
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssociation between metabolic profile and microbiomic changes in rats with functional dyspepsia †‡
Liang Luo§
a,
Minghua Hu§b,
Yuan Lia,
Yongxiong Chena,
Shaobao Zhangc,
Jiahui Chend,
Yuanyuan Wangb,
Biyu Lua,
Zhiyong Xiec and
Qiongfeng Liao *a
*a
aSchool of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, Guangzhou, 510006, China. E-mail: liaoqf2075@yahoo.com; Fax: +86 20 3958081; Tel: +86 20 3958081
bInfinitus (China) Company Ltd, Guangzhou, 510623, China
cSchool of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Guangzhou, 510006, China
dKey Laboratory of State Administration of Traditional Chinese Medicine, Sunshine Lake Pharma Company Ltd, Dongguan, 523867, China
First published on 4th June 2018
Abstract
Functional dyspepsia (FD) is one of the most prevalent functional gastrointestinal disorders (FGIDs). Accumulated evidence has shown that FD is a metabolic disease that might relate to gut microbiota, but the relationship between microbiome and the host metabolic changes is still uncertain. To clarify the host–microbiota co-metabolism disorders related to FD, an integrated approach combining 1H NMR-based metabolomics profiles, polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) and 16S rRNA gene sequencing was used to investigate the relationship among FD, metabolism of gut microbiota and the host. 34 differential urinary metabolites and 19 differential fecal metabolites, which affected the metabolism of energy, amino acids, nucleotides and short chain fatty acids (SCFAs), were found to have associated with FD. Based on the receiver operating characteristic (ROC) analysis, 10 biomarkers were screened out as diagnostic markers of FD. Meanwhile, the concentrations of Flintibacter, Parasutterella, Eubacterium and Bacteroides significantly increased in the FD group, whereas Eisenbergiella, Butyrivibrio, Intestinimonas, Saccharofermentans, Acetivibrio, Lachnoanaerobaculum and Herbinix significantly decreased. Furthermore, the above altered microbiota revealed a strong correlation with the intermediate products of the tricarboxylic acid (TCA) cycle, amino acids and SCFAs. In our study, it suggested that the energy metabolism was mainly disturbed in FD rats. Our findings also demonstrated that FD might be the result of gut microbiota and metabolism disorders, which was potentially valuable to enrich our understanding of the pathogenesis of FD.
Introduction
Functional dyspepsia (FD) is a widely prevalent gastrointestinal disorder throughout the world.1 FD is not life-threatening but can adversely influence the life quality of patients. Clinical observation has shown that patients with FD suffer from gastrointestinal motility obstacles, epigastric pain, abdominal distension, poor appetite, Helicobacter pylori infection, fatigue, emaciation and other symptoms.2 However, the precise etiology of FD remains uncertain because of complicated multiple factors. Only understanding how FD interferes with the normal physiological function of organisms, can we identify objective diagnostic indicators and find new therapies.Several lines of evidence indicate that gut microbiota may involve the pathogenesis and pathophysiology of FD. For instance, small intestinal bacterial overgrowth is associated with the etiology of FGIDs.3 The overgrowth of coliform bacteria causes the abnormal fermentation of carbohydrates, resulting in luminal distension, increased intestinal permeability and immune response perpetuation in predisposed hosts, which may further manifest as FD symptoms.4 A causal link between small intestinal bacterial and FD might exist, but little attention has paid to it. Some researchers speculated that the co-metabolites produced from the interaction of host and the gut microbiota might affect the potential mechanisms of intestinal microflora associated with FD. Thus, figuring out the relationship between gut microbiota and metabolites may help to understand the potential hazard of FD.
In the past few decades, metabonomic characterization of disease through metabolic profiling revealed the potential to generate novel noninvasive diagnostics and to understand the disease mechanism. Metabonomics broadly aims to measure the systemic, dynamic metabolic response of living systems to biological stimuli and simply represents the recognition and measurement of the entire metabolic reaction of an organism in response to an internal or external influence.5,6 1H NMR-based metabonomics has the potential to identify biomarkers and enhance clinical diagnosis. It has been successfully applied in searching for novel biomarkers in cardiovascular diseases,7 neuropsychiatric diseases,8 cancer,9 and many other diseases. FD is a typical systematic and metabolic disease.10 Likewise, metabolomics can help to identify potential biomarkers of FD. In addition, polymerase chain reaction (PCR) and denaturing gradient gel electrophoresis (DGGE) of the V3 region of the 16S ribosomal ribonucleic acid (16S rRNA) gene sequences, commonly employed for description of the substantial diversity of the gut microbiome, are powerful techniques for identifying and quantifying the complex microbial communities.11 Therefore, the association of metabonomics and microbiology analysis is a well-suited tool to investigate the correlation between the host and gut microbes.
In the current study, the microbiological and metabonomic response of FD at the systematic level was performed by combining 16S rRNA gene sequencing and 1H NMR metabolomics techniques (Fig. 1). The aim of our study was to evaluate the effects of FD on the microbiota and its metabolic profiles in feces and urine. Analyzing the altered microbes and metabolic pathways, we can identify the biomarkers of FD. Moreover, we investigated the correlation between gut microbiome and metabolites. Exploring this information is particularly significant for revealing the systematic pathogenesis and the physiological responses associated with FD, which will provide a foundation for more accurate diagnosis and better treatment of FD.
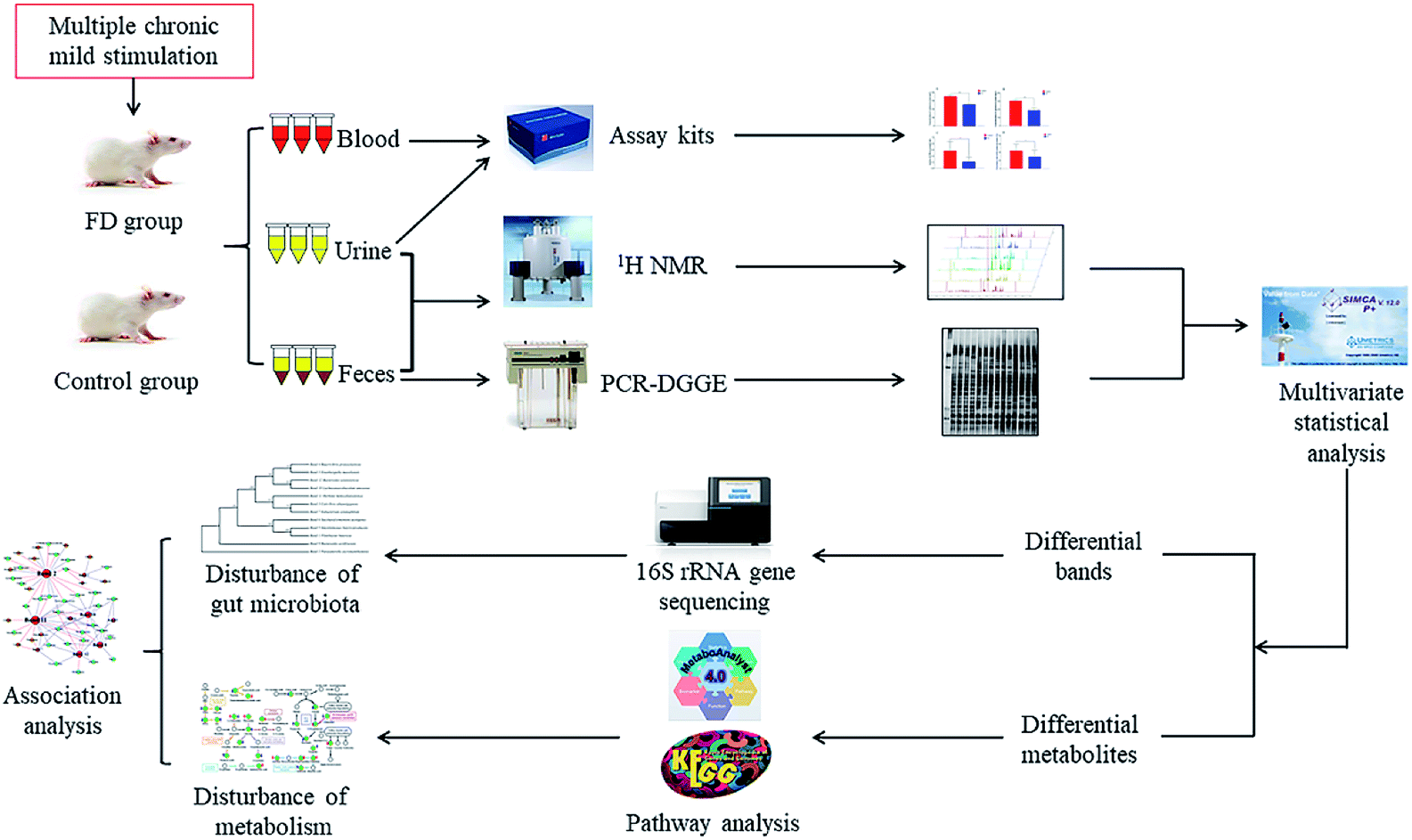 | ||
| Fig. 1 Scheme of the study. | ||
Materials and methods
Reagents and materials
The assay kits for detecting the concentration of motilin (MTL), gastrin (GAS), the activity of creatine phosphokinase (CPK) and the excretion rate of D-xylose were purchased from Jiancheng Bioengineering Institute (Nanjing, China). The TIANamp Stool DNA Kit was purchased from Tiangen Biotechnology Corporation (Beijing, China). D2O with 0.05% sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 (TSP) was purchased from Sigma-Aldrich Corporation (St. Louis, USA). Rhubarb (Radix et Rhizoma Rhei) was purchased from Zhixin Pharmaceutical Corporation (Guangzhou, China) and then extracted with distilled water and concentrated into the 1 g mL−1 rhubarb extract. Other chemicals were all analytical grade.Animal experiment
14 male Sprague-Dawley (SD) rats (180–220 g) were purchased from the Laboratory Animal Centre of Guangdong (Guangzhou, China, no. SCXK (Guangzhou) 2013-0002). The rats were kept in a SPF animal facility (temperature, 24–26 °C; humidity, 50–70%; light–dark cycle, 12–12 h) at Guangzhou University of Chinese Medicine (Guangzhou, China, no. SYXK (Guangzhou) 2013-0085). All rats had free access to a commercial rodent diet and water throughout the experimental period. All protocols involving animals were approved by the Institutional Animal Care and Use Committee of Guangzhou University of Chinese Medicine. All procedures of animal treatment were in accordance with the National Guidelines for Experimental Animal Welfare (MOST, China, 2006) at the Centre for Animal Experiments. Maximum efforts were exerted to minimize experimental animal suffering and the number of animals was necessary for the obtainment of reliable data.After 7 day acclimatization, the rats were randomly grouped into a FD group (n = 7) and a control group (n = 7). Every three or four rats in the same group were kept in a cage. The FD rat model was established through a multiple stimulation (diarrhea, fasting and swimming-induced fatigue) once a day for 14 days, which was an optimized method reported in our previous study.12 While the FD group was intragastrically given 1 g mL−1 cold rhubarb extract at a dose of 10 mL kg−1, the control group received the same volume of distilled water. The FD group suffered from alternate-day fasting and the control group was feed normally. While the control group receive no treatment, the FD group rats had to accept a loading swimming until they exhausted themselves. The model establishment lasted for 14 days. We measured body weight, food and water intake of rats once a week.
Sample collection and biochemistry assays
The urine, feces, blood and thymus samples were collected on day 14. The metabolic cages were used to collect urine and feces samples. The urine samples were collected from 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :00 to 16:00 and feces samples were collected at 8:00. All urine samples were centrifuged (6000g, 4 °C) for 10 min. In order to separate serum and plasma, the blood samples were collected and centrifuged at 1400g for 15 min at 4 °C. The thymus samples were snap-frozen in liquid nitrogen immediately. All samples were collected in Eppendorf tubes on ice and stored at −80 °C in a freezer. The biochemistry indexes, including serum MTL, plasma GAS, serum CPK and the excretion rate of urine D-xylose, were measured according to the instructions of enzymatic kits on day 14. The urine samples collection of D-xylose test processed as follow: after 12 h fasting, the rats were intragastrically given 4% D-xylose at a dose of 20 mL kg−1. The collected urine samples were immediately keep on ice in the whole collecting period.
:00 to 16:00 and feces samples were collected at 8:00. All urine samples were centrifuged (6000g, 4 °C) for 10 min. In order to separate serum and plasma, the blood samples were collected and centrifuged at 1400g for 15 min at 4 °C. The thymus samples were snap-frozen in liquid nitrogen immediately. All samples were collected in Eppendorf tubes on ice and stored at −80 °C in a freezer. The biochemistry indexes, including serum MTL, plasma GAS, serum CPK and the excretion rate of urine D-xylose, were measured according to the instructions of enzymatic kits on day 14. The urine samples collection of D-xylose test processed as follow: after 12 h fasting, the rats were intragastrically given 4% D-xylose at a dose of 20 mL kg−1. The collected urine samples were immediately keep on ice in the whole collecting period.
NMR spectra process and analysis
Samples stored at −80 °C were thawed on ice first. Urine samples were prepared by blending 600 μL urine and 60 μL phosphate buffer (K2HPO4/NaH2PO4, 1.5 M, pH 7.4, 100% D2O) contained 0.05% TSP for chemical shift reference (δ 0.00) and 0.1% NaN3 for bacteriostasis. The mixtures were centrifuged at 16000g, 4 °C for 10 min to remove sediment. 600 μL supernatant was transferred into 5 mm NMR tubes for NMR analysis.13
Feces samples were extracted using the method as follow: mix 100 mg thawed feces samples with 800 μL phosphate buffer (0.1 M, K2HPO4/NaH2PO4, pH 7.4, 100% D2O) containing 0.05% TSP and 0.01% NaN3. After eddying for 1 min, the mixture was carried on 3 rounds of freeze–thaw treatment and follows with 10 rounds of ultrasonication (20 s)-eddying (10 s)-standing (30 s) cycles. Then the mixture was centrifugated at 16000g, 4 °C for 10 min. 600 μL supernatant was pipetted into 5 mm NMR capillary tubes for NMR analysis (detailed in ESI‡). Urea and H2O signals were carefully discarded in NMR analysis to obtain pure endogenous metabolite changes. Discarded regions included d 4.56–4.86 (for H2O) and d 5.20–6.00 (for urea) in urine samples and d 4.56–4.86 (for H2O) in feces samples. Afterwards, all regions were normalized to the total sum of integral regions before pattern recognition analysis.
Microbiological analysis
Total bacterial DNA was extracted from 98–102 mg feces samples using the TIANamp Stool DNA Kit (Tiangen Biotechnology Corporation, Beijing, China) according to the manufacturers' instructions. The isolated DNA was stored at −20 °C for further analysis. PCR amplification was performed using universal primers 357f_GC clamp and 518r, which can target the variable V3 region of the 16S rRNA gene of the predominant bacteria. The PCR mixture (25 μL) contained 12.5 μL Tap PCR master mix, 1 μL upstream primer, 1 μL downstream primer, 2 μL DNA template and 8.5 μL double-distilled water. The amplification began with a 5 min initial denaturation at 95 °C, and then followed with 10 rounds circulation of 1 min denaturation at 95 °C, 1 min anneal at 65 °C, and 1 min extension at 72 °C. The annealing temperature was subsequently decreased by 1 °C every cycle until a touchdown at 55 °C. Another 20 rounds circulation of 1 min denaturation at 95 °C, 1 min anneal at 55 °C, and 1 min extension at 72 °C were performed later. Finally, a 10 min extension at 72 °C ended the whole amplification. The PCR products were verified with 1.7% (wt/vol) agarose gel electrophoresis to identify our target bands.The PCR products were analyzed with a linear gradient from 38% to 55%. Initial electrophoresis was conducted under 150 V constant voltage at 60 °C for 15 min in 1× Tris-acetate-EDTA (TAE) buffer using the DCode universal mutation detection system (Junyi, Beijing, China), then condition was changed into 70 V constant voltage for 12 h at 60 °C. Polyacrylamide gels were stained with silver nitrate and then visualized with a digital camera (Canon, Japan). DGGE images were converted to black-and-white with Adobe Photoshop (version CC 2017) and digitized by Quantity One (version 4.6.2).
Multivariate statistical analysis and correlation analysis
As a non-supervised pattern, principal component analysis (PCA) can reveal the intrinsic similarity or dissimilarity within each data set. In addition, a supervised model, partial least squares (PLS-DA) and orthogonal projection to latent structure discriminant analysis (OPLS-DA), were further employed in data analysis. (detailed in ESI‡)The correlation between NMR spectra and DGGE data was modeled by using Pearson's correlation coefficient and OPLS regression. In our study, a cutoff value (|r| > 0.755) was chosen as discrimination significance based on the test for significance of the Pearson's product-moment correlation coefficient. After matching with Human Metabolome Database (HMDB, http://www.hmdb.ca/) and Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/), the metabolic pathways related to the differential metabolites could be screened out and further analyzed. And the resultant sequences were searched in Basic Local Alignment Search Tool (BLAST, https://blast.ncbi.nlm.nih.gov/), followed by function analysis.
Cloning and sequencing of specific bands
According to the multivariate data analysis results, the bands that VIP > 1 in OPLS-DA pattern and P < 0.05 in t-test were differential bands. The differential bands in the two groups were excised and subjected to further identification by sequencing. Rinsed DNA bands were dissolved in 30 μL sterile double-distilled water for 16 h at 4 °C, and then amplified with universal V3 primers without GC clamp. The positive clones were purified and verified as described in previous research.14 The differential bands were separated again with DGGE and then excised with a scalpel, retreated in 30 μL sterile double-distilled water for 16 h at 4 °C, and then amplified with universal V3 primers without GC clamp. The above PCR products were performed with 1.7% (wt/vol) agarose gel electrophoresis and the targeted bands were accurately removed and purified with SanPrep Spin Column (Sangon Biotech, Shanghai, China) for further determination of concentration and purity. The purified PCR products were cloned into Escherichia coli DH5α using pUCm-T Vector (Sangon Biotech, Shanghai, China). The positive clones were purified and verified as described in previous research.14 For each differential band, we randomly chose 5 positive clones for sequencing. The sequencing was conducted by Sangon Biotech (Shanghai, China). Then, we selected the most matching sequence with MEGA software (version 7) and the results were assembled with DNAMAN software for homology searches in NCBI GenBank databases (http://www.ncbi.nlm.nih.gov/BLAST/). Based on BLAST results, reference sequences of phylogenetic neighbor species (up to 92% similarity) were included to confirm the allocation of the purified band sequences to the most probable species. The results with over 92% similarity were reliable.Results
Influence of FD on body weight, food and water intake, thymus index
During the model establishing, diarrhea and fatigue occurred in FD rats and no animals died. The representative changes of body weight, food and water intake were presented in Fig. S1.‡ The body weights in the FD group were significant lower than the control group from day 7 to day 14 during modeling. Since day 7, the food and water intake in FD group was lower than control group.Thymus, the important immune organ in mammals, is also where immunological cells grow and proliferate. The thymus index can partly reflect the immune function. The thymus index in FD group was significantly lower than that in the control group (P < 0.05, shown in Fig. S1D‡), which indicated the low the immune function in FD group.
Biochemical indexes
The biochemical indexes of experimental animals were summarized in Fig. 2. The concentration of MTL, GAS, the activity of CPK and the excretion rate of D-xylose in FD group were significant lower than those in control group (P < 0.05). The above results can evidence that the FD model was successfully established.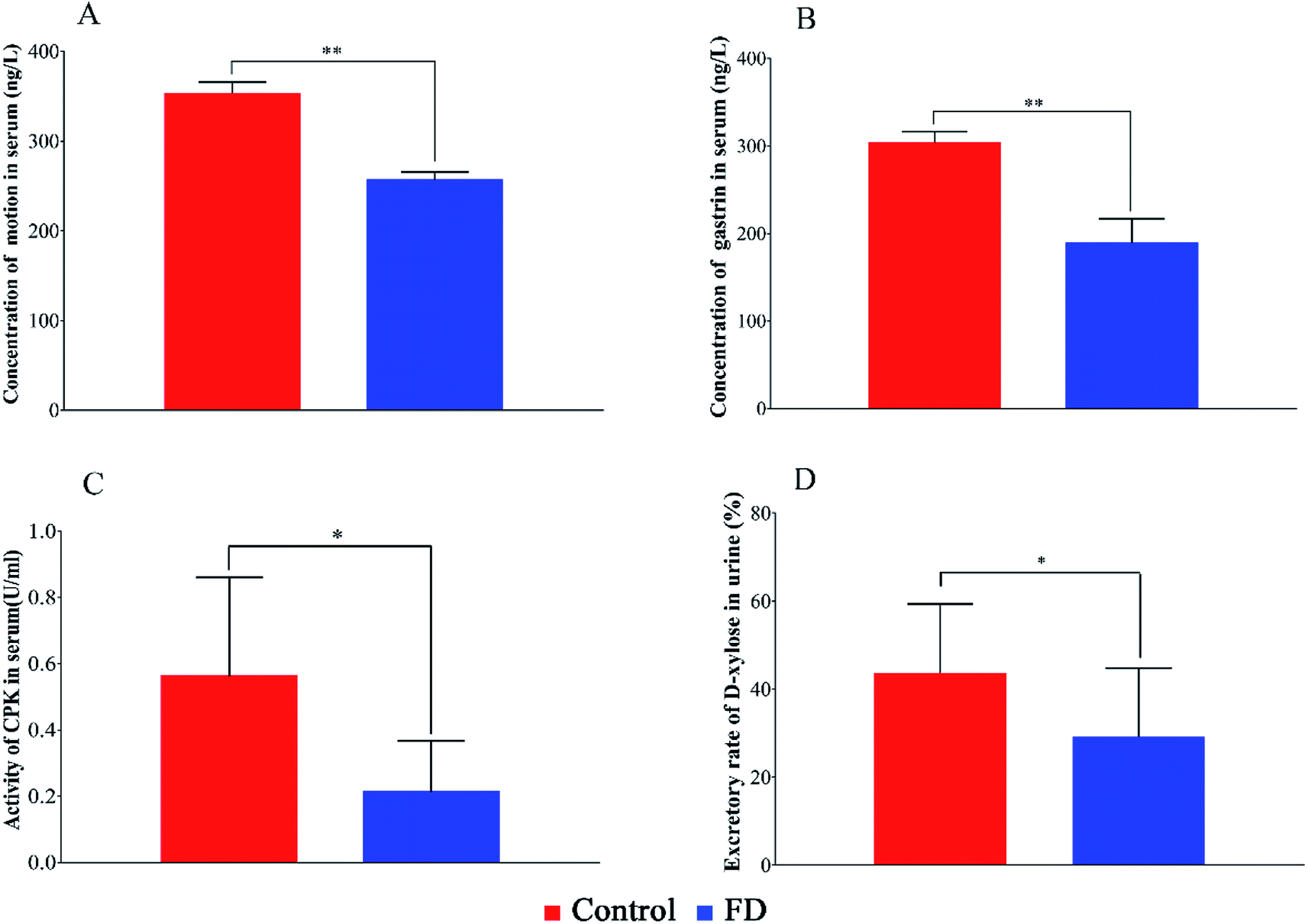 | ||
| Fig. 2 The concentration MTL (A), GAS (B), the activity of CPK (C) in serum samples and the excretion rate of D-xylose (D) in urine samples on day 14. (n = 14) *: P < 0.05, **: P < 0.01. | ||
Metabolites assignment with 1H NMR spectroscopy
The typical 1H NMR spectra of feces and urine samples on day 14 were shown in Fig. 3. 133 specific metabolites (60 feces-derived metabolites and 73 urine-derived metabolites) were identified from 1H NMR data with the reference of literature data15,16 (Tables S1 and S2‡). The endogenous metabolites in feces samples mainly comprised of glucose, amino acids (threonine, glycine, serine), short chain fatty acids (formic acid, acetic acid, propionic acid, butyric acid), bile acids (cattle sulfonated, taurocholic acid), etc. The endogenous metabolites in urine samples included amino acids (histidine, proline, succinate, creatine), carbohydrates (sucrose, glucose, trehalose), amine (methylamine, dimethylamine (DMA), trimethylamine (TMA), trimethylamine oxide (TMAO)), aromatic compounds (hippuric acid, phenylalanine, hydroxyphenylacetic acid), etc. These spectra exhibited obvious differences between the two groups. In order to obtain more details about the metabonomic changes in FD, multivariate data analyses were further conducted on the NMR data of urine and feces samples.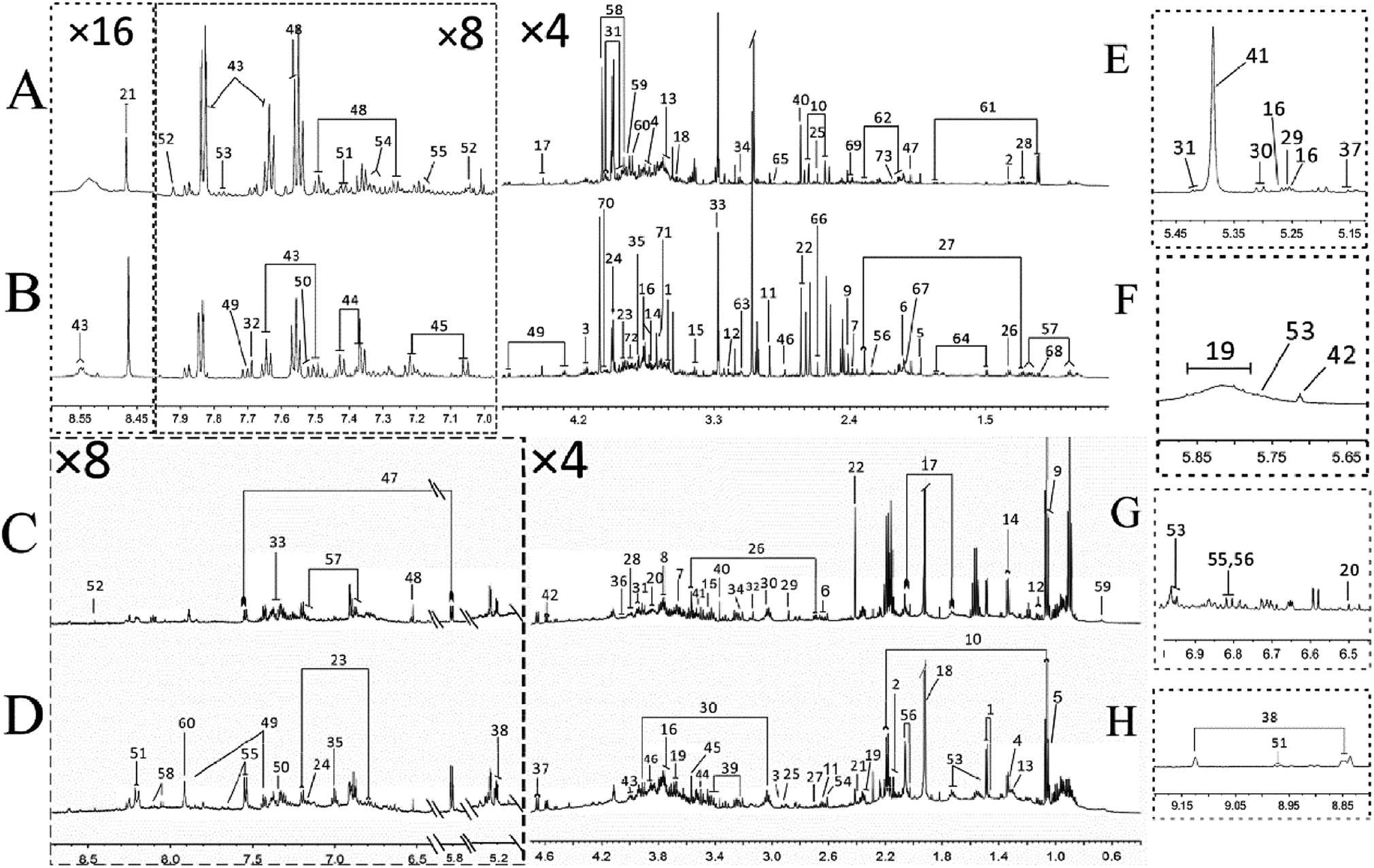 | ||
| Fig. 3 Typical 600 MHz 1H NMR spectra of urine from FD group (A), control group (B) and feces extracts from FD group (C), control group (D) on day 14. The expansion of δ 5.15–5.45 (E), the expansion of δ 5.65–5.85 (F), the expansion of δ 6.50–6.90 (G), the expansion of δ 8.85–9.15 (H) of spectra of urine samples. Metabolite numbers are shown in Tables S1 and S2.‡ | ||
Fecal and urinary metabolites changes
PCA was performed on the pareto variance (Par)-scaled 1H NMR data collected from feces and urine samples of two groups. In the PCA score plots (Fig. S2‡), the FD and control groups were well separated, revealing obvious difference between the FD and control groups in fecal and urinary metabolites on day 14. PLS-DA and OPLS-DA were further carried out. The R2 and Q2 values of PCA, PLS-DA and OPLS-DA confirmed good qualities of all models suggesting that our models were reliable (Table S3‡). And CV-ANOVA (P < 0.05) can confirm the validities of these models. The clear classifications between FD and control group on day 14 were shown in the OPLS-DA score plots (Fig. 4). The corresponding loadings plots illustrate the significantly differential metabolites related to FD. The color-coded correlation coefficients indicate the variable contributions to class separation, those contributing the most to the prediction of the response are shown in red, whereas those with slight or no association with the response are shown in blue. Therefore, we can screen out the significantly different metabolites between two groups. The verification with fold changes (FC) and Student's t-test were used to analyze authentically differential metabolites by comparing FD and control group on day 14. The significant metabolites (P < 0.05 in t-test, FC > 1.2 or < 0.83) were considered to be the differential metabolites. Finally, 19 differential metabolites in feces samples and 34 differential metabolites in urine samples were sorted out (Table 1). In addition, the VIP value and fold change of 2-ketoglutarate (U8) were very high in our study (P < 0.01, VIP > 2, FC > 2 or < 0.5), indicating 2-ketoglutarate significantly in FD rats.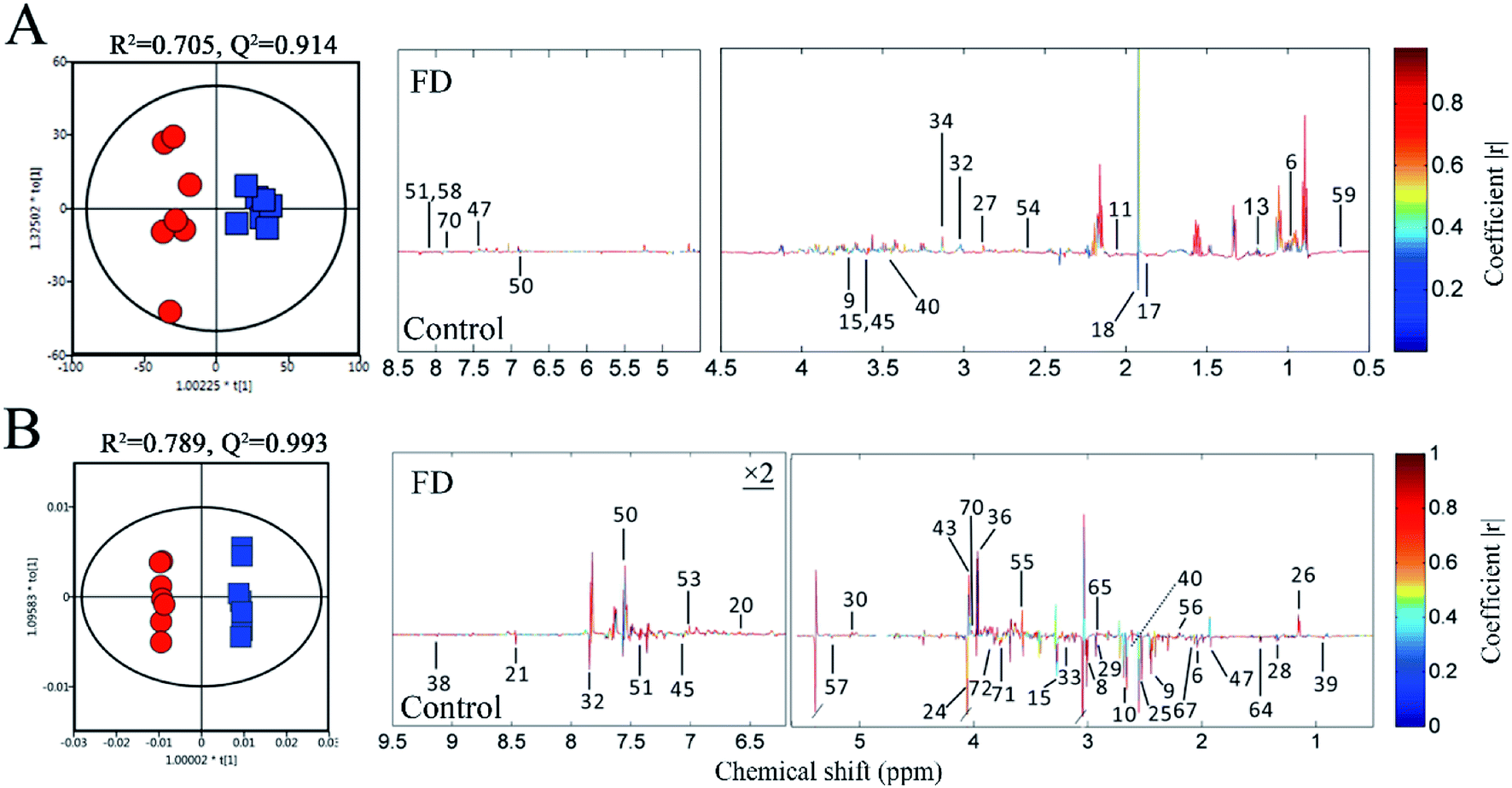 | ||
| Fig. 4 OPLS-DA score plots (left panel) and the correlation coefficient loading plots (right panel) of feces (A) and urine (B) metabolites derived from control group (red circles) and FD group (blue squares) on day 14. The corresponding metabolite numbers can be found in Tables S1 and S2.‡ (n = 14). | ||
| Numbers | Metabolites | VIP | log2(FC) | −log10(P) | Variations |
|---|---|---|---|---|---|
| a F: feces sample; U: urine sample; *: P < 0.05; **: P < 0.01. | |||||
| F6 | α-Ketoisocaproate | 0.1598 | 0.1660 | 1.7437 | ↓* |
| F9 | Valine | 1.7664 | −0.3346 | 2.5514 | ↑** |
| F11 | Methyl succinate | 1.0383 | 0.2236 | 2.5751 | ↓** |
| F13 | N-Heptanoate | 1.0659 | 0.4895 | 4.4989 | ↓** |
| F15 | Threonine | 2.0838 | −0.4569 | 1.6288 | ↑* |
| F17 | Glutarate | 0.9965 | −0.2680 | 1.8679 | ↑* |
| F18 | Acetate | 3.8133 | −0.3487 | 1.5481 | ↑* |
| F27 | Dimethylamine | 0.2581 | 0.2543 | 2.1465 | ↓** |
| F32 | Malonate | 0.4205 | 0.2566 | 1.5368 | ↓* |
| F34 | Ethanolamine | 0.6717 | 0.1676 | 1.5045 | ↓* |
| F40 | Methanol | 0.6396 | −0.5594 | 1.9771 | ↑* |
| F45 | Glycine | 0.7499 | −0.3597 | 2.0817 | ↑** |
| F47 | Uracil | 0.8178 | 0.5575 | 2.2330 | ↓** |
| F50 | Imidazole | 0.3904 | −0.3144 | 2.0348 | ↑** |
| F51 | Adenine | 0.9873 | 0.9356 | 3.8959 | ↓** |
| F54 | Methylamine | 0.4221 | 0.2438 | 2.2560 | ↓** |
| F58 | Hypoxanthine | 0.8925 | 0.9585 | 3.9999 | ↓** |
| F59 | Tauro-chenodeoxycholate acid | 0.4535 | 0.4486 | 1.5901 | ↓* |
| F60 | Xanthine | 0.8225 | 0.8325 | 3.6533 | ↓** |
| U6 | Nag C | 0.4493 | 0.3444 | 1.6153 | ↓* |
| U8 | 2-Ketoglutarate | 2.0754 | 1.1393 | 5.3066 | ↓** |
| U9 | Succinate | 2.0754 | 1.1393 | 5.3066 | ↓* |
| U10 | Citrate | 2.0624 | 0.8387 | 7.3014 | ↓** |
| U15 | Taurine | 1.9803 | 0.5613 | 2.9519 | ↓** |
| U20 | Fumarate | 0.1553 | −0.6047 | 2.8030 | ↑** |
| U21 | Formate | 0.2442 | 0.4181 | 1.6940 | ↓* |
| U24 | Creatinine | 2.4528 | 0.6340 | 6.1353 | ↓** |
| U25 | Methylamine | 0.4396 | −0.8112 | 4.3922 | ↑** |
| U26 | 2-Hydroxyisobutyrate | 0.2754 | 0.2696 | 4.1536 | ↓** |
| U28 | Methylmalonate | 0.7947 | 0.2816 | 7.5477 | ↓** |
| U29 | Succinimide | 0.2181 | 0.2804 | 5.3001 | ↓** |
| U30 | Allantoin | 0.1947 | −0.5172 | 3.6337 | ↑** |
| U32 | Guanine | 0.4074 | 0.6365 | 3.2909 | ↓** |
| U33 | TMAO | 1.1662 | 0.9073 | 5.9476 | ↓** |
| U36 | L-Allothreonine | 1.0202 | −0.2893 | 6.7548 | ↑** |
| U38 | Trigonelline | 0.3179 | 0.3802 | 3.7349 | ↓** |
| U39 | Pantothenate | 2.3272 | 0.6986 | 7.0094 | ↓** |
| U40 | Dimethylamine | 0.8258 | 0.3619 | 6.2099 | ↓** |
| U43 | Hippurate | 3.8191 | −0.8198 | 7.1457 | ↑** |
| U45 | P-Cresol glucuronide | 0.9815 | 0.5318 | 4.5894 | ↓** |
| U47 | Acetamide | 0.4674 | 0.3161 | 2.5757 | ↓** |
| U50 | Indole-3-acetate | 0.5173 | −0.5747 | 2.3636 | ↑** |
| U51 | Niacinamide | 0.5997 | 0.3968 | 2.0829 | ↓** |
| U53 | P-Hydroxybenzoate | 1.1710 | −1.3485 | 14.4780 | ↑** |
| U55 | P-Hydroxyphenylacetate | 0.5644 | −0.3012 | 4.6164 | ↑** |
| U56 | P-Cresol | 0.8966 | −0.5732 | 6.6598 | ↑** |
| U57 | Triglycerides | 1.4797 | 0.2934 | 6.3655 | ↓** |
| U64 | Lysine | 1.6916 | 0.7450 | 7.2615 | ↓** |
| U65 | TMA | 0.2550 | −0.4948 | 4.5971 | ↑** |
| U67 | N-Acetylglycoproteins | 0.4493 | 0.3444 | 1.6153 | ↓** |
| U70 | Isopropanol | 1.7075 | −0.9656 | 6.6282 | ↑** |
| U71 | N-Acetylglycine | 1.1923 | 0.3268 | 7.1409 | ↓** |
| U72 | Glutamine | 1.6409 | 0.4722 | 5.6382 | ↓** |
In the feces samples, the levels of 12 metabolites in FD group were obvious lower than control group and 7 metabolites were significantly higher. Compared with control group, the levels of α-ketoglutarate, valine, methyl succinate, DMA, malonate, ethanolamine, uracil, adenine, methylamine, hypoxanthine, tauro-chenodeoxycholate acid and xanthine significantly decreased in FD group, whereas the levels of valine, threonine, glutarate, acetate, methanol, glycine and imidazole significantly increased in fecal metabolites.
In the urine samples, the levels of 23 metabolites in FD group were significantly lower than control group and 11 metabolites were significantly higher. The down-regulated metabolites included: Nag C, 2-ketoglutarate, succinate, citrate, taurine, formate, creatinine, 2-hydroxyisobutyrate, methylmalonate, succinimide, guanine, TMAO, trigonelline, pantothenate, DMA, P-cresol glucuronide, acetamide, niacinamide, triglycerides, lysine, N-acetylglycoproteins, N-acetylglycine and glutamine. The up-regulated metabolites included: fumarate, methylamine, allantoin, L-allothreonine, hippurate, indole-3-acetate, P-hydroxybenzoate, P-hydroxyphenylacetate, P-cresol, TMA and isopropanol. The correlation analysis between feces and urine differential metabolites was shown in Fig. 5.
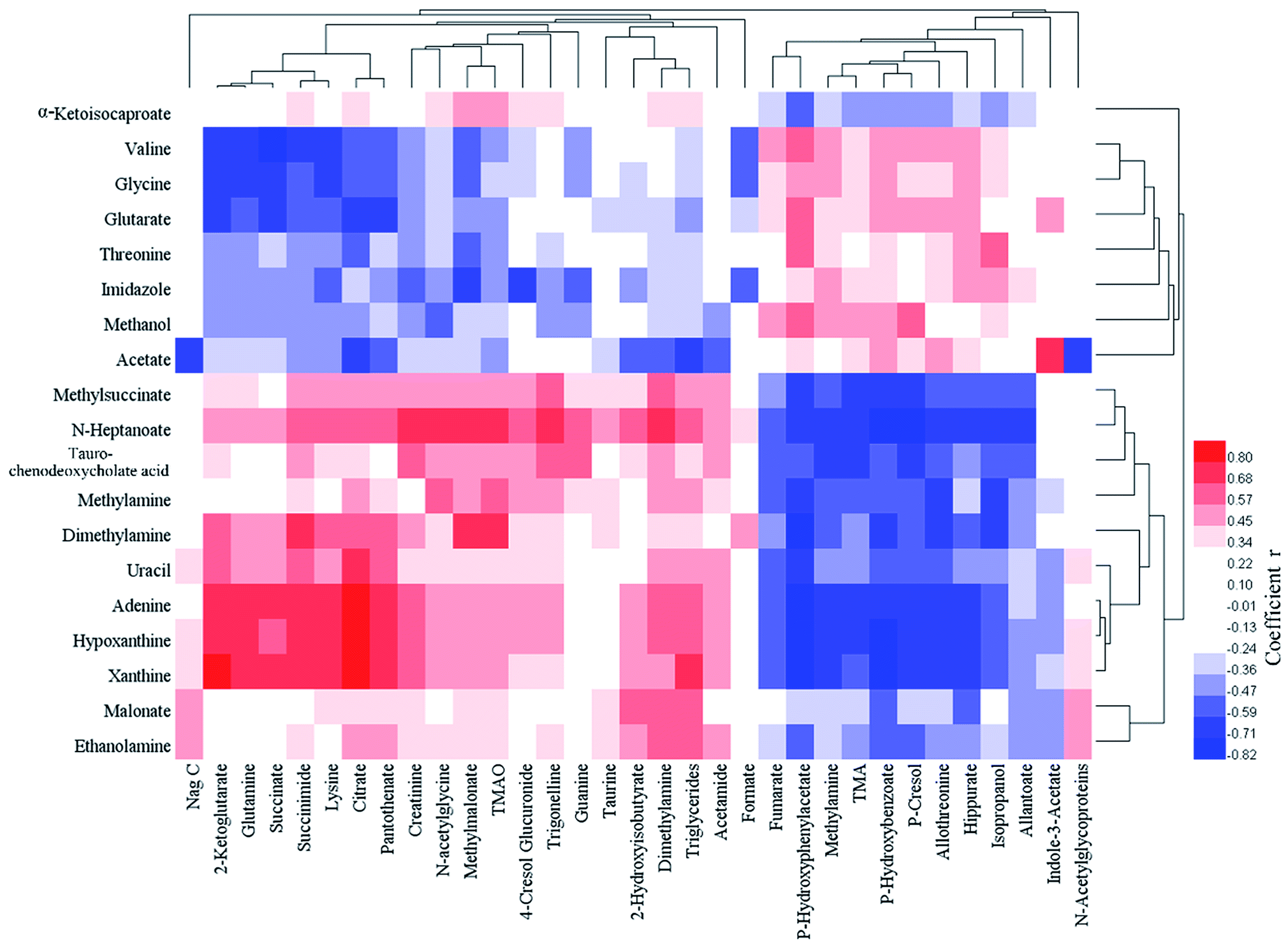 | ||
| Fig. 5 Pearson correlation coefficient heat map between feces (abscissa) and urine (ordinate) differential metabolites. | ||
ROC curve analysis was further employed to confirm the potential utility of the 53 differential metabolites with significant correlations with FD. The area under the curve (AUC) is used to evaluate the results of the ROC analysis. The higher AUC value means good quality of the diagnostic ability. The ROC prediction is in a low accuracy when AUC ranges in 0.5–0.7, a medium accuracy in 0.7–0.9, and high in over 0.9.17 As a result, the top 10 ranked metabolites including: fumarate, allantoin, P-hydroxyphenylacetate, methylamine, allothreonine, hippurate, P-hydroxybenzoate, P-cresol, TMA and isopropanol, achieved good AUC values (AUC > 0.9) in distinguishing the FD group and control group (Fig. 6), indicating that they could be identified as potential biomarkers related to FD.
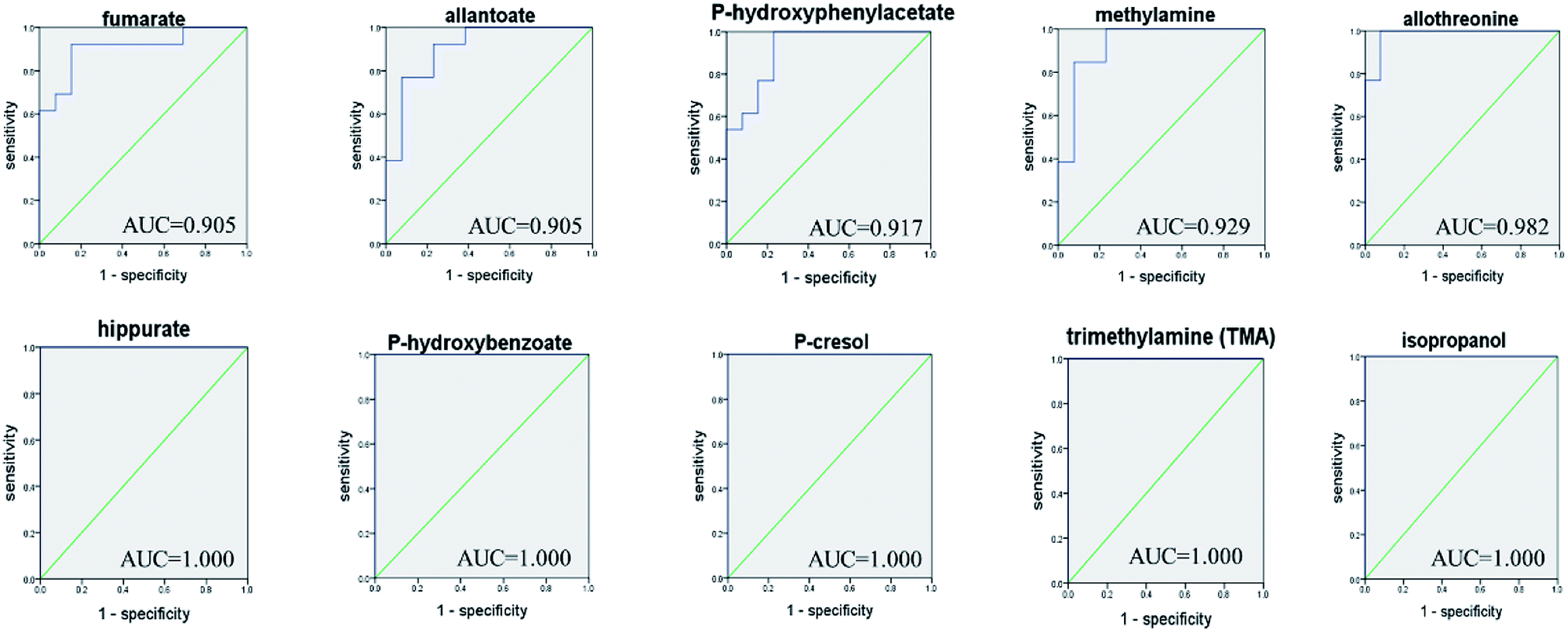 | ||
| Fig. 6 ROC curve analysis for the predictive power of top 10 ranked biomarkers relating to FD model. | ||
Metabolic pathway changes related to FD
MetaboAnalyst 4.0 (http://www.metaboanalyst.ca/), a web server designed for comprehensive metabolomic data analysis and visualization,18,19 was used to perform pathway analysis. In our study, the combination of metabolomics pathway analysis (MetPA) and metabolite set enrichment analysis (MSEA) were used to find out the pathway changes related to FD. The meaningful pathways were shown in Fig. 7. According to the MetPA and MSEA results (Tables S4–7‡), we mainly focused on the significant pathways including energy metabolism (TCA cycle, pyruvate metabolism), amino acids metabolism (valine, leucine and isoleucine metabolism, tryptophan metabolism, D-glutamine and D-glutamate metabolism, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, tryptophan metabolism), nucleotides metabolism (pyrimidine metabolism, purine metabolism), SCFAs metabolism (butanoate metabolism, propanoate metabolism), and primary bile acid biosynthesis. The pathways in feces reflect the changes in host–microbiota co-metabolism while the pathways in urine mainly reflect the host metabolism. In our study, urinary and fecal pathways had an intersection in energy metabolism. Pyruvate metabolism, the core of TCA cycle, changed significantly in the fecal pathways while TCA cycle showed differences in urinary pathways.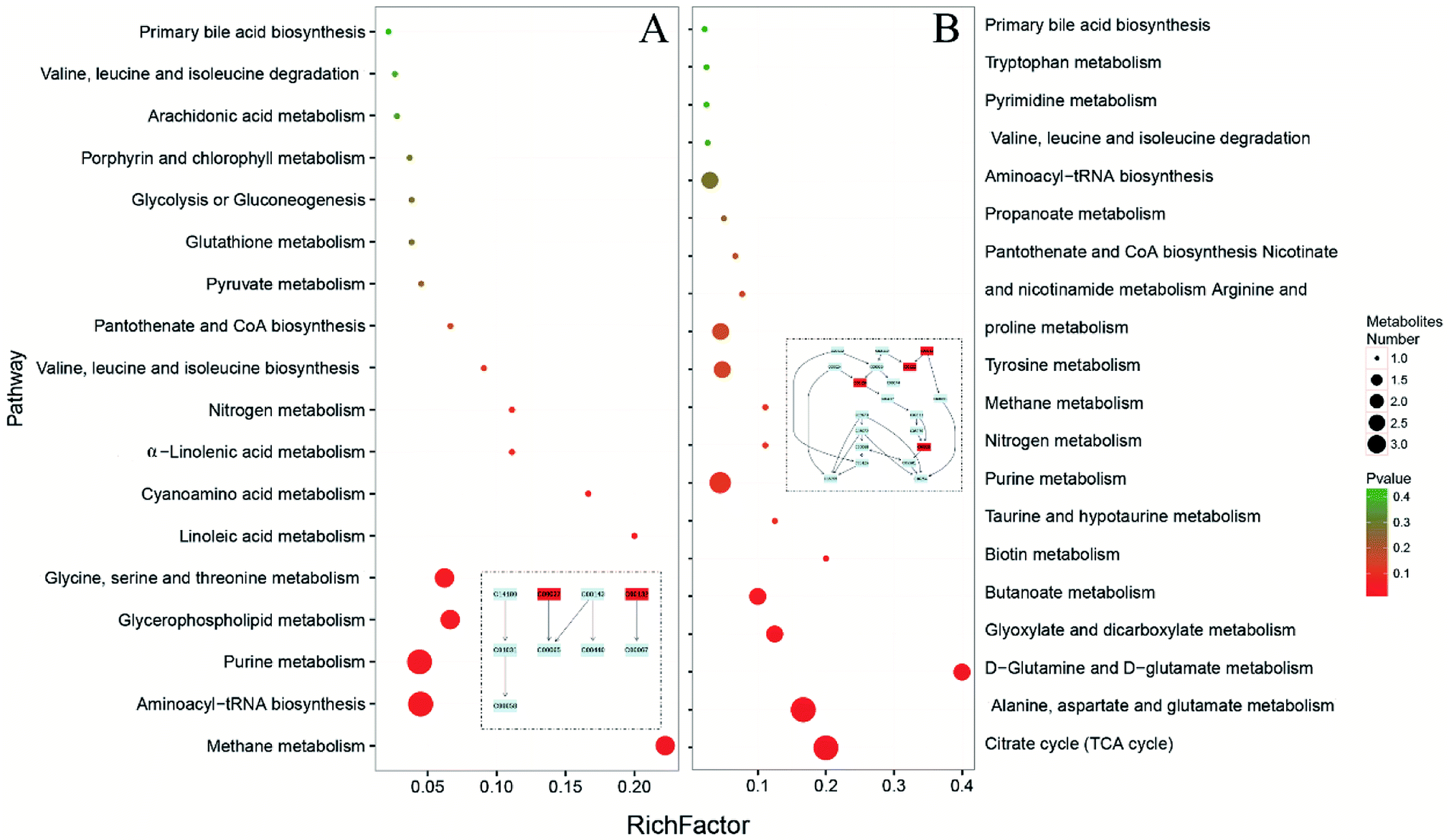 | ||
| Fig. 7 Meaningful metabolic pathways of feces (A) and urine (B) from metabolomics pathway analysis. | ||
Microbial profiles of feces samples in DGGE analysis
To demonstrate the disturbances of gut microbiota in FD group, the microbial structure on day 14 was represented by analyzing the V3-region 16s rRNA gene of predominant bacteria with DGGE. Compared with the control group, the bacterial diversity of FD group was reduced. Then the multivariate pattern analysis was conducted on DGGE data. The PCA scores plots, PLS-DA permutation test plots, OPLS-DA score plots and corresponding coefficient loading plots were shown in Fig. 8. Totally 12 differential bands were identified by cloning and sequencing of 16S rRNA gene V3 regions (Table 2, Fig. S3‡). The sequences are deposited in the GenBank with access numbers of KY792535–KY792545.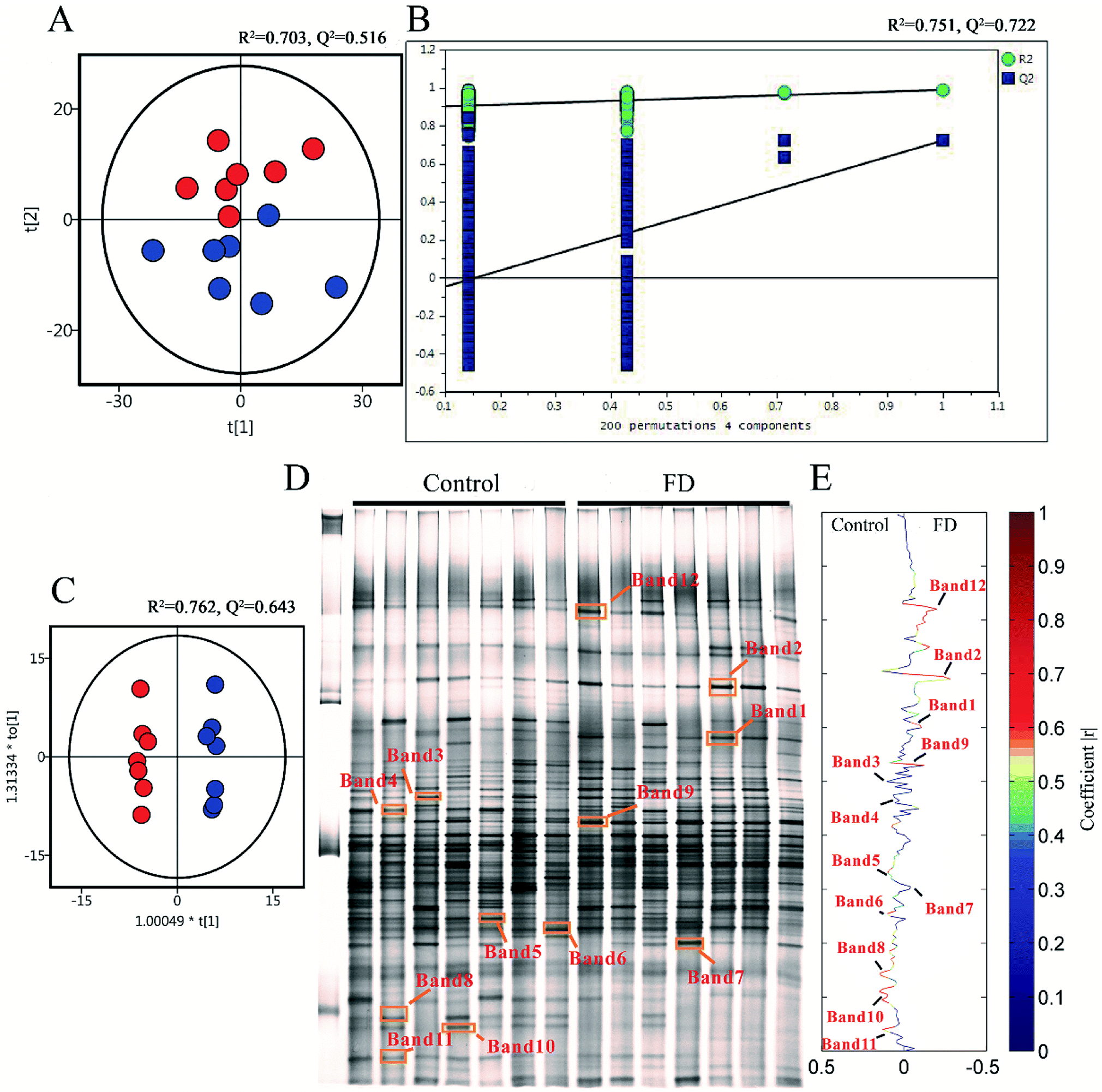 | ||
| Fig. 8 PCA scores plots (A), PLS-DA permutation test plots (B), OPLS-DA score plots (C), PCR-DGGE profiles (D) and correlation coefficient loading plots (E) derived from 16S rRNA gene V3 region, indicating the discrimination between control group (red) and FD group (blue) on day 14. The corresponding bacterial species can be found in Table 2. (n = 14). | ||
| DGGE band | Seq. length (bp) | Domain | Phylum | Class | Order | Family | Genus | Species | S_ab score | Accession numbers |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 183 | Bacteria | Firmicutes | Clostridia | Clostridiales | Clostridium | Flintibacter | Flintibacter butyricus | 1.000 | KY792535 |
| 2 | 194 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Sutterellaceae | Parasutterella | Parasutterella excrementihominis | 0.940 | KY792536 |
| 3 | 191 | Bacteria | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Eisenbergiella | Eisenbergiella massiliensis | 0.980 | KY792537 |
| 4 | 189 | Bacteria | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Butyrivibrio | Butyrivibrio proteoclasticus | 0.920 | KY792538 |
| 5 | 194 | Bacteria | Firmicutes | Clostridia | Clostridiales | Clostridium | Intestinimonas | Intestinimonas butyriciproducens | 0.980 | KY792539 |
| 6 | 184 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Saccharofermentans | Saccharofermentans acetigenes | 0.930 | KY792540 |
| 7 | 190 | Bacteria | Firmicutes | Clostridia | Clostridiales | Eubacteriaceae | Eubacterium | Eubacterium xylanophilum | 0.990 | KY792541 |
| 8 | 190 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Acetivibrio | Acetivibrio ethanolgignens | 0.990 | KY792542 |
| 9 | 191 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | Bacteroides acidifaciens | 1.000 | KY792543 |
| 10 | 193 | Bacteria | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnoanaerobaculum | Lachnoanaerobaculum umeaense | 0.980 | KY792544 |
| 11 | 181 | Bacteria | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Herbinix | Herbinix hemicellulosilytica | 0.920 | KY792545 |
| 12 | 193 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | Bacteroides xylanolyticus | 0.960 | KY792535 |
We analyzed the 12 differential bands in the level of the genus. Flintibacter (band 1), Parasutterella (band 2), Eubacterium (band 7) and Bacteroides (band 9, band 12) significantly increased in FD group, whereas Eisenbergiella (band 3), Butyrivibrio (band 4), Intestinimonas (band 5), Saccharofermentans (band 6), Acetivibrio (band 8), Lachnoanaerobaculum (band 10) and Herbinix (band 11) significantly decreased. Firmicutes, which contained nine bands, was main community in the feces of control group. One band from Proteobacteria (band 2) and two bands from Bacteroidetes (band 9, band 12) had a higher abundance in FD group (Fig. 8). Interestingly, seven Firmicutes families (band 3, 4, 5, 6, 8, 10, 11), classified into the same order of Clostridiales, significantly decreased in FD group.
Integration of 1H NMR spectral data and DGGE data
To ascertain the interaction between host and gut microbes, we integrated 1H NMR spectral data and DGGE microbial data to demonstrate the mechanism of FD. DGGE and 1H NMR spectral data were correlated by using orthogonal projection to latent structure (OPLS) regression and Pearson's correlation coefficient (|r| > 0.755). The association between differential DGGE bands and endogenous metabolites was shown in Fig. 9. Lots of the correlated compounds were host–gut microbial co-metabolites, such as SCFAs, DMA, TMA and hippurate. Hippurate (U43) showed a strong positive correlation with Acetivibrio (band 8), Lachnoanaerobaculum (band 10), Herbinix (band 11) and Bacteroides (band 12) and a negative correlation with Proteobacteria (band 2). In addition, TMA (U65) only showed a positive link to Herbinix (band 11).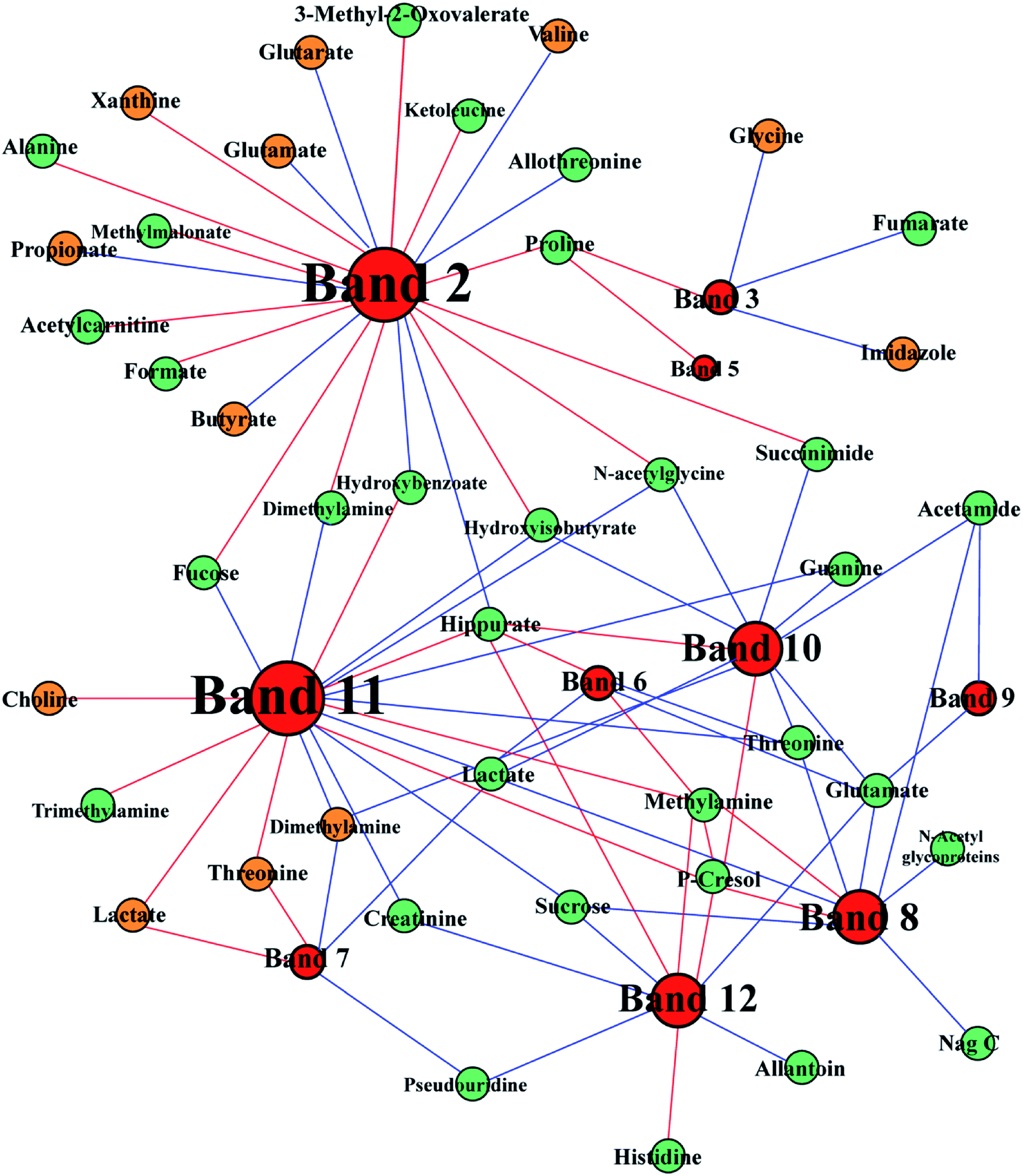 | ||
| Fig. 9 Correlation network between metabolites in urine (green) and feces (brown) and altered DGGE bands visualized with Gephi. Node size denotes the highest effect size for each metabolite or band comparing FD group with control group. Red lines correspond to positive correlations, whereas blue lines correspond to negative correlations. | ||
Discussion
Model validation
Researchers have reported that multiple pathological processes are involved in the development of FD, including physiological, psychological and biological factors.10 As FD is caused by multiple factors, the FD rat model was established by multiple chronic mild stimulation every day. In the animal experiment, cold rhubarb would cause diarrhea, loading swimming would cause fatigue and fasting would cause gastrointestinal disorders. Combining the above modeling method, the characteristics of FD could be well simulated. In addition, our pre-experiment showed that the models were well established in the 7 mice of FD group, scoring 100% in success rate.Most FD patients have no obvious organic cause for their symptoms after evaluation. Objective diagnosis of FD is ambiguous in clinical practice, as well as the FD model validation in the FD researches. In our study, the main symptoms of FD patients were observed in FD rats after modeling, such as the decreased body weight, diarrhea, inappetence, fatigue and abdominal distension. Reduced food intake is a common manifestation of poor appetite that resulted from FD. Because of the reduction of food and water intake, the FD rats had to consume the nutrients stored in the body to maintain normal physiological activity, which might cause the loss of body weight. Moreover, biochemical indexes could evidence that the model rats were suffering chronic gastritis. Therefore, the FD rat model had been successfully established.
The concentration of MTL and GAS in FD group decreased significantly (P < 0.05). Motilin is called “housekeeper of the gut” because it accelerates gastric emptying and improves peristalsis in the small intestine.20 The low motilin might indicate the insufficient gastrointestinal motility. Gastrin is a peptide hormone that stimulates the parietal cells of the stomach involved in gastric motility to secrete gastric acid (HCl).21 The low gastrin can weaken antral muscle mobility, stomach contractions and gastric emptying.22 It seemed that the gastrointestinal motility went down in FD model, which might be the cause of digestive disorders.
CPK is an enzyme expressed by the tissues and cells that consume ATP rapidly.23 The activity of CPK in FD rats was significant lower than normal rats, suggesting that the energy supplying was inhibited.
D-Xylose excretion test is a medical test performed to diagnose the malabsorption of small intestine. D-Xylose is a monosaccharide that does not require any enzymes to digest and is only absorbed by intestinal mucosa. A decreased excretion of D-xylose in urine is seen in diseases related to gastrointestinal mucosa, such as small intestinal bacterial overgrowth and Whipple's disease. The D-xylose excretion rate in FD group was significant lower than control group (P < 0.05), which suggested that FD might associate with the malabsorption in intestine and stomach.
Disturbance of TCA cycle
Pathway analysis showed that TCA cycle was the most significant pathways (P < 0.05, impact > 0.1, shown in Fig. 7 and Table S4‡), suggesting that there was a remarkable difference in energy metabolism between FD group and control group. Reduced intermediate products of TCA cycle, including citrate (U10), succinate (U9), 2-oxoglutarate (U2) and glutamine (U72), were observed in the urine samples, and increased intermediate products of TCA cycle, including fumarate (U20) and 2-oxoglutarate (F6) were observed in the urine and feces samples (Table 1). These results indicated that the energy metabolism in model rats was disturbed. In the D-glutamine and D-glutamate metabolism, the glutamine can transform into 2-oxoglutarate (Fig. 10). In the current study, both the levels of glutamine (U72) and 2-oxoglutarate (U8) decreased significantly in the urine samples, which indicated that the D-glutamine and D-glutamate metabolism was inhibited. Glutamate (U72) is the contributor of energy generation in the intestine and glutamate metabolism is associated with central fatigue.24 The decreased glutamate probably caused the fatigue symptom in FD. As the ultimate pathway of the oxidation of carbohydrates, amino acids and fatty acids, TCA cycle connects with numerous other pathways. Thus, the disturbed TCA cycle has a huge impact on host metabolism.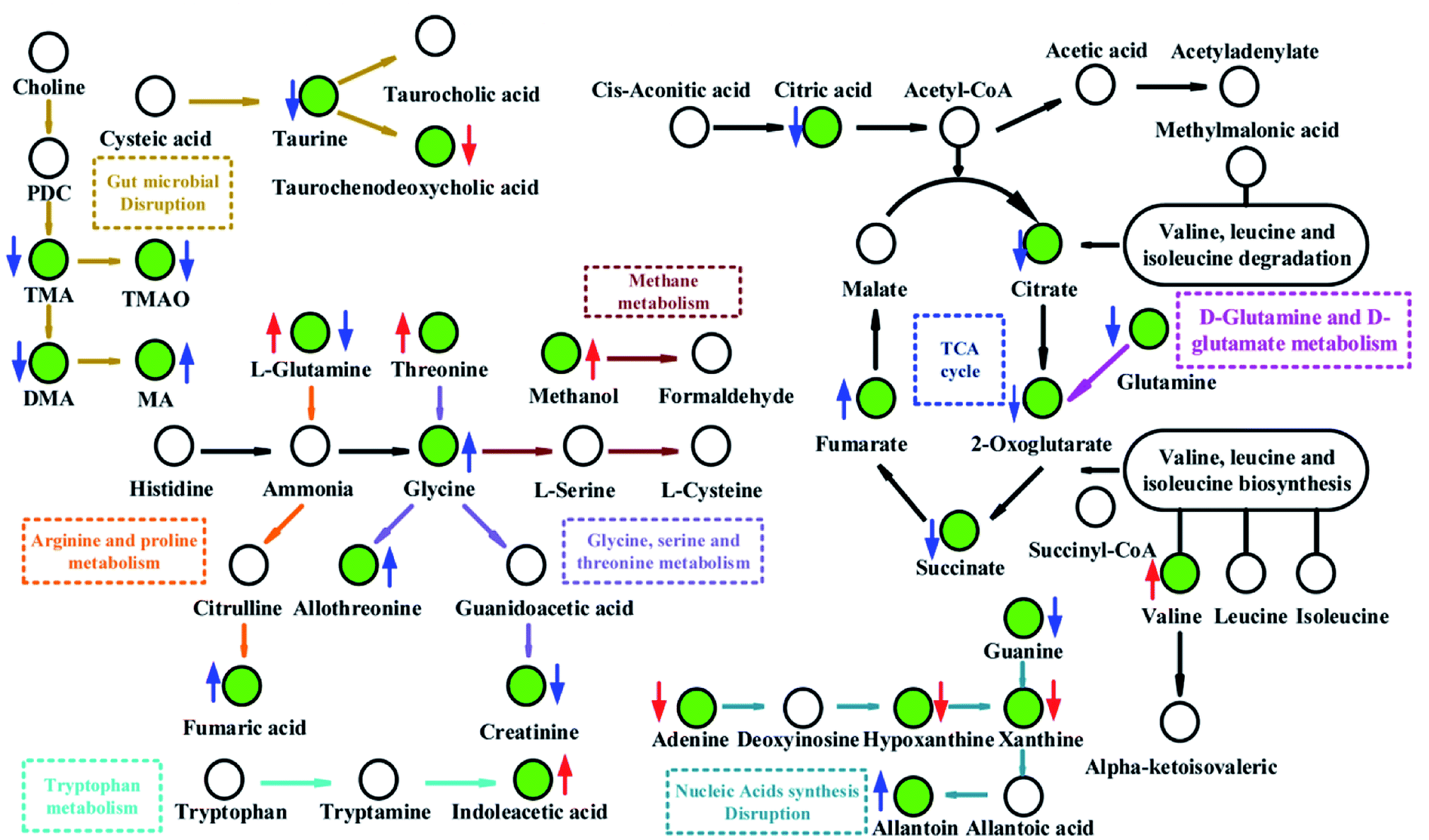 | ||
| Fig. 10 Metabolic pathways altered by FD. Green point: differential metabolite; ↓: significant decrease; ↑: significant increase; red colour: feces; blue colour: urine. | ||
Moreover, we had found that the contents of glutamate (U72) and 2-oxoglutarate (U8) were negatively correlated with the abundance of Parasutterella (band 2) in Fig. 9, which suggested that Parasutterella might play an important role in energy metabolism. Study25 has shown that the elevated abundance of Parasutterella is related to sugar consumption. In addition, the abundance of Eisenbergiella (band 3) reduced in FD group and negatively correlated with fumarate (U20). These results indicated that the disturbance of TCA cycle was related to the interactions between the gut microbiota and host metabolism.
Compared with the control group, the lower CPK in FD group indicated an inefficient energy production, which also supported our assumption. Therefore, we found that FD is related to a net depression in energy metabolism and the altered intermediate metabolites of TCA cycle could serve as potential biomarkers for the diagnosis of FD.
Disturbance of creatinine metabolism
Compared with control group, remarkable decreased creatinine (U24) was observed in urine of FD group, which indicated that the metabolism pathway of creatinine was depressed. In addition, creatinine was negatively correlated with Herbinix (band 11) and Bacteroides (band 12) in Fig. 9. Creatine is a waste product formed by the slow spontaneous degradation of creatine-phosphate which functions as a battery that stores the energy of excess ATP.26 Creatinine is synthesized in the body from creatine, which is produced from creatine phosphate during muscle contractions, and excreted in the urine.27 The decreased creatinine meant that the energy provided to skeletal muscle correspondingly decreased. The above results could be demonstrated by the decreased in the low level of TMAO (U33). TMAO, which participates in the methane metabolism, can promote the proliferation of muscle cells and the growth of muscle tissue. These results also verified the inhibited energy supply.Disturbance of amino acids metabolism
Glycine, serine and threonine metabolism, arginine and proline metabolism, tryptophan metabolism and valine, leucine and isoleucine metabolism were significantly altered in FD rats (Fig. 10). Besides, valine (F9), glycine (F45) and threonine (F15) increased obviously in FD group and lysine (U64) decreased significantly. Notably, amino acids metabolism alters greatly after the FD model intervention. Valine, glycine and threonine are all glucogenic amino acids which can be converted into glucose through gluconeogenesis. The increased concentration of valine (F9), glycine (F45) and threonine (F15) might be a signal of enhanced gluconeogenesis. Moreover, glycine, serine, threonine metabolism and valine, leucine, isoleucine degradation provides important energy metabolism precursors for TCA cycle.28 The gluconeogenesis has enhanced to keep the balance of energy metabolism since the TCA cycle is inhibited.Interestingly, the altered amino acids metabolism pathways were highly correlated with perturbed gut microbes. The arginine and proline metabolism changed significantly in FD rat (Fig. 10). Meanwhile, Bacteroides (band 9, 12) had negatively correlation with glutamate which is the intermediate product of arginine and proline metabolism (Fig. 9). Research29 has proved that Bacteroides plays an important role in converting mucoitin into proline which is enriched in glycoprotein of intestinal epithelial cells. The altered proline metabolism might reflect the damaged morphology and functions of intestinal epithelial cells. Moreover, Parasutterella (band 2) were positively correlated with threonine (U3), leucine (U61), proline (U58) and alanine (U4), negatively correlated with valine (F9), shown in Fig. 9. Our findings are supported by other studies. Nagai et al.30 isolated Parasutterella from human feces and found this strain is largely saccharolytic. Parasutterella can also inhibit the branched chain amino acids biosynthesis. Thus, the altered amino acids metabolism pathways related with FD had a closed relationship with perturbed gut microbiota.
The analysis of amino acids metabolism provides an additional support for our notion that the inhibited energy metabolism is closely related to FD. The disturbed amino acids metabolism inhibits protein synthesis, which can probably explain the body weight loss in the current study.
Disturbance of nucleotides metabolism
The nucleotides biosynthesis in FD rats was affected by the disturbed amino acids metabolism. For example, glycine (F45) can be transformed into one carbon unit that is indispensable for the synthesis of purine and pyridine.31 So the increased glycine (F45) in our study could provide abundant precursors for the purine and pyridine synthesis. The increased allantoin (U30), guanine (U32) and decreased uracil (F47), adenine (F51), hypoxanthine (F58) and xanthine (F60) were observed in FD rats. These fluctuant concentration of nucleotide metabolites indicated that nucleotides metabolism was disturbed in FD rat. The absence of allantoin (U30) in the urine can be an indicator of low reactive oxygen.32 In the presence of xanthine oxidase, hypoxanthine can transform into xanthine (Fig. 10), and then degrades into urate. And guanine can also transform into xanthine (Fig. 10). Thus, the decreased levels of purine derivatives suggested that the FD related to net suppression of purine metabolism. In Fig. 9, xanthine (F60) had positively correlation with Parasutterella (band 2). In addition, recent studies33,34 revealed that hypoxanthine could serve as an indicator of the bacterial abundance, because enzymatic conversion, which can transform nucleotides into inosine and hypoxanthine, increases with increasing bacterial abundance. Therefore, we tentatively hypothesized that the increased Parasutterella acted as one of the factors which could explain the disturbed purine metabolism in FD rats.Disturbance of SCFAs synthesis
SCFAs, such as propionate and butyrate, are produced from fermentation of nondigestible dietary fibers by gut microbiota. SCFAs are also an important energy source for the host, providing 5–15% of energy requirements in human.35–37 The pathway analysis showed that the propanoate metabolism and the butyrate metabolism changed significantly (Fig. 7). Interestingly, propionate and butyrate were closely related to Parasutterella (band 2 in Fig. 9). It seemed that the increased abundance of Parasutterella might cause the disturbance of propanoate and butanoate metabolism and then partly suppressed the energy supply of the host.Compared with the control group, SCFAs-producing bacteria decreased in the FD group, such as the decreased abundances of Eisenbergiella (band 3), Intestinimonas (band 5), Eubacterium (band 7), Bacteroides (band 9, 12), Lachnoanaerobaculum (band 10) and Herbinix (band 11). Research by Biesterveld et al.38 has shown that Bacteroides can transform sugar into formate, acetate and ethanol in the broiler chicken. Koeck et al.39 found that Herbinix (band 11) can produce acetate, propionate and ethanol as major end products in cellulose metabolism. Amir et al.40 found that Eisenbergiella can produce butyrate, acetate, lactate and succinate as major metabolic products. Kläring et al.41 proved that Intestinimonas are butyrate-producing bacteria in intestine. Schwab et al.42 demonstrated that Eubacterium can produce butyrate from lactate and acetate, and convert 1,2-propanediol to propionate. Hedberg et al.43 isolated Lachnoanaerobaculum from the human small intestine and found that Lachnoanaerobaculum can produce butyrate, acetate, H2S and NH3 as major metabolic products. Butyrate can provide nutrients for the tissue and protect the integrity of the intestinal mucosa. In our study, the decreased abundance of butyrate-producing bacteria Eisenbergiella (band 3), Intestinimonas (band 5), Eubacterium (band 7), Lachnoanaerobaculum (band 10) might hamper the production of butyrate, which probably leaded to unprotect intestinal mucosa.
Disturbance of gut microbiota
Hippurate, a common small molecular urotoxin, is considered as a biomarker in assessing the balance of gut microbial community.44 The increased hippurate (U48) was previously reported as a consequence of the perturbation in gut microbiota.45 In our study, hippurate (U43) increased significantly, which indicated the imbalance of host metabolism caused by microbiota disturbance in FD rats. In Fig. 9, we observed that hippurate displayed closely correlations with Firmicutes including Saccharofermentans (band 6), Lachnoanaerobaculum (band 10) and Herbinix (band 11). Previous research46 has demonstrated that Firmicutes make up the largest portion of the mice and human gut microbiome. And Firmicutes have been shown to be involved in energy resorption, and potentially related to the development of diabetes and obesity.47–49 Therefore, the energy deficiency of host may relate to the decrease in Saccharofermentans (band 6), Lachnoanaerobaculum (band 10) and Herbinix (band 11). Interestingly, the increased hippurate displayed negative correlation with Parasutterella (band 2) as shown in Fig. 9. In our previous discussion, the increased abundance of Parasutterella could cause an inefficient energy production in host. It was conceivable to suggest that the increased hippurate caused the inhibition of energy metabolism through gut microbiota.Further associations between disturbance of gut microbes and differential metabolites were the alterations of TMA, TMAO and DMA. The decreased levels of TMA (U65), TMAO (U33) and DMA (F27) were observed in rats with FD. Previous research50 has shown that urinary TMA is produced from choline under the action of gut microbiota, then TMA is detoxified in the liver through flavine monooxygenase forming TMAO.51,52 These results provided another evidence for the disturbance of gut microbes in FD rats. A reduced level of TMAO associated with the disturbance of gut microbiota signified the availability of more choline for producing phosphocholine, leading to the observed reduction of lipids in the liver.53
Another evidence for disturbance of gut microbiota was the disturbance of primary bile acid biosynthesis. Bile acids are synthesized in the liver and conjugated with taurine or glycine before secreting along with bile into the intestine. It was reported that the biosynthesis of bile acid is closely related to gut microbiota. We found taurine (U15) and tauro-chenodeoxycholate acid (F59) decreased in FD rats, which also supported our notion.
These results support our notion that the metabolic biomarkers are highly correlated with perturbed gut bacterial families. In conclusion, FD is related to the disturbed host–microbiota co-metabolism.
Conclusion
This work combined 16S rRNA gene sequencing and metabolomics profiling to study the impact of gut microbiome and variations in fecal and urinary metabolites of FD. We analyzed the potential biomarkers of FD and discussed the effect in physiology. Our results indicated that FD was mainly related to energy metabolism and the inhibited energy supply might be associated with some symptoms of FD such as fatigue, emaciation and loss of body weight. The altered intestinal microbial communities in FD rats implied FD disturbed the balance of gut microbiota. The disturbance of microbes closely associated with abnormal metabolism including energy metabolism, amino acids metabolism, nucleotides metabolism and SCFAs synthesis. Taken together, these data proved strong correlations between metabolites and the abundances of Parasutterella, Herbinix, Intestinimonas, Eubacterium, Lachnoanaerobaculum and Bacteroides were observed to guide further research. Our work demonstrated substantial insight into the effects regarding microbiology and metabolomics response to FD. Future work can focus on the energy metabolism pathway analysis from the perspective of target metabolomics and analyze microbial structure applying more advanced technology. The systematic view on the holistic metabolic profiling and microbial structure can be regarded as a commendable strategy for studying the host–microbiota co-metabolism of FD.Conflicts of interest
There are no conflicts to declare.Abbreviations
| AUC | The area under the curve |
| CPK | The activity of creatine phosphokinase |
| DMA | Dimethylamine |
| FD | Functional dyspepsia |
| FGIDs | Functional gastrointestinal disorders |
| FC | Fold changes |
| GAS | The concentration of gastrin |
| MTL | The concentration of motilin |
| MetPa | Metabolomics pathway analysis |
| MSEA | Metabolite set enrichment analysis |
| NMR | Nuclear magnetic resonance |
| OPLS-DA | Orthogonal signal correction partial least squares-discriminant analysis |
| PCA | Principal Component Analysis |
| PLS-DA | Partial least squares discriminant analysis |
| PCR-DGGE | Polymerase chain reaction-denaturing gradient gel electrophoresis |
| SCFAs | Short chain fatty acids |
| TMA | Trimethylamine |
| TMAO | Trimethylamine oxide |
| TCA | Tricarboxylic acid cycle |
| TSP | Sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 |
| VIP | Variable importance to the projection |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81473319 and No. 81473540), the Key Projects of Guangdong Natural Science Foundation (No. 2017A030311022), the Guangdong Science and Technology Program (No. 2015A030401031, 2014A020209026), the Guangdong Natural Science Foundation (No. 2015A030313123), the Zhongshan Science and Technology Program (No. 2016C1015), and the Science Program for Overseas Scholar of Guangzhou University of Chinese Medicine (Torch Program) (No. XH20170111).References
- R. J. Saad and W. D. Chey, Aliment. Pharmacol. Ther., 2006, 24, 475–492 CrossRef PubMed.
- H. B. El-Serag and N. J. Talley, Aliment. Pharmacol. Ther., 2004, 19, 643–654 CrossRef PubMed.
- G. Barbara, C. Feinle-Bisset, U. C. Ghoshal, J. Santos, S. J. Vanner, N. Vergnolle, E. G. Zoetendal and E. M. Quigley, Gastroenterology, 2016, 150, 1305–1318 CrossRef PubMed.
- G. Tziatzios, E. J. Giamarellos-Bourboulis, I. S. Papanikolaou, M. Pimentel, G. D. Dimitriadis and K. Triantafyllou, Med. Hypotheses, 2017, 106, 26–32 CrossRef PubMed.
- J. K. Nicholson and J. C. Lindon, Nature, 2008, 455, 1054–1056 CrossRef PubMed.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef PubMed.
- Y. L. Wang, E. Holmes, J. K. Nicholson, O. Cloarec, J. Chollet, M. Tanner, B. H. Singer and J. Utzinger, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12676–12681 CrossRef PubMed.
- A. W. Nicholls, E. Holmes, J. C. Lindon, J. P. Shockcor, R. D Farrant, J. N. Haselden, S. J. Damment, C. J. Waterfield and J. K. Nicholson, Chem. Res. Toxicol., 2001, 14, 975–987 CrossRef PubMed.
- A. W. Nicholls, R. J. Mortishire-Smith and J. K. Nicholson, Chem. Res. Toxicol., 2003, 16, 1395–1404 CrossRef PubMed.
- N. B. Vakil, C. W. Howden, P. Moayyedi and J. Tack, Clin. Gastroenterol. Hepatol., 2017, 15, 1191–1194 CrossRef PubMed.
- S. O. Noor, K. Ridgway, L. Scovell, E. K. Kemsley, E. K. Lund, C. Jamieson, I. T. Johnson and A. Narbad, BMC Gastroenterol., 2010, 10, 134 CrossRef PubMed.
- L. Luo, J. Chen, Y. Wang, X. Zhang, X. Yin, B. Lu, Y. Li, H. Zheng, Z. Xie and Q. Liao, Chin. Pharmacol. Bull., 2017, 33, 1363–1370 Search PubMed.
- M. Lin, Z. Xie, Y. Zhou, Y. Li, J. Ren, X. Peng, M. Yao, Z. Yang and Q. Liao, RSC Adv., 2015, 5, 65415–65426 RSC.
- H. Lu, J. He, Z. Wu, W. Xu, H. Zhang, P. Ye, J. Yang, S. Zhen and L. Li, Microb. Ecol., 2013, 65, 781–791 CrossRef PubMed.
- X. Shi, C. Xiao, Y. Wang and H. Tang, J. Proteome Res., 2013, 12, 991–1006 CrossRef PubMed.
- Y. Zhao, J. Wu, J. V. Li, N. Y. Zhou, H. Tang and Y. Wang, J. Proteome Res., 2013, 12, 2987–2999 CrossRef PubMed.
- D. Liu, T. Li, H. Zheng, X. Yin, M. Chen, Z. Liao, M. Hu, Z. Luo, B. Lu, Y. Li, Q. Liao, Z. Xie and F. Wu, RSC Adv., 2017, 7, 48136–48150 RSC.
- J. G. Xia, I. V. Sinelnikov, B. Han and D. S. Wishart, Nucleic Acids Res., 2015, 43, 251–257 CrossRef PubMed.
- J. G. Xia and D. S. Wishart, Bioinformatics, 2010, 26, 2342–2344 CrossRef PubMed.
- A. Andersson and L. Mäler, J. Biomol. NMR, 2002, 24, 103–112 CrossRef PubMed.
- J. Feng, C. D. Petersen, D. H. Coy, J. K. Jiang, C. J. Thomas, M. R. Pollak and S. A. Wank, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17791–17796 CrossRef PubMed.
- Y. Kariya, K. Kato, Y. Hayashizaki, S. Himeno, S. Tarui and K. Matsubara, Gene, 1986, 50, 345–352 CrossRef PubMed.
- S. M. Bong, J. H. Moon, H. K. Nam, K. S. Lee, Y. M. Chi and K. Y. Hwang, FEBS Lett., 2008, 582, 3959–3965 CrossRef PubMed.
- M. Swiatkiewicz, M. Fiedorowicz, J. Orzel, M. Welniak-Kaminska, P. Bogorodzki, J. Langfort and P. Grieb, Front Physiol., 2017, 8, 19 Search PubMed.
- E. E. Noble, T. M. Hsu, R. B. Jones, A. A. Fodor, M. I. Goran and S. E. Kanoski, J. Nutr., 2017, 147, 20–28 CrossRef PubMed.
- M. Wyss and R. Kaddurah-Daouk, Creatine, Physiol. Rev., 2000, 80, 1207–1213 CrossRef PubMed.
- J. T. Brosnan, R. P. da Silva and M. E. Brosnan, Amino Acids, 2011, 40, 1325–1331 CrossRef PubMed.
- J. Cui, Y. Liu, Y. Hu, J. Tong, A. Li, T. Qu, X. Qin and G. Du, J. Pharm. Biomed. Anal., 2017, 132, 77–86 CrossRef PubMed.
- P. Corfield, S. A. Wagner, J. R. Clamp, M. S. Kriaris and L. C. Hoskins, Infect. Immun., 1992, 60, 3971–3978 Search PubMed.
- F. Nagai, M. Morotomi, H. Sakon and H. R. Tanaka, Int. J. Syst. Evol. Microbiol., 2009, 59, 1793–1797 CrossRef PubMed.
- X. Cha and C. Zhou, in Biochemistry, ed. N. Cheng, and A. Zhou, People's Medical Publishing House, Beijing, 2008, pp. 179–206 Search PubMed.
- T. Asano, S. Aida, S. Suemasu, K. Tahara, K. Tanaka and T. Mizushima, Sci. Rep., 2015, 5, 17519 CrossRef PubMed.
- K. L. B. Chang, J. J. Chang, C. Y. Shiau and B. S. Pan, J. Agric. Food Chem., 1998, 46, 682–686 CrossRef PubMed.
- N. Osman, D. Adawi, G. Molin, S. Ahrne, A. Berggren and B. Jeppsson, BMC Gastroenterol., 2006, 6, 31 CrossRef PubMed.
- E. N. Bergman, Physiol. Rev., 1990, 70, 567–590 CrossRef PubMed.
- D. L. Topping and P. M. Clifton, Physiol. Rev., 2001, 81, 1031–1064 CrossRef PubMed.
- L. V. Hooper, T. Midtvedt and J. I. Gordon, Annu. Rev. Nutr., 2002, 22, 283–307 CrossRef PubMed.
- P. W. J. J. van der Wielen, S. Biesterveld, S. Notermans, L. J. A. Lipman and F. van Knapen, Appl. Environ. Microbiol., 2000, 6, 2536–2540 CrossRef.
- D. E. Koeck, W. Ludwig, G. Wanner, V. V. Zverlov, W. Liebl and W. H. Schwarz, Int. J. Syst. Evol. Microbiol., 2015, 65, 2365–2371 CrossRef PubMed.
- I. Amir, P. Bouvet, C. Legeay, U. Gophna and A. Weinberger, Int. J. Syst. Evol. Microbiol., 2014, 64, 907–914 CrossRef PubMed.
- K. Kläring, L. Hanske, N. Bui, C. Charrier, M. Blaut, D. Ha-ller, C. M. Plugge and T. Clavel, Int. J. Syst. Evol. Microbiol., 2013, 63, 4606–4612 CrossRef PubMed.
- C. Schwab, H. J. Ruscheweyh, V. Bunesova, V. T. Pham, N. Beerenwinkel and C. Lacroix, Front Microbiol., 2017, 8, 95 Search PubMed.
- M. E. Hedberg, E. R. B. Moore, L. Svensson-Stadler, P. Horstedt, V. Baranoy, O. Hernell, S. N. Wai, S. Hammarstrom and M. L. Hammarstrom, Int. J. Syst. Evol. Microbiol., 2012, 62, 2685–2690 CrossRef PubMed.
- I. K. Yap, J. V. Li, J. Saric, F. P. Martin, H. Davies, Y. Wang, I. D. Wilson, J. K. Nicholson, J. Utzinger, J. R. Marchesi and E. Holmes, J. Proteome Res., 2008, 7, 3718–3728 CrossRef PubMed.
- J. K. Nicholson, E. Holmes, J. Kinross, R. Burcelin, G. Gibson, W. Jia and S. Pettersson, Science, 2012, 336, 1262–1267 CrossRef PubMed.
- R. E. Ley, D. A. Peterson and J. I. Gordon, Cell, 2006, 124, 837–848 CrossRef PubMed.
- R. E. Ley, P. J. Turnbaugh, S. Klein and J. I. Gordon, Nature, 2006, 444, 1022–1023 CrossRef PubMed.
- R. E. Ley, F. Bäckhed, P. Turnbaugh, C. A. Lozupone, R. D. Knight and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11070–11075 CrossRef PubMed.
- A. L. Komaroff, J. Am. Med. Assoc., 2017, 317, 355–356 CrossRef PubMed.
- M. Al-Waiz, M. Mikov, S. C. Mitchell and R. L. Smith, Metabolism, 1992, 41, 135–136 CrossRef PubMed.
- J. L. Smith, J. S. Wishnok and W. M. Deen, Toxicol. Appl. Pharmacol., 1994, 125, 296–308 CrossRef PubMed.
- V. Tremeroli and F. Bäckhed, Nature, 2012, 489, 242–249 CrossRef PubMed.
- M. E. Dumas, R. H. Barton, A. Toye, O. Cloarec, C. Blancher, A. Rothwell, J. Fearnside, R. Tatoud, V. Blanc, J. C. Lindon, S. C. Mitchell, E. Holmes, M. I. McCarthy, J. Scott, D. Gauguier and J. K. Nicholson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12511–12516 CrossRef PubMed.
Footnotes |
| † Accession codes: The GenBank submission accession numbers are KY792535–KY792545. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01432a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |