
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeniposide attenuates Aβ25–35-induced neurotoxicity via the TLR4/NF-κB pathway in HT22 cells
Xiu-Fang Huang†
 a,
Jian-Jun Li†a,
Yan-Gu Taoa,
Xie-Qi Wangc,
Ru-Lan Zhanga,
Jia-Lin Zhangc,
Zu-Qing Sub,
Qi-Hui Huang*a and
Yuan-Hui Deng*b
a,
Jian-Jun Li†a,
Yan-Gu Taoa,
Xie-Qi Wangc,
Ru-Lan Zhanga,
Jia-Lin Zhangc,
Zu-Qing Sub,
Qi-Hui Huang*a and
Yuan-Hui Deng*b
aSun Yat-Sen Memorial Hospital of Sun Yat-sen University, Guangzhou, Guangdong 510220, China. E-mail: hqhui84181833@sina.com; Tel: +86 2081332016
bGuangdong Provincial Hospital of Traditional Chinese Medicine, Guangzhou, Guangdong 511400, China. E-mail: yhdeng62@126.com
cDermatology Hospital of Southern Medical University, Dermatology Hospital of Guangdong Province, Guangzhou, Guangdong, China
First published on 23rd May 2018
Abstract
Alzheimer's disease (AD), a neurodegenerative disorder, is marked by the accumulation of amyloid-β (Aβ) and neuroinflammation which promote the development of AD. Geniposide, the main ingredient isolated from Chinese herbal medicine Gardenia jasminoides Ellis, has a variety of pharmacological functions such as anti-apoptosis and anti-inflammatory activity. Hence, we estimated the inflammatory cytotoxicity caused by Aβ25–35 and the neuroprotective effects of geniposide in HT22 cells. In this research, following incubation with Aβ25–35 (40 μM, 24 h) in HT22 cells, the methylthiazolyl tetrazolium (MTT) and lactate dehydrogenase (LDH) release assays showed that the cell survival rate was significantly decreased. In contrast, the reactive oxygen species (ROS) assay indicated that Aβ25–35 enhanced ROS accumulation and apoptosis showed in both hoechst 33342 staining and annexin V-FITC/PI double staining. And then, immunofluorescence test revealed that Aβ25–35 promoted p65 to transfer into the nucleus indicating p65 was activated by Aβ25–35. Moreover, western blot analysis proved that Aβ25–35 increased the expression of nitric oxide species (iNOS), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2) and interleukin-1β (IL-1β). Simultaneously, Aβ25–35 also promoted the expression of toll-like receptor 4 (TLR4), p-p65 and p-IκB-α accompanied with the increase in the level of beta-secretase 1 (BACE1) and caspase-3 which further supported Aβ25–35 induced apoptosis and inflammation. Fortunately, this up-regulation was reversed by geniposide. In conclusion, our data suggest that geniposide can alleviate Aβ25–35-induced inflammatory response to protect neurons, which is possibly involved with the inhibition of the TLR4/NF-κB pathway in HT22 cells. Geniposide may be the latent treatment for AD induced by neuroinflammation and apoptosis.
Introduction
Alzheimer's disease (AD), a common neurodegenerative disorder that emerges as a major threat to elder health, is clinically characterized by progressive memory defects and cognitive impairment in relation to chronic inflammation induced by amyloid-β (Aβ) precipitation.1 It has been proved that the risk of AD in middle-aged men who used nonsteroidal anti-inflammatory drugs was evidently reduced.2 Furthermore, excessive inflammatory factors were found in AD brain tissue.3,4 It is well known that Aβ deposition is involved in neuroinflammation responses which can cause neuronal apoptosis and memory and cognitive deficits.5,6 Aβ probably acts as an immune initiator in the brain tissue of AD patients and activates gliocyte to release excessive inflammatory mediators such as TNF-α, IL-1β, iNOS and COX-2, which were proved to contribute to AD progression.7,8 Meanwhile, the increase of the above-mentioned inflammatory factors can enhance Aβ deposition leading to a vicious circle of inflammatory response. On the other hand, progressive Aβ deposition further increases oxidative stress to aggravate the inflammatory response to bring about progressive damage to neurons. These findings demonstrate that neuroinflammation possibly promotes the development and progression of AD. Although the mechanism between the inflammatory response and the pathogenesis of AD is still unclear, it is certain that neuroinflammation induced by Aβ plays a critical role in promoting the progression of AD. Aβ25–35, a shorter fragment stems from amyloid precursor protein, performs the similar neurotoxic function to Aβ and it is the main responsibility for neurotoxicity of Aβ.9Toll-like receptors (TLRs), the Aβ-recognized pattern recognition receptors, play central roles not only in the beginning of macrophages and immune reactions, but also in the occurrence of inflammatory diseases, for example AD.10,11 What is more, most TLRs have been proved to enhance the expression of inflammation-related nuclear factor-κappa B (NF-κB).12 Coincidentally, TLR4, a member of the TLRs family, can activate the NF-κB signaling pathway to produce a series of inflammatory factors.13 P65, the most common subunit of NF-κB, usually stays in cytoplasm combining with inhibitor of NF-κB, alpha (IκB-α) in a quiet status, but p65 will transfer to the nucleus to start a series of transcriptional reactions once IκB-α is phosphorylated.14 Hence, the TLR4/NF-κB signaling pathway activated by Aβ may play a central role in the neuroinflammation.
Geniposide (the chemical structure was shown in Fig. 1) is the main ingredient of Chinese herbal medicine Gardenia jasminoides Ellis. Modern pharmacological and clinical studies have shown that geniposide has neuroprotective effects especially for neurodegenerative diseases, such as AD, indicating geniposide may be a new therapeutic agent for AD.15,16 It has been confirmed that geniposide can pass through the blood–brain barrier to protect hippocampal neurons from inflammatory neurotoxicity and oxidative stress in vivo and in vitro.17–20 On the other hand, geniposide reduces neuronal apoptosis accompanied with decreasing caspase-3 activity.17 More importantly, geniposide down-regulates the expression of p65 in the nucleus to reduce the production of inflammatory factors.17 The results suggest that geniposide plays a key role in regulating the NF-κB pathway. We assume that geniposide might down-regulate TLR4/NF-κB signal channel to keep neurons from neurodegeneration caused by Aβ25–35-induced neuroinflammation. TAK 242 (the chemical structure was shown in Fig. 2), an inhibitor of the TLR4, was used as a positive control drug in this study.
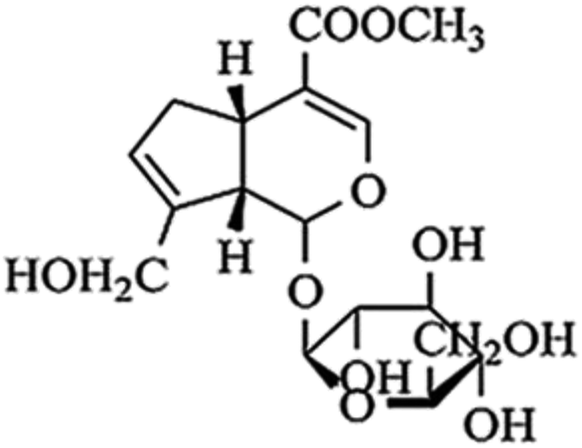 | ||
| Fig. 1 Chemical structure of geniposide. | ||
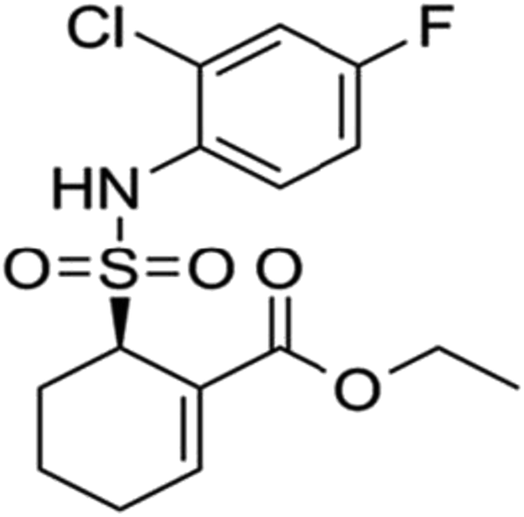 | ||
| Fig. 2 Chemical structure of TAK 242. | ||
Accumulating evidence suggests that microglia and astrocyte are activated by Aβ deposition to release inflammatory mediators causing damage to hippocampal neurons. However, we found that hippocampal neurons can also secrete inflammatory factors through the TLR4/NF-κB pathway suggesting that hippocampal neurons are both passive and active in inflammatory responses.21,22 Consequently, we explored whether geniposide could attenuate the inflammation responses induced by Aβ25–35 through TLR4/NF-κB signal pathway in HT22 cells.
Results and discussion
The appropriate concentrations of drugs
MTT assay was used to examine both the suitable neuroprotective concentrations of geniposide and the neurotoxic concentrations of Aβ25–35. Fig. 3 and 4 indicate that Aβ25–35 can inhibit cell viability and reduce the number of cells, injure cellular morphology, decrease the connection between neuronal synapses in a dose-dependent manner, especially Aβ25–35 remarkably impairs cell viability and cellular morphology at a dose of 40 μM for 24 h compared with the control group, suggesting that concentration of 40 μM for 24 h of Aβ25–35 is the effective concentration at 50% inhibition. Hence, Aβ25–35 at a dose of 40 μM was used to establish cellular model of AD in the subsequent research. In contrast, Fig. 3 and 5 also show that treatment with geniposide alone has no neuroprotective effect on cell vitality except the concentration at 160 μM for 24 h promotes HT22 cells growth and improves the cell density and cellular morphology. Therefore it was chosen as the neuroprotective concentration in this study.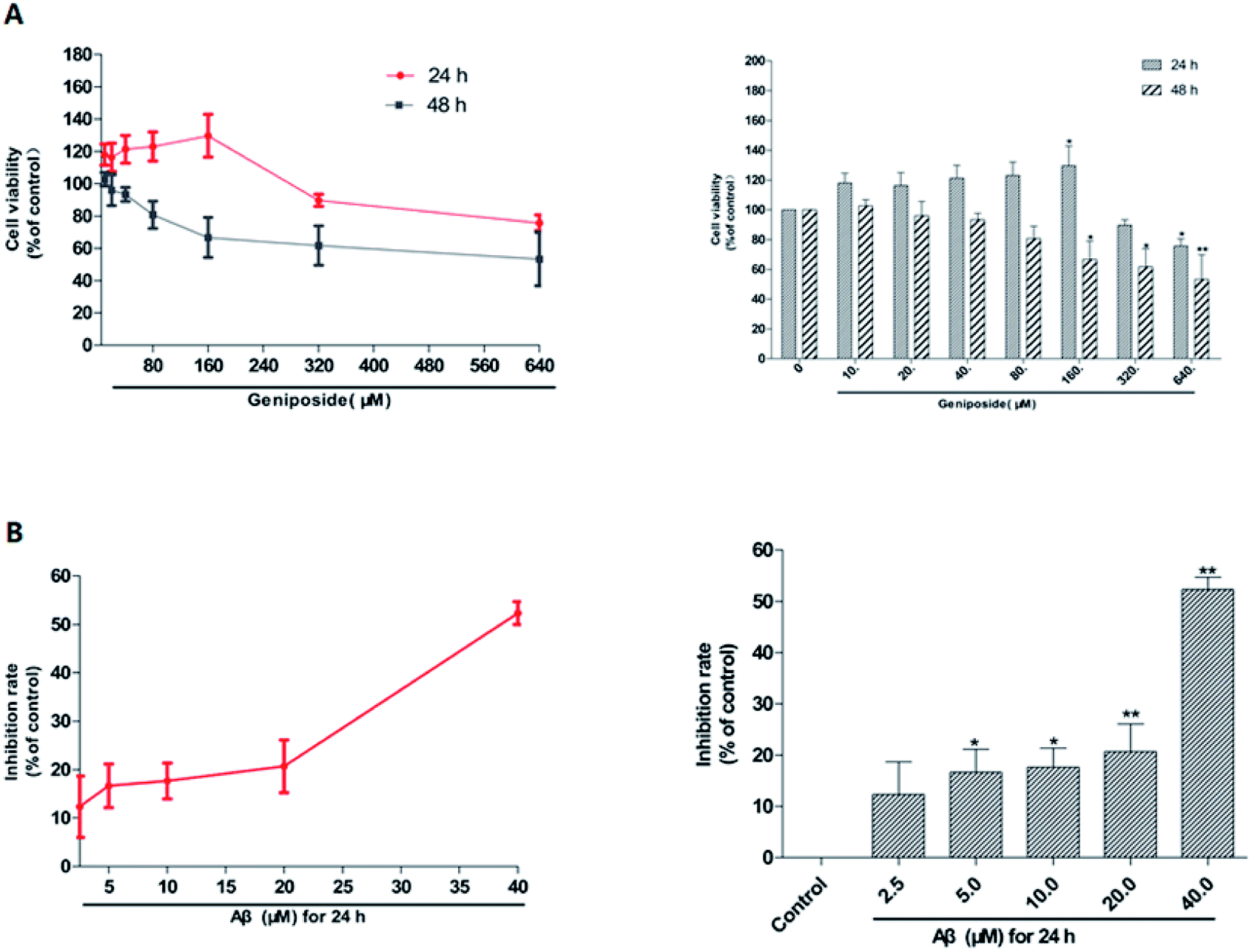 | ||
| Fig. 3 The stimulative concentrations of geniposide and Aβ25–35 on HT22 cells were measured by MTT assay. (A) HT22 cells were treated with various concentrations of geniposide (10–640 μM) for 24 h and 48 h respectively. (B) HT22 cells were incubated with different concentrations of Aβ25–35 (2.5–40 μM) for 24 h. The results are represented as means ± SEM from three independent experiments. (n = 3, *P < 0.05, **P < 0.01 vs. control, the values were represent as the percentage of the control). | ||
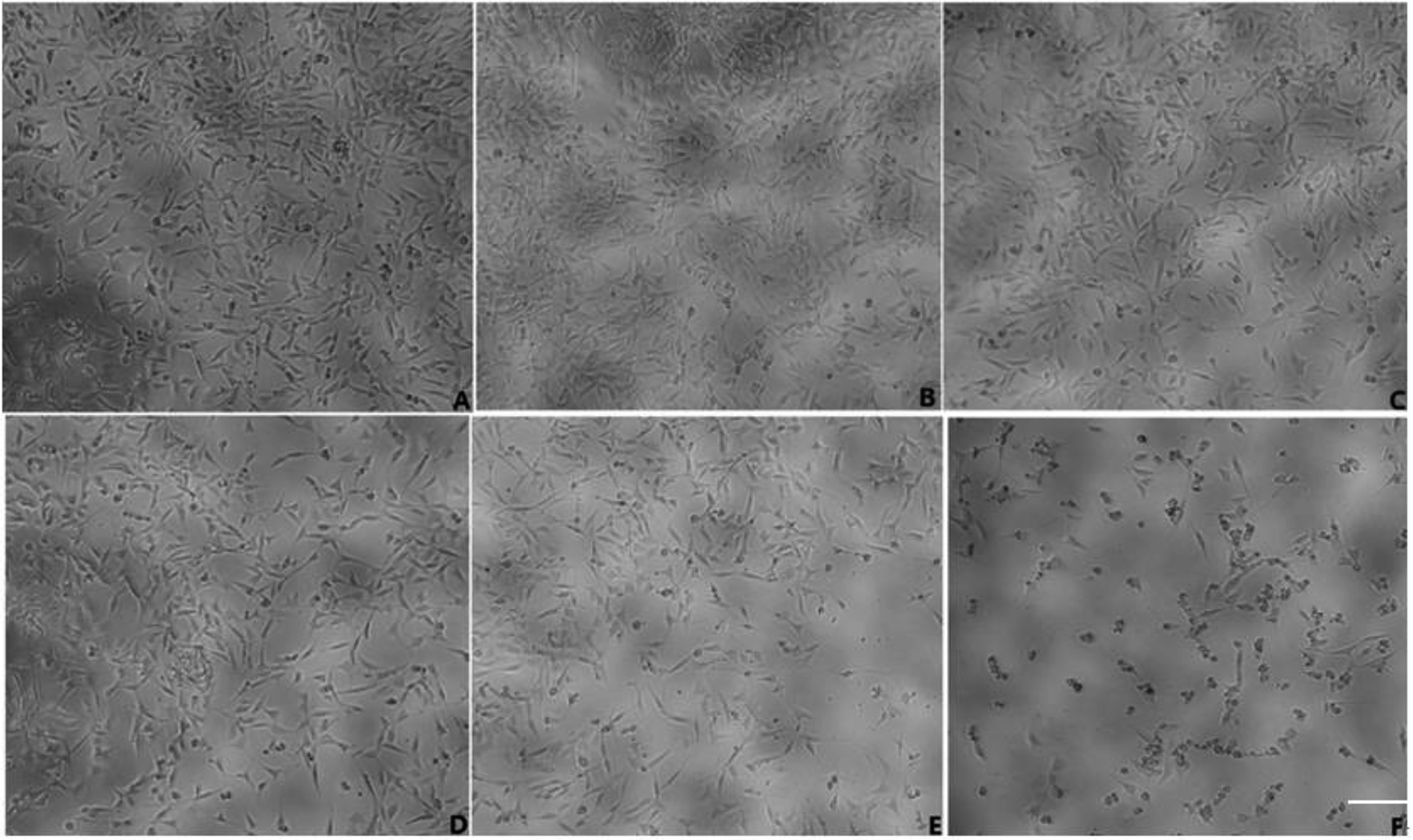 | ||
| Fig. 4 Various concentrations of Aβ25–35 on morphology of HT22 cells (scale bar = 50 μm). (A) Control group; (B) 2.5 μM Aβ25–35 for 24 h; (C) 5 μM Aβ25–35 for 24 h; (D) 10 μM Aβ25–35 for 24 h; (E) 20 μM Aβ25–35 for 24 h; (F) 40 μM Aβ25–35 for 24 h. | ||
 | ||
| Fig. 5 Various concentrations of geniposide on morphology of HT22 cells (scale bar = 50 μm). (A): (A) control group; (B) 10 μM geniposide for 24 h; (C) 20 μM geniposide for 24 h; (D) 40 μM geniposide for 24 h; (E) 80 μM geniposide for 24 h; (F) 160 μM geniposide for 24 h; (G) 320 μM geniposide for 24 h; (H) 640 μM geniposide for 24 h; (B): (A2) control group; (B2) 10 μM geniposide for 48 h; (C2) 20 μM geniposide for 48 h; (D2) 40 μM geniposide for 48 h; (E2) 80 μM geniposide for 48 h; (F2) 160 μM geniposide for 48 h; (G2) 320 μM geniposide for 28 h; (H2) 640 μM geniposide for 48 h. | ||
Geniposide protected HT22 cells against neurotoxicity induced by Aβ25–35
MTT and LDH release tests were applied to investigate the possible effect of geniposide against Aβ25–35-induced cytotoxicity on HT22 cells. As shown in Fig. 6, exposure to Aβ25–35 (40 μM) for 24 h reduces the number of cells, injures cellular morphology, decreases the connection between neuronal synapses, and inhibits cell viability. The results indicated that Aβ25–35 (40 μM) inhibited HT22 cells growth. The results demonstrated that pretreatment with geniposide (160 μM) and TAK 242 (1 μM) remarkably improved the cell density, cellular morphology, the connection between neuronal synapses, cell viability and reduced LDH release. TAK 242, an inhibitor of the TLR4, restrains the downstream neuroinflammation induced by activating TLR4. Our results illustrated that geniposide might protect HT22 cells from Aβ25–35-induced neurotoxicity via inhibiting the TLR4 to down-regulate the downstream neuroinflammation similar to TAK 242. More meaningfully, TAK 242 could protect HT22 cells against Aβ25–35-induced neurotoxicity, which demonstrated that Aβ25–35 might activate TLR4 to cause inflammatory damage to HT22 cells.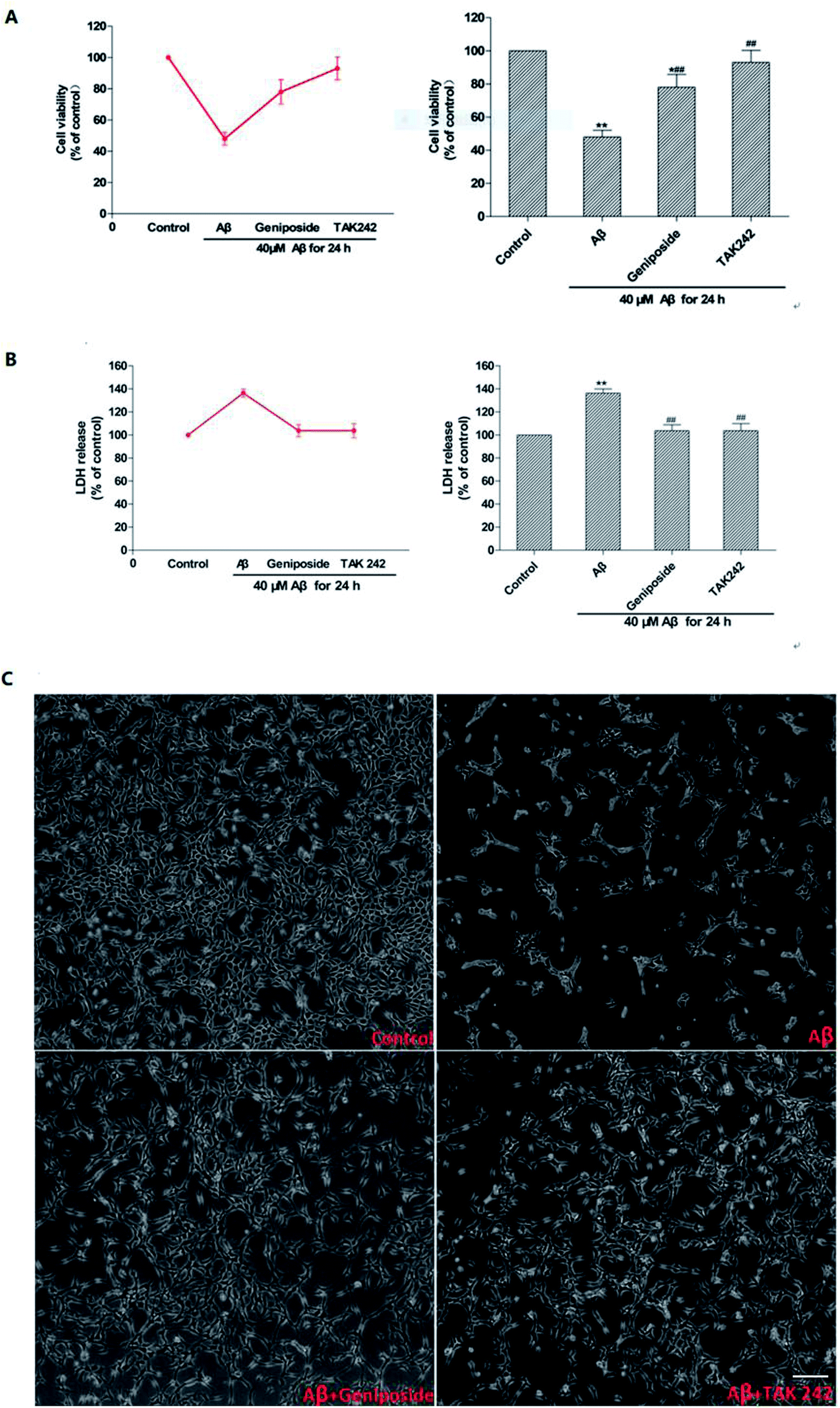 | ||
| Fig. 6 Effects of geniposide on morphology and neurotoxicity in Aβ25–35-induced HT22 cells. HT22 cells were pretreated with 1 h of geniposide (160 μM) and TAK 242 (1 μM), followed by 24 h treatment of Aβ25–35 (40 μM). Neurotoxicity was measured by MTT and LDH assays kits and morphology of HT22 cells were obtained by inverted microscope (scale bar = 50 μm). (A) Geniposide enhanced HT22 cells viability through reducing the toxicity caused by Aβ25–35; (B) geniposide attenuated the Aβ25–35-induced LDH release; (C) control: treated with medium alone; Aβ: 40 μM Aβ25–35; Aβ + geniposide: 160 μM geniposide + 40 μM Aβ25–35; Aβ + TAK 242: 1 μM TAK 242 + 40 μM Aβ25–35. The results are represented as means ± SEM from three independent experiments. (n = 3, **P < 0.01 vs. control, ##P < 0.01 vs. Aβ25–35 treated alone group, the values were represent as the percentage of the control). | ||
Though recent researches conjecture that Aβ is an immune stress responsed to brain infection. Aβ is a savior in the brain tissue rather than a killer. It wraps the viruses, bacteria and fungi that invade the brain and lumps harmful pathogens into pieces to prevent pathogens to damage the brain.23 But it is undeniable that exceeding Aβ leads to the death of nerve cells as the results showed in Fig. 6. In addition, geniposide not only promoted HT22 cells growth, but also protected HT22 cells from Aβ25–35-induced neurotoxicity. Coincidentally, just aiming to protect neurons from damage alone is also effective in the treatment of AD, which means the use of nerve cell protection drugs like geniposide can effectively prevent brain damage caused by AD.24
Aβ25–35-induced apoptosis were reversed by geniposide
To further identify the elevation of apoptosis induced by Aβ25–35, hoechst 33342 staining and annexin V/PI staining were performed. Fig. 7 shows that complete karyomorphism and little apoptosis are found in the control group. However, administration with Aβ25–35 (40 μM) for 24 h causes apoptosis with representative morphological characteristics such as chromatin aggregation, karyopyknosis and increased apoptotic bodies. As expected, treatment with geniposide (160 μM) and TAK 242 (1 μM) significantly inhibit the cell apoptosis. To define the status of geniposide-treated cells, we performed annexin V/PI staining assay to further exam cell apoptosis. As showed in Fig. 7, exposure to 40 μM Aβ25–35 causes cell apoptosis practically at early stage, which reaches to 51.85%. While treatment with geniposide or TAK 242 reduce the apoptosis rate to 31.66% or 25.48% respectively. On the other hand, cleaved caspase-3 remarkably increased after exposure to Aβ25–35. While pretreatment with geniposide and TAK 242 down-regulated the expression of caspase-3 (Fig. 10D). These data revealed that geniposide and TAK 242 pretreatment significantly inhibited Aβ25–35-induced apoptosis of HT22 cells.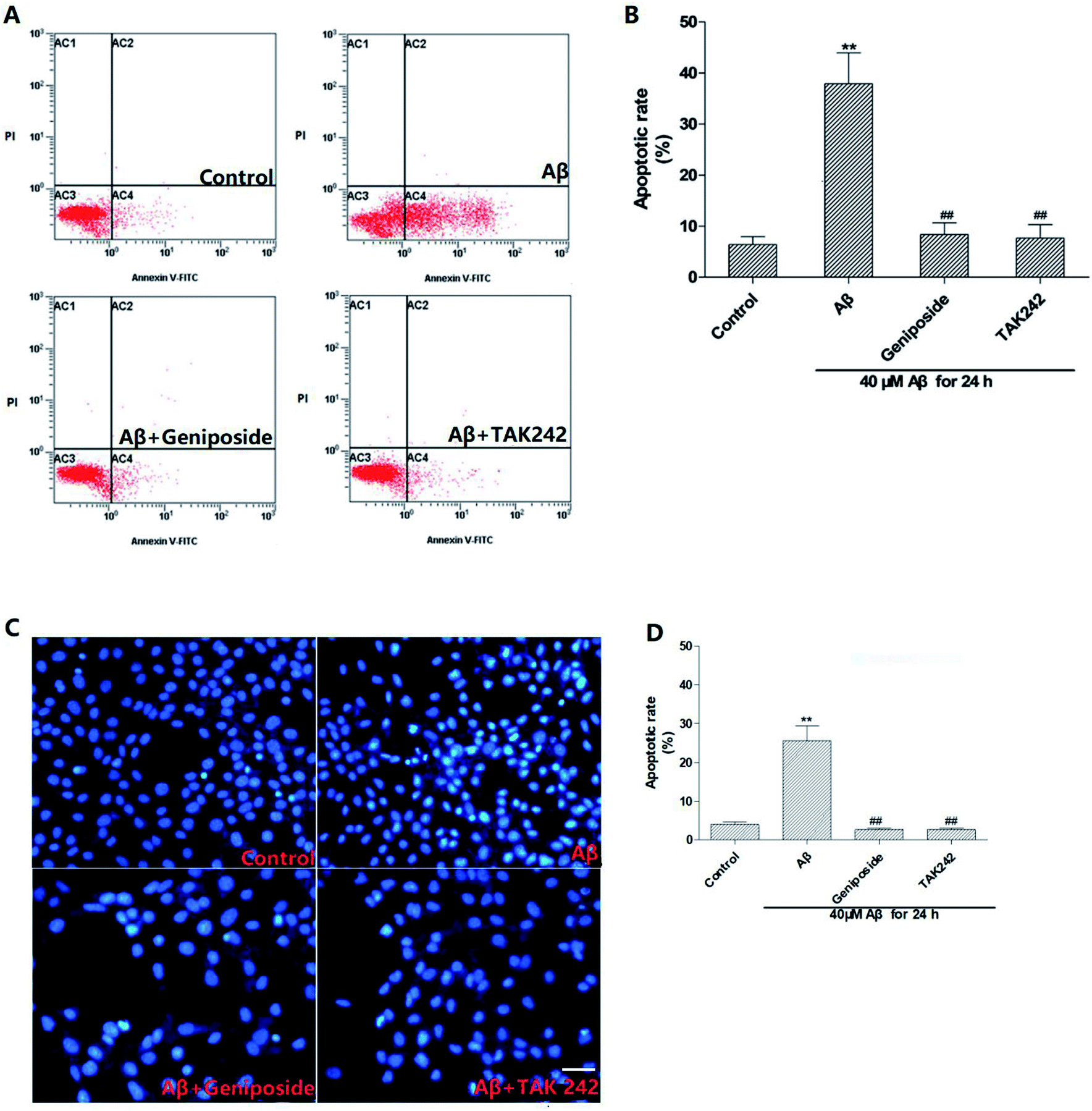 | ||
| Fig. 7 Effects of geniposide on Aβ25–35-induced apoptosis in HT22 cells (scale bar = 20 μm). HT22 cells were pretreated with 1 h of geniposide (160 μM) and TAK 242 (1 μM), followed by 24 h treatment of Aβ25–35 (40 μM). The cells were then stained with annexin V-FITC/PI and hoechst 33342. (A and C) Control: treated with medium alone; Aβ: 40 μM Aβ25–35; Aβ + geniposide: 160 μM geniposide + 40 μM Aβ25–35: Aβ + TAK 242: 1μM TAK 242 + 40 μM Aβ25–35. (B and D) The rate of apoptosis in response to Aβ25–35 stimulation showed in annexin V-FITC/PI double staining and hoechst 33342 staining were calculated. (n = 3, **P < 0.01 vs. control, ##P 0.01 vs. Aβ25–35 treated alone group, the values were represent as mean ± SEM). | ||
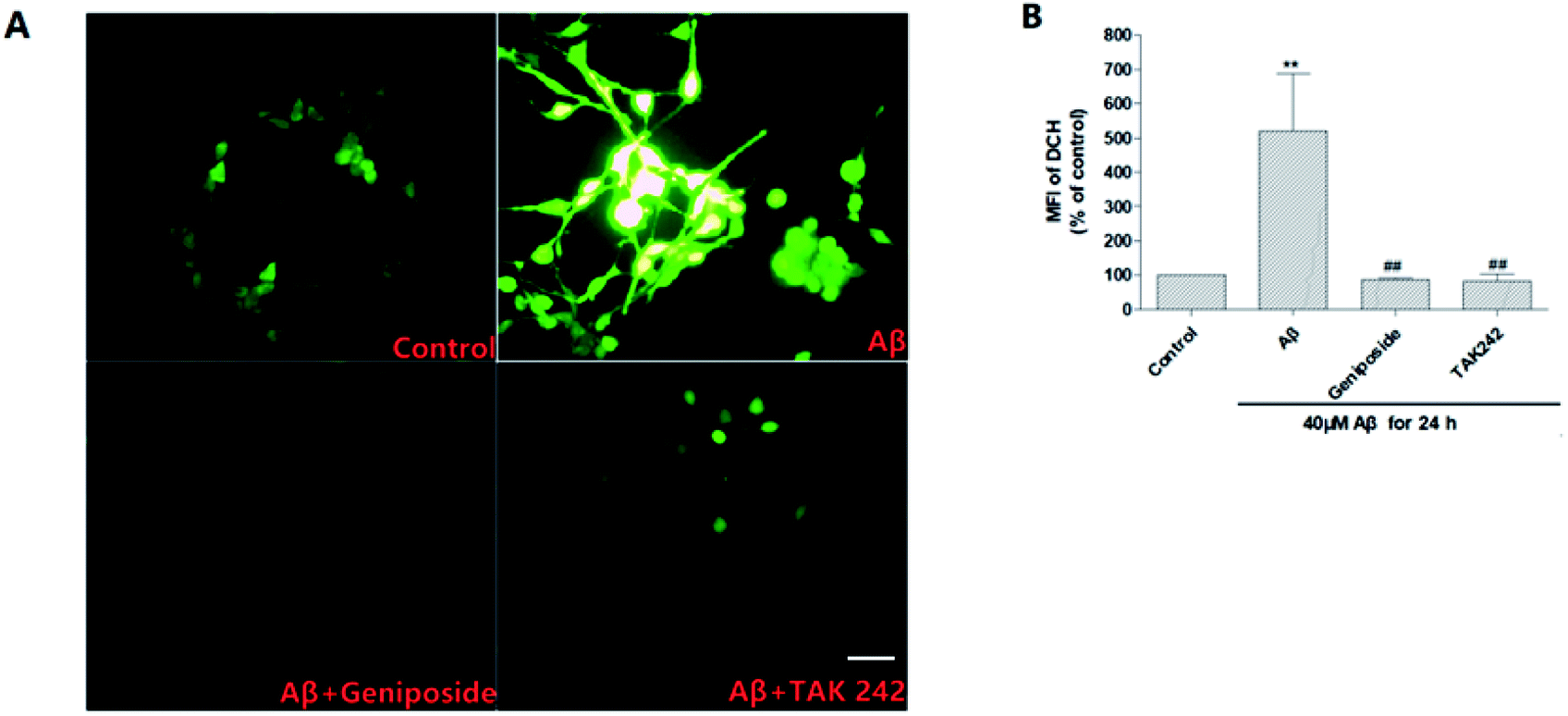 | ||
| Fig. 8 Effects of geniposide on ROS generation induced by Aβ25–35 in HT22 cells (scale bar = 20 μm). HT22 cells were pretreated with 1 h of geniposide (160 μM) and TAK 242 (1 μM), followed by 24 h treatment of Aβ25–35 (40 μM). The cells were then stained with DCFH-DA and the ROS was quantified by Image 3 software in the intensity of fluorescence. (A) Control: treated with medium alone; Aβ: 40 μM Aβ25–35; Aβ + geniposide: 160 μM geniposide + 40 μM Aβ25–35; Aβ + TAK 242: 1 μM TAK 242 + 40 μM Aβ25–35. (B) Quantitative analysis of the mean fluorescence intensity (MFI) of DCF. Values are the mean ± SEM (n = 3, **P < 0.01 vs. control, ##P < 0.01 vs. Aβ25–35 treated alone group, the values were represent as the percentage of the control). | ||
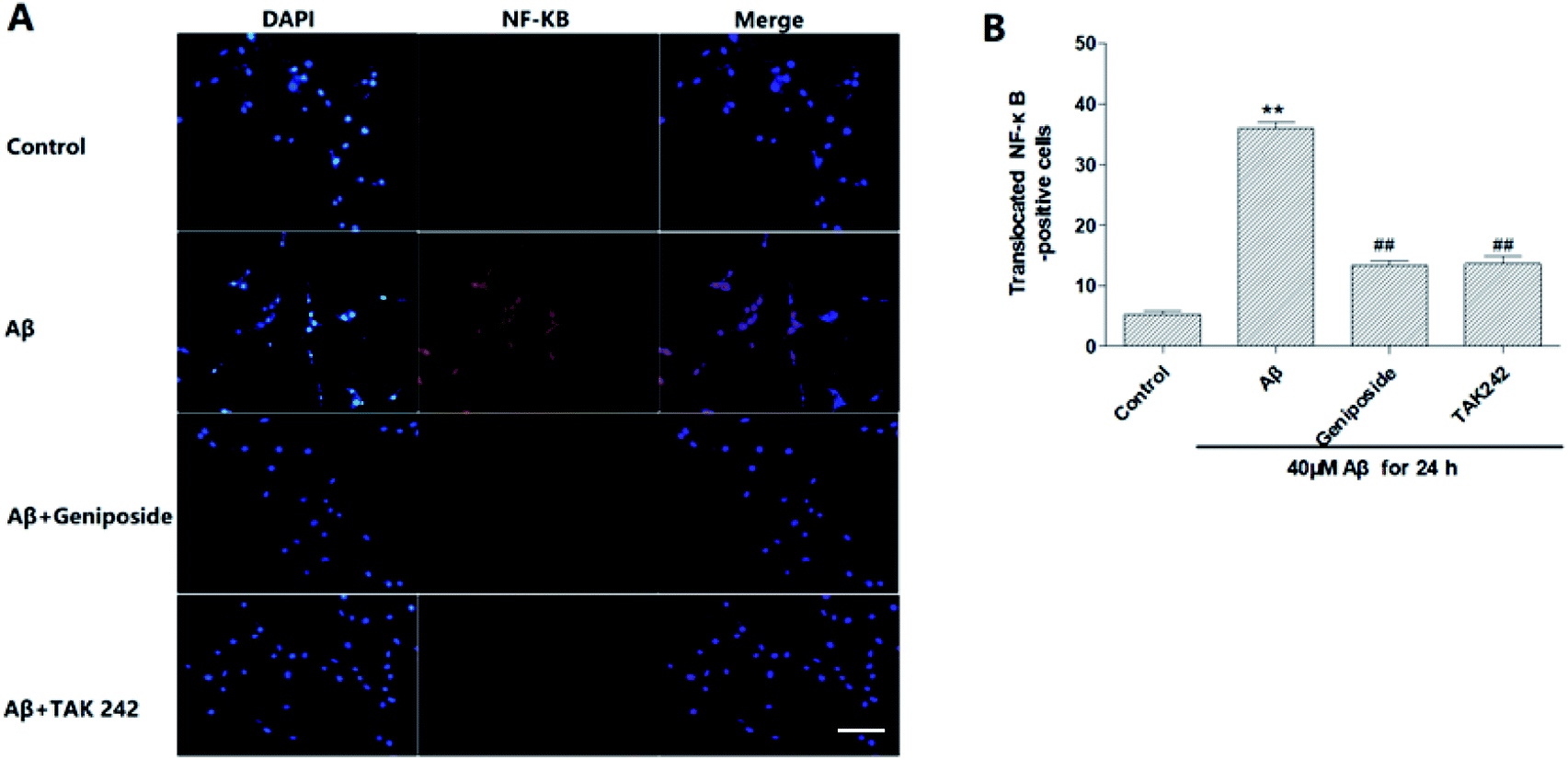 | ||
| Fig. 9 Effects of geniposide on Aβ25–35-induced NF-κB translocation in HT22 cells (scale bar = 20 μm). (A) Immunohistochemical staining for NF-κB with DAPI. Activated NF-κB immunoreactivity in HT22 cells was predominantly localized in the nucleus. (B) The number of translocated NF-κB-positive cells into nucleus is shown. (n = 3, **P < 0.01 vs. control, ##P < 0.01 vs. Aβ25–35 treated alone group, the values were represent as mean ± SEM). | ||
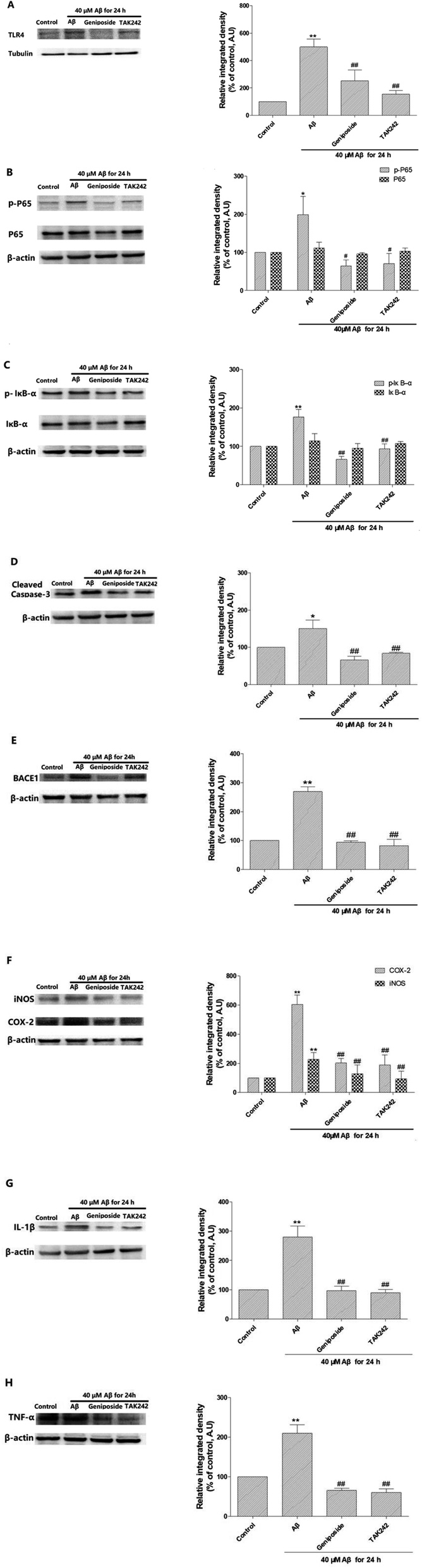 | ||
| Fig. 10 Anti-inflammatory mechanism of geniposide against Aβ25–35-induced neuroinflammation in HT22 cells. (A) Shown is representative western blot probed with TLR4 antibody in HT22 cells. (B) Shown are representative western blots probed with p-P65 and P65 antibodies in HT22 cells. (C) Shown are representative western blots probed with p-IκB-α and IκB-α antibodies in HT22 cells. (D) Shown is representative western blots probed with cleaved caspase-3 antibody in HT22 cells. (E) Shown is representative western blot probed with BACE1 antibody in HT22 cells. (F) Shown are representative western blots probed with iNOS and COX-2 antibodies in HT22 cells. (G) Shown is representative western blot probed with cleaved IL-1β antibody in HT22 cells. (H) Shown is representative western blot probed with cleaved TNF-α antibody in HT22 cells. (n = 3, *P < 0.05, **P < 0.01 vs. control; #P < 0.05, ##P < 0.01 vs. Aβ25–35 treated alone group, the values were represent as the percentage of the control). | ||
Apoptosis is widely involved in the hypoxic-ischemic injury of the cardiovascular system and the progress of neurodegenerative diseases, for example AD.25 But above all, Aβ25–35-induced excessive oxidative stress and inflammation response can cause hippocampal neurons apoptosis which is a major pathologic factor of AD.25,26 The results showed that exposure to 40 μM Aβ25–35 for 24 h significantly induced cell apoptosis characterized by karyotin condensation and apoptotic bodies, which were reversed by pretreatment with geniposide and TAK 242. More meaningfully, caspase-3, the most important terminal enzymes resulting in apoptosis, remarkably increased after incubation with Aβ25–35. However, pretreatment with geniposide and TAK 242 down-regulated the expression of caspase-3, which further supported that geniposide possessed the neuroprotective effect against Aβ25–35-induced apoptosis. These results indicate that cell apoptosis closely connects with Aβ25–35-induced neurotoxicity and geniposide possibly performs an interesting neuroprotective effect against Aβ25–35-induced apoptosis through inhibiting the caspase cascade. Further research will study the exact neuroprotective mechanism of geniposide. TAK 242, an inhibitor of the TLR4, can restrain the neuroinflammation induced by activated TLR4. Geniposide and TAK 242 not only improved the cell viability and morphology, but also reduced the cell apoptosis indicating that the neuroprotective effect of geniposide possibly is involved in its anti-inflammation activity.
Geniposide reduced Aβ25–35-induced intracellular ROS accumulation
It is well known that ROS, a series of reactive oxygen species produced in aerobic cells during the metabolism including O2−, H2O2, HO2, OH and so on, mainly derives from electronic reveal of the respiratory chain. ROS may trigger subsequent cell death through cell apoptosis and cause damage to neurons. Presently, there is more and more evidence suggests that ROS makes a difference to aggravate the progress of AD.27 Growing evidence reveals that progressive Aβ25–35 deposition further increases ROS generation through activating TLR4 to aggravate the inflammatory response to bring about progressive damage to neurons.27–29Thus ROS accumulation in HT22 cells was measured by using the DCFH-DA to further confirm the inhibitory effect of geniposide on Aβ25–35-induced ROS generation. Fig. 8 shows that HT22 cells are administrated with Aβ25–35 (40 μM) alone for 24 hours perform an evident increase in intracellular ROS level compared to control group indicating that Aβ25–35 can induce oxidative stress. Interestingly, treatment with geniposide (160 μM) and TAK 242 (1 μM) remarkably attenuate ROS level in HT22 cells. The results suggested that the inhibition of ROS generation could be contributed to the neuroprotective effect of geniposide. Moreover, TAK 242 treatment probably attenuated the level of ROS via inhibiting TLR4 to reduce the oxidative stress. Perhaps geniposide inhibited ROS generation in a similar way as TAK 242 treatment. These experimental data show that geniposide can alleviate the Aβ25–35-induced oxidative stress through the inhibition of ROS to protect HT22 cells.
Geniposide attenuated Aβ25–35 – induced nuclear transport of NF-κB/p65
NF-κB/p65 is a transcription factor that plays a key role in the expression of various inflammatory genes. It also has been reported that over-expression of NF-κB accompanied with an increase in Aβ accumulation were found in AD patients, which further supports that NF-κB activation promotes the progress of AD.30 Moreover, recent studies have shown that Aβ25–35 can significantly promote the nuclear translocation of p65 subunit leading to p65 activation and over-expression of inflammatory mediators including TNF-α, IL-1, COX-2 and iNOS.31–33Hence, the nuclear transport of NF-κB/p65 was observed by immunofluorescence test under the fluorescent microscope to discriminate the effect of geniposide treatment on nuclear transport of NF-κB/p65 induced by Aβ25–35. Fig. 9 shows that NF-κB/p65 mainly appears in the cytoplasm of HT22 cells in control group. On the contrary, Aβ25–35 evidently promotes NF-κB/p65 to transfer into nucleus representing NF-κB/p65 is activated by Aβ25–35. The effect of Aβ25–35-induced nuclear transport of NF-κB/p65 is reversed when HT22 cells are treated with geniposide (160 μM) and TAK 242 (1 μM). TAK 242 interdicts the TLR4/NF-κB (p65) signaling pathway to down-regulate a series of inflammatory factors. Correspondingly, treatment with TAK 242 prevented NF-κB/p65 from transferring into the nucleus induced by Aβ25–35. The results indicated that geniposide inhabited nuclear transport of NF-κB/p65 to reduce inflammatory response.
Geniposide decreased the expression of BACE-1
Recent studies prove that high level of AD markers such as Aβ and BACE-1 can cause cognitive dysfunction and Aβ-induced inflammation urged a dendritic pathology with an increase in BACE-1 expression in vitro. Further, it is demonstrated that a decrease in the expression of BACE-1 performed an anti-AD function in adult mouse hippocampus.34 What's more, it has been suggested that NF-κB can combine with the promoter site of BACE-1 indicating NF-κB plays a key role in regulating the level of BACE-1.34 Therefore, we inspected the effect of geniposide on the protein level of BACE-1, which is the pathological marker of AD. Western blot assay indicated that Aβ25–35 administration markedly elevated BACE-1 expression compared to control group. However, geniposide and TAK 242 treatment evidently reversed Aβ25–35-induced the increase in BACE-1 expression. The results revealed that geniposide and TAK 242 treatment exhibited an effective decrease in BACE-1 protein level to against AD (Fig. 10E).The effect of geniposide against Aβ25–35-induced inflammation response in HT22 cells
In recent years, it has been found that the chronic inflammatory response in the brain may be one of the important pathological features of neurodegenerative diseases for example AD. AD, characterized by neuroinflammation in the brain of patients, includes astrocyte proliferation, microglial activation and an increase in pro-inflammatory cytokines such as TNF-α and IL-1β.35 In these hypotheses, some studies have shown that TLR4 activation in the hippocampal neurons regulates the occurrence of neuroinflammation via myeloid differentiation factor 88 (MyD88)-dependent pathway and MyD88-independent pathway.36,37 This theory reminds that hippocampal neurons can secrete inflammatory factors to exacerbate AD. Hence, the intervention of excessive inflammatory response in hippocampal neurons can also reduce the damage to the central nervous system. Thus, there is a consensus that inhibition of excessive inflammation in hippocampal neurons may have the latent capacity to improve central nervous system function of AD patients.Increasing researches confirm that TLR family plays a key role in AD pathology.38 TLR family is mainly expressed in astrocyte or microglia in central nervous system. TLR4 is one of the most important members in TLRs for Aβ neurotoxicity. It has been proved that TLR4 can be activated by Aβ25–35 to up-regulate apoptosis.32,33 Furthermore, TLR4 enhances the generation of inflammatory factors via the NF-κB signal pathway.32 NF-κB/p65 usually binds to the inhibitory protein IKB-α in an inactive form in the cytoplasm of resting cells, but IKB-α phosphorylation will result in this complex dissociation leading to NF-κB/p65 activation once it is stimulated by some specific factors to regulate the expression of several inflammatory genes.34 Interestingly, Aβ25–35-induced the activation of TLR4 activates many cellular signaling pathways including TLR4/NF-κB.
COX-2, which expresses at high levels in the brain, usually locates in excitatory nerve cells especially hippocampal neurons and plays a key role in neuronal apoptosis.39 It is confirmed that the decrease in COX-2 expression significantly ameliorates memory defect in vivo.40 Moreover, increasing researches reveal that IL-1β not only activates astrocytes, but also enhances hemato encephalic barrier permeability to promote hemameba soakage and the expression of TNF-α.41 Excessive expression of IL-1β and TNF-α in AD patients and AD mice were proved to contribute to memory deficit.41 In central nervous system, it is confirmed that TNF-α inhibits the activity of cdk5 to generate the nitric oxide (NO) in hippocampal neurons which is tightly related to cell apoptosis.42 iNOS-produced NO is connected with autoimmune and inflammatory disease, over-expression of iNOS can bring about the hyperphosphorylation of tau proteins and the deposition of Aβ.43 These cell factors are crucial to the inflammatory response, which not only is vital in resisting pathogens, but also promoting the progress of AD.
Therefore, western blot assay was carried out to further confirm whether the anti-AD mechanism of geniposide is involved with the inhibition of inflammatory response in HT22 cells, the expression of TNF-α, IL-1β, COX-2 and iNOS was detected to further assess the effect of geniposide on inflammatory factors. Results showed that Aβ25–35 enhanced the expression of TNF-α, IL-1β, COX-2 and iNOS compared to the control group. While a significant decrease was observed in HT22 cells treated with geniposide (160 μM) and TAK 242 (1 μM). The experimental achievement approves that Aβ25–35 could trigger HT22 cells to secrete inflammatory factors. What's more, geniposide and TAK 242 can alleviate the inflammatory response via decreasing the level of TNF-α, IL-1β, COX-2 and iNOS (Fig. 10F–H).
On the other hand, the expression of TLR4, an Aβ25–35 signal receptor, was markedly amplified after exposure to Aβ25–35 compared to the control group and geniposide (160 μM) and TAK 242 (1 μM) treatment significantly decreased the Aβ25–35-induced higher TLR4 expression. Some researches prove that Aβ25–35-activated TLR4 signal pathway regulates the phosphorylation and nuclear transfer of NF-κB/p65 44. IκB-α phosphorylation is mainly related with the NF-κB activation that is initiated by several inflammation irritations. The results revealed that incubation with Aβ25–35 for 24 h obviously enhanced IκB-α phosphorylation leading to NF-κB/p65 phosphorylation compared to the control group. Meaningfully, the results also demonstrated that geniposide and TAK 242 inhibited Aβ25–35-induced IκB-α phosphorylation and NF-κB/p65 activation in HT22 cells (Fig. 10A–C).
Overall, geniposide and TAK 242 pretreatment not only notably down-regulated the expression of TLR4, but also inhibited IKB-α and NF-κB/p65 phosphorylation caused by Aβ25–35. In addition, geniposide and TAK 242 pretreatment inhibited Aβ25–35-mediated NF-κB transfer into the nucleus, thus leading to the decrease in level of IL-1β, TNF-α, COX-2 and iNOS. These experimental achievements support that Aβ25–35 deposition is related to the activation of the TLR4/NF-κB signal pathway in HT22 cells and geniposide can reverse it.
Experimental section
Reagents and drugs
Dulbecco's modified eagle medium (DMEM), fetal bovine serum (FBS) and trypsin (0.25%) were purchased from Gibco (USA). Phosphate buffer saline (PBS) was purchased from Hyclone (USA). Lactate dehydrogenase (LDH) cytotoxicity assay kit, hoechst 33342 staining solution and nuclear transfer of NF-κB assay kit were purchased from Biyuntian (China). AnnexinV-EGFP/PI apoptosis detection kit was purchased from LiankeBio (China). Reactive oxygen species (ROS) assay kit was purchased from NanJingJianCheng (China). Aβ25–35, methylthiazolyl tetrazolium (MTT), dimethyl sulfoxide (DMSO) were purchased from sigma (USA). β-actin and TLR4 antibodies, p65 and phosphorylation-p65 (p-p65) antibodies, TNF-α, COX-2, iNOS, IκB-α and p-IκB-α antibodies, BACE1 and caspase-3 antibodies were purchased from proteintech (USA), IL-1β antibody was purchased from Cell Signaling Technology (USA). TAK 242 (purity > 99%) was obtained from MCE (USA). Geniposide (purity > 98%) was obtained from solarbio (USA) and contains no endotoxin.HT22 cells culture and drugs preparation
HT22 cell, obtained from Sun Yat-Sen Memorial Hospital Laboratory, is a sub-line derived from parent HT4 cells that were originally immortalized from primary mouse hippocampal neuronal culture. HT22 cell is a good model for studying AD pathology in vitro. HT22 cells were grown in DMEM containing 10% FBS at 37 °C in the incubator with 5% CO2. Aβ25–35 was dissolved in double distilled water and were filtered with a 0.22 μm filter, then Aβ25–35 solution were polymers formed from incubating at 37 °C in the incubator for 7 days and stored at −20 °C in darkness before use. Simultaneously, geniposide and TAK 242 were dissolved in double distilled water and DMSO, respectively and were filtered with a 0.22 μm filter and stored at −20 °C in darkness until use.Assessment of the appropriate concentrations of drugs
The appropriate concentrations of geniposide and Aβ25–35 were measured by the MTT assay. Briefly, HT22 cells were grown at a density of 2.5 × 103 cells per well in 96-well plates in 200 μL medium. After 12 h, HT22 cells were treated with various concentrations of Aβ25–35 (0, 5, 10, 20, 40 μM) for 24 h and geniposide (0, 10, 20, 40, 80, 160, 320, 640 μM) for 24 h and 48 h. Subsequently, the cells were incubated with 20 μL MTT solution (5 mg mL−1) for 4 h followed by the medium was removed and added with 150 μL DMSO. At last, the absorbance at 490 nm was assessed using a Biotek-Eon microplate reader. The MTT assay was repeated three times. In the meantime, the effect of various concentrations of Aβ25–35 and geniposide on morphology of HT22 cells was observed by inverted microscope.Cell treatment
In all experiments, HT22 cells in treated groups were pretreated with geniposide (160 μM) or TAK 242 (1 μM) for 1 h,34 followed by incubation with Aβ25–35 (40 μM) for 24 h. While cells in control group or Aβ group were incubated with medium or Aβ25–35 (40 μM) alone for 24 h.Cell viability assay
The effect of geniposide on HT22 cell viability was measured by the MTT and LDH assays. HT22 cells were grown at a density of 2.5 × 103 cells per well in 96-well plates in 200 μL medium for 12 h. Then HT22 cells were pretreated with geniposide (160 μM) or TAK 242 (1 μM) for 1 h, followed by incubation with Aβ25–35 (40 μM) for 24 h. While cells in control group or Aβ group were incubated with medium or Aβ25–35 (40 μM) alone for 24 h. Following this, the MTT and LDH assays were performed according to the product's instructions. For LDH assay, the supernatant of all wells was taken out to detect the level of LDH according to the previous study.45 At the same time, the morphology of HT22 cells in each group were observed by inverted microscope.ROS measurement
The effect of geniposide on the level of ROS in HT22 cells was determined by using a ROS assay kit. HT22 cells were seeded at a density of 1.5 × 104 cells per well in 24-well plates and cultured for 12 h, then cells were pretreated with geniposide (160 μM) or TAK 242 (1 μM) for 1 h prior to exposure to Aβ25–35 (40 μM) for 24 h. The cells in each well were stained with DCFH-DA solution (4 μM) and incubated for 15 min followed by being washed twice with PBS. Fluorescent signals were caught employing a fluorescence microscope linked to an image-forming system. Three visual fields were stochastically chosen to gauge the average fluorescence intensity and interpretated by Image J software.Cell apoptosis assay
Immunofluorescence test
To confirm the effect of geniposide on translocation of NF-κB, HT22 cells were planted at a density of 4 × 103 cells per well in 48-well plates and cultured for 12 h. Then cells were pretreated with geniposide (160 μM) or TAK 242 (1 μM) for 1 h before incubation with Aβ25–35 (40 μM) for 24 h, then cells were washed with PBS once and fixed with fixative solution for 15 min at room temperature (RT). HT22 cells were incubated with immunostaining sealing fluid for 1 h at RT to suppress the nonspecific reaction after being washed with PBS three times. Then cells were incubated with anti-NF-κB for 1 h at RT. After being washed with PBS three times, cells were covered with anti-rabbit Cy3 for 1 h and then nuclei were incubated with DAPI staining solution for 5 min after being washed with PBS twice. At last, the fluorescence signals of cells were caught with the fluorescence microscope. All nuclear translocation of NF-κB-positive cells in three visual fields were calculated to quantizate and compare between groups.Western blot analysis
To confirm the effect of geniposide on anti-inflammation and anti-AD in relation to TLR4/NF-κB (p65) signal pathway in HT22 cells, HT22 cells were cultured at a density of 10 × 104 cells per well in 6-well plates and cultured for 12 h. Then cells were pretreated with geniposide (160 μM) or TAK 242 (1 μM) for 1 h before exposure toAβ25–35 (40 μM) for 24 h, western blot analysis was performed as described previously,46 in addition to the dilution ratios for the different first antibodies used: TLR4, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500; IKB-α, 1:1000; p-IKB-α, 1:2000; p65, 1:2000; p-p65, 1:500; COX2, 1:500; iNOS, 1:500; TNF-α, 1:4000; IL-1β, 1:1000; caspase-3, 1:1000; BACE1, 1:1000; β-actin, 1:2000.
:500; IKB-α, 1:1000; p-IKB-α, 1:2000; p65, 1:2000; p-p65, 1:500; COX2, 1:500; iNOS, 1:500; TNF-α, 1:4000; IL-1β, 1:1000; caspase-3, 1:1000; BACE1, 1:1000; β-actin, 1:2000.
Statistical analysis
The total data were expressed as mean ± SEM. Differences between groups were assessed using one-way analysis of variance followed by least significant difference test using the Statistical Program for Social Sciences 22.0 software. P < 0.05 was recognized as statistically significant.Conclusion
In general, the results confirm that geniposide is a potential treatment approach for AD, which are related to anti-neuroinflammation and anti-apoptosis. Our study supplies a novel strategy to target TLR4/NF-κB signal pathway in hippocampal neurons for the therapy of AD. The study revealed that geniposide inhibited Aβ25–35-induced neuroinflammatory, oxidative-stress and apoptosis in relation to TLR4/NF-κB signal pathway. Nevertheless, more study is necessary for exploring the further mechanism of geniposide in all kinds of neurodegenerative disorders in vivo and in vitro.Author contributions
Qi-Hui Huang and Yuan-Hui Deng designed the study; Xiu-Fang Huang and Jian-Jun Li wrote the manuscript. Xiu-Fang Huang, Yan-Gu Tao and Xie-Qi Wang performed apoptosis and western blot assays. Zu-Qing Su, Ru-Lan Zhang and Jia-Lin Zhang performed MTT and immunofluorescence tests.Conflicts of interest
All authors stated that they have no conflict interests.Abbreviations
| AD | Alzheimer's disease |
| Aβ | Amyloid β-protein |
| MTT | Methylthiazolyl tetrazolium |
| LDH | Lactate dehydrogenase |
| ROS | Reactive oxygen species |
| iNOS | Nitric oxide species |
| TNF-α | Tumor necrosis factor-α |
| COX-2 | Cyclooxygenase-2 |
| IL-1β | Interleukin-1β |
| TLRs | Toll-like receptors |
| TLR4 | Toll-like receptor 4 |
| NF-κB | Nuclear factor-κappaB |
| IκB-α | Inhibitor of NF-κB, alpha |
| BACE1 | Beta-secretase 1 |
| DMEM | Dulbecco's Modified Eagle Medium |
| FBS | Fetal bovine serum |
| PBS | Phosphate-buffered saline |
| DMSO | Dimethyl sulfoxide |
Acknowledgements
This work was supported by the Free application project of Guangdong Provincial Natural Science Foundation, No. 2014A030313147.References
- N. Origlia, C. Criscuolo, O. Arancio, S. S. Yan and L. Domenici, J. Neurosci., 2014, 34, 8749–8760 CrossRef PubMed.
- B. A. in t' Veld, A. Ruitenberg, A. Hofman, L. J. Launer, C. M. van Duijn, T. Stijnen, M. M. Breteler and B. H. Stricker, N. Engl. J. Med., 2001, 345, 1515–1521 CrossRef PubMed.
- P. L. McGeer, S. Itagaki, B. E. Boyes and E. G. McGeer, Neurology, 1988, 38, 1285–1291 CrossRef PubMed.
- J. Rogers, S. Webster, L. F. Lue, L. Brachova, W. H. Civin, M. Emmerling, B. Shivers, D. Walker and P. McGeer, Neurobiol. Aging, 1996, 17, 681–686 CrossRef PubMed.
- H. Liu, J. Wang, J. Wang, P. Wang and Y. Xue, Brain Res., 2015, 1618, 149–158 CrossRef PubMed.
- M. Serpente, R. Bonsi, E. Scarpini and D. Galimberti, NeuroImmunoModulation, 2014, 21, 79–87 CrossRef PubMed.
- H. Akiyama, S. Barger, S. Barnum, B. Bradt, J. Bauer, G. M. Cole, N. R. Cooper, P. Eikelenboom, M. Emmerling, B. L. Fiebich, C. E. Finch, S. Frautschy, W. S. Griffin, H. Hampel, M. Hull, G. Landreth, L. Lue, R. Mrak, I. R. Mackenzie, P. L. McGeer, M. K. O'Banion, J. Pachter, G. Pasinetti, C. Plata-Salaman, J. Rogers, R. Rydel, Y. Shen, W. Streit, R. Strohmeyer, I. Tooyoma, F. L. Van Muiswinkel, R. Veerhuis, D. Walker, S. Webster, B. Wegrzyniak, G. Wenk and T. Wyss-Coray, Neurobiol. Aging, 2000, 21, 383–421 CrossRef PubMed.
- J. W. Guo, P. P. Guan, W. Y. Ding, S. L. Wang, X. S. Huang, Z. Y. Wang and P. Wang, Biomaterials, 2017, 145, 106–127 CrossRef PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef PubMed.
- S. Gordon, Cell, 2002, 111, 927–930 CrossRef PubMed.
- M. Fukata, A. S. Vamadevan and M. T. Abreu, Semin. Immunol., 2009, 21, 242–253 CrossRef PubMed.
- M. Hashimoto, S. Hossain, Y. Tanabe, A. Kawashima, T. Harada, T. Yano, K. Mizuguchi and O. Shido, J. Nutr. Biochem., 2009, 20, 965–973 CrossRef PubMed.
- Y. F. Qing, Q. B. Zhang, J. G. Zhou and L. Jiang, Rheumatol. Int., 2014, 34, 213–220 CrossRef PubMed.
- L. A. Stark and M. G. Dunlop, Mol. Cell. Biol., 2005, 25, 5985–6004 CrossRef PubMed.
- K. N. Nam, Y. S. Choi, H. J. Jung, G. H. Park, J. M. Park, S. K. Moon, K. H. Cho, C. Kang, I. Kang, M. S. Oh and E. H. Lee, Int. Immunopharmacol., 2010, 10, 493–499 CrossRef PubMed.
- J. Lu, J. S. Wang and L. Y. Kong, J. Ethnopharmacol., 2011, 134, 911–918 CrossRef PubMed.
- C. Lv, L. Wang, X. Liu, X. Cong, S. S. Yan, Y. Wang and W. Zhang, Curr. Alzheimer Res., 2014, 11, 430–440 CrossRef PubMed.
- C. Lv, L. Wang, X. Liu, S. Yan, S. S. Yan, Y. Wang and W. Zhang, Neuropharmacology, 2015, 89, 175–184 CrossRef PubMed.
- H. J. Koo, Y. S. Song, H. J. Kim, Y. H. Lee, S. M. Hong, S. J. Kim, B. C. Kim, C. Jin, C. J. Lim and E. H. Park, Eur. J. Pharmacol., 2004, 495, 201–208 CrossRef PubMed.
- T. Q. Pham, F. Cormier, E. Farnworth, V. H. Tong and M. R. Van Calsteren, J. Agric. Food Chem., 2000, 48, 1455–1461 CrossRef PubMed.
- J. Mao, Y. Hu, A. Zhou, B. Zheng, Y. Liu, Y. Du, J. Li, J. Lu and P. Zhou, Neural Regener. Res., 2012, 7, 2333–2339 Search PubMed.
- G. X. Zhang, A. L. Zhou, G. P. Zhang, Y. E. Hu and J. H. Mao, Zhongguo Yingyong Shenglixue Zazhi, 2013, 29, 42–46 Search PubMed.
- S. J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hyman, M. A. Burton, L. E. Goldstein, S. Duong, R. E. Tanzi and R. D. Moir, PLoS One, 2010, 5, e9505 Search PubMed.
- A. A. Pieper, S. Xie, E. Capota, S. J. Estill, J. Zhong, J. M. Long, G. L. Becker, P. Huntington, S. E. Goldman, C. H. Shen, M. Capota, J. K. Britt, T. Kotti, K. Ure, D. J. Brat, N. S. Williams, K. S. MacMillan, J. Naidoo, L. Melito, J. Hsieh, J. De Brabander, J. M. Ready and S. L. McKnight, Cell, 2010, 142, 39–51 CrossRef PubMed.
- A. Degterev, Z. Huang, M. Boyce, Y. Li, P. Jagtap, N. Mizushima, G. D. Cuny, T. J. Mitchison, M. A. Moskowitz and J. Yuan, Nat. Chem. Biol., 2005, 1, 112–119 CrossRef PubMed.
- F. G. Gervais, D. Xu, G. S. Robertson, J. P. Vaillancourt, Y. Zhu, J. Huang, A. LeBlanc, D. Smith, M. Rigby, M. S. Shearman, E. E. Clarke, H. Zheng, L. H. Van Der Ploeg, S. C. Ruffolo, N. A. Thornberry, S. Xanthoudakis, R. J. Zamboni, S. Roy and D. W. Nicholson, Cell, 1999, 97, 395–406 CrossRef PubMed.
- W. L. Fang, D. Q. Zhao, F. Wang, M. Li, S. N. Fan, W. Liao, Y. Q. Zheng, S. W. Liao, S. H. Xiao, P. Luan and J. Liu, CNS Neurosci. Ther., 2017, 23, 428–437 CrossRef PubMed.
- R. F. Zanin, L. S. Bergamin, F. B. Morrone, R. Coutinho-Silva, A. T. de Souza Wyse and A. M. Battastini, Purinergic Signalling, 2015, 11, 463–470 CrossRef PubMed.
- A. Singh, B. Koduru, C. Carlisle, H. Akhter, R. M. Liu, K. Schroder, R. P. Brandes and D. M. Ojcius, Sci. Rep., 2017, 7, 14346 CrossRef PubMed.
- I. Blasko, A. Apochal, G. Boeck, T. Hartmann, B. Grubeck-Loebenstein and G. Ransmayr, Neurobiol. Dis., 2001, 8, 1094–1101 CrossRef PubMed.
- Y. C. Liu, X. X. Gao, L. Chen and X. Q. You, Neuroscience, 2017, 355, 188–199 CrossRef PubMed.
- H. L. Yu, X. Y. Li, X. Zhou, L. H. Yuan, W. W. Ma, Y. D. Xi, X. Zhao, J. Wu and R. Xiao, J. Mol. Neurosci., 2013, 51, 771–778 CrossRef PubMed.
- S. C. Tang, J. D. Lathia, P. K. Selvaraj, D. G. Jo, M. R. Mughal, A. Cheng, D. A. Siler, W. R. Markesbery, T. V. Arumugam and M. P. Mattson, Exp. Neurol., 2008, 213, 114–121 CrossRef PubMed.
- H. Badshah, T. Ali and M. O. Kim, Sci. Rep., 2016, 6, 24493 CrossRef PubMed.
- A. R. Kamer, R. G. Craig, A. P. Dasanayake, M. Brys, L. Glodzik-Sobanska and M. J. de Leon, Alzheimer's Dementia, 2008, 4, 242–250 CrossRef PubMed.
- A. Aderem and R. J. Ulevitch, Nature, 2000, 406, 782–787 CrossRef PubMed.
- F. Vazquez, S. Ramaswamy, N. Nakamura and W. R. Sellers, Mol. Cell. Biol., 2000, 20, 5010–5018 CrossRef PubMed.
- T. Kawai and S. Akira, Nat. Immunol., 2010, 11, 373–384 CrossRef PubMed.
- Y. Shen, R. Li and K. Shiosaki, J. Biol. Chem., 1997, 272, 3550–3553 CrossRef PubMed.
- K. P. Townsend and D. Pratico, FASEB J., 2005, 19, 1592–1601 CrossRef PubMed.
- U. Solomon and E. A. Taghogho, Brain Res. Bull., 2017, 131, 133–141 CrossRef PubMed.
- L. E. Rojo, J. A. Fernandez, A. A. Maccioni, J. M. Jimenez and R. B. Maccioni, Arch. Med. Res., 2008, 39, 1–16 CrossRef PubMed.
- L. Zhou and D. Y. Zhu, Nitric Oxide, 2009, 20, 223–230 CrossRef PubMed.
- X. Zhou, L. Yuan, X. Zhao, C. Hou, W. Ma, H. Yu and R. Xiao, Nutrition, 2014, 30, 90–95 CrossRef PubMed.
- X. Gou, Q. Wang, Q. Yang, L. Xu and L. Xiong, J. Drug Targeting, 2011, 19, 86–95 CrossRef PubMed.
- Z. Suo, M. Wu, S. Ameenuddin, H. E. Anderson, J. E. Zoloty, B. A. Citron, P. Andrade-Gordon and B. W. Festoff, J. Neurochem., 2002, 80, 655–666 CrossRef PubMed.
Footnote |
| † Xiu-Fang Huang and Jian-Jun Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |