
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant-assisted synthesis of Mo–V mixed oxide catalysts for upgraded one-step conversion of glycerol to acrylic acid†
Letícia F. Rasteiro ,
Luiz H. Vieira,
Celso V. Santilli and
Leandro Martins*
,
Luiz H. Vieira,
Celso V. Santilli and
Leandro Martins*
Instituto de Química, Unesp – Universidade Estadual Paulista, Rua Prof. Francisco Degni 55, CEP 14800-900 Araraquara, SP, Brazil. E-mail: leandro@iq.unesp.br
First published on 27th March 2018
Abstract
The catalytic properties of Mo–V mixed oxides hydrothermally synthetized in the presence of ionic surfactants (SDS and CTAB) were investigated in the gas-phase oxidative dehydration of glycerol. The presence of surfactants promoted a change in morphology of MoV2O8 phase, directing to formation of rod-shaped crystals, and, consequently, an increase in macroporosity of materials, generated by intercrystallite spaces, when compared to a reference sample. Rod-like morphology stabilized the MoV2O8 mixed oxide phase during glycerol conversion, avoiding migration of vanadium from crystalline to amorphous phase, like observed in the reference sample, favoring the dynamic of reduction/reoxidation of vanadium and, consequently contributing to an increase in efficiency and stability of the catalyst. Both SDS and CTAB catalysts presented higher productivity of acrylic acid and good catalytic stability, with no coke formation and considerable decrease in COX evolution during 6 h of reaction. SDS presented the best catalytic results with 100% of conversion, 57% of acrylic acid selectivity and 36% of COX selectivity.
Introduction
Due to the increase in biodiesel production and, consequently, the coproduced glycerol, the search for new uses for it becomes indispensable.1,2 Many studies have been reported for glycerol valorisation in the production of different substances using various processes like dehydration,3–5 oxidative dehydration,6–8 acetalization,9 acetylation10 and hydrogenolysis.11 A high value-added product that can be obtained from glycerol oxidative dehydration and that has attracted attention because of the increase in its market demand is acrylic acid.12 Nowadays, acrylic acid is industrially produced from a two-step oxidation of propylene, a non-renewable route, since propylene is derived from petroleum.12,13In the last years, several studies demonstrated the viability of the catalytic conversion of glycerol to acrylic acid by means of a two-stage reaction depicted in Scheme 1. In the first stage, glycerol is dehydrated to 3-hydroxypropanal on acid sites, followed by subsequent dehydration and formation of acrolein. In the second stage, the oxidation of acrolein to acrylic acid occurs on metal–oxygen redox sites.14 Therefore, for the selective synthesis of acrylic acid from glycerol, the catalysts need to possess acid and oxidant sites.12 This route of glycerol valorization can be performed with one or two catalysts, but the use of one catalyst with bifunctional properties is getting more attention due to several advantages.15 Mixed oxides based on molybdenum and vanadium, MoxVyOz, are prominent catalysts to perform the one-step conversion of glycerol due to the moderate acidity and well-known oxidative potential of vanadium in these structures, conducted by the Mars–van-Krevelen mechanism.8,16 However, the main disadvantages of these catalysts are the textural properties, like low specific areas and porosity,8,17–19 and the instability of the mixed oxide phases in the reaction conditions.20 The solid-state reaction and/or spreading of mixed oxide phases when applied in catalytic systems under high temperatures and oxidant atmospheres can lead to a restructuration of phases20 to form single component oxides and/or migration of active species to amorphous phase.
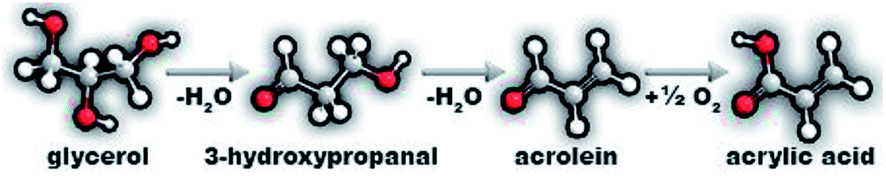 | ||
| Scheme 1 Two-step glycerol oxydehydration: dehydration to acrolein followed by oxidation of acrolein to acrylic acid. | ||
The use of surfactants in the synthesis of catalysts is a method to create porous structures in the material and/or to alter surface characteristics.21,22 The addition of surfactants in the synthesis of MoxVyOz can bring new textural properties and lead to a stabilization of the mixed oxide phases in these catalysts, not yet studied.
In a previous work, we studied the effect of synthesis and calcination atmospheres in the formation of Mo–V mixed oxides crystalline phases prepared by a hydrothermal method and evaluated their performance in the one-pot glycerol oxydehydration to acrylic acid.6 We identified the MoV2O8 and Mo4.65V0.35O14 as the most active phases in the reaction but with low stability during reaction. Herein, we describe the surfactant-assisted synthesis and structural characterization of MoxVyOz catalysts and explore how the presence of SDS and CTAB surfactants in the synthesis mixture influences the composition of phases, porosity, crystal morphology, and catalytic activity of these materials in the one-step oxidative dehydration of glycerol.
Experimental
Hydrothermal synthesis of the Mo–V mixed oxides
The synthesis of the mixed oxides was performed based on a hydrothermal method described in our previous work.6 In a typical synthesis, vanadyl sulfate (VOSO4) and ammonium paramolybdate ((NH4)6Mo7O24·4H2O) were dissolved separately in distilled water. Both mixtures were maintained under stirring for 10 min at 25 °C and after the complete dissolution, they were mixed and stirred for another 10 min at 25 °C, resulting in a solution with a pH of 2.8 and a Mo/(Mo + V) mole ratio of 0.60.6,8 The mole proportion of the reagents was 1 VOSO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 (NH4)6Mo7O24·4H2O:300 H2O. Afterwards, sodium dodecyl sulfate (SDS) or cetyltrimethylammonium bromide (CTAB) were added to the solution and maintained under stirring for 40 min at 25 °C. The mole ratio surfactant/(Mo + V) was 0.10. Later, the solution was transferred to a 50 mL Teflon vessel contained in a stainless steel autoclave reactor, occupying approximately 70% of the vessel capacity. The reactor was hermetically closed and then purged with pure O2 in successive cycles and finally was pressurized with O2 to 6 bar. The reactor was placed in a glycerol bath heated at 160 °C and kept for 48 h under static conditions. The precipitate was separated by centrifugation and dried at 60 °C for 12 h. The resulting catalyst precursors were heat treated in oxidant atmosphere at 500 °C during 2 h. The sample synthesized in the absence of surfactant was named reference and samples synthesized in the presence of SDS and CTAB were named SDS-0.10 and CTAB-0.10, respectively.
:0.2 (NH4)6Mo7O24·4H2O:300 H2O. Afterwards, sodium dodecyl sulfate (SDS) or cetyltrimethylammonium bromide (CTAB) were added to the solution and maintained under stirring for 40 min at 25 °C. The mole ratio surfactant/(Mo + V) was 0.10. Later, the solution was transferred to a 50 mL Teflon vessel contained in a stainless steel autoclave reactor, occupying approximately 70% of the vessel capacity. The reactor was hermetically closed and then purged with pure O2 in successive cycles and finally was pressurized with O2 to 6 bar. The reactor was placed in a glycerol bath heated at 160 °C and kept for 48 h under static conditions. The precipitate was separated by centrifugation and dried at 60 °C for 12 h. The resulting catalyst precursors were heat treated in oxidant atmosphere at 500 °C during 2 h. The sample synthesized in the absence of surfactant was named reference and samples synthesized in the presence of SDS and CTAB were named SDS-0.10 and CTAB-0.10, respectively.
Characterization
The amounts of V and Mo present in the synthesis solutions (mother liquors) were determined by chemical analysis by using an Optima 8000 ICP-OES spectrometer. The amounts of V and Mo present in the solid catalysts were determined by mass balance between the initial quantities fed into the hydrothermal reactor and those obtained by chemical analysis.Crystalline phases of the catalysts, before and after the catalytic reaction, were assessed by X-ray diffraction (XRD) using a Rigaku Miniflex 600 diffractometer with CuKα radiation (0.15418 nm) selected with a curved graphite monochromator. Data were collected in the 2θ (°) range from 5 to 60, with a scan step of 0.02 and counting time of 1 s. All the crystalline phases in the samples were identified with Crystallographica Search Match software and quantified by the Rietveld refinement method using TOPAS® 4.2 software. The occupancy and temperature factors of all atoms were fixed, unit cell parameters, scale factor and atomic positions (except for special positions: 0, 1/4, 1/3, 1/2 etc.) were refined. Sample displacement, zero error and intensity corrections (Lorentz and polarization factors were fixed in 26.4, characteristic value for graphite monochromators) were refined. The background was fitted using a sixth-order Chebyshev polynomial function. Fundamental parameters profile fitting (FPPF) were used for the peak profile refinements.
Specific BET area of the samples were determined by nitrogen sorption measurements performed at liquid nitrogen temperature (−196 °C), with relative pressure interval between 0.001 and 0.998, using a Micromeritics ASAP 2010 system. The samples were pre-treated under vacuum (∼10 × 10−6 Pa) for 12 h at 200 °C.23
Density of the samples was evaluate by helium pycnometry using a Micromeritics AccuPyc 1330 and then transferred to a Micromeritics GeoPyc 1360 to evaluate the total porosity of the samples.
Hg intrusion porosimetry was used to evaluate the textural characteristics of the materials in the macro- and mesopore ranges. The measurements were carried out in a Micromeritics Autopore III equipment, operating for the determination of pores with a minimum diameter of 3 nm. The pore diameter was calculated by means of the Washburn equation (r = −2γcosθ/P), using surface tension and contact angle values of 0.489 N m−1 and 135°, respectively.
Particle size and shape of the catalysts were observed by SEM images on a FEI Magellan 400L microscope. Samples were previously deposited on an aluminum sample holder and subsequently sputtered with gold for 30 s.
Thermogravimetric analysis (TGA) of the spent catalysts was conducted under a flow rate of 100 mL min−1 of O2, using an SDT Q600 TGA/DSC thermobalance, with a heating rate of 10 °C min−1 from 30 to 600 °C.
Acid sites of the Mo–V mixed oxides were measured by temperature programmed desorption of ammonia (TPD-NH3). In these experiments, a mass of 200 mg of sample was placed under a He flow (60 mL min−1) at 300 °C for 1 h, followed by cooling to 100 °C. At this temperature, the sample was exposed to a 60 mL min−1 flow of 1% NH3 in He for 1 h. The excess and physically adsorbed NH3 was then purged at 100 °C under a flow of He during 60 min. Finally, ammonia was desorbed in a He flow of 60 mL min−1, with heating from 100 to 500 °C at a rate of 10 °C min−1. The amount of desorbed ammonia per gram of catalyst was calculated from measurements made using a mass spectrometer (PrismaPlus QMG 220, Pfeiffer).
XPS measurements of the catalysts were performed using a UNI-SPECS UHV spectrometer with Mg-Kα source (λ = 1253.6 eV), pass energy of 10 eV, and 0.2 eV energy step. The pelleted samples were first left overnight under vacuum (<3.7 × 10−11 Pa), after which the analysis was performed at a residual pressure below 3.7 × 10−11 Pa inside the chamber. The binding energies of the chemical elements were determined by fitting the measured spectra and referencing to the C1s (285.1 eV) peak, with accuracy of ±0.1 eV. The fitting of the curves was performed with CasaXPS software, using the Shirley baseline function and Voigt profile (70% Gaussian and 30% Lorentzian).
The critical packing parameters or shape factors (g) of CTAB and SDS surfactants were calculated according to eqn (1):
| g = v/a0lc | (1) |
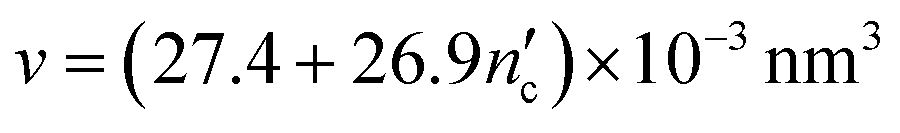 | (2) |
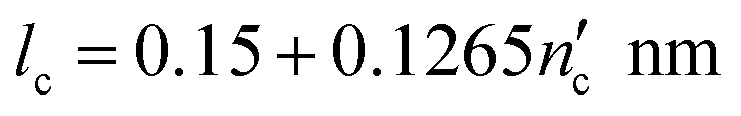 | (3) |
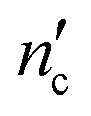 is one less than the total number of carbons in the surfactant tail, nc, since the first carbon after the head group is highly solvated and is considered part of it.26
is one less than the total number of carbons in the surfactant tail, nc, since the first carbon after the head group is highly solvated and is considered part of it.26
Glycerol oxydehydration
The catalytic reaction was conducted in the gas phase at 320 °C, under atmospheric pressure and using a fixed bed reactor. Firstly, 200 mg of the calcined catalyst was supported on a glass wool placed inside the reactor. The reactor was heated to the reaction temperature under a flow of 30 mL min−1 of 100% of O2 and maintained at this temperature for 15 min before initiating the reaction. An aqueous solution containing 10 wt% of glycerol (Sigma-Aldrich, 99%) was fed to the reactor at a flow rate of 0.05 mL min−1. The gas composition was glycerol/oxygen/water = 2/28/70 (% mole). The products outflowing the reactor were condensed in a gas–liquid separator kept at 1 °C and aliquots were withdrawn at every 1 h. A previous test was performed to confirm the efficiency of the condensation and the full recovery of the condensable products outflowing the reactor, by injecting an aqueous solution containing 10 wt% of acetaldehyde (i.e. the most volatile product detected, with a boiling point of 20 °C) into the reactor and determining the mass balance in the gas–liquid separator. The products were quantified using a gas chromatograph (Shimadzu GC-2014) equipped with a capillary column (RTX-1, 30 m, 0.32 mm, 1 μm) and a flame ionization detector (FID). As part of the products was converted to CO and CO2 and the FID detector is not able to detect them, the produced COX (CO + CO2) was determined by mass balance with the liquefied products. The quantification of the products was done by adding a known mass of n-butanol as an internal standard before each injection and comparing with calibration curves of the products.The glycerol conversion (Xgl) and selectivity of products (Si) were calculated according to eqn (4) and (5), respectively.
| Xgl (%) = [(ninputgl − noutputgl)/ninputgl] × 100 | (4) |
| Si (%) = [ni/(ninputgl − noutputgl)] × (zi/zgl) | (5) |
Results and discussion
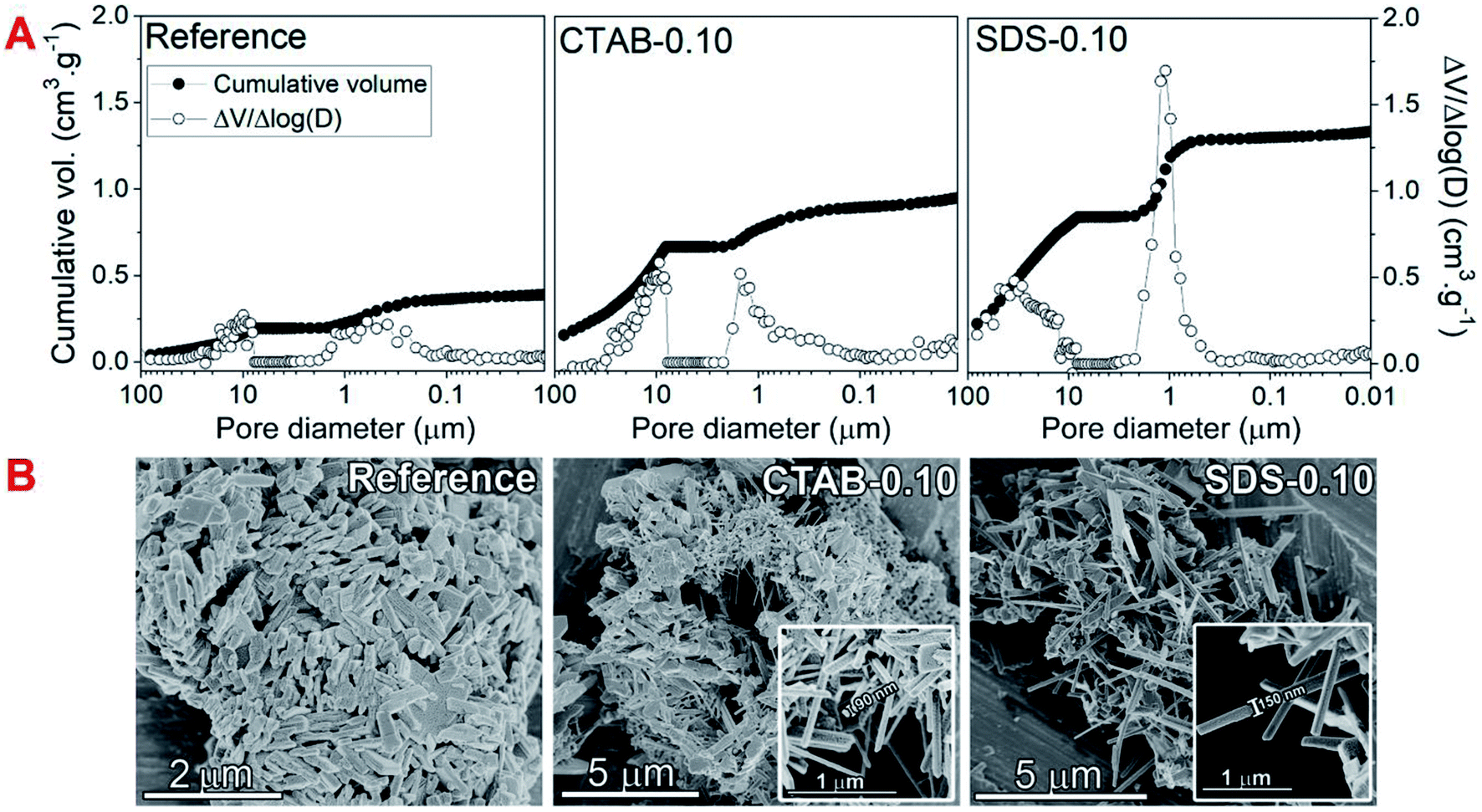
The hydrothermal synthesis procedure allowed a very high solid yield to be obtained, with consequently a high assimilation of molybdenum of 98.8% and vanadium of 97.3% in the catalysts.6 In fact, by visual observation, the mother liquor was very clear and therefore qualitatively free from reactants.The diffraction patterns (Fig. 1) show that despite the use of the surfactants in the syntheses, the formed MoxVyOz crystalline phases are essentially the same, i.e. Mo4.65V0.35O14, MoV2O8 and MoO3 (Table S1†). The results obtained by X-ray diffraction suggests that vanadium in crystalline phase was incorporated in the mixed oxide structures, since no peaks referent to crystalline V2O5 were identified. Table 1 shows the textural and surface properties of the mixed oxides, BET area, pore volume, porosity and surface acidity. The use of both surfactants increased the porosity and the open pore volume as evidenced by He-pycnometry and Hg-porosimetry measurements, but no changes in the BET area of catalysts were noted by N2 physisorption measurements, indicating that the presence of surfactants during synthesis are considerable influencing the porosity in a macropore scale but not generating mesoporosity in the materials. The catalyst SDS-0.10 had the best improvement in porosity from 69.3% in the reference sample to 82.7%. In fact, the Hg-intrusion curves in Fig. 2A show that SDS-0.10 presented a higher pore volume and a narrower pore size distribution when compared with CTAB-0.10 and the reference samples, with a maximum point at, approximately, 1 μm.
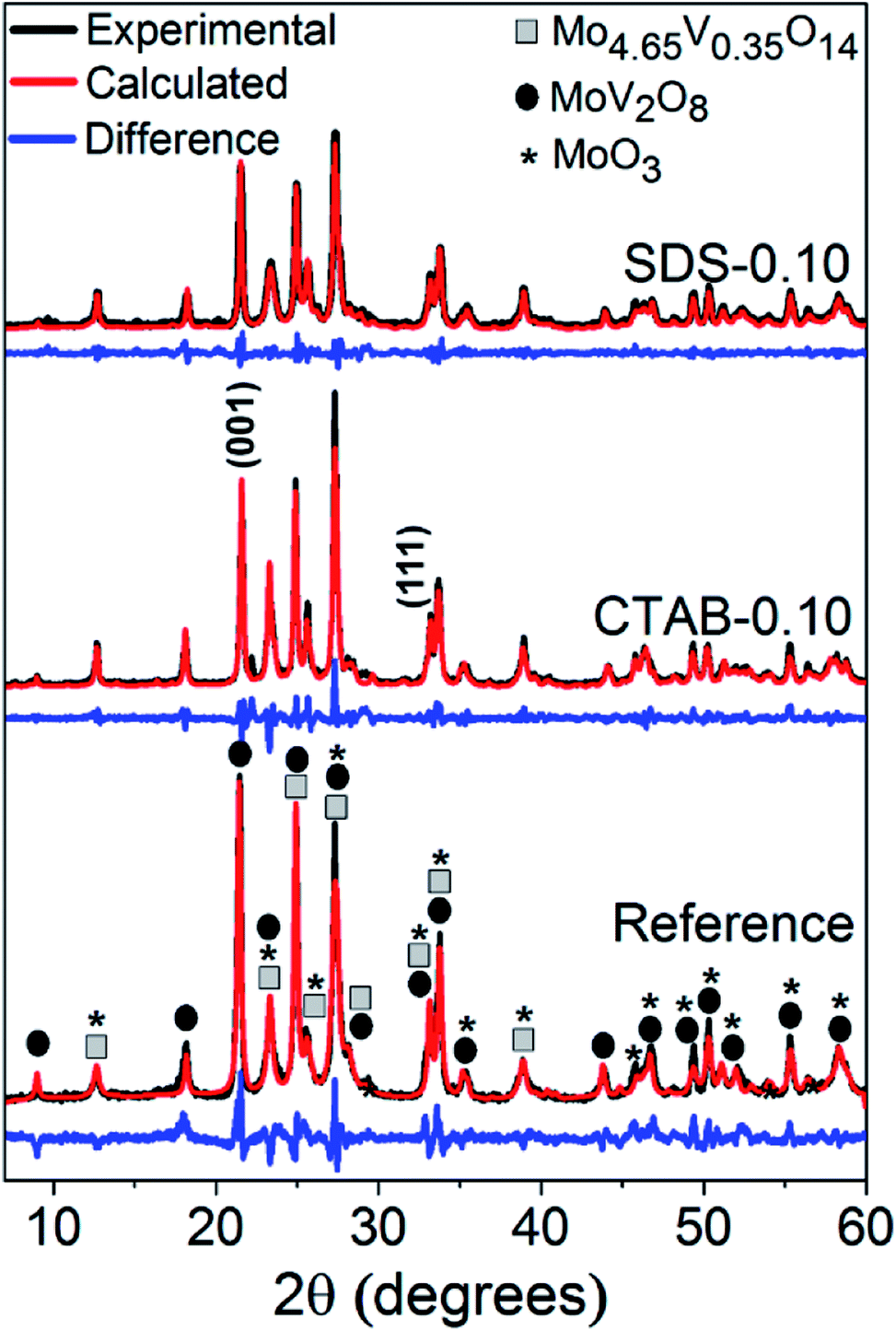 | ||
| Fig. 1 XRD patterns and identification of phases for the Mo–V mixed oxides. The net results of the Rietveld analyses are shown in red and the deviations from the measured diffractograms are depicted in blue. | ||
| Catalyst | Porositya (%) | Pore volumeb (cm3 g−1) | BET area (m2 g−1) | Chemisorbed NH3 (μmol g−1)c | XRD intensityd (I001/I111) | MoV2O8 crystallite sizee (nm) | Critical packing parameterf (g) |
|---|---|---|---|---|---|---|---|
| a Determined by He-pycnometry.b Determined by Hg-porosimetry.c Determined by TPD-NH3.d Intensity ratio of (001) and (111) crystalline plans from MoV2O8 phase.e Determined by LVol-IB of full peak profile from Rietveld analysis performed on TOPAS V4.2 software.f Critical packing parameter values are referent to surfactant micelles formed in the synthesis mixture. | |||||||
| Reference | 69.3 | 0.22 | 11.4 | 38.9 | 3.51 | 41.0 ± 1.3 | — |
| CTAB-0.10 | 77.9 | 0.51 | 5.9 | 33.9 | 5.65 | 72.1 ± 5.4 | 0.33 |
| SDS-0.10 | 82.7 | 1.21 | 1.4 | 27.0 | 5.37 | 88.5 ± 7.7 | 0.34 |
 | ||
| Fig. 2 (A) Representation of ionic surfactant preferentially adsorbed on the side planes of MoxVyOz crystal leading the nanorod morphology and (B) organization of CTAB and SDS micelles around the crystallites. | ||
Additional differences have arisen from scanning electron microscopy images in Fig. 2B, in which nanorod crystals could be clearly observed from very dispersed fragments in CTAB-0.10 and SDS-0.10 samples. The use of both surfactants drastically changed the shape of crystals during synthesis when compared to the reference sample, leading to the formation of randomly stacked nanorods. Considering surfactant-assisted synthesis of materials, the kind and shape of the crystals obtained are directly dependent from type and concentration of surfactant present in the synthesis solution.28 We tried to perform the syntheses with lower concentration of surfactants, below the critical micelle concentration (CMC), however, in this condition, changes in the crystal shape were not observed, probably explained by the non-effective micelle formation, and the presence of surfactant only as isolated molecules, as already reported.28 In this work, the surfactant concentration used in the synthesis mixture was, approximately, 44 mM, a value higher than the CMC of CTAB and SDS in water, of 0.9–1.0 and 8.2 mM, respectively.29,30 The formation of rod-like crystals in the presence of CTAB and SDS ionic surfactants was already reported in the synthesis of other materials.31 The shape factor or critical packing parameter (g) values for these surfactants (Table 1), calculated from head group area, tail length and volume of hydrophobic portion (Scheme 2A and eqn (1)), are in the range expected for the structure aggregation to form cylindrical or rod-shaped micelles,26 and the result fits well with that observed experimentally. The presence of the rod-shaped micelles limits the crystal nucleation on lateral crystalline plans, which favours a preferential growth in the direction normal to the surfactant free surface (Scheme 2A).32 Carefully analyzing the contributions of each phase in the diffraction patterns, a consistent relation between the increasing in the crystallite size and the peak intensity ratio referent to (001) and (111) crystalline plans of MoV2O8 phase with the formation of nanorods was observed (see Fig. S1† and Table 1). These observations lead us to believe that the presence of surfactants are basically affecting the crystal morphology of MoV2O8 phase, and not considerably influencing in the formation of MoO3 and Mo4.65V0.35O14 phases, when compared to the reference sample. The nanorod morphology were already detected and correlated to MoV2O8 phase in previous studies.33,34 Differences in the ratio of intensity between (001) and (111) crystalline plans are very coherent with the thickness of nanorods observed in the Fig. 2B. The catalyst CTAB-0.10, that presented the higher I(001)/I(111) ratio have the crystallites with smaller thickness (approximately, 90 nm), suggesting a fast growth in the direction of (111) facets and a limited growth in the direction normal to the side plans, like (001). The ratio I(001)/I(111) slightly decreased for the catalyst SDS-0.10 leading to the formation nanorods with, approximately, 150 nm of thickness. The differences in crystal thickness in the presence of CTAB and SDS surfactants are probably associated with the limitations in the transfer of Mo–V active phase to lateral plans due to the different aggregation of surfactant monomers to form the micelles (Scheme 2B). The aggregation number increases linearly with the increase of surfactant concentration for CTAB and SDS surfactants in water.35 The concentration of CTAB used in the synthesis was about 44 times the CMC, and an aggregation number above 80 is expected for this compound.35 On the other hand, the concentration of SDS was about 6 times the CMC and an aggregation number of, approximately, 54 is expected.35 The presence of a high number of monomers in CTAB micelles are more effective in preventing the penetration of Mo–V atoms in the sides of micelle and, consequently, the growth perpendicular to the crystal side plans, decreasing the thickness of nanorods, when compared to SDS micelles. Apparently, the presence of SDS promoted the formation of a larger amount of the MoV2O8 rod-like particles when compared with catalyst synthesized in the presence of CTAB, as observed by SEM images (Fig. 2B) and by increase in macroporosity, attributed to the intercrystallite spaces generated between the nanorods confirmed by Hg-porosimetry (Fig. 2A).
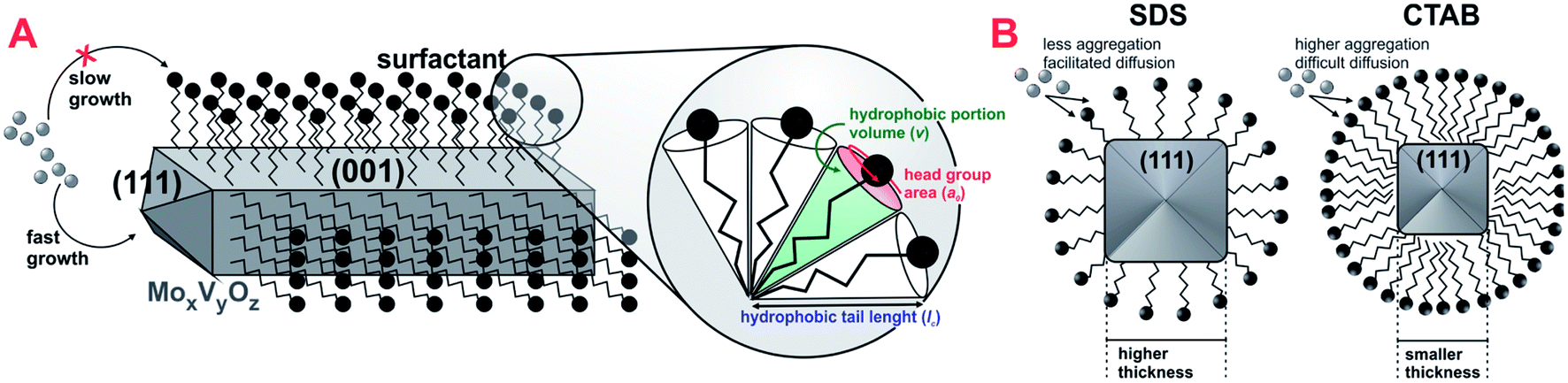 | ||
| Scheme 2 (A) Representation of ionic surfactant preferentially adsorbed on the side planes of MoxVyOz crystal leading the nanorod morphology and (B) organization of CTAB and SDS micelles around the crystallites. | ||
The XPS analyses provided information concerning the surface composition and the oxidation states of vanadium and molybdenum in the samples (Fig. S2† and Table 2). The chemical speciation showed that molybdenum oxidation state remained invariant in the samples produced by surfactant-assisted synthesis compared to reference, and that V4+ was present in low quantity in all samples. According to the stoichiometry of the detected Mo and V crystalline phases described in Table S1,† in MoV2O8, vanadium bears the valence 5+. However, in Mo4.65V0.35O14 vanadium has a mixed valence, that can be the first reason for detection of V4+ in these samples. Even presenting a mixed valence of 4+ and 5+, the low molar concentration of vanadium atoms in Mo4.65V0.35O14 phase are not sufficient to justify the presence of 13–15% of V4+ in the samples, so some V4+ can also be present in the framework of MoV2O8 mixed oxide because, despite the larger atomic radius of molybdenum, the lower valence of vanadium (V4+) makes the metal–oxygen bond distances closely similar for both elements in the MoxVyOz structures, leading to its partial reduction.8 Finally, these reduced forms of vanadium could be present in small quantities of amorphous phases, not detectable by XRD but, previously reported in the synthesis of vanadium-molybdenum based mixed oxides.6,8,36 Another important fact noted is that the catalysts synthetized with surfactants showed higher quantities of O–Mo bonds compared to the reference sample, what can be associated with the higher quantities of MoO3 crystalline phase formed in these samples. The quantity of surface O–H bonds decreased in CTAB-0.10 and SDS-0.10 samples, influencing the total acidity of these catalysts (Table 1). This observation are probably related to the decrease of surface defects on crystal surfaces formed in the presence of the surfactants. The decrease in low temperature peaks (100–200 °C) of NH3-TPD profiles (Fig. S3†) observed in the CTAB-0.10 and SDS-0.10 in relation to the reference catalyst, are very coherent with the amount of surface hydroxyl bonds provided by XPS, suggesting that these peaks are related to weakly bonded ammonia adsorbed on hydroxyl groups of surface.
| Catalyst | Speciation of V and Mo (%) | Surface oxygen atoms (%) | ||||
|---|---|---|---|---|---|---|
| V4+ | V5+ | Mo6+ | O–H | O–V | O–Mo | |
| Reference | 15 | 85 | 100 | 10.0 | 32.4 | 57.6 |
| CTAB-0.10 | 13 | 87 | 100 | 5.0 | 30.8 | 64.2 |
| SDS-0.10 | 13 | 87 | 100 | 5.6 | 21.6 | 72.8 |
The results by applying the catalysts in the glycerol oxydehydration reaction are depicted in Fig. 3. The optimal reaction conditions for these materials are determined using the reference sample from our previous study.6 For comparison, the catalytic activities of pure vanadium and molybdenum oxides were determined (Fig. S4†). The MoO3 and V2O5 showed very low selective for acrylic acid formation, 13 and 11%, with conversions of 95 and 98%, and high COX production, 72 and 70%, respectively. The presence of mixed oxide phases considerably enhanced selectivity when compared to the pure oxide samples.6 The reference sample showed a gradual increase in the conversion and selectivity to acrylic acid until the fourth hour of reaction, when the conversion reached 100%. After the fifth hour, the acrylic acid selectivity started to decrease in this sample. The catalysts prepared with surfactants presented a considerable increase in the acid acrylic formation compared to the reference sample, with selectivities of 36–45% for CTAB-0.10, and 45–57% for SDS-0.10 catalyst. The catalysts showed a conversion of 100% with no deactivation after 6 h on stream and no coke was detected by elemental analysis of CNHS (not shown) or by thermogravimetry (Fig. S5†), only a weight loss in 100–200 °C range due to elimination of adsorbed water and an increase above 400 °C corresponding to the capture of molecular oxygen to site reoxidation from V4+ to V5+ were observed. Both CTAB-0.10 and SDS-0.10 showed 100% of glycerol conversion and presented lower COX formation when compared to the reference catalyst. The COX production considerable decreased in the presence of CTAB-0.10 and SDS-0.10 catalysts with selectivities in the range of 49–53% and 36–45%, respectively. It is important to note that there was a significant decrease in the amount of COX compared to the reference sample (60–66%). The catalyst SDS-0.10 had the best activity in terms of higher acrylic acid selectivity and low COX formation, with 57% and 36%, respectively, in the sixth hour of reaction.
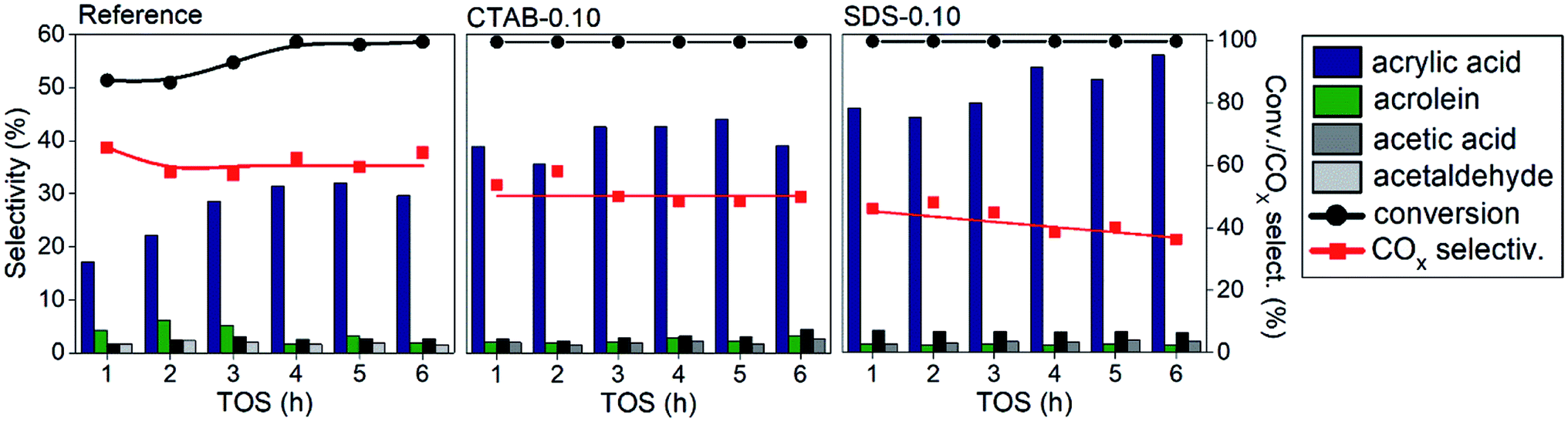 | ||
| Fig. 3 Catalytic results of glycerol oxydehydration performed during 6 h at 320 °C under a flow of 100% of O2. | ||
The crystalline phases present in the catalysts identified before and after reaction (Fig. 4A) were carefully analyzed and quantified by Rietveld refinement (Fig. 1 and S6†). Before the reaction, all catalysts showed the phases Mo4.65V0.35O14, MoV2O8 and MoO3. After reaction, the formation of an additional phase, MoVO5, was observed. The presence of this phase, that contains basically reduced forms of vanadium (V4+), is associated with the V5+/V4+ equilibrium established during reaction by reduction/oxidation dynamics where acrolein is oxidized into acrylic acid, generating oxygen vacancy in the catalyst structure, and subsequent filling of the vacancy with O2 as predicted by Mars-van-Krevelen mechanism.37
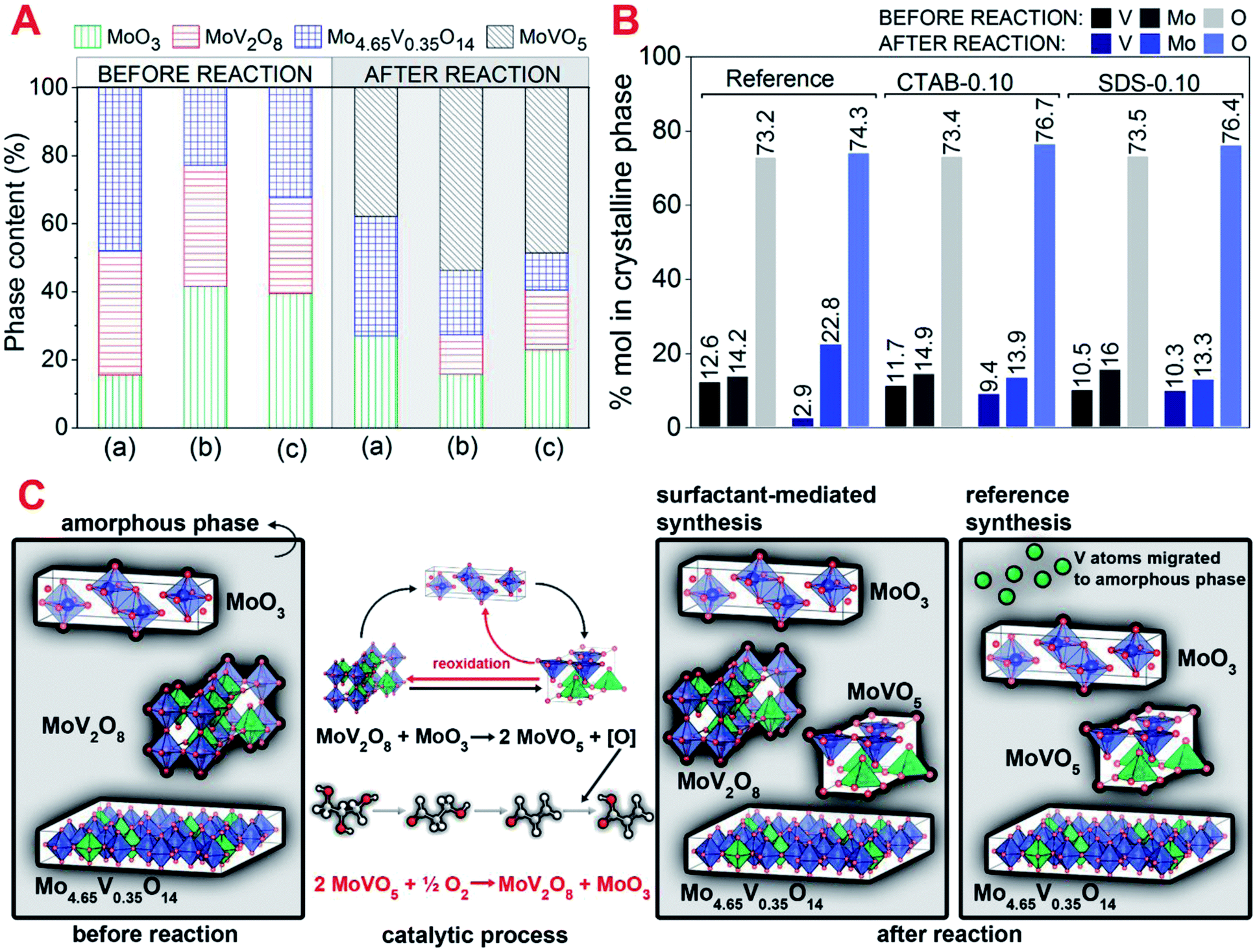 | ||
| Fig. 4 (A) Quantification of the crystalline phases determined by Rietveld refinement of fresh and spent catalysts (a – reference, b – CTAB-0.10 and c – SDS-0.10), (B) % in mole of vanadium, molybdenum and oxygen present in fresh and spent catalysts calculated through Rietveld quantification (only crystalline phases identified in XRD patterns were considered in the calculation) and (C) schematic representation of modifications in the catalyst structure during catalytic reaction. | ||
Significant changes were observed in the phase composition after reaction suggesting the occurrence of solid-state reactions between the single and bicomponent oxide phases. These reactions were already observed for MoVTe-based oxides in high temperatures and oxidant atmospheres.20 Considering the reference sample, MoV2O8 phase disappeared and molybdenum oxide phase, MoO3, considerably increased (from 15 to 30%) in the spent catalyst. Analysing the mole percentage of vanadium, molybdenum and oxygen in the crystalline phases of fresh and spent catalysts (Fig. 4B) for reference sample, a significant decrease in the mole percentage of vanadium (from 12.6 to 2.9%), followed by an increase in the amount of molybdenum (from 14.2 to 22.8%) was observed. These observations strongly suggest that MoV2O8 is very unstable in this sample, and the most part of vanadium originally present in this mixed oxide structure is transferred to amorphous phase as shown in eqn (6). Most part of the low amount of vanadium that remained in the crystalline phase is present in the reduced phase MoVO5.
| MoV2O8 → MoO3 + V2O5(amorphous) | (6) |
CTAB-0.10 and SDS-0.10 catalysts showed decreases of 2.3% (from 11.7 to 9.4%) and 0.2% (from 10.5 to 10.3%) in the mole percentage of vanadium present in crystalline phases, respectively. The lower migration of vanadium to amorphous phase in the catalyst SDS-0.10, compared to CTAB-0.10, are probably associated with the presence of higher MoV2O8 phase in nanorod morphology in this sample, as observed by SEM images. In these catalysts MoV2O8 phase are still present after reaction, and the reduced MoVO5 phase appears as a result of the vanadium redox cycle. The percentage of MoO3 phase is considerably low in CTAB-0.10 and SDS-0.10 catalysts, suggesting a restructuration of phases according to reaction in eqn (7), where vanadium is being transferred from MoV2O8 to MoO3 to form MoVO5 phase and an oxygen remaining from the structure that are reacting with acrolein to produce acrylic acid. Feeding molecular oxygen to the reaction media and considering the restructuration of phases a reversible process, the MoV2O8 phase are being re-established as shown in eqn (8), and this cycle generates the equilibrium between MoV2O8/MoVO5 phases observed in CTAB-0.10 and SDS-0.10 spent catalysts. The decrease in acrylic acid selectivity observed for reference sample, between the fifth and sixth hour of reaction could be associated with the migration of vanadium from MoV2O8 crystalline phase to amorphous phase, but the presence of Mo4.65V0.35O14 phase in the catalysts is contributing to the oxidation of acrolein during reaction, since it was already reported as active in the glycerol oxydehydration,38 and is, probably, maintaining some activity in the reference catalyst. Fig. 4C shows a schematic representation of key structural points observed for fresh and spent catalysts prepared in the presence of CTAB, SDS and in the absence of surfactant, and a proposed mechanism of phase restructuration during reaction.
| MoV2O8 + MoO3 → 2MoVO5 + [O]structure | (7) |
| 2MoVO5 + 1/2O2(g) → MoV2O8 + MoO3 | (8) |
Conclusions
In summary, Mo–V mixed oxides were successfully synthetized in the absence and presence of ionic surfactants by a facile hydrothermal method and applied as catalysts in the glycerol oxydehydration to acrylic acid. Addition of surfactants in the synthesis mixture not significantly changed the nature of phases present in the catalyst but strongly influenced the crystal morphology of MoV2O8 phase, orienting the nucleation across specific plans during synthesis and leading to the formation of nanorod-type crystals. Rod-like morphology stabilized the MoV2O8 crystalline phase, favouring the redox process, through the MoV2O8/MoVO5 equilibrium, during the reaction, avoiding the migration of vanadium atoms to amorphous phase, as observed for the reference sample, resulting in a significant improvement in the performance of the catalysts. The SDS-0.10 sample achieved the best catalytic result with 100% of conversion, a maximum of 57% of selectivity to acrylic acid and a considerable decrease in COX production from 66 to 36%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Brazilian agencies CNPq (grant #304698/2014-8) and FAPESP (grants #2013/10204-2, #2014/20116-6 and #2016/10597-2). Authors also thank Dr Peter Hammer (LEFE/IQ-UNESP) for XPS measurements and data analysis.References
- M. Anitha, S. K. Kamarudin and N. T. Kofli, Chem. Eng. J., 2016, 295, 119 CrossRef CAS.
- B. Katryniok, S. Paul and F. Dumeignil, ACS Catal., 2013, 3, 1819 CrossRef CAS.
- L. H. Vieira, K. T. G. Carvalho, E. A. Urquieta-González, S. H. Pulcinelli, C. V. Santilli and L. Martins, J. Mol. Catal. A: Chem., 2016, 422, 148 CrossRef CAS.
- M. V. Rodrigues, C. Vignatti, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2015, 495, 84 CrossRef CAS.
- L. G. Possato, R. N. Diniz, T. Garetto, S. H. Pulcinelli, C. V Santilli and L. Martins, J. Catal., 2013, 300, 102 CrossRef CAS.
- L. F. Rasteiro, L. H. Vieira, L. G. Possato, S. H. Pulcinelli, C. V. Santilli and L. Martins, Catal. Today, 2017, 296, 10 CrossRef CAS.
- L. G. Possato, W. H. Cassinelli, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2015, 492, 243 CrossRef CAS.
- L. G. Possato, W. H. Cassinelli, C. I. Meyer, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2017, 532, 1 CrossRef CAS.
- L. H. Vieira, L. G. Possato, T. F. Chaves, S. H. Pulcinelli, C. V. Santilli and L. Martins, Mol. Catal., 2017 DOI:10.1016/j.mcat.2017.11.027 , in press.
- P. Okoye, A. Abdullah and B. Hameed, Renewable Sustainable Energy Rev., 2017, 74, 387 CrossRef CAS.
- L. Zheng, X. Li, W. Du, D. Shi, W. Ning, X. Lu and Z. Hou, Appl. Catal., B, 2017, 203, 146 CrossRef CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Green Chem., 2017, 19, 3186 RSC.
- R. K. Grasselli and F. Trifirò, Rendiconti Lincei, 2017, 28, 59 CrossRef.
- L. G. Possato, T. F. Chaves, W. H. Cassinelli, S. H. Pulcinelli, C. V Santilli and L. Martins, Catal. Today, 2016, 289, 20 CrossRef.
- J. Deleplanque, J. L. Dubois, J. F. Devaux and W. Ueda, Catal. Today, 2010, 157, 351 CrossRef CAS.
- C. Doornkamp and V. Ponec, J. Mol. Catal. A: Chem., 2000, 162, 19 CrossRef CAS.
- S. Ishikawa, M. Tashiro, T. Murayama and W. Ueda, Cryst. Growth Des., 2014, 14(9), 4553 CAS.
- S. Ishikawa, X. D. Yi, T. Murayama and W. Ueda, Catal. Today, 2014, 238, 35 CrossRef CAS.
- S. Ishikawa and W. Ueda, Catal. Sci. Technol., 2016, 6, 617 CAS.
- L. Chen, J. Liang, W. Weng, Y. Wang, H. Wan and J. C. Védrine, Catal. Commun., 2004, 5(11), 697 CrossRef CAS.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207 CrossRef CAS.
- J. H. de Boer, A. van den Heuvel and B. G. Linsen, J. Catal., 1964, 3, 268 CrossRef CAS.
- F. Zhao, Y.-K. Du, J.-K. Xu and S.-F. Liu, Colloid J., 2006, 68(6), 784 CrossRef CAS.
- H.-J. Butt, K. Graf and M. Kappl, Physics and Chemistry of Interfaces, Wiley-VHC Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 Search PubMed.
- D. Myers, Surfactant Science and Technology, John Wiley & Sons, Hoboken, United States, 2006 Search PubMed.
- W. Suprun, M. Lutecki, T. Haber and H. Papp, J. Mol. Catal. A: Chem., 2009, 309, 71 CrossRef CAS.
- B. Naik and N. N. Ghosh, Recent Pat. Nanotechnol., 2009, 3, 213 CrossRef CAS PubMed.
- J. M. Goronja, A. M. J. Ležaić, B. M. Dimitrijević and A. M. Malenović, Hem. Ind., 2016, 70, 485 CrossRef.
- R. F. Ali, A. H. Nazemi and B. D. Gates, Cryst. Growth Des., 2017, 17, 4637 CAS.
- C. Ni, P. A. Hassan and E. W. Kaler, Langmuir, 2005, 21(8), 3334 CrossRef CAS PubMed.
- M. S. Bakshi, Cryst. Growth Des., 2016, 16, 1104 CAS.
- M. Shahid, J. Liu, Z. Ali, I. Shakir and M. F. Warsi, J. Power Sources, 2013, 230, 277 CrossRef CAS.
- Z. Yin, Y. Xiao, X. Wang, W. Wang, D. Zhao and M. Cao, Nanoscale, 2016, 8, 508 RSC.
- M. Pisárčik, F. Devínsky and M. Pupák, Open Chem., 2015, 13, 922 Search PubMed.
- A. Chieregato, J. M. López Nieto and F. Cavani, Coord. Chem. Rev., 2015, 301–302, 3 CrossRef CAS.
- P. Mars and D. W. Van Krevelen, Chem. Eng. Sci., 1954, 3, 41 CrossRef CAS.
- L. Liu, B. Wang, Y. Du, Z. Zhong and A. Borgna, Appl. Catal., B, 2015, 174–175, 1 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01443d |
| This journal is © The Royal Society of Chemistry 2018 |