
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermo-responsive poly(N-isopropylacrylamide)-block-poly(ionic liquid) of pyridinium sulfonate immobilized Pd nanoparticles in C–C coupling reactions†
Soheila Ghasemi * and
Zahra Amini Harandi
* and
Zahra Amini Harandi
Department of Chemistry, College of Sciences, Shiraz University, Shiraz, Iran. E-mail: ghasemis@shirazu.ac.ir
First published on 18th April 2018
Abstract
A thermo-responsive poly(N-isopropylacrylamide)-block-poly(ionic liquid) (PNIPAM-b-PIL) of pyridinium-type was prepared. Initially, controlled synthesis of PNIPAM was performed via RAFT method. Subsequently, PNIPAM as macromolecular chain transfer agent (macro-CTA) was used for fabrication of PNIPAM-b-PIL through reaction with a synthesized IL monomer i.e. 4-vinyl pyridinium propane sulfonate. The Pd catalyst was produced throughout palladium nanoparticles' anchoring into this block copolymer. The catalyst was characterized using ICP, FT-IR, NMR, UV-Vis, TGA, XRD, SEM and EDX techniques. The catalyst's TEM image proved nearly fine dispersion of PdNPs with negligible agglomeration. The catalyst was used in the production of a variety of substituted alkenes and biaryl compounds (Heck and Suzuki coupling) in organic and aqueous media and under solvent free conditions. Additionally, the results signified extreme reusability of the catalyst with a simple recycling procedure.
1. Introduction
Block copolymers are an appealing type of macromolecule with significant profit in technological applications and theoretical exploration.1 Outstanding electrical, mechanical, magnetic, and acoustic properties of block copolymers are the result of their micro phase separation and self-assembly into 3D highly ordered nanostructures. Among diverse polymerization systems i.e. free radical, anionic, cationic, coordination, group transfer, etc for the synthesis of block copolymers, controlled radical polymerization (CRP) is the most extensively used approach.2,3 The most applied CRP strategies for the production of well-defined (co)polymers are atom transfer radical polymerization (ATRP),4,5 nitroxide-mediated polymerization (NMP),6 and reversible addition–fragmentation chain transfer (RAFT) polymerization.7–10 However, employing NMP is restricted to the monomer type and condition of polymerization and a bit of metal catalyst persisting in the ultimate product has limited the utilization of ATRP in biomedical and electronic applications. In this regard, RAFT has demonstrated as exceedingly successful technique for the synthesis of block copolymers.11 For instance, block copolymers of 1,2,4-triazolium-based polymers with remarkable ion conductivity,12 lipid-based hydrophilic-hydrophobic block copolymer as nano drug carrier13 and thermo-responsive star block copolymers of N,N-dimethyl and diethyl acrylamide (DMA and DEA)14 were synthesized through RAFT polymerization.Monomers of ionic liquid (IL) produces a remarkable kind of polyelectrolytes, which are entitled poly(ionic liquid)s (PILs) with enormous potential applications.15–18 PIL emerged the property of IL and polymer in order to generate novel characters and functions. The significant benefits of PILs compare to ILs are enhanced mechanical stability, processability and strength and structural control over the IL groups.
Due to the extraordinary characteristics of block copolymers and PILs, the preparation of block copolymers include at least single PIL segment has merit of remarkable notice.19 However, by means of choosing one thermo-responsive block, it is possible to study thermal phase behavior of these systems.20,21 Generally, PIL block copolymers were synthesized through CRP techniques such as RAFT and ATRP.22,23 However, the reactivity of IL monomer ought to be adapted with these polymerization techniques. Texter et al. was announced the use of ATRP for the production of ABA triblock terpolymer with symmetric side chains PILs blocks of imidazolium type using bifunctional poly(propylene oxide) by macro ATRP initiator termini.24 Triblock copolymers with short cationic PIL end blocks and a PNIPAM middle block were prepared using ATRP.25 Gu and Lodge was announced the utilization of RAFT for the preparation of ABA triblock terpolymer with the middle imidazolium based PIL block.26 Thermo responsive diblock copolymers consist of a PNIPAM and PIL segments have been synthesized via RAFT method.27 Moreover, NMP was employed appropriately for the production of imidazolium and phosphonium class of PIL diblock copolymers or triblock terpolymers.28,29
PILs can complex and stabilize catalytically active metal nanoparticles strongly and perpetually due to the excessive charge density and polymer architecture.30–32 In addition, PIL-based systems assisted the recovery and further use of the supports.
Pd-catalyzed carbon–carbon cross coupling as Heck and Suzuki in ILs was achieved remarkable progress in the past decade.33–35 A great number of Pd coupling assisted in ILs i.e. tetraalkyl-ammonium, phosphonium, imidazolium and pyridinium-based systems can be found in the following reviews.36–39 In addition, supported ILs are developed in order to provide robust, reusable and efficient catalysts.40,41 Selected examples are; 1-aminoethyl-3-vinylimidazolium bromide ([VAIM]Br) attached on the macromolecular network supported PdNPs42 and Pd immobilized on poly(1-aminoethyl-3-vinylimidazolium bromide) coupled with magnetic nanoparticles (Pd/Fe3O4@PIL-NH2) for solvent-free Heck reaction,43 palladium catalysts anchored on gel-supported ionic liquid-like phases (g-SILLPs) in Heck coupling,44 assembly of PdCl2 in IL brushes for coupling of iodoarenes with acrylic acid in water,45 IL/Pd(OAc)2 on the mesoporous SBA-15 for Heck reaction,46 N-heterocyclic carbene (NHC)/Pd supported on IL-functionalized graphene oxide for Suzuki coupling,47 NHC/Pd complex supported IL-modified SBA-16 for the Suzuki and Heck reactions,48 functional IL modified Fe3O4/PdNPs for biaryl formation via Suzuki coupling49 and PIL entrapped magnetic nanoparticles/Pd for the solvent-free Heck reaction.50
In continuation of our previous report on the preparation of different type of heterogeneous Pd catalysts,51–53 and considering the outstanding characteristics of block copolymers and PILs, herein we reported a proper strategy for the preparation of thermo-responsive PNIPAM-b-PIL via RAFT method. The corresponding Pd catalyst was produced through the immobilization of PdNPs onto this block copolymer. Afterwards, the potency of the Pd catalyst was examined in C–C coupling (Mizoroki–Heck and Suzuki–Miyaura).
2. Experimental
2.1. General remarks
2.2. Poly(N-isopropylacrylamide-b-ionic liquid) (PNIPAM-b-PIL) supported PdNPs
2.3. Cross-coupling reactions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (10) as eluent to afford the coupling product. The aryl-alkene products characterization was carried out by comparing their spectral data with the authentic specimens.
:ethyl acetate (10) as eluent to afford the coupling product. The aryl-alkene products characterization was carried out by comparing their spectral data with the authentic specimens.2.3.3.1. In organic media (DMF). After a Heck reaction in DMF, the suspension was chilled to ambient temperature and diethyl ether was added to the mixture. The product was remained in organic phase and the catalyst was precipitated. After solvent decanting, the recovered catalyst was reused in the subsequent reaction run with a pristine amount of substrates with no pretreatment. The Pd catalyst was used more than 20 cycles and maintained its catalytic function in the repeating sequences.
2.3.3.2. In aqueous media. Following a Heck or Suzuki reaction in water, the reaction mixture was chilled to ambient temperature and diethyl ether was added to the suspension. The product extracted to organic phase and the catalyst remained in water. Two phases were isolated and the aqueous phase was washed with diethyl ether three times. Afterward, to a mixture of recycled Pd catalyst in H2O was added new quantity of reactants. The Pd catalyst was used for 10 cycles in coupling reactions and preserved its activity in the repeating runs.
3. Result and discussion
3.1. Thermo-responsive PNIPAM-b-PIL containing PdNPs via RAFT polymerization; preparation and characterization
PNIPAM-b-PIL as a thermo-responsive block polymeric support was prepared through RAFT polymerization system (Scheme 1). Originally, NIPAM, DDMAT and AIBN with the molar ration of 100:1:0.2 in the act of monomer, CTA and initiator respectively were used for the preparation of PNIPAM in dioxane with the phase transition ≃32 °C. PNIPAM (DPth = 100) was identified using multiple techniques i.e. FT-IR, NMR, TGA and SEM.
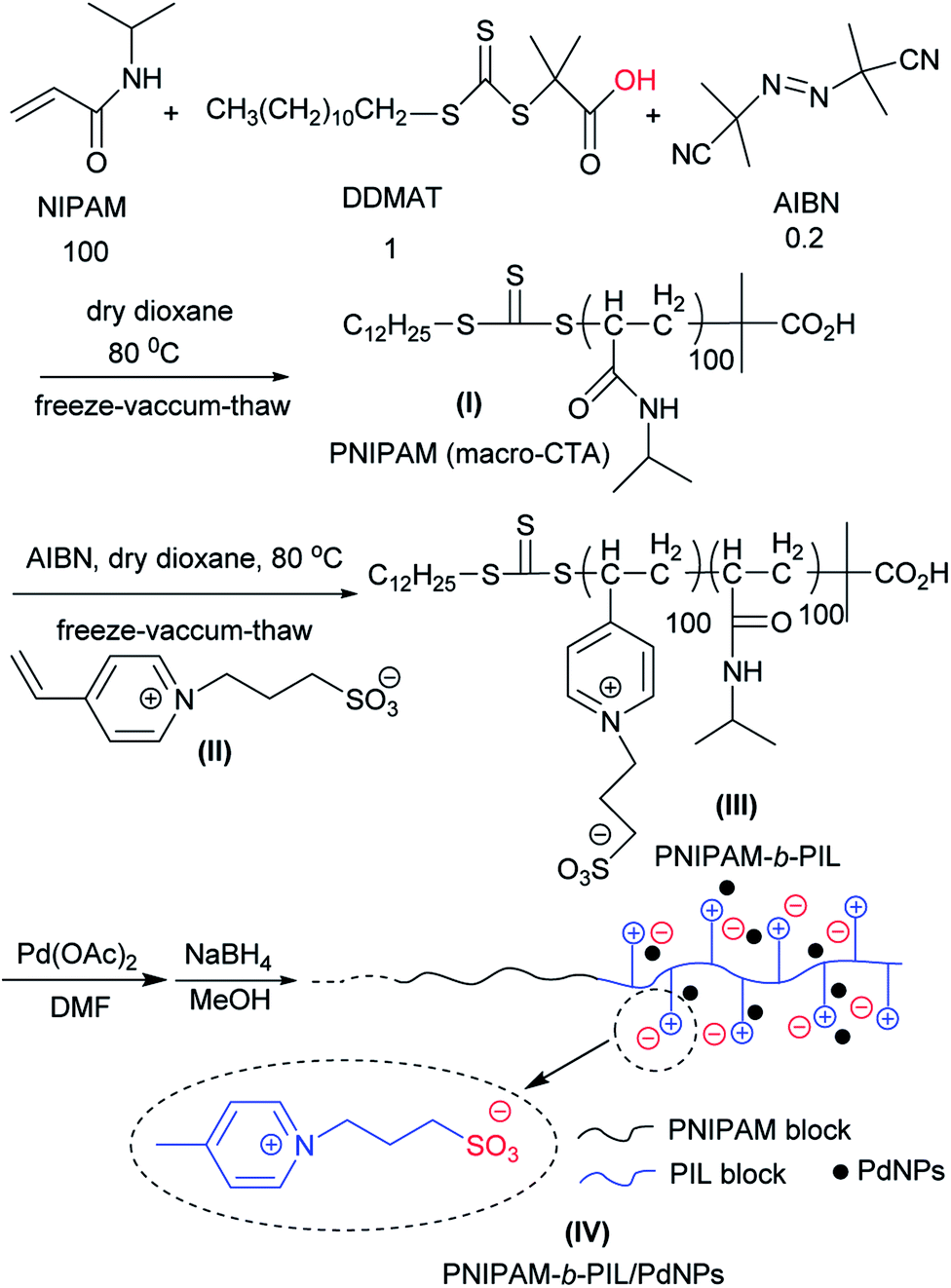 | ||
| Scheme 1 Schematic representation for the preparation of thermo-responsive PNIPAM-b-PIL/PdNPs. | ||
The PNIPAM's IR spectrum represented the absorption peak of amide group (CO–NHR), NH bending, carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and C–N stretching at 3311, 1542, 1654 and 1459 cm−1 respectively (Fig. A1, ESI†). The existence of the trithiocarbonate functionality at the PNIPAM's end group was demonstrated using 1H-NMR spectroscopy through the depiction of a peak at 3.7 ppm (CH3–(CH2)10–CH2–S–), 0.86 ppm (CH3–(CH2)10–CH2–S–) and 11.3 ppm (–C(CH3)2–CO2H) (Fig. A2, ESI†). In addition, average degree of polymerization (DPn, NMR) derived by comparing signal intensity at 3.7 ppm of the dodecylthiocarbonate group with integration at 4 ppm (–NH–CH(CH3)2) or 7.1 ppm (–NH–CH(CH3)2) of the NIPAM repeating group (Fig. A2, ESI†). The results expressed proper accord between experimental DP and theoretical ones.
O) and C–N stretching at 3311, 1542, 1654 and 1459 cm−1 respectively (Fig. A1, ESI†). The existence of the trithiocarbonate functionality at the PNIPAM's end group was demonstrated using 1H-NMR spectroscopy through the depiction of a peak at 3.7 ppm (CH3–(CH2)10–CH2–S–), 0.86 ppm (CH3–(CH2)10–CH2–S–) and 11.3 ppm (–C(CH3)2–CO2H) (Fig. A2, ESI†). In addition, average degree of polymerization (DPn, NMR) derived by comparing signal intensity at 3.7 ppm of the dodecylthiocarbonate group with integration at 4 ppm (–NH–CH(CH3)2) or 7.1 ppm (–NH–CH(CH3)2) of the NIPAM repeating group (Fig. A2, ESI†). The results expressed proper accord between experimental DP and theoretical ones.
Thermo gravimetric (TG) data of the PNIPAM (I) (heat rate of 10 °C min−1 from 25 to 800 °C in N2/O2 atmosphere) is depicted in Fig. 1a. The thermal deterioration of PNIPAM was took place in one main pyrolysis step. The beginning minor amount of weight loss was ascribed to the elimination of adsorbed small molecules and water. The PNIPAM manifested good thermal stability up to 300 °C. The weight markedly reduced around 350 °C up to 400 °C and organic components were decomposed completely over 410 °C. In addition, the scanning electron microscopy (SEM) image demonstrated the macroscopic structure, morphology and size of PNIPAM (Fig. 1b). The PNIPAM's SEM photograph emerged almost amorphous particles with nearly 2 μm dimension.
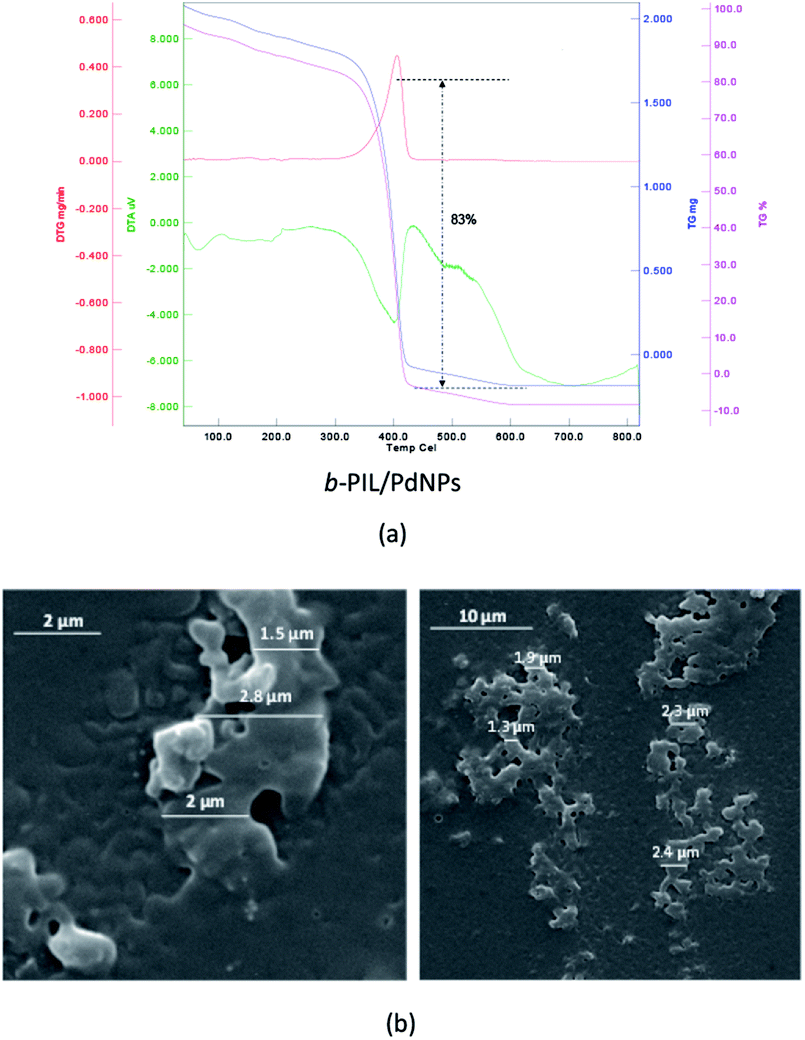 | ||
| Fig. 1 (a) TG, DTG and DTA data and (b) SEM images of PNIPAM (I). | ||
PNIPAM as macro-CTA was used to synthesize PNIPAM-b-PIL (III) through the reaction with ionic liquid monomer (II) (Scheme 1). 4-Vinyl pyridinium propane sulfonate (II) was produced formerly throughout the reaction of 4-vinyl pyridine and 1,3-propanesultone (Scheme 2). FT-IR spectrum of IL monomer (II) demonstrated the required characteristic peaks i.e. stretching vibration absorption of CN (1519 cm−1) and CC bond of the pyridine rings (1472 cm−1 and 1644 cm−1) and SO asymmetric and symmetric stretching absorption of the –SO3− group (1184 cm−1 and 1036 cm−1) (Fig. A3, ESI†). In addition, 1H-NMR spectrum of the IL monomer (II) is revealed the signals of the aromatic hydrogens of the pyridine ring at 7.5–9 ppm (2H appeared at 7.7 ppm and the other 2H ones at 8.6 ppm), vinyl hydrogens at 5.8, 6.3 and 6.8 ppm and aliphatic hydrogens at 1.7 ppm appropriately (Fig. A4, ESI†).
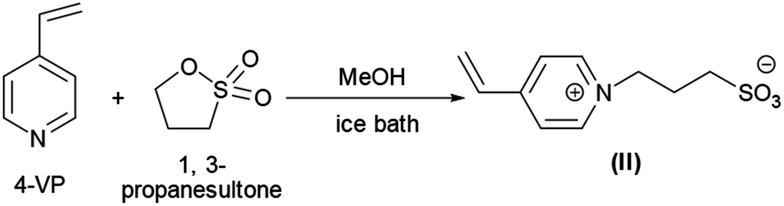 | ||
| Scheme 2 Illustrative preparation of ionic liquid monomer (II). | ||
In order to disclose the phase transition of PNIPAM-b-PIL (III), the transmittance of the block copolymer solution in deionized water was measured as a function of temperature using a UV-Vis spectrophotometer at 330 nm (Fig. 2). Cloud point of PNIPAM-b-PIL (III) occurred at around 40 °C that showed a shift compare to the homopolymer of PNIPAM (≃32 °C). It is expected that PIL as hydrophilic co-monomer increases the PNIPAM's phase transition. Above the lower critical solution temperature (LCST), the hydrophobic interactions become dominant and the cloudiness of the solution is clearly visible, owing to the collapse of the polymer structure. In addition, conversion of PNIPAM (I) to PNIPAM-b-PIL (III) is accompanied by the change in zeta potential from −93.4 to + 21.4.
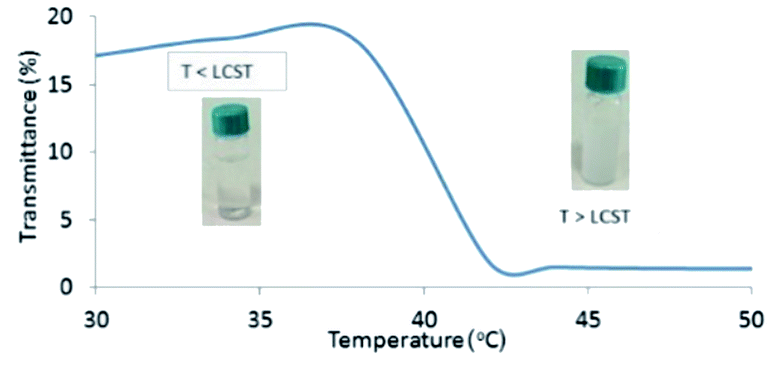 | ||
| Fig. 2 UV-Vis spectrum of PNIPAM-b-PIL (III) respecting temperature changes based on transmittance percentage. | ||
FT-IR spectrum of PNIPAM-b-PIL (III) expressed absorption of amide group (CO–NHR) at 3322 cm−1, (NH) bending at 1541 cm−1, carbonyl functionality (CO) at 1643 cm−1, CC bond of aromatic ring at 1467 cm−1 and SO asymmetric and symmetric absorption of the –SO3− functionality at 1181 cm−1 and 1036 cm−1 respectively (Fig. A5, ESI†). Furthermore, 1H-NMR spectrum of PNIPAM-b-PIL (III) indicated the signal of the hydrogens of the pyridine ring at 7–7.5 ppm and the signal of hydrogen of isopropyl of PNIPAM at 3.7 ppm accordingly (Fig. A6, ESI†). The ratio integration for peaks of pyridine ring of PIL to isopropyl of PNIPAM is 1.1:1; therefore, we conclude that the DP of the PNIPAM block (DP = 100) is nearly quadruple than the DP of the PIL block (DP = 27). This may be due to the lower activity of the IL monomer (II) compare to NIPAM in this RAFT polymerization condition.
TGA diagram of PNIPAM-b-PIL (III) under the same condition as PNIPAM is represented in Fig. 3a. The little amount of weight loss around 100 °C was corresponded completely to the removal of adsorbed H2O. The TGA graph displayed two pyrolysis stages due to the presence of two different blocks. The PNIPAM block decomposed nearly at 350 °C and PIL block at around 400 °C. The integration of PIL block is less than of PNIPAM block that matched to 1H-NMR data. Furthermore, the SEM image of PNIPAM-b-PIL (III) is outlined in Fig. 3b and illustrated the particles with quasi-spherical morphology and size range of 0.12–0.33 μm.
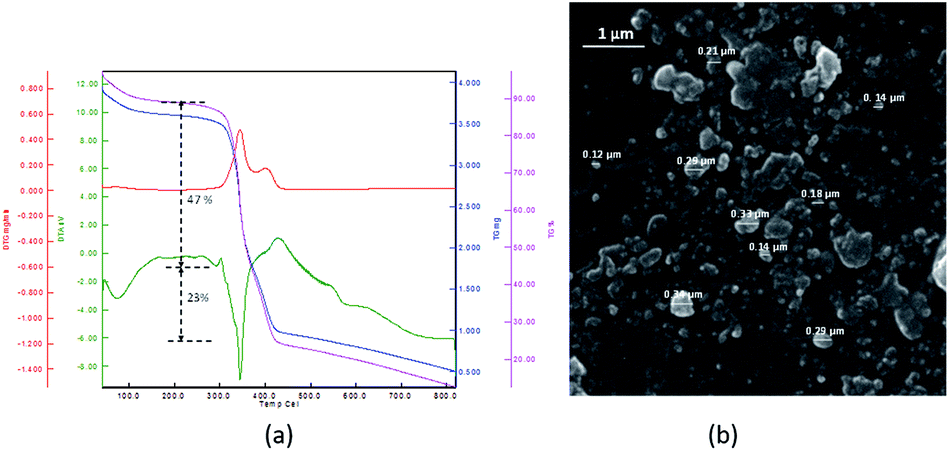 | ||
| Fig. 3 (a) TG, DTG and DTA data and (b) SEM image of PNIPAM-b-PIL (III). | ||
Finally, PNIPAM-b-PIL/Pd catalyst (IV) was generated by treating PNIPAM-b-PIL (III) with palladium acetate salt and subsequent reducing of Pd(II) to Pd(0) with NaBH4 (Scheme 1). ICP analysis disclosed 0.196 mmol g−1 Pd loading. DLS experiment indicated approximate hydrodynamic diameter of this catalyst (IV) before, after and at its LCST (Fig. 4a). At ambient temperature of 25 °C (before its LCST) average size of 100 nm is observed, while at its LCST (40 °C) and after its LCST (65 °C) aggregates of approximately 140 and 220 nm were found respectively. Moreover, wide angle powder X-ray diffraction (XRD) motif of Pd catalyst is displayed in Fig. 4b. Crystallographic planes of Pd0 ((111), (200), (220) and (311)) was emerged at 2θ = 40.00, 46.49, 67.90 and 81.85 respectively and the mean size of the PdNPs (roughly calculated from the Scherrer equation) was found to be nearly 27 nm. In addition, the UV-Vis spectra of Pd(OAc)2, PNIPAM-b-PIL (III), PNIPAM-b-PIL/PdII and PNIPAM-b-PIL/Pd0 catalyst (IV) are represented in Fig. 4c. The spectrum of Pd(OAc)2 exhibited a peak at 328 nm while no peak was observed for PNIPAM-b-PIL (III). Binding the Pd to the block copolymer result in a peak appearance at 294 nm which is attributed to Pd(II). After reduction step, the peak is disappeared which confirmed the Pd(II) to Pd(0) conversion.
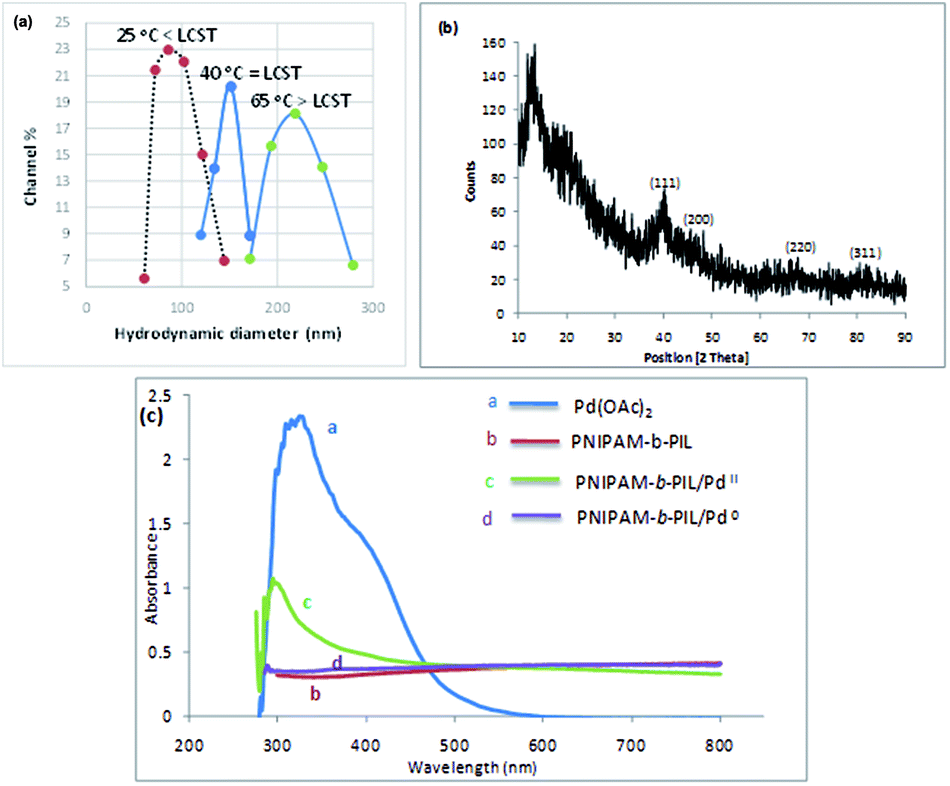 | ||
| Fig. 4 (a) Hydrodynamic size distribution plot of Pd catalyst (IV) before, after and at its LCST, (b) XRD spectrum of Pd catalyst (IV) and (c) UV-Vis illustration of Pd(OAc)2, PNIPAM-b-PIL (III), PNIPAM-b-PIL/PdII and PNIPAM-b-PIL/Pd0 catalyst (IV). | ||
Fig. 5a is depicted the SEM photograph of PNIPAM-b-PIL/Pd catalyst (IV). The morphology (supposed spherical) and size (0.15 to 0.2 μm) of the catalyst do not revealed significant change compare to the block copolymer. Additionally, EDX spectrum of catalyst (IV) demonstrated 1.98% Pd loaded on the block copolymer. TEM picture of the Pd catalyst (IV) is rendered in Fig. 5b. The catalyst has a core (PdNPs)-corona (block copolymer) structure with the PdNPs' average size of 20 nm. No agglomeration of PdNPs occurred due to the electrostatic stabilization of PIL segments and steric stabilization of polymer chains.54
 | ||
| Fig. 5 (a) SEM image and (b) TEM images of Pd catalyst (IV). | ||
3.2. Mizoroki–Heck and Suzuki–Miyaura coupling reactions using PNIPAM-b-PIL/Pd catalyst
|
|||||
|---|---|---|---|---|---|
| No | Solvent | Base | Pd catalyst (mol%) | Time (min) | Conversionb (%) |
| a Optimization reactions are accomplished using 1.0 eq. iodobenzene, 1.2 eq. n-butyl acrylate, 2.0 eq. base and 2 mL of solvent at 85 °C.b According to iodobenzene conversion.c 0.5 mL H2O.d TBAB (tetra-n-butylammonium bromide) as an additive. | |||||
| 1 | DMF | K2CO3 | 1.5 | 10 | 98 |
| 2 | DMF | K2CO3 | 1 | 20 | 97 |
| 3 | DMF | K2CO3 | 0.5 | 20 | 97 |
| 4 | DMF | K2CO3 | 0.25 | 25 | 97 |
| 5 | DMF | K2CO3 | 0.1 | 35 | 95 |
| 6 | DMF | K2CO3 | 0.05 | 45 | 95 |
| 7 | DMF | K2CO3 | 0.01 | 45 | 90 |
| 8 | DMF | Na2CO3 | 1 | 15 | 95 |
| 9 | DMF | NaOH | 1 | 15 | 96 |
| 10 | DMF | KOH | 1 | 10 | 98 |
| 11 | DMF | Cs2CO3 | 1 | 25 | 65 |
| 12 | DMF | Et3N | 1 | 15 | 97 |
| 13 | DMF | Base free | 1 | 180 | — |
| 14 | H2O | Et3N | 1 | 120 | 95 |
| 15 | Dioxane | Et3N | 1 | 50 | 65 |
| 16 | Ethanol | Et3N | 1 | 40 | 90 |
| 17 | CH2Cl2 | Et3N | 1 | 40 | 10 |
| 18 | H2O/ethanol | Et3N | 1 | 100 | 50 |
| 19 | H2O/DMF | Et3N | 1 | 20 | 97 |
| 20c | H2O | Et3N | 1 | 20 | 98 |
| 21d | H2O | Et3N | 1 | 15 | 95 |
| 22 | Solvent free | Et3N | 1 | 15 | 94 |
The influence of catalyst quantity on the coupling of phenyl iodide and n-butyl acrylate exhibited that the catalyst loading can be minimized up to 0.01 mol% of Pd catalyst while the reaction time extended to 45 min (Table 1, entry 1–7). Performing the model reaction in different bases (using DMF as solvent and 1 mol% Pd catalyst) revealed Et3N as the best possibility (Table 1, entry 2 and entries 8–12). No coupling product was formed without base (Table 1, entry 13). Among various solvents and mixture of solvents, DMF was selected as the most appropriate one (Table 1, entry 12 and entries 14–19). Applying water as a green reaction medium is extremely important, in spite of the fact that, performing the reaction in H2O was generated the product after 120 min (Table 1, entry 14). Moreover, utilizing fewer amount of water as solvent (0.5 mL) or employing TBAB as additive yielded the superior result (Table 1, entries 20, 21). In addition, the reaction was carried out under solvent free condition promisingly (Table 1, entry 22).
|
||||||
|---|---|---|---|---|---|---|
| No | Ar/X | R | Time (min) | TONb | TOF (min−1)c | Isolated yieldd (%) |
| a All coupling reactions were accomplished with a molar ratios of ArX:olefin:NEt3:Pd catalyst (IV) = 1.0:1.2:2.0:0.001 using 2 mL of DMF at 85 °C.b TON = mmol of product/mmol of Pd in the catalyst.c TOF = TON/time (min).d Product identification was performed via their FT-IR and NMR spectra. |
||||||
| 1 | Ph/I | CO2Bun | 30 | 960 | 32 | 96 |
| 2 | 4-OHPh/I | CO2Bun | 160 | 940 | 5.9 | 94 |
| 3 | 4-CH3Ph/I | CO2Bun | 15 | 930 | 62 | 93 |
| 4 | 2-CH3Ph/I | CO2Bun | 20 | 970 | 48.5 | 97 |
| 5 | 4-NO2Ph/I | CO2Bun | 60 | 940 | 15.7 | 94 |
| 6 | 4-BrPh/I | CO2Bun | 30 | 970 | 32.3 | 97 |
| 7 | 4-ClPh/I | CO2Bun | 30 | 960 | 32 | 96 |
| 8 | 4-OCH3Ph/I | CO2Bun | 12 | 900 | 75 | 90 |
| 9 | 4-OHPh/Br | CO2Bun | 420 | 950 | 2.3 | 95 |
| 10 | 4-NO2Ph/Br | CO2Bun | 540 | 850 | 1.6 | 85 |
| 11 | 2-OHPh/Br | CO2Bun | 420 | 800 | 1.9 | 80 |
| 12 | Ph/Br | CO2Bun | 360 | 800 | 2.2 | 80 |
| 13 | Ph/I | Ph | 35 | 950 | 27.1 | 95 |
| 14 | 4-OHPh/I | Ph | 150 | 800 | 5.3 | 80 |
| 15 | 4-CH3Ph/I | Ph | 120 | 910 | 7.6 | 91 |
| 16 | 2-CH3Ph/I | Ph | 300 | 800 | 2.7 | 80 |
| 17 | 4-NO2Ph/I | Ph | 120 | 820 | 6.8 | 82 |
| 18 | 4-BrPh/I | Ph | 120 | 960 | 8 | 96 |
| 19 | 4-ClPh/I | Ph | 120 | 920 | 7.7 | 92 |
| 20 | 4-OCH3Ph/I | Ph | 120 | 800 | 6.7 | 80 |
Since numerous beneficial features of water as an alternative to organic solvents, we resolved to examine the Heck coupling reaction in water as green reaction media. Diverse iodoarenes include substrates with electron-withdrawing and electron donating functionalities are coupled with n-butyl acrylate or styrene utilizing Pd catalyst (IV). The results are organized in Table 3. Furthermore, 2-iodothiophene is reacted with n-butyl acrylate successfully and provided the corresponding product in 96% yield after 240 min.
|
||||||
|---|---|---|---|---|---|---|
| No | Ar | R | Time (min) | TON | TOF (min−1) | Isolated yieldb (%) |
| a All coupling reactions were accomplished with a molar ratios of ArI:olefin:NEt3:Pd catalyst (IV) = 1.0:1.2:2.0:0.001 using 0.5 mL of H2O at 85 °C.b Product identification was performed via their FT-IR and NMR spectra. |
||||||
| 1 | Ph | CO2Bun | 35 | 940 | 26.8 | 94 |
| 2 | 2-CH3Ph | CO2Bun | 360 | 930 | 2.6 | 93 |
| 3 | 4-OCH3Ph | CO2Bun | 210 | 300 | 1.4 | 30 |
| 4 | 4-OHPh | CO2Bun | 240 | 800 | 3.3 | 80 |
| 5 | 4-CH3Ph | CO2Bun | 420 | 800 | 1.9 | 80 |
| 6 | 4-NO2Ph | CO2Bun | 150 | 950 | 6.3 | 95 |
| 7 | 4-BrPh | CO2Bun | 240 | 970 | 4 | 97 |
| 8 | Ph | Ph | 180 | 440 | 2.4 | 45 |
| 9 | 4-OCH3Ph | Ph | 180 | 500 | 2.8 | 50 |
| 10 | 4-OHPh | Ph | 120 | 500 | 4.2 | 50 |
| 11 | 4-NO2Ph | Ph | 240 | 960 | 4 | 96 |
| 12 | 4-BrPh | Ph | 30 | 200 | 6.7 | 20 |

Additionally, some Heck reactions are performed under solvent free condition in order to expand the capacity of our catalytic methodology (Scheme 3).
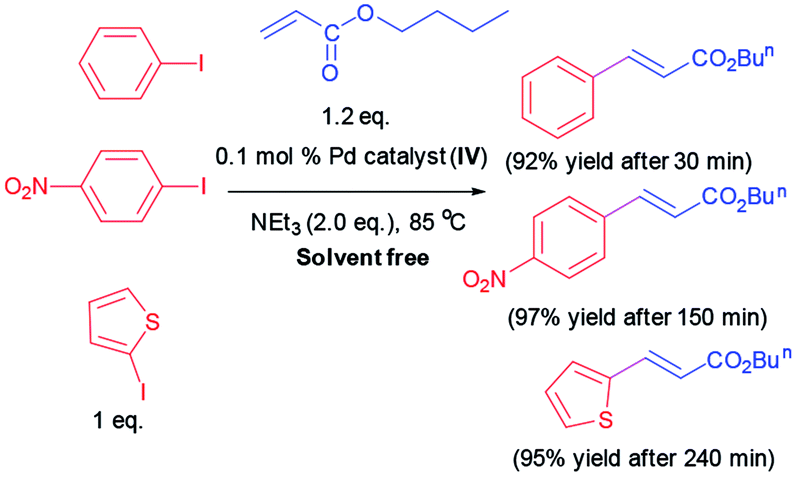 | ||
| Scheme 3 Heck reaction under solvent free condition. | ||
|
||||||
|---|---|---|---|---|---|---|
| No | Ar | X | Time (min) | TON | TOF (min−1) | Isolated yieldb (%) |
| a All coupling reactions were accomplished with a molar ratios of ArX:phenyl boronic acid:NEt3:Pd catalyst (IV) = 1.0:1.2:2.0:0.001 using 0.5 mL of H2O at 85 °C.b Products identification was performed via their FT-IR and NMR spectra. |
||||||
| 1 | Ph | I | 20 | 950 | 47.5 | 95 |
| 2 | 4-OCH3Ph | I | 40 | 960 | 24 | 96 |
| 3 | 4-CH3Ph | I | 80 | 950 | 11.9 | 95 |
| 4 | 2-CH3Ph | I | 90 | 960 | 10.7 | 96 |
| 5 | 4-ClPh | I | 15 | 970 | 64.7 | 97 |
| 6 | 4-NO2Ph | I | 40 | 940 | 23.5 | 94 |
| 7 | 4-CH3Ph | Br | 120 | 920 | 7.7 | 92 |
| 8 | 2-CH3Ph | Br | 180 | 900 | 5 | 90 |
| 9 | 4-OHPh | Br | 180 | 700 | 3.9 | 70 |
| 10 | 2-OHPh | Br | 720 | 700 | 1 | 70 |
| 11 | 4-NO2Ph | Br | 180 | 920 | 5.1 | 92 |
3.3. The recyclability experiments of the catalyst
The ability of precious metal catalyst's recycling with simple procedure and low leaching is very crucial from commercial and economical standpoint. To probe the reusability of the catalyst, the coupling Heck reaction of phenyl iodide with n-butyl acrylate in DMF was investigated for catalyst (IV). For this purpose, after fulfilment of the reaction, the suspension was chilled to room temperature and diethyl ether was added to the corresponding mixture. The product was remained in organic phase and the catalyst was precipitated (Fig. 6). After solvent decanting, the recovered catalyst was used in 20 subsequent reactions successfully without appreciable change in its efficiency (Fig. 7).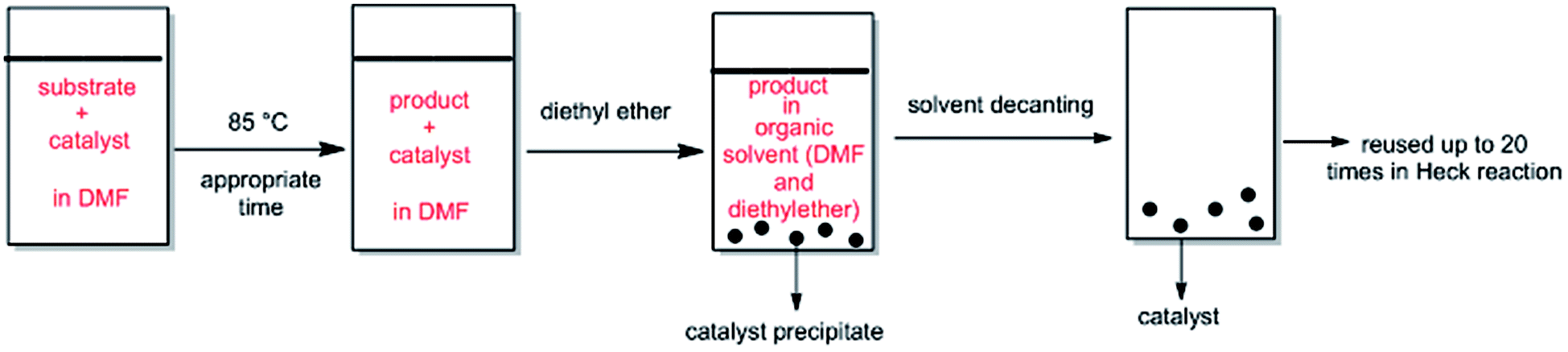 | ||
| Fig. 6 Schematic procedure of catalyst recycling in DMF. | ||
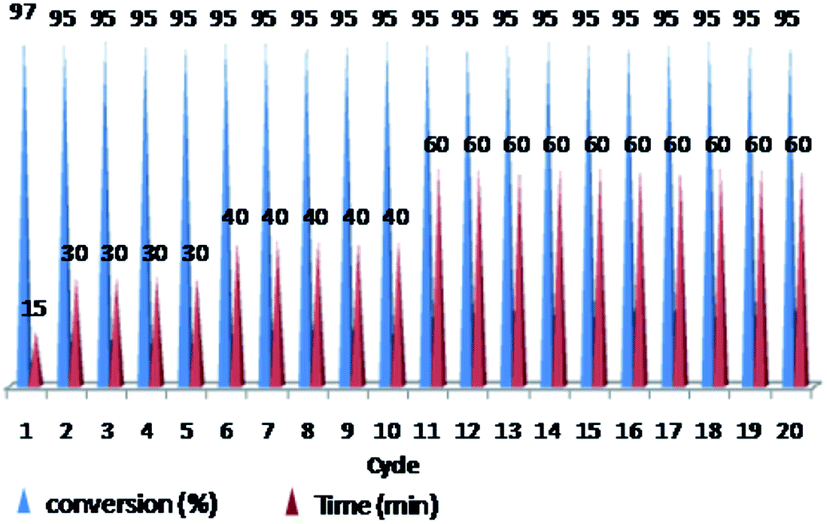 | ||
| Fig. 7 Reusability of PNIPAM-b-PIL/Pd catalyst (IV) in DMF (iodobenzene: n-butyl acrylate: Et3N: catalyst (IV) with a molar ratios of 1.0:1.2:2.0:0.01 at 85 °C). | ||
Furthermore, we probed the reusability of the Pd catalyst (IV) in H2O. In this regard, after completion of a Heck reaction between phenyl iodide and n-butyl acrylate, diethyl ether was added to the suspension. The product extracted into organic phase and the catalyst remained in water. Aqueous and organic phases are separated and the recycled catalyst in water reused eleventh times in successive reactions without appreciable loss of its catalytic activity in these repeating cycles (Fig. 8 and 9).
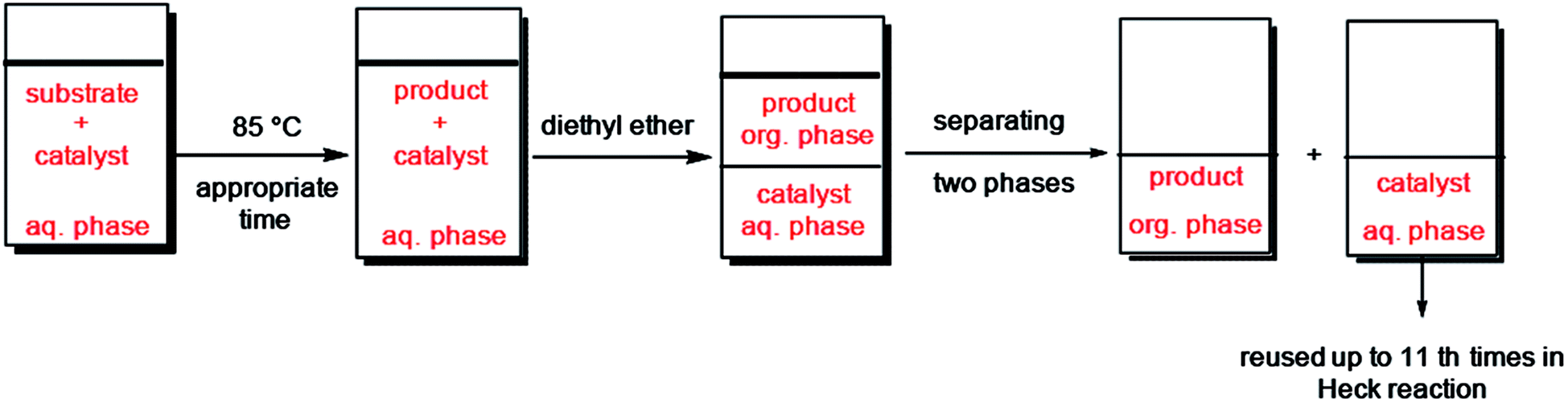 | ||
| Fig. 8 Schematic procedure of catalyst recycling in H2O. | ||
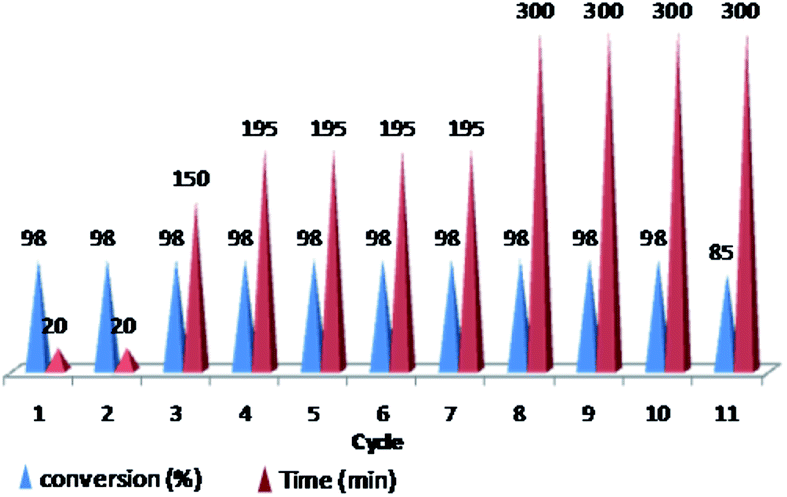 | ||
| Fig. 9 Reusability of PNIPAM-b-PIL/Pd catalyst (IV) in H2O (iodobenzene: n-butyl acrylate: Et3N: catalyst (IV) with a molar ratios of 1.0:1.2:2.0:0.01 at 85 °C). | ||
Effective catalytic activity of PNIPAM-b-PIL/PdNPs in water at 85 °C i.e. the temperature above the LCST of PNIPAM is related to the diffusion of the reactants into collapsed PNIPAM's chain due to its micellization.55,56 Actually, PNIPAM-b-PIL/PdNPs as a nanoreactor offer an extremely high concentration of substrates around the PdNPs as shown in Fig. 10.
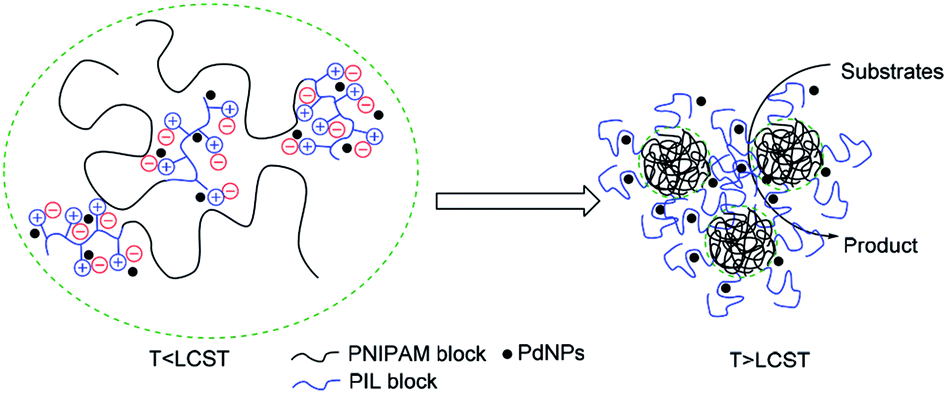 | ||
| Fig. 10 Schematic illustration of the Heck reaction within the PNIPAM-b-PIL/PdNPs in water. | ||
Investigation the catalyst recycling for the Suzuki coupling of iodobenzene and phenyl boronic acid undergoing optimized reaction condition also revealed successful reusing of the catalyst at the minimum of ten times in successive reactions without remarkable depletion of its activity i.e. no decrease of product yields and time increasing from 20 min to 40 min.
In addition, to explore the amount of palladium leaching from the support, the Pd content (mmol g−1) of the catalyst was determined after 10 repeating cycles in Heck coupling reaction and compared with Pd content of fresh catalyst. It is reduced from 0.196 mmol g−1 to 0.176 mmol g−1 and therefore, indicated low Pd leaching from the support.
4. Conclusion
In the present research, the synthesis of thermo-responsive PNIPAM-b-PIL based on modified vinylpyridine-type ionic liquid via RAFT technique is reported. RAFT polymerization imparted a facile process to prepare block copolymer with foreseeable molecular weights. PNIPAM-b-PIL supported PdNPs is prepared via Pd(OAc)2 treatment. The catalyst was identified thoroughly by ICP, UV-Vis spectrophotometer, TGA, XRD, SEM and TEM analysis. The PNIPAM-b-PIL/PdNPs was employed in the Heck and Suzuki coupling for the production of various substituted alkenes and biaryls in a moderate reaction condition. The coupling reactions can be performed in organic, aqueous or even under solvent free condition. Effective catalytic activity of PNIPAM-b-PIL/PdNPs in water at the temperature above the LCST of PNIPAM is related to the diffusion of the reactants into collapsed PNIPAM's chain. The catalyst displayed extreme recyclability with simple procedure and low leaching.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are appreciated from Research Council of Shiraz University to a great extent for partial supporting of the present study.References
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- K. Matyjaszewski, Controlled/living radical polymerization: progress in ATRP, NMP, and RAFT, American Chemical Society, Washington DC, 2000 Search PubMed.
- Y. Xu, L. Shi, R. Ma, W. Zhang, Y. An and X. X. Zhu, Polymer, 2007, 48, 1711–1717 CrossRef CAS.
- W.-C. Wu, C.-Y. Chen, W.-Y. Lee and W.-C. Chen, Polymer, 2015, 65, A1–A16 CrossRef CAS.
- A. Britze, J. Jacob, V. Choudhary, V. Moellmann, G. Grundmeier, H. Luftmann and D. Kuckling, Polymer, 2010, 51, 5294–5303 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37(5), 312–325 CrossRef CAS PubMed.
- C. E. Lipscomb and M. K. Mahanthappa, Macromolecules, 2009, 42(13), 4571–4579 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- K. Nakabayashi, A. Umeda, Y. Sato and H. Mori, Polymer, 2016, 96, 81–93 CrossRef CAS.
- M. Arshad, R. A. Pradhan and A. Ullah, Materials Science and Engineering: C, 2017, 76, 217–223 CrossRef CAS PubMed.
- C. Herfurth, A. Laschewsky, L. Noirez, B. von Lospichl and M. Gradzielski, Polymer, 2016, 107, 422–433 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- A. S. Shaplov, D. O. Ponkratov and Y. S. Vygodskii, Polym. Sci., Ser. B, 2016, 58, 73–142 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- A. Eftekhari and T. Saito, Eur. Polym. J., 2017, 90, 245–272 CrossRef CAS.
- K. Vijayakrishna, S. K. Jewrajka, A. Ruiz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Macromolecules, 2008, 41, 6299–6308 CrossRef CAS.
- Z. Wang, H. Lai and P. Wu, Soft Matter, 2012, 8, 11644–11653 RSC.
- E. Karjalainen, N. Chenna, P. Laurinmaki, S. J. Butcher and H. Tenhu, Polym. Chem., 2013, 4, 1014–1024 RSC.
- K. Vijayakrishna, D. Mecerreyes, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5167–5174 CrossRef CAS.
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS.
- J. Texter, V. A. Vasantha, R. Crombez, R. Maniglia, L. Slater and T. Mourey, Macromol. Rapid Commun., 2012, 33, 69–74 CrossRef CAS PubMed.
- E. Karjalainen, V. Khlebnikov, A. Korpi, S.-P. Hirvonen, S. Hietala, V. Aseyev and H. Tenhu, Polymer, 2015, 58, 180–188 CrossRef CAS.
- Y. Gu and T. P. Lodge, Macromolecules, 2011, 44, 1732–1736 CrossRef CAS.
- H. Mori, M. Yahagi and T. Endo, Macromolecules, 2009, 42, 8082–8092 CrossRef CAS.
- W. Yang, X. He, J. Gao, H. Guo, X. He, F. Wan, X. Zhao, Y. Yu and B. Pei, Chin. Sci. Bull., 2010, 55, 3562–3568 CrossRef CAS.
- S. Cheng, F. L. Beyer, B. D. Mather, R. B. Moore and T. E. Long, Macromolecules, 2011, 44, 6509–6517 CrossRef CAS.
- Z. He and P. Alexandridis, Adv. Colloid Interface Sci., 2017, 244, 54–70 CrossRef CAS PubMed.
- J. W. Lee, J. Y. Shin, Y. S. Chun, H. B. Jang, C. E. Song and S.-g. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef CAS PubMed.
- Y. Wang, G. Wei, F. Wen, X. Zhang, W. Zhang and L. Shi, J. Mol. Catal. A: Chem., 2008, 280, 1–6 CrossRef CAS.
- L. Yuan, Y. Xu, X. Hu, G. Yang and Y. Wu, J. Mol. Catal. A: Chem., 2015, 396, 55–60 CrossRef CAS.
- P. Nehra, B. Khungar, K. Pericherla, S. C. Sivasubramanian and A. Kumar, Green Chem., 2014, 16, 4266–4271 RSC.
- A. D. Sawant, D. G. Raut, N. B. Darvatkar, U. V. Desai and M. M. Salunkhe, Catal. Commun., 2010, 12, 273–276 CrossRef CAS.
- F. Bellina and C. Chiappe, Molecules, 2010, 15, 2211–2245 CrossRef CAS PubMed.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441–3461 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 15, 2419–2440 Search PubMed.
- D. Elhamifar, B. Karimi, J. Rastegar and M. Hossain Banakar, ChemCatChem, 2013, 5, 2418–2424 CrossRef CAS.
- Z. Hou and B. Han, in Coupling Reactions with Supported Ionic Liquid Catalysts, ed. R. Fehrmann, A. Riisager and M. Haumann, Wiley-VCH, 2014 Search PubMed.
- G. Liu, M. Hou, J. Song, T. Jiang, H. Fan, Z. Zhang and B. Han, Green Chem., 2010, 12, 65–69 RSC.
- W. Liu, D. Wang, Y. Duan, Y. Zhang and F. Bian, Tetrahedron Lett., 2015, 56, 1784–1789 CrossRef CAS.
- M. I. Burguete, E. Garcia-Verdugo, I. Garcia-Villar, F. Gelat, P. Licence, S. V. Luis and V. Sans, J. Catal., 2010, 269, 150–160 CrossRef CAS.
- X. Shi, X. Han, W. Ma, J. Fan and J. Wei, Appl. Organomet. Chem., 2012, 26, 16–20 CrossRef CAS.
- J.-Y. Jung, A. Taher, H.-J. Kim, W.-S. Ahn and M.-J. Jin, Synthesis, 2009, 1, 39–42 Search PubMed.
- S. Kazemi Movahed, R. Esmatpour salmani and A. Bazgir, RSC Adv., 2014, 4, 14586–14591 RSC.
- H. Yang, X. Han, G. Li and Y. Wang, Green Chem., 2009, 11, 1184–1193 RSC.
- J. Wang, B. Xu, H. Sun and G. Song, Tetrahedron Lett., 2013, 54, 238–241 CrossRef CAS.
- W. Liu, D. Wang, Y. Duan, Y. Zhang and F. Bian, Tetrahedron Lett., 2015, 56, 1784–1789 CrossRef CAS.
- F. Farjadian, M. Hosseini, S. Ghasemi and B. Tamami, RSC Adv., 2015, 5, 79976–79987 RSC.
- B. Tamami and S. Ghasemi, Collect. Czech. Chem. Commun., 2011, 76, 1967–1978 CrossRef CAS.
- B. Tamami, F. Farjadian, S. Ghasemi, H. Allahyari and M. Mirzadeh, J. Braz. Chem. Soc., 2015, 26, 1591–1598 Search PubMed.
- S. Ghasemi, F. Farjadian and B. Tamami, Appl. Organomet. Chem., 2016, 30, 818–822 CrossRef CAS.
- Y. Lee, M. Chan Hong, H. Ahn, J. Yu and H. Rhee, J. Organomet. Chem., 2014, 769, 80–93 CrossRef CAS.
- G. Wei, W. Zhang, F. Wen, Y. Wang and M. Zhang, J. Phys. Chem. C, 2008, 112, 10827–10832 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Include FT-IR spectra of PNIPAM (I), ionic liquid monomer (II) and PNIPAM-b-PIL (III) and 1H-NMR spectra of PNIPAM (I) (in acetone-d6), ionic liquid monomer (II) (in D2O) and PNIPAM-b-PIL (III) (in D2O). See DOI: 10.1039/c8ra01303a |
| This journal is © The Royal Society of Chemistry 2018 |