
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(N,N-dimethylacrylamide-octadecyl acrylate)-clay hydrogels with high mechanical properties and shape memory ability†
Feng Wanga,
Xueyong Yongabc,
Jianping Deng bc and
Youping Wu*ac
bc and
Youping Wu*ac
aState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: wuyp@mail.buct.edu.cn; Fax: +86-10-64456158; Tel: +86-10-64442621
bA State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
cCollege of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 8th May 2018
Abstract
As a type of important intelligent materials, shape memory hydrogels (SMHs) have gathered a lot of interest due to their promising applications. However, preparing SMHs with excellent mechanical properties still remains a big challenge. In this study, a new type of SMHs with excellent mechanical strength is created. The SMHs were prepared by free radical micellar polymerization of hydrophobic monomer (octadecyl acrylate) with gelatin as emulsifier in a aqueous system containing hydrophilic monomer (N,N-dimethylacrylamide, DMA) and clay as reinforcing filler. The polymerization provided physically cross-linked network structures constructed by two non-covalent interactions, i.e. hydrophobic association formed by monomer units and multiple H-bonds among inorganic clay, gelatin and DMA. A judicious combination of the two physically cross-linked networks significantly improved the mechanical strength of hydrogels. More interestingly, the hydrogels demonstrated shape memory behavior due to the hydrophobic poly(octadecyl acrylate) domains. The novel SMHs are expected to find practical applications as biomaterials.
1. Introduction
Hydrogels with three-dimensional network structure have gathered a lot of interest due to their high-water content, low friction coefficient, and good biocompatibility.1,2 These features make hydrogels quite similar to human tissues; accordingly, hydrogels have found a wide range of applications as biological materials, such as bio-adhesives,3 tissue engineering4 and drug delivery carriers.5 However, for most hydrogels, mechanical strength is poor and cannot meet the requirement for practical applications.6,7 Therefore, a variety of hydrogels with high strength and toughness have been developed, for instance, double network hydrogels,8–10 topological hydrogels,11,12 macromolecular microsphere composite hydrogels,13,14 and hydrophobically modified hydrogels.15–21 The essential idea in developing these hydrogels is to dissipate the energy and thereby to improve the strength and toughness of hydrogels.22Hydrophobically modified hydrogels are physically cross-linked hydrogels incorporating hydrophobic blocks into a hydrophilic polymer. Due to the reversible non-covalent bonds, such hydrogels exhibit remarkable mechanical properties.16,17 To date, various hydrophobically modified hydrogels have been established, demonstrating improved mechanical strength. Liu et al.17 prepared hydrophobic association hydrogels with quite large elongations at break (1828%); nonetheless, the strength of these hydrogels (0.21 MPa) could be further improved. Okay et al.18–21 prepared tough hydrogels based on hydrophobic interactions which even provided some other fascinating properties, e.g. self-healing and shape memory properties. However, small molecular emulsifier, sodium dodecyl sulfate (SDS) was used and may produce some unfavorable influence on practical applications. To overcome this problem, some biomacromolecules can stabilize the oil phase in water, and so may act as emulsifier to replace SDS, such as gelatin.23 Our preceding study has justified this hypothesis, in which gelatin was used as emulsifier for preparing hydrogels.24
In another work,25 clay, which can be easily dispersed into nano-sheets in aqueous medium, was used as reinforcing filler for rubbers in latex form. Considering the characteristics of hydrogels containing water, clay was also selected as reinforcing filler in this study. High mechanical strength hydrogels were successfully prepared through simultaneous free radical polymerization of hydrophilic monomer N,N-dimethylacrylamide (DMA) and free radical micellar polymerization of hydrophobic monomer octadecyl acrylate (C18) in the presence of inorganic clay in aqueous medium at 60 °C, using gelatin as emulsifier and ammonium persulfate (APS) as initiator. The preparation strategy is illustratively presented in Scheme 1. The structure of the as-prepared hydrogels is schematically illustrated in Fig. 1, which are constructed by multiple physically cross-linked networks. Hydrophobic interactions among C18-derived polymer chains acted as the physical crosslinking regions; clay acted as a reinforcing filler through forming H-bonds with other components.26–28 In addition, gelatin is not only used as emulsifier for performing the micellar polymerization, but also is expected to endow hydrogels with strength.
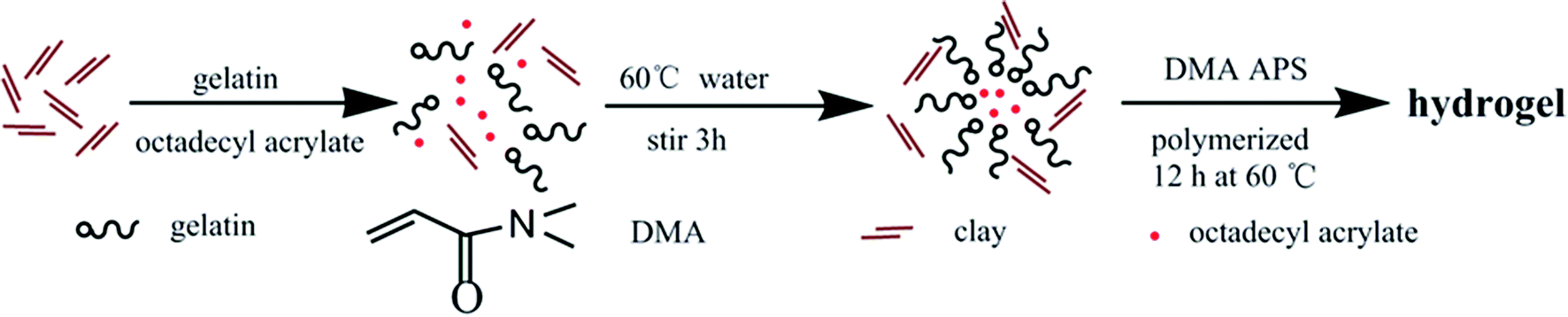 | ||
| Scheme 1 The strategy for preparing hydrogels. | ||
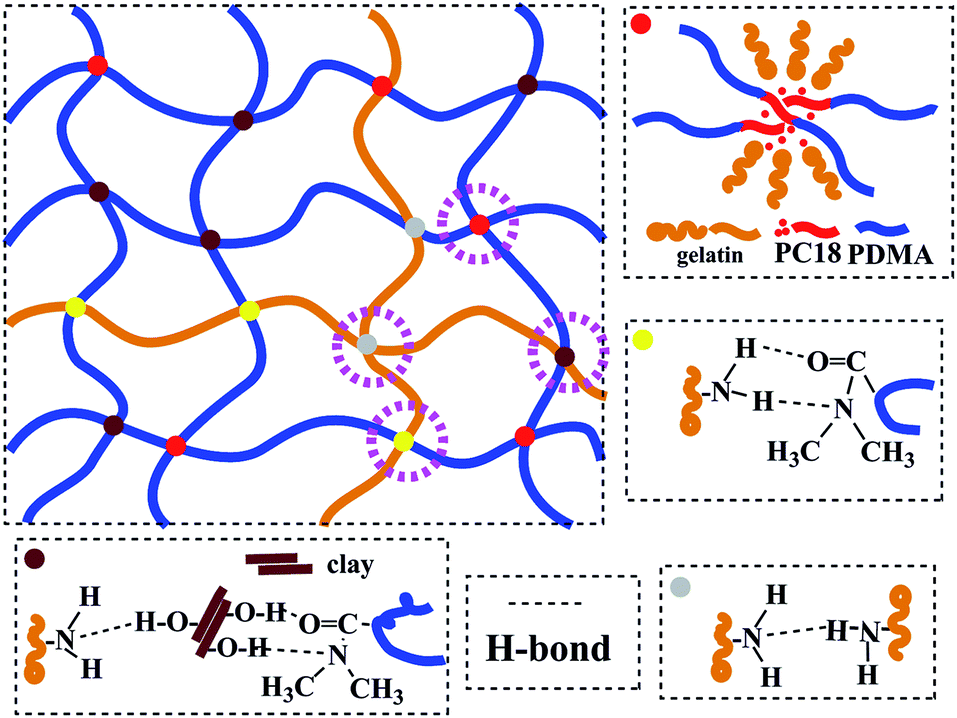 | ||
| Fig. 1 Schematic illustration of the hydrogel network structure. | ||
2. Experimental
2.1 Materials
N,N-Dimethylacrylamide (DMA) was purchased from Beijing HWRK Chem. Co, Ltd., China. Gelatin (type A, from porcine skin with gel strength about 300 g Bloom) and octadecyl acrylate (C18) were purchased from Sigma-Aldrich Company. Ammonium persulfate (APS) was purchased from Beijing Chemical Reagents Company. Clay was obtained from Siping Liufangzi Aska bentonite Co, Ltd., China. Murine osteoblast MC3T3-E1 were provided by Beijing Jishuitan Hospital, China. Fetal bovine serum (FBS), α-modified minimum essential medium (α-MEM), and penicillin–streptomycin were purchased from Hyclone, America. CCK-8 and phosphate buffer solution (PBS) were purchased from Shanghai Biyuntian Bio-Technology Co, Ltd., China. All the chemicals were used without further purification.2.2 Hydrogel preparation
Hydrogels were prepared by micellar copolymerization of DMA in water containing 3 mol% C18 and varied amount of clay at 60 °C for 12 h, using gelatin as emulsifier and APS as initiator. This study focused on the effect of clay content on hydrogel properties, so the total solid content kept constant (0.317 g ml−1), and the mass ratio of clay to monomers (DMA and C18) was 0%, 1%, 3% and 5%. The corresponding hydrogels are denoted as NC-0, NC-1, NC-3 and NC-5, respectively. In order to explore the effect of gelatin on the mechanical properties of hydrogels, the hydrogel without gelatin was synthesized and designated as Ref-1. The recipes for preparing the five hydrogels are listed in Table 1. The detailed synthetic procedure of the hydrogels was briefly described below, taking NC-5 as example. 0.0503 g clay was dispersed in 4 ml deionized water under stirring at room temperature, and then 0.8 g gelatin was added at 60 °C and stirred until it was completely dissolved. Hydrophobic monomer C18 (0.277 mmol) was then added in the aqueous system. Gelatin–C18 formed a uniform emulsion after stirring for 3 h, and then DMA and APS were added in the emulsion. After that, the mixture was transferred to glass tubes of 8 mm internal diameter (for tensile test) or 24 mm internal diameter (for compression test) quickly and polymerized 12 h at 60 °C. Typical photographs of the hydrogels are presented in Fig. S1 (ESI,† the same below).| Hydrogels | DMA/mmol | C18/mmol | Clay/g | Gelatin/g | APS/mmol |
|---|---|---|---|---|---|
| NC-0 | 9.7 | 0.291 | 0 | 0.8 | 0.097 |
| NC-1 | 9.6 | 0.288 | 0.0105 | 0.8 | 0.096 |
| NC-3 | 9.4 | 0.283 | 0.0308 | 0.8 | 0.094 |
| NC-5 | 9.2 | 0.277 | 0.0503 | 0.8 | 0.092 |
| Ref-1 | 9.2 | 0.277 | 0.0503 | 0 | 0.092 |
2.3 Characterizations
FT-IR, DSC, and TEM measurements were accomplished using dried gels in order to analyze their composition and structure. Dried gels were prepared as follows. To remove unreacted monomer, all hydrogels were first immersed in an excess of deionized water for 1 day at room temperature and then in an excess of ethanol for 2 days, with renewing ethanol every day, and finally were dried for 2 days in an oven at 60 °C.FT-IR spectra were recorded with a Nicolet 8700 spectrophotometer (KBr tablet). All powder and liquid materials were measured by TR mode, and dried hydrogels were measured by ATR mode. DSC measurement was conducted on STAR system by heating samples from 0 to 200 °C at a heating rate of 20 °C min−1 under nitrogen atmosphere. Melting temperatures were determined by endothermal peaks. Transmission electron microscopy (TEM: Tecnai G2 20, Hong Kong FEI, China.) was used to observe the clay dispersion in the hydrogels, for which the thickness of the dried hydrogel samples was less than 50 nm by slicing with a microtome. X-ray diffraction patterns (XRD, Shimadzu, Japan) of wet and soft hydrogels were tested using a high power Cu Kα source (λ = 0.154 18 nm) operating at 40 kV/200 mA, in 2θ range of 5–90° at a scan rate of 10 °C min−1.
2.4 Swelling ratio
Approximately 0.1 g dried hydrogel was immersed in deionized water at room temperature. At predetermined time intervals, the hydrogel was taken out; the surface water on the hydrogel was wiped by filter paper, and then the hydrogel was weighed. This procedure was repeated until the weight of the hydrogel reached a constant value. The swelling ratio was calculated according to Ws/Wd, where Ws and Wd are the weight of swollen hydrogels and the corresponding dried hydrogels, respectively. For each hydrogel, three samples were measured, and the error bars mean the standard deviation in this study.2.5 Mechanical properties
Uniaxial tensile tests were performed on electric tensile tester (SANS, China) with a 500 N load cell at room temperature. The hydrogel samples were about 45 mm in length and 8 mm in diameter. The sample length between the jaws was 25 mm and the crosshead speed was 200 mm min−1. Compressive test was performed on CMT4204 system (SANS Test Machine Co., Ltd., China) at room temperature using hydrogel samples of 25 mm thickness and 24 mm diameter at a speed of 5 mm min−1.Fig. 2 presents the compression process of NC-5, for which more details will be reported later on. The hardness of the hydrogels was measured on a Shore A0 Hardness Tester (Instron, American).
 | ||
| Fig. 2 The compressing process of NC-5 hydrogels. | ||
2.6 Shape memory property
Cylindrical hydrogels of about 8 mm in diameter and 25 mm in length were placed at 60 °C for 10 min, and then they were stretched to 60 mm and fixed for 3 h at room temperature. After that, the deformed hydrogels were placed again at 60 °C and the length of the hydrogels L was measured at certain time intervals. The shape recovery ratio was calculated according to (60 − L)/(60–25) × 100%.To further explore the shape memory behavior of the hydrogels, the recovery process of spirally shaped NC-5 hydrogel was recorded as model. Firstly, the cylindrical NC-5 hydrogel was placed at 60 °C for 10 min, and then it was twisted to form a spiral shape. The temporary spiral shape was fixed for 3 h by external force at room temperature. After that, the deformed hydrogel with spiral shape was placed in a self-sealed bag and completely immersed into a 60 °C water bath. The recovery state of the hydrogel was photographed using a digital camera at different time intervals.
2.7 Cytotoxicity assay
MC3T3-E1 cells were cultured in complete medium (α-MEM with 10% FBS and 1% penicillin–streptomycin) at 37 °C in a humidified atmosphere with 5% CO2. The cytotoxicity of hydrogels was evaluated using MC3T3-E1 cells by CCK-8 method.29,30 The test samples were prepared as follows. First, the hydrogels (about 0.06 g) were disinfected for 15 min in gradient alcohol (75%, 95%, and absolute ethanol) and phosphate buffer solution (PBS) using a track vibrator at 200 rpm, respectively. Then, they were sterilized for 1.5 h under UV lamp. After that, the sterilized hydrogels were immersed in 1 ml α-MEM at 37 °C for 24 h to obtain hydrogel extracts. The hydrogel extracts were diluted with complete medium at volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to form the test sample solutions.
:1 to form the test sample solutions.
MC3T3-E1 cells at density of 1 × 103 cells per well were seeded in a 96-well plate for 24 h, followed by replacing the test sample solutions and incubation for a predetermined time (24, 48, and 72 h). Other wells filled with complete medium were taken as control test. After the predetermined incubation period, the cultures were examined to observe the morphology of the cells under inverted microscope (magnification 10 × 10 times). Then the test and blank solutions were aspirated, and in the wells were added 100 μl of α-MEM and 10 μl of CCK-8; and then cells were incubated for 4.5 h. The optical density (OD) of each well was read using a Biorad microplate reader (model 680) at 450 nm. According to the linear correlation between absorbance and cell number at the low concentration range, the cell viability of samples compared to the control sample was calculated from the following equation: cell viability (%) = ODsample/ODcontrol × 100%.
3. Results and discussion
3.1 Strategy for preparing hydrogels
In our previous studies, we have successfully prepared a series of (chiral) hydrogels via micellar polymerization approach.24,31 These hydrogels showed pH sensitivity, optical activity, and chiral release ability. Unfortunately, the mechanical strength of the hydrogels was not satisfying enough. In the present study, the primary purpose is to synthesize shape memory hydrogels with excellent mechanical strength. Such hydrogels were prepared by free radical (micellar) polymerization. The materials used for preparing the hydrogels include hydrophilic monomer N,N-dimethylacrylamide (DMA), hydrophobic monomer, octadecyl acrylate (C18), emulsifier gelatin, initiator APS, and reinforcing filler inorganic clay. The formation principle of hydrophobic associated structure is as follows. Gelatin emulsifies C18 and forms micelles. APS decomposes to form primary free radicals, initiating DMA to polymerize and forming PDMA. The primary free radicals and PDMA macromolecular free radicals diffuse into the micelles, in which initiating polymerization of C18. This produces the hydrophobic regions inside the hydrogels. In addition, hydrogen bonds occur among clay, gelatin, PDMA, etc. The unique structure endows the hydrogels with interesting properties. The preparation strategy and the proposed structure of the hydrogels are illustratively shown in Scheme 1 and Fig. 1. The resulting hydrogels were constructed by crosslinking points formed by hydrophobic association and multiple H-bond interactions among clay, PDMA and gelatin. The clay can improve the strength of hydrogels as reinforcing filler.Four hydrogels were prepared mainly by changing the clay amount (see Table 1). Photos are presented in Fig. S1,† to show the formation of micellar structure. To preliminarily identify the successful formation of the hydrogels, NC-5 was taken as a representative for measuring FT-IR, together with the major starting materials. Before the measurement, the hydrogel samples were purified by a thorough washing process, to completely exclude the residual monomers. The FT-IR spectra are illustrated in Fig. 3. A comparison among the spectra of C18 (spectrum a), DMA (spectrum b), clay (spectrum c), gelatin (spectrum d), and hydrogel NC-5 (spectrum e) provides evidence for the successful formation of the hydrogels. Specifically, the peaks at 1630 cm−1 (Fig. 3a, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in C18) and 1603 cm−1 (Fig. 3b, CC double bonds in DMA) vanished completely in NC-5 (Fig. 3e), demonstrating that the monomers polymerized and formed the hydrogel. In addition, due to the disappearance of the CC conjugation effect, the peak at 1648 cm−1 (C(O)–N) in spectrum b shifted to 1633 cm−1 in NC-5.
C double bonds in C18) and 1603 cm−1 (Fig. 3b, CC double bonds in DMA) vanished completely in NC-5 (Fig. 3e), demonstrating that the monomers polymerized and formed the hydrogel. In addition, due to the disappearance of the CC conjugation effect, the peak at 1648 cm−1 (C(O)–N) in spectrum b shifted to 1633 cm−1 in NC-5.
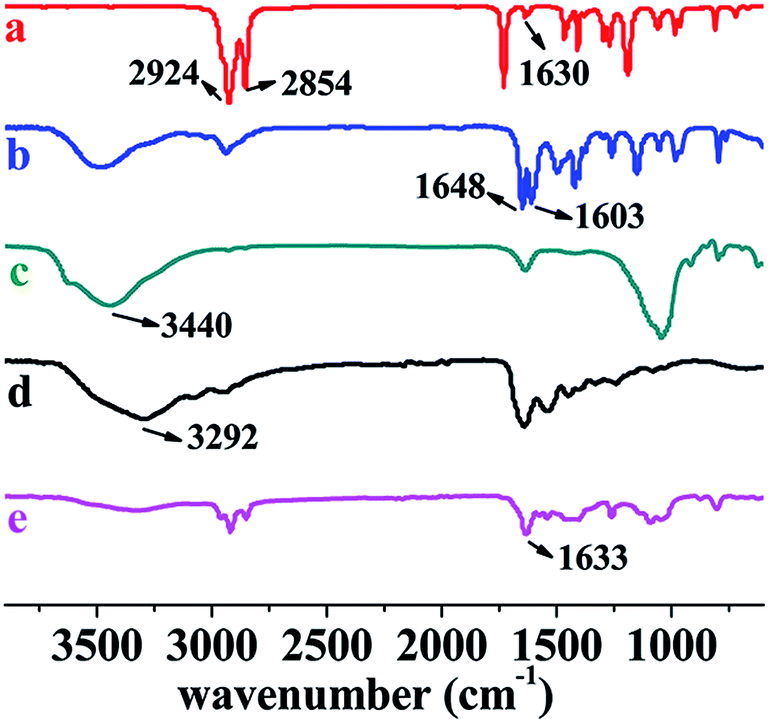 | ||
| Fig. 3 Typical FT-IR spectra of (a) C18, (b) DMA, (c) clay, (d) gelatin, and (e) NC-5 (as representative for the hydrogels). All powder and liquid materials were measured by TR mode; dried hydrogels were measured by ATR mode. | ||
The X-ray diffraction patterns (XRD) for the four hydrogels were measured, as presented in Fig. S2.† No X-ray diffraction peak appeared around for 2θ = 7° for all the hydrogels, while the clay showed a strong diffraction peak at 2θ = 7°. This indicates that clay was sufficiently exfoliated in the hydrogels, since the X-ray diffraction peak corresponding to a layer spacing of 1.3 nm for regularly stacked clay sheets25 was not observed. Nonetheless, there was no diffraction peak also possibly attributed to small amount of clay in hydrogels. We further observed the clay dispersion inside the three dried hydrogels by TEM, as shown in Fig. 4. Nano-dispersed clay sheets were observed for the three hydrogels; clay sheets increased with increasing clay content, indicating that the clay was exfoliated and dispersed at nano-level in the hydrogels.25 This observation is in good agreement with the phenomena obtained in XRD tests, as discussed above.
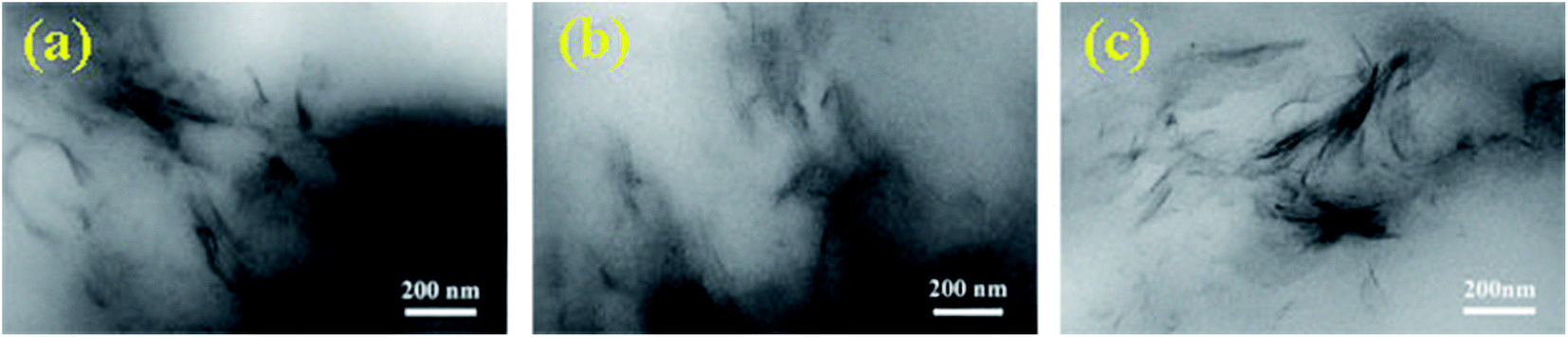 | ||
| Fig. 4 TEM images of the dried hydrogels: (a) NC-1, (b) NC-3 and (c) NC-5. Scale bar, 200 nm. | ||
3.2 Swelling ratio of hydrogels
Swelling behavior of the hydrogels was carried out by immersing the hydrogels in deionized water at room temperature, for which the results are presented in Fig. 5. It can be found that all the hydrogels exhibited the same trend; namely, the swelling ratio reached equilibrium within 8 h and then changed little. The hydrogels with different clay contents showed the equilibrium swelling ratio between 10 and 11.5 times. As we know, the swelling behavior of hydrogels is closely correlated with their cross-linking density. As the clay content increased, the swelling ratio of the hydrogels reduced in theory. This can be attributed to the H-bond interactions among clay, PDMA and gelatin, resulting in the increased cross-linking density. Nonetheless, the physical interactions originated in clay just made partial contribution to the whole hydrogel networks. Accordingly, the swelling ratio caused by changing clay content changed not remarkably.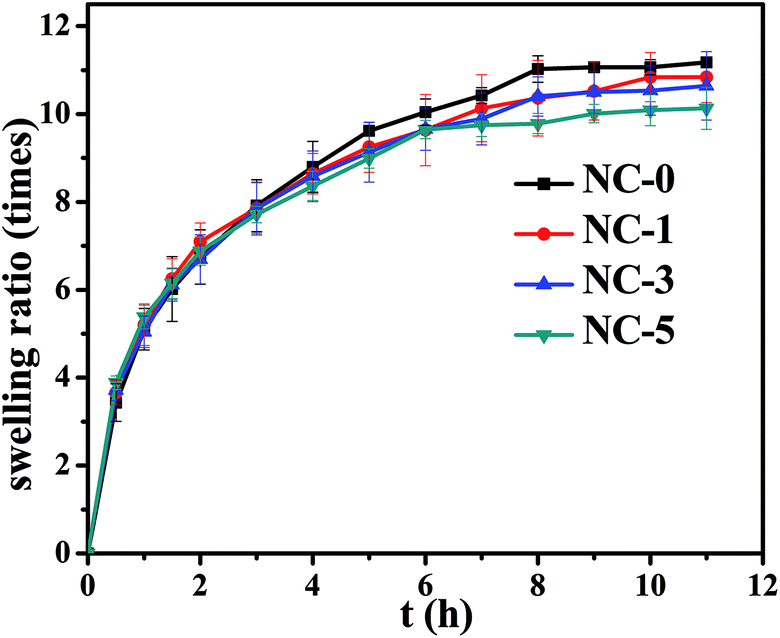 | ||
| Fig. 5 Curves of swelling ratio vs. swelling time of the four hydrogels. | ||
3.3 Mechanical properties of hydrogels
Fig. 6a shows the stress–strain curves of the five hydrogels (NC-0, NC-1, NC-3, NC-5 and Ref-1). The hydrogels, especially NC-5, exhibited outstanding mechanical strength compared to typical physically cross-linked hydrogels reported in literature.17–19 With increasing clay content, stress increased and reached the maximum value of 0.31 MPa, and the strain could also keep the same high level, indicating that clay also increased the toughness of hydrogels. This can be attributed to the combination of multiple physical crosslinking effects, i.e. H-bonds occurring among clay nano-sheets, PDMA and gelatin, and hydrophobic association regions of PC18. When the hydrogels were stretched, physically cross-linked points could slip, break and reform, dissipating the mechanical energy and thus increasing the hydrogels' tensile strength and toughness. Compared to hydrogel Ref-1, the other hydrogels showed largely increased tensile strength. The tensile stress of Ref-1 only reached 0.018 MPa, much lower than those of gelatin-containing hydrogels. This result indicates that gelatin has the ability to improve mechanical properties of hydrogels. Gelatin not only helped C18 form hydrophobic association structure, but also formed H-bonds with other components, which resulted in increased crosslinking density, and strain of the hydrogels containing gelatin was lower than Ref-1. To investigate whether polymers were grafted to gelatin chains, C18 was particularly polymerized in the presence of gelatin (mass ratio = 4:3). However, hydrogels remained liquid even after polymerization for 48 h, indicating no obvious grafting occurring to the present systems.
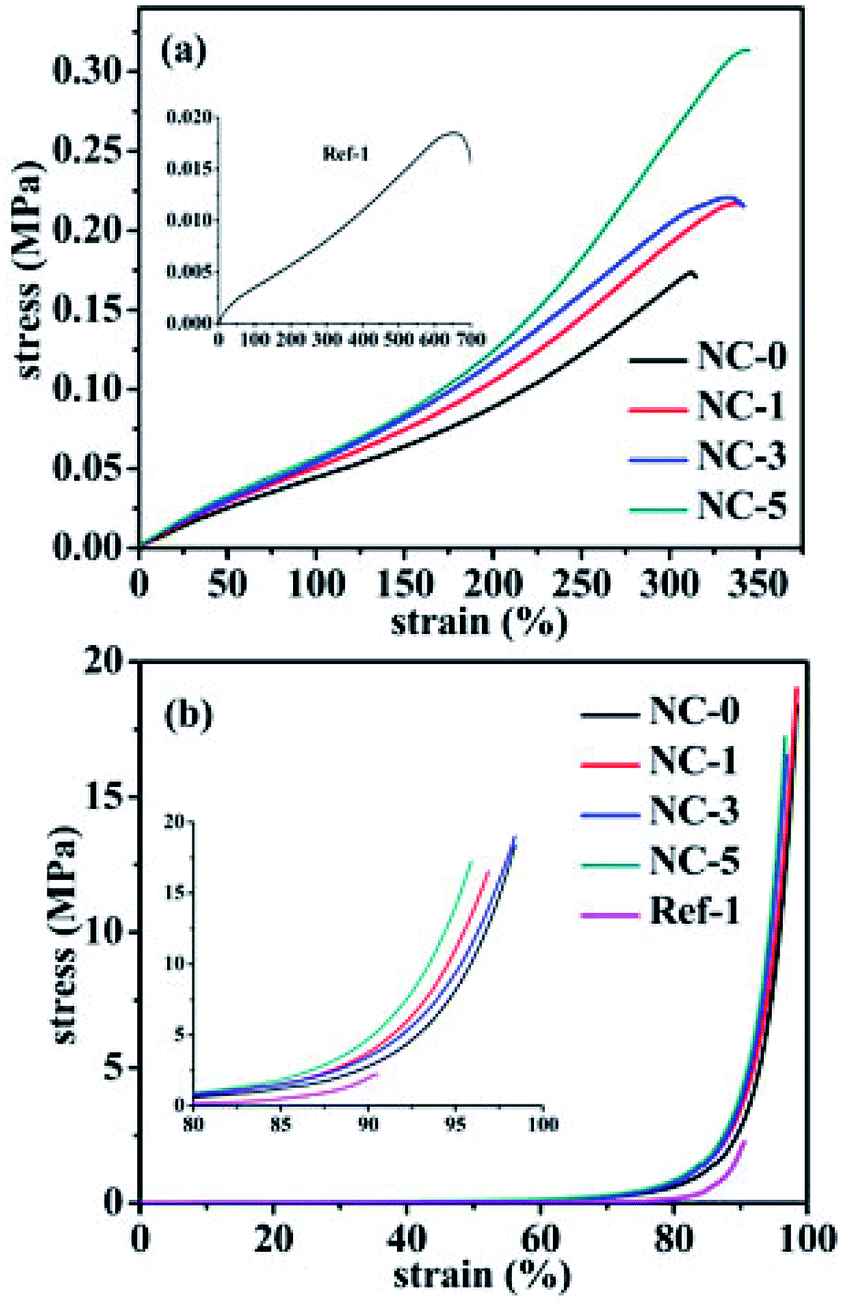 | ||
| Fig. 6 (a) Tensile stress–strain curves and (b) compressive stress–strain curves of the five hydrogels. | ||
Compressive strength was measured and the results are presented in Fig. 6b. The stress of all hydrogels increased at a lower rate up to 85%, from 0 MPa to 1.7 MPa, and increased more quickly between 85 to 97% strain, from 1.7 MPa to 17 MPa. The compressive strength was also enhanced with an increase in clay content. At 90% strain, the compressive stress of NC-1, NC-3, and NC-5 was 3.5, 3.8, and 4.0 MPa, respectively, much higher than that of NC-0 (2.8 MPa) and Ref-1 (2.0 MPa). It is noteworthy that the structure of hydrogels still remained intact without obvious damage (see Fig. 2), which is due to two reasons. One is that the slip of the reversible physical crosslinks in hydrogels can dissipate the energy; the other is that polymer chain segments remain high mobility in water.32
The hardness of the four hydrogels was also measured. The results are shown in Fig. S3.† Similar to the aforementioned tensile and compressive strengths, the hardness of hydrogels also increased with increasing the clay content. The shore A0 hardness of NC-5 was 32, 1.14 times of NC-0 hydrogel. Therefore, clay indeed exhibited evident reinforcement effects for the as-prepared hydrogels, just as expected. The cross-linking network becomes more compact, resulting from the fact that the cross-linking points increase after clay is added to hydrogels, so both the compressive strength and the hardness of hydrogels (NC-1, NC-3, and NC-5) are higher than the NC-0 hydrogel.
The hydrogels stability was further investigated. NC-0 decomposed to small pieces after immersing in PBS at 37 °C for 5 days, while the hydrogels with clay remained structurally intact even after remaining for 30 days under the same conditions. This phenomenon shows that clay can helpfully maintain the shape of hydrogels. The pictures of hydrogels after 30 days' decomposition test are presented in Fig. S4.† The hydrogels initially decomposed fast, degraded about 25–30% over a week, and then degraded at a lower rate (see Fig. S5†). The slow degradation ratio and the sustainable structure of hydrogels is more suitable for cell adhesion and proliferation, meeting the requirements for scaffolds, compared to hydrogels with faster degradation.33,34
As discussed in the previous section, all the hydrogels could swell in water, so water can easily penetrated into hydrogels and hydrolyzed gelatin. To support the hypothesis of the hydrogel degradation mechanism, the degraded hydrogels and the hydrogel degradation solution were measured by ATR-FTIR and TR-FITR spectra (the film on KBr plates were cast from hydrogel degradation solution). The results are presented in Fig. 7. A comparison among NC-5 (spectrum a), degraded hydrogel (spectrum b), and hydrogel degradation solution (spectrum c) demonstrates gelatin degraded first. The peaks at 2920 (–CH2, spectrum a and b), 2860 (–CH2), 1633 (C(O)–N), and 1260 cm−1 (C–N) appeared in NC-5 and degraded hydrogel, indicating PDMA and PC18 existed in the degraded hydrogel. The peak at 1644 cm−1 (–NH2, spectrum c) indicates there was a large amount of gelatin in hydrogels degradation solution. The degradation ratio of hydrogels reached 30% after 30 days in PBS, indicating that some gelatin (13 wt% out of total 43 wt%) still remained in hydrogel samples.
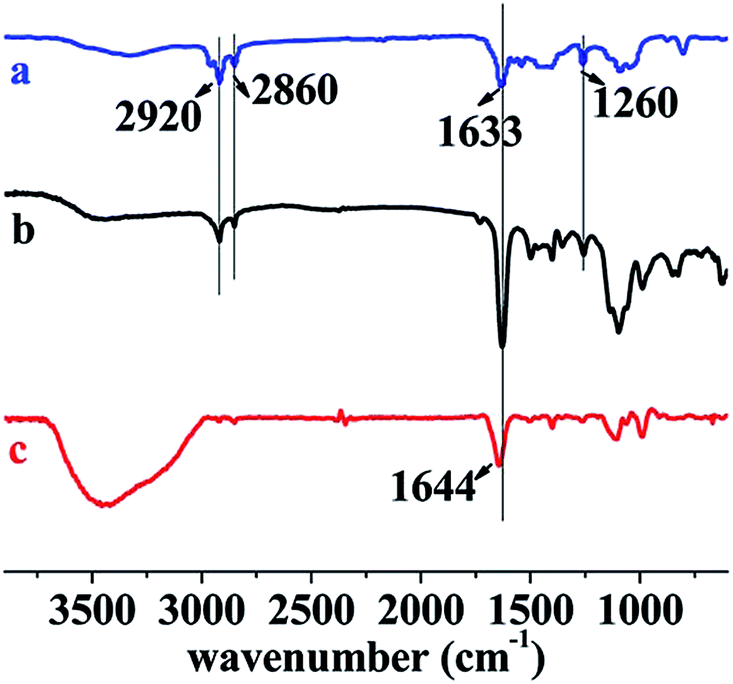 | ||
| Fig. 7 Typical FT-IR spectra of (a) NC-5, (b) degraded hydrogel (NC-5, as representative for the degraded hydrogels) and (c) hydrogel degradation solution (NC-5), NC-5 hydrogel and the degraded hydrogel were measured by ATR mode, and degradation solution was measured by TR mode. | ||
3.4 Shape memory performance of the hydrogels
DSC measurement results of the hydrogels are shown in Fig. S6.† PC18 crystalline structure melted at 50 °C. X-Ray diffraction peak corresponding to C18 crystallization was not observed in Fig. S2.† The reason for this phenomenon is the little content of C18 in hydrogels. In our designed hydrogels, PC18 served as the reversible phase in terms of shape memory property, while the physical cross-linking structure formed by other components acted as the stationary phase. Fig. 8 shows the shape recovery ratio of the hydrogels at different time intervals. At 1.0 min, the shape recovery ratio reached up to about 60%. All the four hydrogels showed high shape recovery ratio (>85%) within 4.0 min. The change in clay content just led to slight variations in the shape recovery ratio. Both the physically crosslinked network structures from hydrophobic association and the multi H-bonds enabled the hydrogels to recover fast.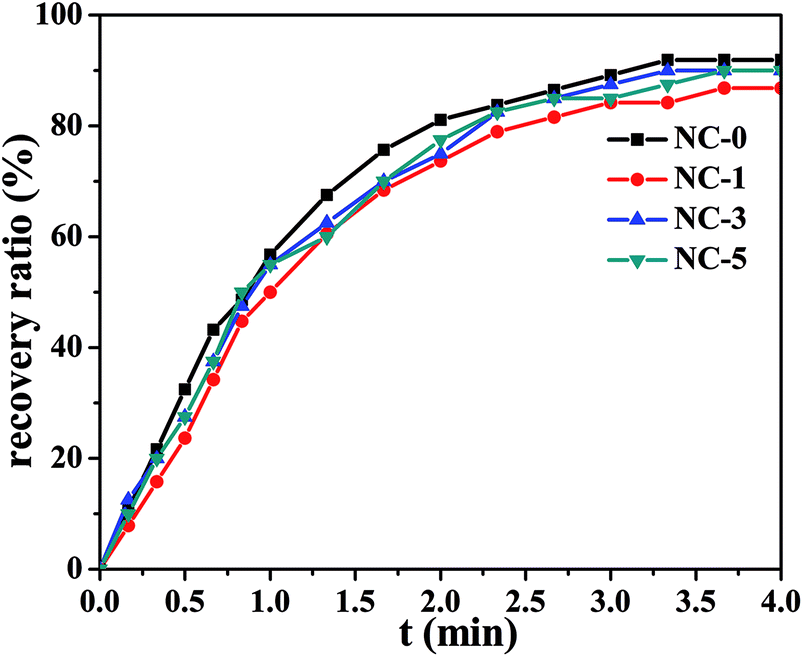 | ||
| Fig. 8 Shape recovery ratio of NC-0, NC-1, NC-3, NC-5 at 60 °C. | ||
The shape memory property of hydrogels was further investigated taking NC-5 hydrogel as an example and the process is depicted in Fig. 9. PC18 crystalline structure can melt at 60 °C. At this temperature the temporary spiral shape of hydrogel was formed under external force. Then, PC18 gradually crystallized at room temperature and the spiral shape would be fixed even after the external force was removed. The spiral NC-5 hydrogel almost restored to its original shape within 1 min in a water bath of 60 °C. This is consistent with the results of shape recovery ratio in Fig. 8. The results in Fig. 9 provide further evidence for the shape-memory property of the hydrogels. The shape memory property of Ref-1 was measured by the same methods, and the results are presented in Fig. S7.† Unfortunately, it cannot form a temporary spiral shape for lacking reversible phase. It further means PC18 cannot form hydrophobic domains in hydrogels without gelatin.
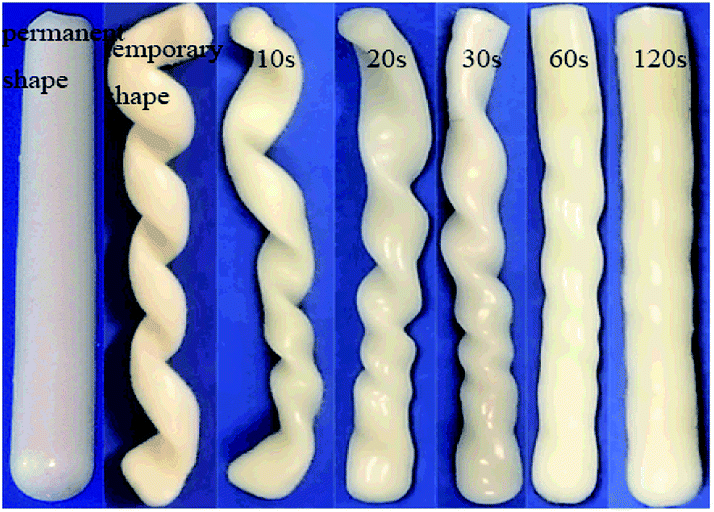 | ||
| Fig. 9 Photographs of shape recovery process of NC-5 from the temporary spiral shape to the permanent rod shape. | ||
3.5 Cytotoxicity of hydrogels
Cytotoxicity is one of the key properties for hydrogels when used as biological materials. Accordingly, we further studied the cytotoxicity of the four hydrogels using MC3T3-E1 cells. The photo micrographs of MC3T3-E1 cells incubated for 1 day are presented in Fig. 10. For all the four hydrogel samples, the shape of MC3T3-E1 cells is spindle or triangular, indicating a good growth of cells.35 In addition, the cell number grew with prolonging incubation time. The relative cell viability is summarized in Fig. 11. For the four hydrogels, the cell viability was higher than 90% within 3 days. Both the cell shape and cell viability show that the hydrogel extracts have no significant cytotoxicity.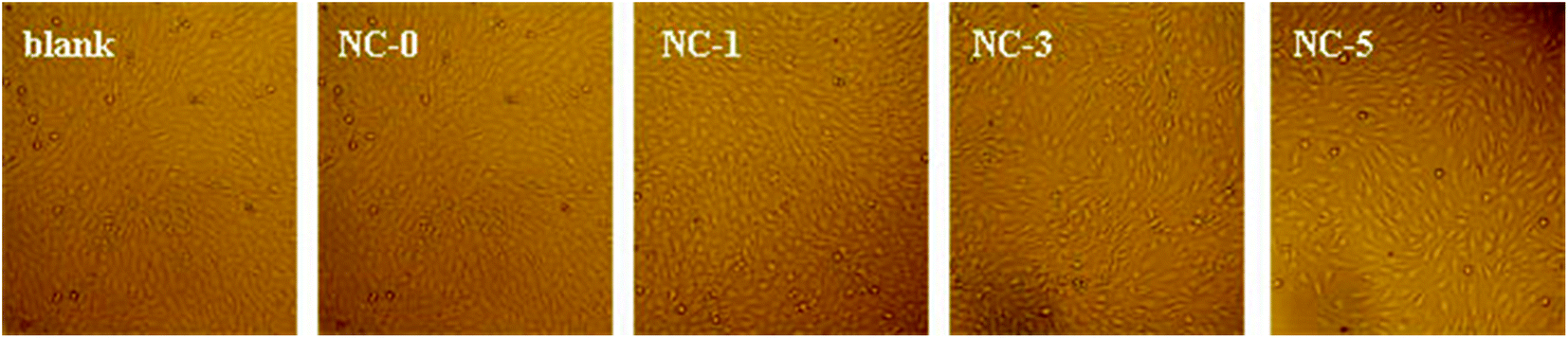 | ||
| Fig. 10 Photomicrographs of MC3T3-E1 cells cultured for 1 day in the extracts of the hydrogels. | ||
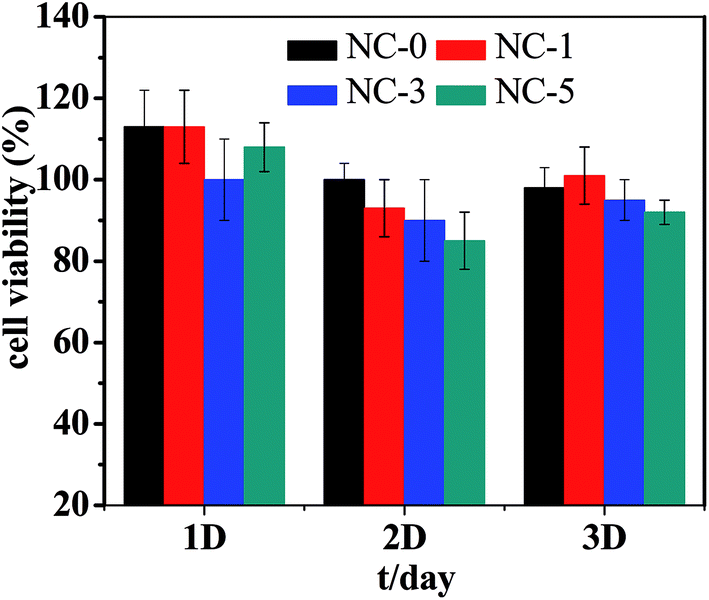 | ||
| Fig. 11 Viability of MC3T3-E1 cells for 1, 2, and 3 days. | ||
4. Conclusions
Shape memory hydrogels were synthesized by micellar free-radical polymerization approach. Multiple physical crosslinks including hydrophobic association and H-bonds endowed the hydrogels with remarkably high tensile strength, compressive strength, toughness and hardness. With increasing clay content which served as reinforcing filler, the shape recovery ratio of the hydrogels still remained over 80%. Cell cytotoxicity tests prove that the hydrogels possess low cytotoxicity. Accordingly, the hydrogels can be potentially used in biological scaffolds due to the high mechanical strength, shape memory property, and low cytotoxicity. Besides, other potential applications are also expected from the novel hydrogels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21474007, 21774009).References
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820–1835 RSC.
- J. F. Xing, M. L. Zheng and X. M. Duan, Chem. Soc. Rev., 2015, 44, 5031–5039 RSC.
- N. K. Singh and D. S. Lee, J. Controlled Release, 2014, 193, 214–227 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- K. R. Shull, Nature, 2012, 489, 36–37 CrossRef CAS PubMed.
- H. J. Zhang, T. L. Sun, A. K. Zhang, Y. Ikura, T. Nakajima, T. Nonoyama, T. Kurokawa, O. Ito, H. Ishitobi and J. P. Gong, Adv. Mater., 2016, 28, 4884–4890 CrossRef CAS PubMed.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed.
- C. Li, M. J. Rowland, Y. Shao, T. Y. Cao, C. Chen, H. Y. Jia, X. Zhou, Z. Q. Yang, O. A. Scherman and D. S. Liu, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS PubMed.
- J. Gao, C. Tang, M. A. Elsawy, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2017, 18, 826–834 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. Zhao, K. Jiao, J. Yang, C. C. He and H. L. Wang, Polymer, 2013, 54, 1596–1602 CrossRef CAS.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622–1626 CrossRef CAS.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- W. B. Li, H. Y. An, Y. Tan, C. G. Lu, C. Liu, P. C. Li, K. Xu and P. X. Wang, Soft Matter, 2012, 8, 5078–5086 RSC.
- G. Q. Jiang, C. Liu, X. L. Liu, Q. R. Chen, G. H. Zhang, M. Yang and F. Q. Liu, Polymer, 2010, 51, 1507–1515 CrossRef CAS.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449–5455 CrossRef CAS.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- C. Bilici and O. Okay, Macromolecules, 2013, 46, 3125–3131 CrossRef CAS.
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287–10293 RSC.
- M. Shibayama, Soft Matter, 2012, 8, 8030–8038 RSC.
- L. Lobo, J. Colloid Interface Sci., 2002, 254, 165–174 CAS.
- Z. Cui, R. Cheng, J. Liu, Y. P. Wu and J. P. Deng, RSC Adv., 2016, 6, 38957–38963 RSC.
- Y. P. Wu, Y. Q. Wang, H. F. Zhang, Y. Z. Wang, D. S. Yu, L. Q. Zhang and J. Yang, Compos. Sci. Technol., 2005, 65, 1195–1202 CrossRef CAS.
- A. Phadke, C. Zhang, B. Arman, C. C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- P. Kanmani and J. W. Rhim, Food Hydrocolloids, 2014, 35, 644–652 CrossRef CAS.
- K. Haraguchi, R. Farnworth, A. Ohbayashi and T. Takehisa, Macromolecules, 2003, 36, 5732–5741 CrossRef CAS.
- Y. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- Z. J. Chen, Y. Wang, T. Ba, Y. Li, J. Pu, T. Chen, Y. S. Song, Y. E. Gu, Q. Qian, J. L. Yang and G. Jia, Toxicol. Lett., 2014, 226, 314–319 CrossRef CAS PubMed.
- R. Cheng, J. Liu, P. Xie, Y. P. Wu and J. P. Deng, Polymer, 2015, 68, 246–252 CrossRef.
- Y. Liu, M. Zhu and X. Liu, et al., Polymer, 2006, 47, 1–5 CrossRef CAS.
- L. C. Li, J. Ge, B. L. Guo and P. X. Ma, Polym. Chem., 2014, 5, 2880–2890 RSC.
- X. J. Han, Z. Q. Dong, M. M. Fan, Y. Liu, J. H. Li, Y. F. Wang, Q. J. Yuan, B. J. Li and S. Zhang, Macromol. Rapid Commun., 2012, 33, 1055–1060 CrossRef CAS PubMed.
- L. Cai, K. Wang and S. Wang, Biomaterials, 2010, 31, 4457–4466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01167b |
| This journal is © The Royal Society of Chemistry 2018 |