
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired temporal supramolecular polymerization
Shikha
Dhiman
 ,
Aritra
Sarkar
and
Subi J.
George
*
,
Aritra
Sarkar
and
Subi J.
George
*
Supramolecular Chemistry Laboratory, New Chemistry Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore, India-560064. Web: http://www.jncasr.ac.in/georgeE-mail: george@jncasr.ac.in; subijg@gmail.com
First published on 22nd May 2018
Abstract
Thriving natural systems precisely regulate their complex chemical organizations in space and time by recruitment of a complex network of fuel-driven, kinetically controlled, out-of-equilibrium transformations. Indeed this provides an active, adaptive and autonomous smart actions & functions. In contrast, synthetic systems exhibit simpler behavior owing to thermodynamically driven supramolecular polymerization with no temporal modulation of spatial organization. Stimulated by an outstanding control that nature demonstrates, a drive towards artificial out-of-equilibrium systems with the ambition to program activation and duration of structural transformations has emerged. To realize this vision, overwhelming efforts across the globe have been initiated to design temporally programmed synthetic supramolecular polymers. In an attempt to contribute to this trending field, our supramolecular chemistry group has thoroughly investigated a structure–property relationship that determines the mechanism of supramolecular polymerization. Exploiting these mechanistic insights, along with a bio-inspired fuel-driven enzyme mediated approach, we further attempted to program supramolecular polymers in both structural and temporal regimes. We believe, nature is the inspiration to the current era challenges and it also provides with the solution, a fuel-driven approach to address these. In this account, we shall discuss the efforts made by our group to build generic concept to create temporally programmable supramolecular polymers.
 Shikha Dhiman | Shikha Dhiman did her BSc in Chemistry from University of Delhi, India in 2014. She then finished her Masters in Chemical Sciences from JNCASR Bangalore, India in 2017. She is now pursuing her PhD under the guidance of Prof. Subi George at the New Chemistry Unit, JNCASR, Bangalore, India. Her research area of interest includes kinetically controlled supramolecular polymerization and non-equilibrium transient supramolecular polymerization using a bio-inspired fuel-driven approach. |
 Aritra Sarkar | Aritra Sarkar received his BSc in Chemistry from Krishnath College, Berhampore under Kalyani University in 2013 and his MSc in Chemical Science from Indian Institute of Technology, Guwahati in 2015. He joined as a PhD student in New Chemistry Unit, in JNCASR, Bangalore in 2015 under the supervision of Prof. Subi George and is working on fundamental investigation on supramolecular polymerization, chirality driven self-sorting and kinetically controlled supramolecular polymerization. |
 Subi J. George | Subi J. George is presently an Associate Professor at the New Chemistry Unit, School of Advanced Materials (SAMat) of JNCASR, Bangalore, India and is leading a supramolecular chemistry group. He has achieved his PhD degree at the National Institute for Interdisciplinary Science and Technology, India in 2005 under the supervision of Prof. A. Ajayaghosh. He pursued his post-doctoral research at the Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, The Netherlands, under Prof. Dr E. W. (Bert) Meijer. His current research interests emphasize on functional organic and hybrid materials, supramolecular chirality, temporally controlled fuel-driven non-equilibrium supramolecular polymers. |
Introduction
Supramolecular polymers are molecular assemblies composed of monomeric units that are held together by dynamic and reversible non-covalent bonds.1 Thus, supramolecular polymers are different from conventional covalent polymers where monomeric or repeating units are connected by covalent bonds. The weak and directional non-covalent interactions such as hydrogen bonding, π–π interaction, metal coordination, and host–guest interaction are highly reversible and thus supramolecular polymers exhibit dynamic and responsive behavior, unlike covalent polymers. During the last two decades, various classes of functional supramolecular polymers have been designed for various applications using non-covalent synthetic strategies and they exhibited unique properties.2As the development goes further, newer challenges have emerged in the field of supramolecular polymers that demand a structural control for better functional performances and more complexity of these supramolecular materials.3 Precisely, the two grand challenges identified are (i) structural control and (ii) temporal control over supramolecular polymers. Structural control aims towards achieving control over length and dispersity of supramolecular polymers and synthesis of complex multicomponent block copolymers as has been achieved in conventional covalent polymers.4 This is very important for controlling the functional outcome of these class of materials. In contrast, temporal control targets a pre-programmability of time duration of a dynamic structural and functional state of supramolecular polymer similar to biological systems.5 This is important for designing life-like non-equilibrium materials with programmed growth and decay with a tunable lifetime of the steady state.
In conventional covalent polymers, a precise structural control has been achieved via living polymerization reactions; a controlled chain-growth polymerization where the ability of the polymers to terminate is minimized.6 Using various living polymerization strategies dispersity control and block copolymer architectures have been designed in covalent polymers, which expanded the functional scope of these polymers to emerging fields like organic photovoltaics and soft-lithography. Inspired by this, the field of living supramolecular polymerization has come into interest to achieve precise structural control and to design novel materials such as supramolecular block copolymers. However, it demands a clear understanding on the mechanistic aspects of supramolecular polymerization7 and later it has been realized that the challenges are quite different from its covalent counterparts, and this will be discussed in detail in the subsequent sections.
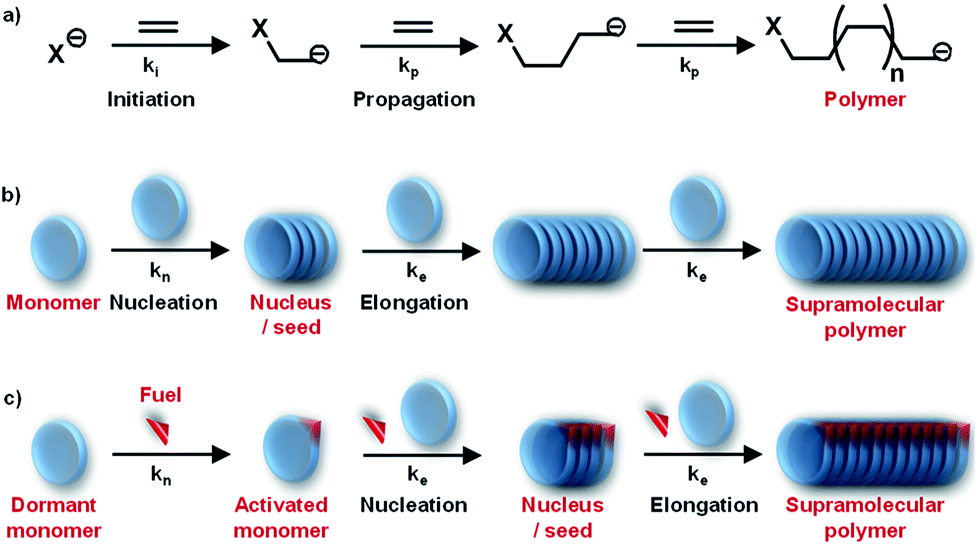
Living beings are highly complex, self-assembled systems composed of cascades of chemical reactions and processes occurring simultaneously and in tandem.8 Fundamental understandings at the cellular level have suggested that structural changes of these assemblies happen out-of-equilibrium with a precise temporal control. This temporal programming is very important for various biological functions such as mobility and signaling. For instance, actin filaments are responsible for cell motility which require structural reorganization, precisely a sequential increase and decrease in the length of these filaments.9 In order to achieve this, nature utilizes G-actin protein monomers that lack in potency to self-assemble and triggers their spontaneous supramolecular polymerization by utilizing a chemical fuel-driven strategy (Fig. 1a). Lack in potency to self-assemble suggests thermodynamic stability of G-actin protein monomers, hence the polymerization process is an energetically uphill process where the polymerized state is a non-equilibrium thermodynamic state. To facilitate this process, the system needs energy uptake that is consumed as energy penalty. This energy is provided by the bio-fuel adenosine triphosphate (ATP) which is dissipated as adenosine diphosphate (ADP). Since the polymerization is enabled by fuel (ATP), its continuous input is necessary to keep the system under non-equilibrium polymerized form. As fuel is completely consumed, the depolymerization occurs, thus, amount of fuel (ATP) determines the time duration of non-equilibrium state. Since the non-equilibrium state occurs temporarily for a particular time, the state is also referred as the transient state. Hence, to achieve a temporal control over supramolecular structure and functional states and build complex smart systems like biological systems, we need to design systems which work under non-equilibrium. Moreover, this would yield a novel class of transient and adaptive materials with pre-programmed functions.10
 | ||
| Fig. 1 Schematic illustration of (a) ATP-fueled living and transient supramolecular polymerization of actin filaments and (b) desired bio-inspired chemical-fuel driven synthetic supramolecular polymer, where temporal programing is mediated by coupling a deactivating agent to the fuel. | ||
The challenges discussed above come from two different origins and address diverse characteristics i.e. structural and temporal control. For these, it requires careful designing of monomers and precise understanding of their supramolecular mechanisms. In this regard, recent research on the fundamental understanding of mechanism of supramolecular polymerization has enormously helped in identifying the appropriate self-assembly mechanisms and molecular characteristics, which is very important in addressing these new challenges.
In this account, we shall discuss the recent efforts made by our group to address these grand challenges in the supramolecular polymerization process. This includes mechanistic investigations of supramolecular polymerization11 and new non-covalent synthetic strategies to obtain structural12 and temporal control over supramolecular polymerization and the resultant structural programmability. We have introduced a unique bio-inspired fuel-driven approach to achieve both living (controlled) and non-equilibrium (transient) supramolecular polymerization which can be extended to diverse monomeric structures (Fig. 1b). Our design strategies along with the future challenges in this area would be discussed in this account.
Mechanistic investigation
By the interplay of various weak non-covalent interactions such as hydrogen bonding, π–π interaction, dipole moment and van der Waals interactions, monomers undergo self-assembly into supramolecular polymeric structures. Generally, these reversible supramolecular interactions result in the formation of thermodynamically most stable assemblies by an energy downhill process. Mechanistic investigations on the supramolecular polymerization have been first investigated by Meijer and coworkers and they defined that the mechanism of supramolecular polymerization can be divided into two major categories namely isodesmic and cooperative mechanism.7 The isodesmic mechanism involves the addition of monomers to short oligomers or to other monomers to form a polymeric structures via an energy downhill process, similar to step-wise polymerization mechanism in covalent polymers (Fig. 2a and c). The Gibbs free energy of each monomer addition is independent of the length of the supramolecular polymerization and hence the process is also referred as the equal-K mechanism. In contrast, cooperative mechanism is a two-step process with the former known as nucleation (Kn) step that involves formation of a critical size oligomer called as nucleus via an energy uphill process and the latter step known as elongation step (Ke) where a further association of monomers to these nucleus occurs via an energy downhill process (Fig. 2b and c).13 This process is also called as nucleation–elongation mechanism analogous to the chain-growth polymerization in covalent polymers (Fig. 3a and b).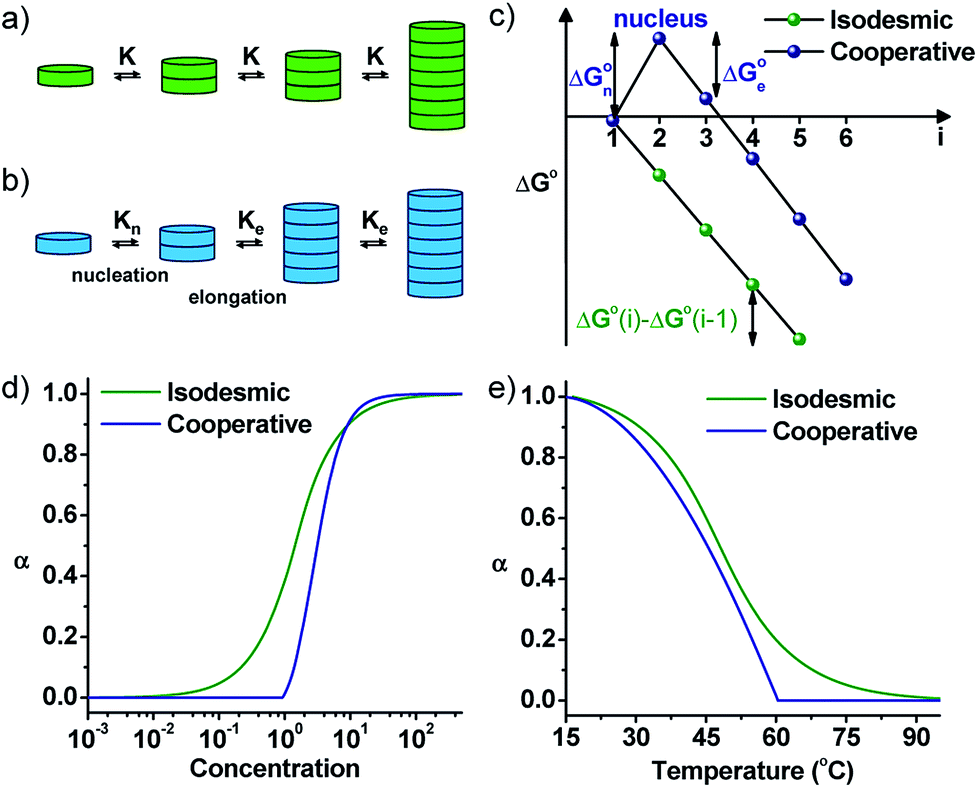 | ||
| Fig. 2 Schematic representations of (a) isodesmic and (b) cooperative mechanism of supramolecular polymerization showing equal K and nucleation–elongation steps, respectively. (c) The standard Gibbs free energy vs. oligomer size, (i) plot representing a constant slope for an isodesmic growth and change in slope at critical nucleus size for a cooperative growth. Fraction of aggregates versus (d) concentration and (e) temperature for isodesmic and cooperative growth depicted by a sigmoidal and non-sigmoidal with critical nucleation step profile, respectively (adapted with permission from ref. 7. Copyright (2009) American Chemical Society). | ||
 | ||
| Fig. 3 (a) Anionic chain-growth polymerization in covalent polymers, (b) cooperative supramolecular polymerization and (c) fuel-driven kinetically controlled cooperative supramolecular polymerization. | ||
Mechanism of supramolecular polymerization that a particular monomer undergoes can be probed via, concentration and temperature dependent changes during supramolecular polymerization process (Fig. 2d and e). A sigmoidal growth suggests an isodesmic pathway, whereas a non-sigmoidal growth with critical point depicts a cooperative mechanism. The cooperative mechanism is described by a parameter called the cooperativity parameter, σ = Kn/Ke, where Kn and Ke are the equilibrium constants for nucleation and elongation, respectively (Fig. 2d and e). In case of isodesmic growth, σ = 1. The mechanistic investigations and understanding of supramolecular polymerization by Meijer and coworkers enables the structural and functional modulation of supramolecular polymers. These mechanisms, the thermodynamics and kinetics of growth, along with many examples have been well-described in excellent reviews by Meijer and coworkers and is out of the scope for this review.1a,7 Similar to chain-growth polymerization, monomers undergoing a cooperative mechanism has a narrow length distribution than molecules undertaking an isodesmic pathway.
Although a number of supramolecular systems have been studied to realize the mechanism of supramolecular polymerization, until a few years ago, no rational design strategy was available to program the mechanism of supramolecular polymerization that it will undergo. Thus considering the above mentioned importance of nucleation–elongation mechanism it was important to rationalize the design strategy or supramolecular interactions that would dictate the monomer to follow an isodesmic or cooperative mechanism of self-assembly. In this context, our group has undertaken a structure–property study on previously reported supramolecular polymers to arrive at a rational design for the monomer structures that follow a cooperative growth.14
Initial reports in literature and the extensive work by Meijer and coworkers suggested the importance of characteristic molecular features like presence of intermolecular hydrogen bond to govern the cooperativity of a supramolecular system. For example, Meijer and coworkers reported N,N′,N′′-trialkyl derivative of benzene-1,3,5-tricarboxamide (BTA), (1) that follows a cooperative mechanism to form long columnar aggregates facilitated by triple helical intermolecular hydrogen bonding between amide groups along the stacking direction aided by hydrophobic interaction (Fig. 4a).15 On the other hand, in BTA derivatives containing large 3,3′-bis(acylamino)-2,2′-bipyridine groups (2) on the amide nitrogen atoms, the hydrogen bonding occurs intramolecularly between bipyridine groups and the amide hydrogen and not in the stacking direction (Fig. 4a). As a result, the self-assembly is mainly aided by hydrophobic interaction and thus the system undergoes an isodesmic pathway of self-assembly.16 The main difference between the two molecules is the orientation of hydrogen bonding, in 1 the intermolecular hydrogen bonding is oriented along the stacking direction and in 2 intramolecular hydrogen bonding along the plane of the monomer is present.17 Apart from these, a plethora of systems that lack in any hydrogen bonding motif is also known to undergo a cooperative mechanism. Hence there must be other molecular features and not explicitly hydrogen bonding that dictates cooperativity. Thus, we envisaged to address this and attribute the mechanism of supramolecular polymerization to specific molecular design.
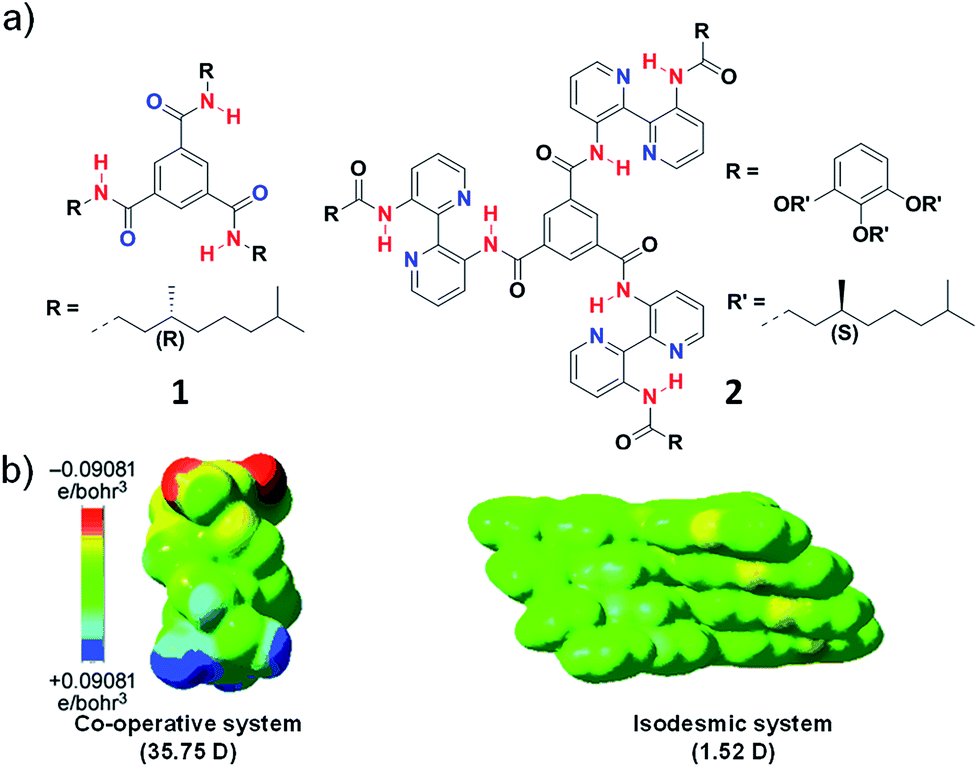 | ||
| Fig. 4 (a) Chemical structure of molecules 1 and 2. (b) Tetramer of the molecules showing electrostatic potential isosurfaces as attained from the BLYP/DZVP//B97D/ccPVDZ level of theory. The dipole moment in stacking direction shows 35.75 D and 1.52 D for molecule 1 and 2 (reprinted with permission from ref. 14. Copyright 2013 Wiley-VCH). | ||
In this context, we have published our perspective on the molecular features that govern the mechanism of supramolecular polymerization and readers may refer it for further understanding.14 We surveyed previously reported monomers that vary in their chemical structure and attempted to rationalize their mechanism of supramolecular polymerization. Based on detailed computational and experimental studies, we have proposed that permanent long-range interactions (any interaction which varies as R−n where R is the distance between the two interacting systems and n ≤ 3) like dipole–dipole interaction or electrostatic interaction governs the cooperative growth of a supramolecular polymer, whereas absence of these long-range interaction leads to isodesmic growth.14 These observations suggest the importance of electronic interactions (hydrogen bonding, electrostatic interaction, dipole–dipole interaction) in determining the cooperativity. Therefore, we believe that permanent dipole moment in the assembling/growing direction permits interaction between neighboring as well as distant molecules via long-range dipole–dipole interactions. As a result, an enrichment in stabilization of stacks occurs. This continuous enhancement in stability of supramolecular polymer with subsequent addition of monomers is the cooperativity in the process of supramolecular polymerization. In absence of these long-range interactions, no enhancement in stability occurs and thus leads to isodesmic mechanism of supramolecular polymerization.
To validate our hypothesis theoretically, we have performed computational studies on BTA monomers 1 and 2, experimentally investigated by Meijer and coworkers and analyzed the long-range interactions in their supramolecular stacks (Fig. 4). The self-assembly was optimized up to tetramers and to identify long-range interactions, calculation of total self-consistent field electron density mapped on to isosurfaces of molecular potential was performed. The calculations suggested a large macrodipole moment of 35.75 D in tetramer of 1, occurring due the charge polarization along the stacking direction. In contrast, tetramer of 2, does not show any significant charge polarization in the stacking direction and hence a low macrodipole moment of 1.52 D (Fig. 4b). Thus, these calculations support our hypothesis that long-range interactions such as dipole moment dictate the mechanism of supramolecular polymerization is justified.
To further validate our results experimentally, we further designed molecules exhibiting dipole–dipole interactions in stacking direction apart from hydrogen bonding interactions. With this objective, we have synthesized three perylene diimide derivatives appended with cholesterol or alkyl groups as self-assembling moiety and linked with either carbonate or ether groups (3–5) (Fig. 5a).18 The molecule 3 contains a cholesterol as rigid assembly moiety along with dipolar carbonate linker. Molecule 5 contains flexible swallow-tail self-assembling moiety and an ether linker that lacks in dipolar nature. In contrast, molecule 4 contains rigid cholesterol self-assembling motif and ether linker. None of the molecules contain hydrogen bonding motifs and hence, we can explicitly investigate the effect of long-range interactions and the effect of the dipolar linker as well as self-assembling motif. Spectroscopic studies in appropriate conditions (concentration and solvent composition) depicted that molecule 3 follows a cooperative pathway to form a helical assembly but molecules 4 and 5 undergo an isodesmic route to yield a helical and achiral assembly, respectively (Fig. 5b).
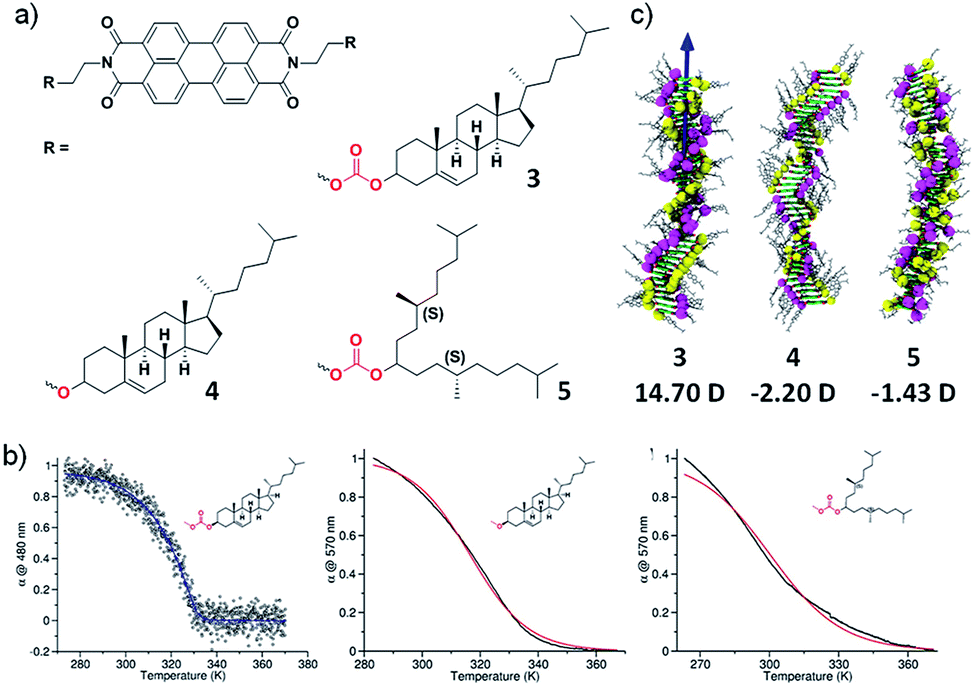 | ||
| Fig. 5 (a) Chemical structure of molecules 3–5. (b) Cooling curves of 3–5 showing a cooperative growth for molecule 3 and isodesmic growth for 4 and 5. (c) Final snapshots of the assemblies of 3–5 after the MD simulations showing 14.90 D, −2.20 D and −1.43 D dipole moment respectively (adapted with permission from ref. 18. Copyright 2015 American Chemical Society). | ||
To elucidate the role of the linker and the self-assembling moiety in dictating the mechanism of self-assembly, computational studies were undertaken. The molecular dynamics simulations on the aggregates of 40 units each of molecule 3–5 in explicit cyclohexane solvent was performed. The final snapshot of molecule 3 and 4 exhibited a helical packing as a result of the ordered organization of peripheral cholesterol groups in agreement with the helical arrangement observed with Circular Dichroism (CD) spectroscopy. In contrast, molecule 5 depicted an ill-defined assembly owing to the presence of flexible chiral swallowtail, also in agreement with the experimental observations. The study of inter-linker distance, carbonate in molecule 3 and 5, suggested closely arranged linker motifs in the assembly of 3 at a distance of 5.5 Å whereas, in molecule 5, the distance was calculated to be 9 Å. In case of molecule 4, the inter-linker distance between adjacent ether groups was 5.1 Å that recommend a close and uniform packing of linkers in the assembly. The consequence of structural organization of these self-assembling moiety and linker on the long-range interaction, i.e. the resultant macrodipole moment along the stacking direction was calculated. Interestingly, the calculations depicted an order higher macrodipole moment in the assembly of molecule 3 (14.7D) than of molecule 5 (1.42D) and molecule 4 shows negligible macrodipole (Fig. 5c). This result revalidates our hypothesis that presence of permanent long-range interaction plays a crucial role in determining the cooperativity of the self-assembled system. The extent of polarization in these supramolecular polymers was experimentally determined using dielectric measurements and molecule 3 displayed 5 times higher magnitude of the dielectric constant than molecule 4 and 5 which rationalize the cooperativity in the self-assembly of 3 and is in agreement with the theoretical calculations.
The studies discussed above has established the molecular origin of cooperativity for the first time and dictates the importance of long-range interactions like dipolar interaction, electrostatic interaction in governing the cooperativity.
Kinetic control on supramolecular polymerization
Detailed understanding of the mechanistic aspects of supramolecular polymerization has provided an opportunity to address the next grand challenges in this field. One of the important challenges in this direction is to have structural control such as control over molecular weight and dispersity. Conventional living polymerization is achieved via a chain-growth polymerization where the polymer chains have minimal propensity to terminate (Fig. 3a). Remarkably, in living radical polymerization, the termination propensity is decreased by introducing a dormant state for propagating monomers. Moreover, a kinetic control over polymerization process is induced where a fast initiation rate controls dispersity of polymers and subsequent slow propagation controls molecular weight.Owing to dynamicity, supramolecular polymers do not have the concern of chain termination, however, a controlled chain-growth polymerization and the introduction of dormant is the challenge. As discussed above, mechanistic studies have suggested a cooperative supramolecular polymerization to be analogous to chain-growth polymerization (Fig. 3a and b). Although we are now capable to rationally design molecules that will follow a cooperative growth and hence should result in a controlled dispersity and length. However, most of these systems lack in structural control owing to its fast nucleation step that results in random initiation of self-assembly. Hence, to address the challenge to precisely modulating dispersity and length and to further build multicomponent complex system, we need additional control over the supramolecular polymerization of the system. This can be achieved via introduction of a dormant state that shall retard/delay the nucleation step.20a
In this context, investigations on molecules with low dynamicity has shown that the molecules may get trapped in energy minimum higher than the thermodynamically most stable state (Fig. 6a). These metastable states with energy barrier ∼kBT often escape the trap and relax to the thermodynamic dormant state. These metastable traps delay the nucleation process for the most stable, thermodynamic self-assembled state and act as a source for slow and constant buffering of monomers. As a result, a control over structural characteristics of supramolecular polymers can be tuned. This pathway complexity of supramolecular polymerization is thoroughly studied by Meijer and coworkers and they have concluded that pathway can be influenced by the chemical structure and other parameters such as solvent and temperature during the supramolecular polymerization process.19 This energy landscape of supramolecular polymerization have been explained in detail by us20a and also by Hermans and coworkers20b and readers may refer it.
 | ||
| Fig. 6 Energetic pathway of supramolecular polymerization. (a) Conventional living supramolecular polymerization via a kinetically trapped dormant state. (b) Bio-inspired fuel-driven living supramolecular polymerization and transient self-assembly (our approach). | ||
With studies so far, it is evident that to address the next level challenges in the field of supramolecular polymerization, we should probably achieve a kinetic control on the supramolecular polymerization process. During the last years, we have attempted to address these grand challenges using a bio-inspired fuel-driven strategy and some of the early breakthroughs are described below.
Fuel-driven-supramolecular polymerization
Although these excellent examples are a breakthrough in the trending field of living supramolecular polymerization, they are highly dependent molecular structure and lack in its modularity. In most of these designs, the dormant states to buffer the monomer concentration is through metastable states. In contrast, natural systems that have an efficient control over their structural and functional properties, utilize a thermodynamic dormant state that lacks in its propensity to assemble. However, when triggered by a specific chemical fuel, the molecule undergoes conformational or chemical changes that activate its spontaneous and controlled growth. As discussed above, G-actin in presence of ATP follows such a kinetically controlled polymerization via a nucleation–elongation mechanism (Fig. 1a). Moreover, it also exhibits seeded characteristics and as a result, a control over length and dispersity is attained.
In this regard, our group has been working on a bio-inspired fuel-driven strategy to build cooperative and seeded supramolecular polymers and to build various thermodynamic dormant states for a generalized design strategy for kinetically controlled growth. Taking this as an inspiration, instead of depending on inherent kinetics or dynamics of monomers that may undergo a living supramolecular polymerization, we thought of designing systems with a thermodynamic dormant state which can be activated via a chemical fuel similar to actin polymerization (Fig. 1 and 6b). Moreover, this will enable us to modulate the kinetics of system by mere change in concentration and other stimuli effecting the fuel. In this account, we concentrate only on our contributions in biomimetic, ATP-fueled temporal supramolecular polymers.
Inspired by the fuel-driven strategy used by actin, we synthesized phosphate receptor group appended chromophoric monomers that can be activated via biofuels like adenosine phosphates. For this, zinc(II) dipicolylamine (ZnDPA) based cationic phosphate receptor was appended to naphthalene diimide chromophore (NDPA) (Fig. 7a). In water, due to cationic charges, the molecule remains in a monomeric state, however, on the addition of a negatively charged phosphate, the molecule should undergo supramolecular polymerization due to charge neutralization along with other hydrophobic and π–π interactions. For this, we utilized adenosine phosphates as the activator/fuel/guest. Although, the addition of phosphate guest facilitated the supramolecular polymerization into 1-dimensional helical assemblies as depicted by CD spectra but the process was instantaneous (Fig. 7a).27 As a result, no control over length and dispersity could be obtained. We also attempted to use single-stranded DNA (ssDNA) as a phosphate guest and utilized perylene diimide (PDPA) as chromophore that forms left-handed double zipper assembly as indicated by CD spectra, but here also the instantaneous growth could not be retarded (Fig. 7a).28 This is due to the fast binding of phosphate guests to the molecule and the molecule's inherent property to spontaneously assemble via π–π interactions.
 | ||
| Fig. 7 (a) Chemical structure of NDPA and its ATP-fueled helical assembly. Corresponding CD spectra showing fuel dependent helical assembly. (b) Chemical structure of PDPA and its ssDNA induced double-zipper helical assembly. CD spectra show the formation of helical assembly (part of Fig. 7b reproduced with permission from ref. 28. Copyright 2016 The Royal Society of Chemistry). | ||
All these molecules are characterized by their fast dynamics of fuel binding and induced supramolecular polymerization. This dynamic and adaptive nature give rise to fast response to a competitive binding of multivalent phosphate guests. Exploiting these features, we presented an unprecedented stereomutation cycle of guest induced helical supramolecular polymers via sequential addition of competitive binding guests.29 Extending this feature, a key biological phenomenon of allosteric regulation was also shown to control supramolecular helicity.30 A homotropic control using an ATP guest and heterotropic control using ADP and pyrophosphate was achieved in a helically dormant ATP bound self-assembly. This phosphate guest induced clipping of chromophores into supramolecular polymers was also utilized to control both ground-state and excited state bimolecular photophysical processes charge-transfer and excimers.31
In an attempt to achieve a fuel-driven, temporally controlled supramolecular polymerization, we envisage to retard the spontaneous nucleation of the monomers by modifying the monomer structure. For this, we have designed a unique, extended π-conjugated monomer, oligo (p-phenylenevinylene) derivative (OPVDPA), functionalized with the previously investigated (ZnDPA) phosphate receptor group (Fig. 8a).32 The molecule is designed to form a distinct acceptor–donor–acceptor (A–D–A) electronic structure that may remain in an intermolecular charge-transfer (CT) slipped state. Moreover, due to cationic terminal receptor group, this state will be highly facilitated due to intermolecular electrostatic repulsions. However, to form an extended supramolecular polymer, the molecule needs to overcome the charge repulsion as well as it needs to slip and convert from an intermolecular slipped CT state to a π–π stacked organization. Thus, the transformation from intermolecular CT state to an extended supramolecular polymer would require an energy barrier to be crossed and hence may prevent its spontaneous nucleation. However, the addition of a phosphate guest/fuel must neutralize the charges and reduces the electrostatic repulsion. As a result, energy barrier should decrease and transformation from intermolecular CT state to extended supramolecular polymer should be facilitated. It was indeed found that OPVDPA monomer doesn't undergo supramolecular polymerization in absence of ATP. However, on addition of ATP, the molecule undergoes a slow and gradual kinetically controlled growth with a finite lag phase, characteristic of a fuel-driven cooperative supramolecular polymerization. Interestingly, the molecule shows high selectivity to ATP for supramolecular polymerization and do not grow in presence of other phosphate fuels such ADP, AMP, GTP etc. similar to ATP-selective and ATP-fueled actin filaments. Detailed spectroscopic characterization confirmed the presence of intermolecular CT state in absence of ATP. Addition of ATP results in subsequent reorganization of chromophore from a slipped CT state to π–π stacked state that enable an elongated self-assembly. Thus binding of ATP reorganizes the conformation of the monomers from a thermodynamically stable dormant state to a π–π stacked extended supramolecular polymer. Additionally, intermolecular hydrogen bonding between adenine bases of ATP molecules facilitate the elongation into a supramolecular polymer and hence an ATP selectivity is observed. The thermodynamic dormant state and the fuel-driven change in the conformation from an inactive state to an active state and its ATP selectivity is also characterized by extensive molecular dynamics simulations in explicit solvent (Fig. 8b).
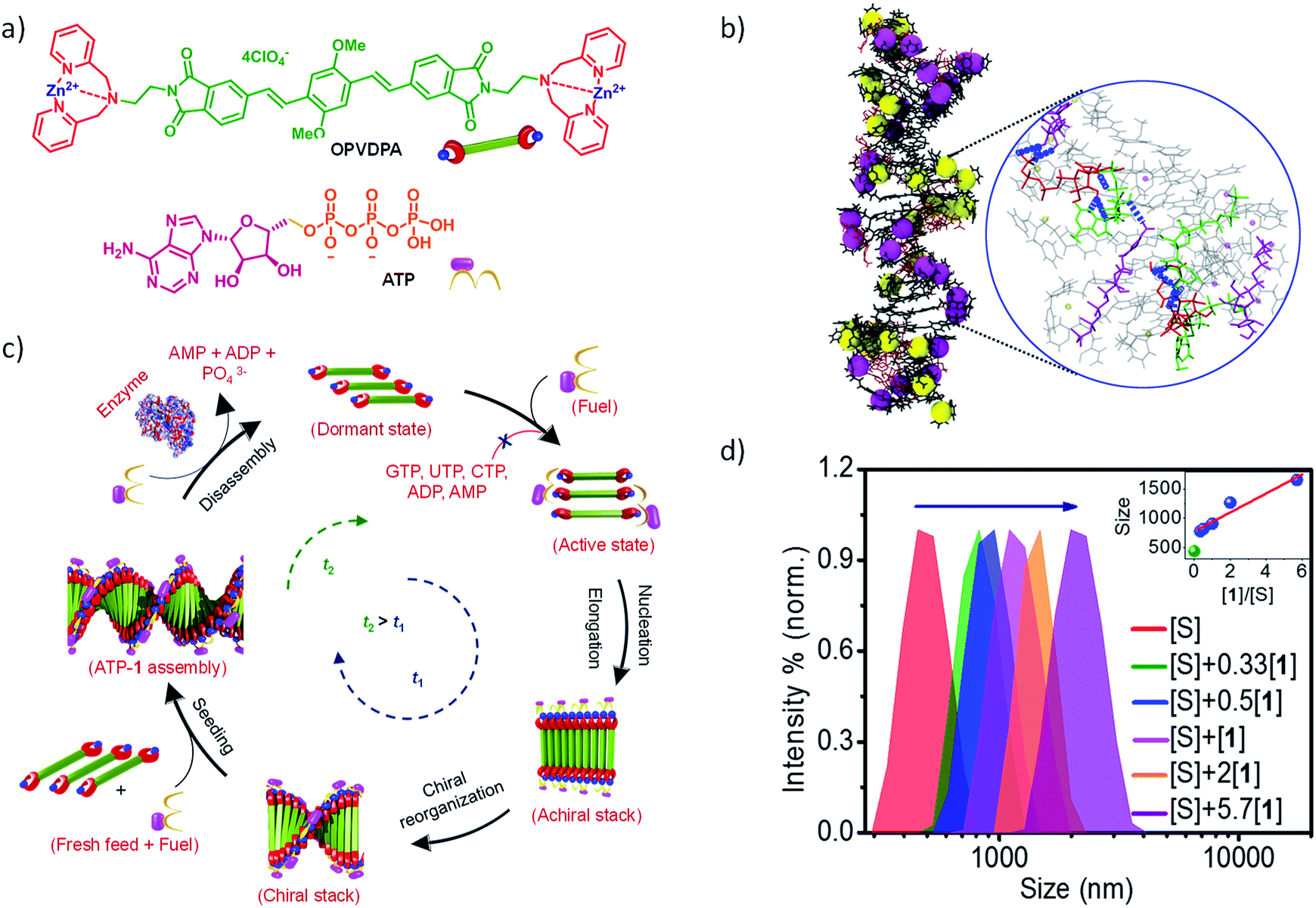 | ||
| Fig. 8 (a) Chemical structure of OPVDPA and ATP. (b) Final snapshots of MM/MD simulations of ATP fuel-driven supramolecular polymers with zoomed in image depicting intermolecular hydrogen bonding between ATP molecules. (c) Schematic illustration of ATP-fueled temporal assembly of OPVDPA into a helical supramolecular polymer via cooperative mechanism exhibiting seeded and transient characteristics. (d) Dynamic light scattering (DLS) data showing a gradual increase in size of supramolecular polymer by increasing seed to monomer ratio. Inset shows linear increase in size with increasing monomer to seed ratio suggesting a fuel-driven living supramolecular polymerization. | ||
More interestingly, this system also showed an ATP-fueled seeded growth similar to actin polymerization (Fig. 8c). Subsequent additions of ATP along with monomers to pre-grown ATP-bound OPVDPA seeds, showed a non-sigmoidal growth confirming the subsequent addition of monomers on pre-grown stacks rather than forming its own nucleus was observed. Again this seeded growth is very selective to ATP and triggers only in presence of ATP. Fuel-driven seeded supramolecular polymerization has been further used to control the degree of polymerization and dispersity of the supramolecular polymerization (Fig. 8d). Hence this study stands out to be the first report of the fuel-driven living supramolecular polymerization, similar to most of the biological self-assembly process.
Yagai, Sugiyasu and Takeuchi's group have recently elaborated this concept for living supramolecular polymerization using light as fuel.33 An initial non-assembling cis azobenzene derivative is subjected to light as fuel to isomerize the inactive cis isomer to an active trans isomer to trigger the supramolecular polymerization.
Remarkably, the above studies have dictated the design strategy of the thermodynamic dormant state as an efficient alternative that doesn't depend on inherent kinetically trapped states. The next level of understanding should search for various stimuli/fuel triggered conversion of non-assembling thermodynamic dormant state to activated monomers which can initiate a controlled supramolecular polymerization, kinetically controlled by the fuel. Once established, the living supramolecular polymerization should be extended towards the creation of unique complex systems such as supramolecular block copolymers.
Non-equilibrium or transient supramolecular polymerization
In the above sections, we discussed the mechanistic insights and strategies to control length and dispersity of supramolecular polymers via kinetically controlled living supramolecular polymerization. In this section, we shall discuss the attempts made by our group towards life-like smarter materials which operate under non-equilibrium. Natural systems not only have a precise control over structural characteristics, but they also have a temporal control over structural and functional states. Circadian rhythms, cell division, etc. are coupled to the biological clock and maintained by a cascade of chemical reactions. When signaled, the proteins undergoes structural changes that activate their functional state. These structural and functional states appear for a predetermined time and subsequently switch back to the original states. These programmed changes as a function of time are referred as transient changes. The strategy here involves a thermodynamically stable state that gets activated on the addition of fuel, as a result of which the system is pushed to a non-equilibrium unstable state (Fig. 1b and 6b). Due to instability, the system undergoes energy dissipation and relaxes back to thermodynamic equilibrium. Here, two processes that govern formation and dissipation/decay of non-equilibrium state act simultaneously. In order to have a finite lifetime of transient (non-equilibrium) state, the rate of formation should be significantly higher than the rate of decay. The key factor that enhances the rate of formation, is the amount of fuel and hence the amount of fuel directly determines the lifetime of the state.During the last 2–3 years, there have been many significant attempts to build synthetic transient systems. The key strategy is to use a monomer that lacks in its potency to assemble and then activates it via a fuel, in general, a chemical species, that alters its chemical structure and as a result, the modified monomer undergoes a spontaneous self-assembly (Fig. 1b). Simultaneously, there occurs an opposite reaction that reverses the changes in the monomer and thereby decreasing its potency to assemble and as a result, the system comes back to equilibrium. Importantly, the assembly and its monomers should be highly dynamic and adaptive so that a direct influence of change in molecular structure and environment in the self-organization is observed.
Based on this design strategy various synthetic approaches have been reported recently. van Esch and coworkers have utilized a methylation–demethylation chemical reaction couple to transiently self-assemble a monomer with carboxylate functional group.34 Carboxylate due to its anionic nature in alkaline media is highly stable, but on addition of methylating agent (chemical fuel), carboxylate is covalently modified to ester that has high propensity to assemble and a supramolecular polymerization is observed. A simultaneous base mediated demethylation (hydrolysis) of ester to carboxylate occurs that brings back the system to equilibrium. Utilizing this strategy of transient covalent modification of non-assembling monomers various reports are published.35 A step ahead with a bioinspired approach, Ulijn and coworkers used enzyme-based transient gels36 and Hermans and coworkers reported transient conformational changes of supramolecular polymeric systems.37
As an alternative, a non-covalent modification of monomers, similar to protein assembly via a fuel addition in presence of a fuel dissipating medium can result in transient changes in the system. Based on this, Prins and coworkers have utilized an amphiphilic molecule appended with zinc(II) based cationic motif that due to electrostatic repulsion remains as monomer in water, but addition of anionic guest such as adenosine triphosphate triggers its charge neutralization mediated self-assembly.38 Thus, in presence of an ATP hydrolyzing enzyme, the molecule undergoes transient non-covalent modification that results in transient self-assembly.
Another factor that can influence change in properties of molecule to self-assemble is their environmental factors. Amongst these, pH has been extensively utilized by Walther and coworkers.39 For this they have utilized temporal change in pH by urease catalyzed hydrolysis of urea into ammonium hydroxide that gradually increases the pH of solution. Alternatively, base mediated lactone hydrolysis decreases the pH of solution temporally. Moreover, they utilized these temporal changes in pH for transient gel formation, transient conformational switching in i-motif DNA and for photonics application. van Hest and coworkers also utilized covalently bonded urease to a block copolymer to build self-regulated breathing microgels.40 Klajn and coworkers have reported out-of-equilibrium assembly of gold nanoparticles.41a All these examples along with other reports have been reviewed by many, for further details readers may refer it.5,10,20,41b,42
Although these examples have led to significant development in the field of non-equilibrium supramolecular polymerization, most of them are different from their biological counterparts. Biological systems have a precise control over their structure and function and utilize cascade of enzymes to facilitate the temporal programming. As we urge towards smarter life-like materials, biological systems being smartest of all are inspiration to us. We believe using a “bio-inspired fuel-driven strategy mediated by cascade of enzyme reactions” can be exploited as a generalized strategy and a higher the complexity of supramolecular systems can be targeted. Also, amongst a plethora of biologically relevant molecules, natural systems employ a very few specific and highly selective fuels to drive transient changes. For instance, actin filaments utilize ATP and microtubules utilize GTP. Additionally, these fuels such as ATP have a range of enzymes that facilitate ATP formation and transformation via orthogonal routes and hence, giving us a control over independent modulation of formation and decay kinetics. Based on this we utilized a bio-inspired fuel-driven strategy to build complex supramolecular systems with kinetic control over formation and decay rates and temporal regulation of transient state lifetimes.
We selected NDPA molecule from our previously studied bio-fuel (phosphate guest) driven self-assembly due to its salient features such as high dynamicity, adaptiveness and phosphate selective self-assembly, necessary to build complex transient supramolecular polymers (Fig. 7a). As discussed above, the molecule assembles into a right-handed helix in presence of ATP and forms left-handed helix in presence of ADP/AMP (Fig. 1b). Moreover, inorganic phosphate lacks in inducing any significant assembly. Utilizing these properties of NDPA, we utilized a bio-inspired “enzymes in tandem” strategy and coupled it to two enzymes in tandem to transiently modulate the NDPA structure (Fig. 9a). First, we attempted to obtain ATP fueled transient conformational switching similar to biologically important proteins such as ATP Binding Cassette (ABC) transporters that undergo conformational switching when fueled by ATP to facilitate the exchange of matter across the cell membrane.43 For this, we selected two complementary phosphoryl transferase enzymes that generate and consume ATP, creatine phosphokinase (CPK) and hexokinase (HK). CPK generates ATP from ADP by consumption of phosphate donor phosphocreatine and enzyme HK hydrolyzes ATP to ADP in presence of its substrate glucose. Although, the two enzymes undergo opposite reactions but via orthogonal pathways and hence formation and decay process of fuel can be easily programmed. A solution containing an appropriate concentration of both the enzymes, glucose and ADP-bound NDPA shows a steady negative CD signal confirming the presence of left-handed helix and no enzyme activity. On introducing, phosphocreatine, the fuel in this case, activates the CPK enzyme for the formation of ATP. The in situ formation of ATP at the expense of ADP converts the left-handed helix into right-handed helix gradually as seen by a change in CD signal from negative to positive (Fig. 9b). As ATP forms in the solution, HK gets activates and a simultaneous hydrolysis of ATP to ADP occurs. As a result, transiently formed right-handed helix converts back to the initial state of ADP bound left-handed helix as depicted by retrieval of negative CD signal. Hence, we obtain transient conformational switching in a supramolecular polymer. Since the kinetics of system is directly determined by enzyme kinetics, we modulated the rates and lifetime of the transient state by changing the concentration of enzymes CPK and HK and their substrates phosphocreatine and glucose. Hence for the first time, a modular temporal change in conformation of helical self-assembly is obtained.
 | ||
| Fig. 9 Bioinspired chemical fuel-driven and enzymes mediated transient changes in supramolecular polymer. (a) Phosphocreatine fueled, creatine phosphokinase and hexokinase in tandem mediated transient helical conformational switching in a supramolecular polymer. (b) Corresponding time dependent changes in CD signal showing transient helical inversion. (c) ATP fueled, alkaline phosphatase (ALP) mediated two non-equilibrium state transient self-assembly. (d) Resultant time dependent changes in CD signal showing helical inversion in transient self-assembly. Enzyme concentration modulated programmable rate of disassembly and lifetime of transient states ((a) and (b), reprinted with permission from ref. 43. Copyright 2017 Wiley-VCH. (c) and (d) Reprinted with permission from ref. 44. Copyright 2017 American Chemical Society). | ||
Going a step ahead towards the higher complexity of transient supramolecular systems, we extended our system to undergo programmable multiple transient/non-equilibrium helical conformational states before the system relaxes to thermodynamic equilibrium via our bioinspired fuel-driven and enzyme mediated strategy (Fig. 9c).44 For this we first utilized phosphate hydrolyzing enzyme alkaline phosphatase (and alternatively apyrase) to convert ATP to Pi via ADP and AMP. In presence of only the enzyme, NDPA shows negligible traces of self-assembly as seen by absorption spectra, no CD signal (Fig. 9d) and dynamic light scattering measurement. However, the addition of chemical fuel ATP results in instantaneous formation of ATP bound right-handed helical assembly of NDPA, as the first transient state indicated by an instantaneous increase in positive CD signal. As ATP gets enzymatically hydrolyzed to ADP/AMP, a helical inversion from right-handed (transient state 1) to left-handed (transient state 2) assembly depicted by a gradual change of CD signal from positive to negative is observed. This occurs due to the phosphate selective self-assembly of NDPA. On successive hydrolysis of ADP/AMP to inorganic phosphate results in complete disassembly of the supramolecular polymer and CD signal becomes zero. Hence, an unprecedented transient self-assembly with two helical transient state is presented. The transient cycles could be efficiently refueled via subsequent addition of fuel (ATP) which follows the same path as the first cycle. Hence, we could obtain multiple cycles by refueling with ATP. Moreover, the rate of disassembly and lifetime of transient states were modulated by variation of phosphatase enzyme that controls the rate of ATP hydrolysis (Fig. 9d).
We further increased the complexity of our system by coupling the NDPA monomers to our previously studied creatine phosphokinase enzyme and alkaline phosphatase (or apyrase). The two enzymes have an orthogonal function, CPK forms ATP from ADP using phosphocreatine and alkaline phosphatase hydrolyze adenosine phosphates. Hence, the bio-inspired “enzymes in tandem” strategy could result in controllable rates and realization of newer transient species via a complex network of enzymatic transformations. We began with the solution containing both the enzymes and NDPA in the monomeric state. Addition of ADP along with phosphocreatine converted NDPA monomers into an ADP induced left-handed helical assembly (transient state 1). The addition of fuel activates the CPK enzyme that slowly converts ADP into ATP and hence a helical inversion from left-handed transient state 1 to right-handed transient state 2. Interestingly, the in situ formed ATP undergoes hydrolysis into ADP (and AMP) via the action of alkaline phosphatase and again a left-handed helical assembly (transient state 3) is obtained. Until this point, no significant disassembly occurs, but further hydrolysis into inorganic phosphate disassembles the supramolecular polymer into the thermodynamic equilibrium state. As a result of the coupling of phosphate selective NDPA with orthogonal “enzymes in tandem”, we could obtain transient self-assembly up to three transient states. Since, this supramolecular system is highly dynamic, adaptive and dependent on enzymatic action, we further envisaged to control the extent of formation of transient states and their lifetimes. For this, we varied enzyme concentration and alternatively employed apyrase enzyme instead of alkaline phosphatase that has higher selectivity towards ATP for hydrolysis than other adenosine phosphates. Indeed, we could selectively enrich particular transient states amongst the three and control their independent lifetimes. This is a unique report exemplifying complex transient self-assembly with multiple non-equilibrium/transient states that can be preprogrammed via appropriate coupling of an intricate loop of enzymatic conversions.
Our group has also extended the field of transient systems by utilizing different chemical fuels and chemical reactions.45 We have recently reported transient conformational changes of a foldamer and morphological switching of a foldamer in an amphiphilic charge-transfer based foldamer monomer using redox fueled modulation of charge-transfer interactions. Since a variety of charge-transfer based supramolecular systems are known in the literature, this strategy can be extended as a general approach to attain a temporal control over their self-assembly. We are further extending the field by using light and other chemical fuel systems.
Conclusions
With the shift of interest from thermodynamically driven supramolecular polymers to a fuel-driven out-of-equilibrium systems, unprecedented bio-mimetic properties have been imparted to synthetic assemblies. With this perspective, our group has made significant advancement to build generic bio-inspired fuel-driven and enzyme mediated strategy to control structural and temporal fate of supramolecular assemblies. Precisely, in an actin-mimetic system, we have demonstrated a control over size and dispersity of supramolecular polymers using an ATP-fueled, kinetically controlled cooperative and seeded growth and also constructed ATP-driven out-of-equilibrium transient self-assemblies that undergo a preprogrammed conformational switching and complex transient state reorganizations prior to disassembly. With this fuel-driven approach, we could temporally program the kinetics of supramolecular polymerization to obtain controlled length and dispersity and alternatively to attain transient structures and functions with modular lifetimes.The field it still under infancy and a wider scope is still unaccomplished. The multiple components needed to acquire a temporal programming are still primitive, a plethora of fuels such as light, chemical, mechanical etc. are underexplored. A significant level of advancement has to be developed to build orthogonal systems that specifically interact and communicate amongst each other. Mesmerizing properties such as self-replicating, self-evolving and learning and thinking behavior are long term goals that urge a clear identification and investigation of complex systems. The successful orchestration of fuel-driven growth in a temporal domain shall pave the way to build multicomponent systems that can form varied architectural supramolecular block co-polymers that can foster towards real-life applications such as soft-lithography.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. C. N. R. Rao, FRS for his support and guidance. We thank JNCASR, Sheikh Saqr Laboratory (SSL), Nanomission, DST (SR/NM/TP-25/2016) and Department of Science and Technology, Government of India, for financial support. A. S. thanks CSIR for a fellowship.Notes and references
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS; (b) L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS; (c) T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171 CrossRef CAS.
- (a) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS; (b) A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS; (c) V. K. Praveen, C. Ranjith, E. Bandini, A. Ajayaghosh and N. Armaroli, Chem. Soc. Rev., 2014, 43, 4222–4242 RSC; (d) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024–16038 CrossRef CAS.
- D. van der Zwaag, T. F. A. de Greef and E. W. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334–8336 CrossRef CAS.
- S. A. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- (a) M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS; (b) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS.
- Spatiotemporal mechanisms of life, Nat. Chem. Biol., 2007, 3, 593 Search PubMed.
- M. Kasai, S. Asakura and F. Oosawa, Biochim. Biophys. Acta, 1962, 57, 22–31 CrossRef CAS.
- (a) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC; (b) G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC; (c) F. della Sala, S. Neri, S. Maiti, J. L. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27–33 CrossRef CAS.
- C. Kulkarni, P. A. Korevaar, K. K. Bejagam, A. R. A. Palmans, E. W. Meijer and S. J. George, J. Am. Chem. Soc., 2017, 139, 13867–13875 CrossRef CAS.
- (a) A. Sarkar, S. Dhiman, A. Chalishazar and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 13767–13771 CrossRef CAS; (b) B. Narayan, K. K. Bejagam, S. Balasubramanian and S. J. George, Angew. Chem., Int. Ed., 2015, 54, 13053–13057 CrossRef CAS.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661–673 CrossRef CAS.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS.
- T. Metzroth, A. Hoffmann, R. Martn-Rapffln, M. M. J. Smulders, K. Pieterse, A. R. A. Palmans, J. A. J. M. Vekemans, E. W. Meijer, H. W. Spiess and J. Gauss, Chem. Sci., 2011, 2, 69–76 RSC.
- C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928–1936 CrossRef CAS.
- C. Kulkarni, K. K. Bejagam, S. P. Senanayak, K. S. Narayan, S. Balasubramanian and S. J. George, J. Am. Chem. Soc., 2015, 137, 3924–3932 CrossRef CAS.
- (a) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS; (b) P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS; (c) T. Haedler, S. C. Meskers, R. H. Zha, M. Kivala, H. W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef; (d) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS; (e) D. van der Zwaag, P. A. Pieters, P. A. Korevaar, A. J. Markvoort, A. J. H. Spiering, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 12677–12688 CrossRef CAS; (f) P. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2013, 26, 576–586 CrossRef.
- (a) S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS; (b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- (a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS; (b) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS.
- A. Aliprandi, M. Mauro and L. de Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS.
- (a) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS; (b) V. Stepanenko, X.-Q. Li, J. Gershberg and F. Würthner, Chem.–Eur. J., 2013, 19, 4176–4183 CrossRef CAS; (c) S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS.
- J. S. Valera, R. Gómez and L. Sánchez, Small, 2017, 1702437 Search PubMed.
- (a) D. S. Pal, H. Kar and S. Ghosh, Chem. Commun., 2018, 54, 928–931 RSC; (b) H. Kar, G. Ghosh and S. Ghosh, Chem.–Eur. J., 2017, 23, 10536–10542 CrossRef CAS PubMed.
- M. Kumar, P. Brocorens, C. Tonnelé, D. Beljonne, M. Surin and S. J. George, Nat. Commun., 2014, 6, 5793 CrossRef.
- J. Rubio-Magnieto, M. Kumar, P. Brocorens, J. Idé, S. J. George, R. Lazzaroni and M. Surin, Chem. Commun., 2016, 52, 13873–13876 RSC.
- M. Kumar, M. D. Reddy, A. Mishra and S. J. George, Org. Biomol. Chem., 2015, 13, 9938–9942 RSC.
- M. Kumar and S. J. George, Chem. Sci., 2014, 5, 3025–3030 RSC.
- M. Kumar, A. O. Ushie and S. J. George, Chem.–Eur. J., 2014, 20, 5141–5148 CrossRef CAS PubMed.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef PubMed.
- M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed.
- (a) J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS; (b) J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–5482 CrossRef CAS; (c) B. G. van Ravensteijn, W. E. Hendriksen, R. Eelkema, J. H. van Esch and W. K. Kegel, J. Am. Chem. Soc., 2017, 139, 9763–9766 CrossRef CAS PubMed.
- L. S. Kariyawasam and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 11949–11955 CrossRef CAS.
- (a) C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS; (b) S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS.
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed.
- (a) S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–781 CrossRef CAS; (b) C. Pezzato and L. J. Prins, Nat. Commun., 2015, 6, 7790 CrossRef CAS.
- (a) T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef PubMed; (b) L. Heinen and A. Walther, Chem. Sci., 2017, 8, 4100–4107 RSC; (c) T. Heuser, R. Merindol, S. Loescher, A. Klaus and A. Walther, Adv. Mater., 2017, 29, 1606842 CrossRef PubMed.
- H. Che, B. C. Buddingh' and J. C. M. van Hest, Angew. Chem., Int. Ed., 2017, 56, 12581–12585 CrossRef CAS PubMed.
- (a) M. Sawczyk and R. Klajn, J. Am. Chem. Soc., 2017, 139, 17973–17978 CrossRef CAS; (b) W. C. Geng, Y. C. Liu, Z. Zheng, D. Ding and D.-S. Guo, Mater. Chem. Front., 2017, 1, 2651–2655 RSC.
- S. De and R. Klajn, Adv. Mater., 2018, 1706750 CrossRef.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed.
- S. Dhiman, A. Jain, M. Kumar and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS.
- K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC.
| This journal is © The Royal Society of Chemistry 2018 |