. Li, C. G. Hu, X. Wang and Y. Xi, Appl. Surf. Sci., 2012, 258, 4370–4376 CrossRef CAS.
- S. Horikoshi, H. Hidaka and N. Serpone, Environ. Sci. Technol., 2002, 36, 1357–1366 CrossRef CAS PubMed.
- Z. L. He, W. X. Que, X. T. Yin and Y. C. He, RSC Adv., 2014, 4, 39678–39683 RSC.
- Y. Gao, C. Hu, W. J. Zheng, S. Yang, F. Li, S. D. Sun, M. Zrinyi, Y. Osada, Z. M. Yang and Y. M. Chen, ChemPhysChem, 2016, 17, 1999–2007 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01855c |
| ‡ Both contributed equally to this publication. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b