
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of α-hydroxyphosphonates as a convenient route to N-tosyl-α-aminophosphonates†
Tomasz Cytlak *ab,
Monika Skibińskaa,
Patrycja Kaczmareka,
Marcin Kaźmierczakab,
Magdalena Rappa,
Maciej Kubickia and
Henryk Koroniaka
*ab,
Monika Skibińskaa,
Patrycja Kaczmareka,
Marcin Kaźmierczakab,
Magdalena Rappa,
Maciej Kubickia and
Henryk Koroniaka
aFaculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89b, 61-614 Poznań, Poland
bCentre for Advanced Technologies, Adam Mickiewicz University in Poznań, Umultowska 89c, 61-614 Poznań, Poland. E-mail: cytlak@amu.edu.pl
First published on 27th March 2018
Abstract
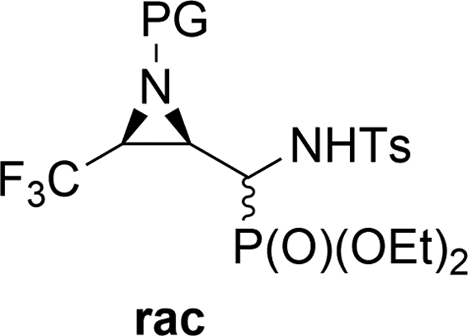
Direct conversion of the α-hydroxyl group by para-toluenesulfonamide to yield α-(N-tosyl)aminophosphonates is reported. α-Aminophosphonates 23a,b–37a,b were obtained from the corresponding α-hydroxyphosphonates 6a,b–21a,b in the presence of K2CO3, via the retro-Abramov reaction of the appropriate aldehydes, 1–5. The subsequent formation of imines with simultaneous addition of diethyl phosphite provided access to the α-sulfonamide phosphonates 23a,b–37a,b with better diastereoselectivity than in the case of the Pudovik reaction. The mechanism for this transformation is proposed herein. When Cbz N-protected aziridine 9a,b and phenylalanine analogue 12a,b were exploited, intramolecular substitution was observed, leading to the corresponding epoxide 38 as the sole product, or oxazolidin-2-one 39 as a minor product. Analogous substitution was not observed in the case of proline 18a,b and serine 21a,b derivatives.
Introduction
α-Sulfonamide phosphonates constitute a very interesting class of compounds. They can be potential candidates for fluorescent-β-lactamase1 and matrix metalloproteinase (MMPs) inhibitors2 such as compounds containing carboxylate and hydroxamate moieties, which are well known MMP inhibitors. Moreover, these compounds are promising substrates for the synthesis of N-deprotected α-aminophosphonates, which are important isosteres of α-amino acids, possessing a wide range of biological activities. They act as antibiotics, herbicides, antifungal agents, enzyme inhibitors, and pharmacological agents.3There are several routes for the synthesis of α-aminophosphonates. One of the most important protocols is the Kabachnik–Fields (or phospha-Mannich) reaction involving the condensation of dialkyl phosphite, carbonyl compounds and primary or secondary amines;4 however, the reaction mechanism is still under investigation.5 There are several other possibilities for the preparation of α-aminophosphonates, among which a very convenient route is the addition of phosphite nucleophiles to imines or enamines.6 The major disadvantage of this approach is the stability of the imine/enamine prepared from the aliphatic amine. Transformation of the α-hydroxyphosphonates seems to provide an encouraging method for achieving α-aminophosphonates. These compounds are easily obtained using the Abramov reaction7 and its modifications,8 giving access to the synthesis of different types of α-functionalized phosphonates.9
α-Hydroxyphosphonates exhibit interesting medicinal properties as potential antibacterial, antiviral and anticancer agents,10 as well as enzyme inhibitors such as protease, EPSP synthase, human rennin, human calpain I and tyrosine-specific protein kinase.11 Among the methods of synthesis of α-aminophosphonates utilizing α-hydroxyphosphonates, Mitsunobu azidation followed by the Staudinger reduction is commonly applied with good yields and is well-known in the literature.12 Unfortunately, using the volatile and highly toxic hydrazoic acid is the main disadvantage of this method. Another type of conversion of α-hydroxyphosphonates into α-aminophosphonates is nucleophilic substitution at C1, but this method is rather difficult due to the hindered hydroxy function,13 although substitution of the hydroxyl group in primary α-hydroxyphosphonates by a good leaving group, e.g. triflate,14 mesylate,15 tosylates,16 or via acid-mediated displacement,17 has been described in the literature. To our knowledge, only one paper reported the substitution of the hydroxyl group by an amine at the secondary centre, under microwave-assisted and solvent-free conditions.18
In the literature there exist methods of conversion of primary α-hydroxyphosphonates to α-amino analogues, such as intramolecular cyclodehydration via the alkoxyphosphonium salt19 or phosphonation via the retro-Abramov reaction,13d which is an effort to explain the Kabachnik–Fields reaction mechanism (Scheme 1).
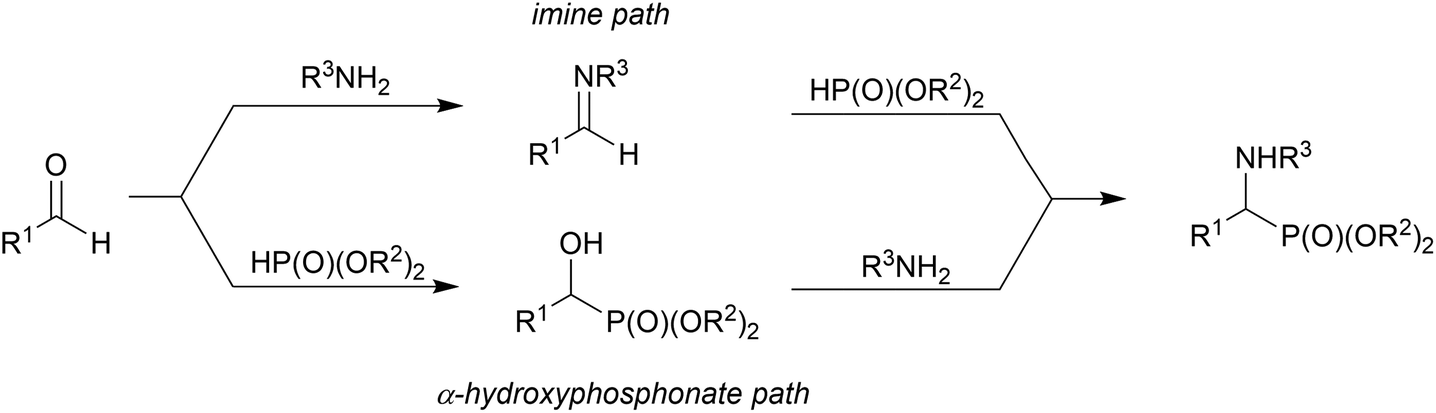 | ||
| Scheme 1 Kabachnik–Fields reaction. | ||
In this mechanism, one possibility is that an imine is formed from the carbonyl compound and primary amine (or iminium salt when a secondary amine is applied). Then, the addition reaction of dialkyl phosphite to the imine leads to α-aminophosphonate.4b The second possibility is based on the amine promoted (especially by the highly basic amines) addition of dialkyl phosphite to the carbonyl group, leading to α-hydroxyphosphonate, in a reversible step, and then nucleophilic substitution of the hydroxyl group by an amine moiety.20 The first approach is based on the reversibility of the addition of dialkyl phosphite to the carbonyl group (retro-Abramov reaction) with subsequent irreversible imine formation followed by immediate dialkyl phosphite addition.21 The second approach argues that α-aminophosphonates are obtained at high temperatures. During heating, the disappearance of the α-hydroxyphosphonate in favour of the α-aminophosphonate formation is postulated.22 On the basis of kinetic studies, it is suggested that the mechanism depends on the nature of the reacting substrates.5a The reaction of benzaldehyde, aniline and dialkyl phosphite supports the imine pathway. The formation of the hydrogen bond between the phosphoryl group of the dialkyl phosphite and amine promotes the formation of imine without additional catalyst.23 On the other hand, aniline is too weak a base to promote the addition of dialkyl phosphite to the carbonyl group. Cherkasov et al. performed the reaction of the more nucleophilic cyclohexylamine with benzaldehyde and dialkyl phosphite and they suggested the hydroxyphosphonate path where the amine was basic enough to interact with the hydrogen of the phosphite to promote the attack of phosphite on the carbonyl carbon.5a,24 Subsequent papers provided more evidence supporting the imine pathway, even when hard nucleophilic amines were applied to the reaction.24,25
Results and discussion
In the course of our studies, we were able to synthesize a wide range of α-hydroxyphosphonates that were subsequently used in reactions with para-toluenesulfonamide towards obtaining α-aminophosphonate derivatives.As the convenient starting materials, the N-protected amino aldehydes such as aziridine 1, as well as aldehydes 2–5 originating from amino acids, possessing various amino protecting groups, e.g. benzyl (Bn) 1a–5a, tert-butoxycarbonyl (Boc) 2b–5b, carboxybenzyl (Cbz) 2c–5c were chosen. All aldehydes were prepared from the corresponding alcohols according to the literature data (see Experimental section). The introduction of a new C–P bond, yielding α-hydroxyphosphonates, was conducted by the three main methodologies. First, aldehydes 1a, 2, 3 were used in the reactions with lithium diethyl phosphite in dry THF at −30 °C. This strategy afforded phosphonates 6a,b, 10a,b–12a,b and 13a,b–15a,b. The yields varied from moderate in the case of (S)-phenylalanine 10a,b–12a,b and (S)-valine analogues 13a,b–15a,b, to very good for aziridines 6a,b, while the diastereoselectivity of this reaction varied from poor in the case of 6a,b, to very good for 10a,b. Moreover, phosphonates 6a,b were transformed to N-unprotected α-hydroxyphosphonates 7a,b with the subsequent introduction of the protecting groups, Boc 8a,b and Cbz 9a,b. In the case of transformations of 7a,b yielding 9a,b, due to steric hindrance between the Cbz group and the phosphonate moiety in 9a, we observed a small predominance of diastereoisomer 9b (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, d.r. 19F, 31P NMR). Thus, the stereochemistry of aziridine 9a was analogous to the major diastereoisomer of 6a and parallel to a study reported previously (for comparison see Fig. 1 and N-benzyl protected aziridines 6a,b).26 In the case of Boc protected aziridine, the reaction gave only one diastereoisomer 8b as the sole product, where 7a was left unreacted in the reaction mixture. Moreover, 8b existed as a mixture of two rotamers (1.9:1 NMR ratio) that could be separated by chromatography techniques. In the second route, the TEA-catalyzed addition of HP(O)(OEt)2 to appropriate aldehydes at room temperature with 0.1 eq. TEA or 0.2 eq. TEA at r.t. or 50 °C led to products 6a,b, 17a,b–21a,b, in yields ranging from moderate for 19a,b to excellent in the case of 20a,b; there was also good diastereoselectivity for 18a,b and excellent diastereoselectivity in the case of aziridines 6a,b and serine analogues 20a,b. Only in case of the synthesis of 16a,b was the application of i-Pr2EtN needed (Table 1).
:1.2, d.r. 19F, 31P NMR). Thus, the stereochemistry of aziridine 9a was analogous to the major diastereoisomer of 6a and parallel to a study reported previously (for comparison see Fig. 1 and N-benzyl protected aziridines 6a,b).26 In the case of Boc protected aziridine, the reaction gave only one diastereoisomer 8b as the sole product, where 7a was left unreacted in the reaction mixture. Moreover, 8b existed as a mixture of two rotamers (1.9:1 NMR ratio) that could be separated by chromatography techniques. In the second route, the TEA-catalyzed addition of HP(O)(OEt)2 to appropriate aldehydes at room temperature with 0.1 eq. TEA or 0.2 eq. TEA at r.t. or 50 °C led to products 6a,b, 17a,b–21a,b, in yields ranging from moderate for 19a,b to excellent in the case of 20a,b; there was also good diastereoselectivity for 18a,b and excellent diastereoselectivity in the case of aziridines 6a,b and serine analogues 20a,b. Only in case of the synthesis of 16a,b was the application of i-Pr2EtN needed (Table 1).
 | ||
| Fig. 1 A perspective view of 9a, showing the numbering scheme. Ellipsoids were drawn at the 30% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
|
||||||
|---|---|---|---|---|---|---|
| Aldehyde | Product | PG | Cond. | Yieldb [%] | d.r.c | |
| a (i) LiP(O)(OEt)2, −30 °C → rt, 16–18 h; (ii) HP(O)(OEt)2, 0.1 or 0.2 eq. TEA, neat, r.t. or 50 °C, 1 d or 7 d; (iii) 6a,b, H2/Pd/C, EtOH; 0 °C → rt; (iv) 7a,b, Boc2O, DMAP, MeCN, r.t., 1 d; (v) 7a,b, CbzCl, NaHCO3, CH2Cl2, 0 °C → rt, 1 d; (vi) HP(O)(OEt)2, 1 eq. i-Pr2EtN, CH2Cl2.b Isolated yield.c Crude reaction mixture (19F NMR and/or 31P NMR).d Configuration (2S).e Configuration (2R). | ||||||
| 1a |  |
6a,b | Bn | i | 84 | 1:1 |
| 1a | 6a,b | Bn | ii | 77 | 20:1 |
|
| 1a | 7a,b | H | iii | 97 | 1:1 |
|
| 1a | 8a,b | Boc | iv | 43 | 1:99 |
|
| 1a | 9a,b | Cbz | v | 74 | 1:1.2 |
|
| 2a |  |
10a,b | Bn2 | i | 52 | 11:1 |
| 2b | 11a,b | Boc | i | 61 | 2.1:1 |
|
| 2c | 12a,b | Cbz | i | 52 | 2.3:1 |
|
| 3a | 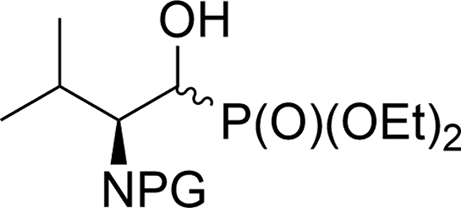 |
13a,b | Bn2 | i | 44 | 1.9:1 |
| 3b | 14a,b | Boc | i | 55 | 3.4:1 |
|
| 3c | 15a,b | Cbz | i | 41 | 2.3:1 |
|
| 4ad | 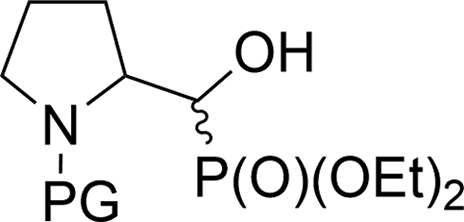 |
16a,bd | Bn | vi | 61 | 1.3:1 |
| 4bd | 17a,bd | Boc | ii | 77 | 3.7:1 |
|
| 4ce | 18a,be | Cbz | ii | 76 | 2.9:1 |
|
| 5a | 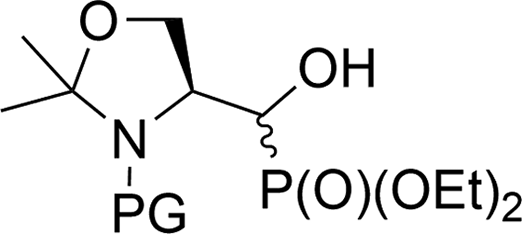 |
19a,b | Bn | ii | 40 | 3.1:1 |
| 5b | 20a,b | Boc | ii | 94 | 95:5 |
|
| 5c | 21a,b | Cbz | ii | 67 | 13:1 |
|
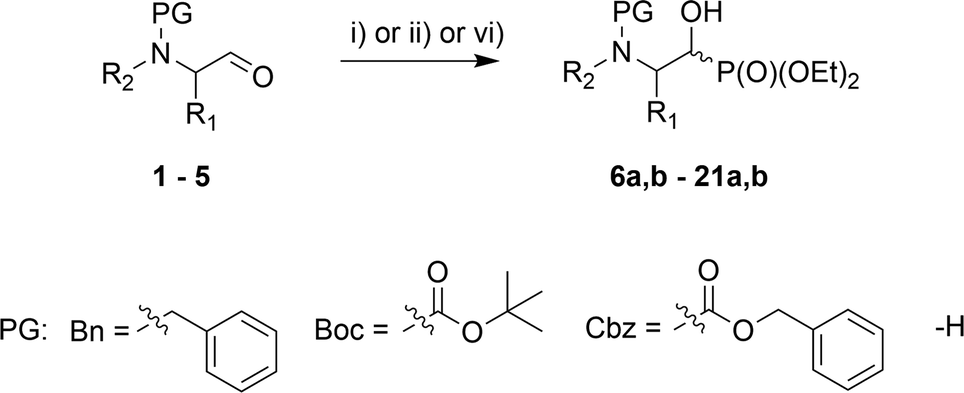
The stereochemistry of the addition of dialkyl phosphite to N-protected (S)-amino aldehydes [or (R)- in the case of 4c] was a consequence of the steric hindrance on the adjacent stereogenic centre. Moreover, the Pudovik reaction conducted on N-protected aldehydes 2, 3 derived from phenylalanine and valine led to anti addition, giving rise to major diastereoisomers (1R,2S) according to the data reported for nucleophilic additions to (N,N-dibenzylamino)aldehydes.27 These assumptions were confirmed by the absolute stereochemistry of compound 10a determined by X-ray diffraction analysis (Fig. 2).
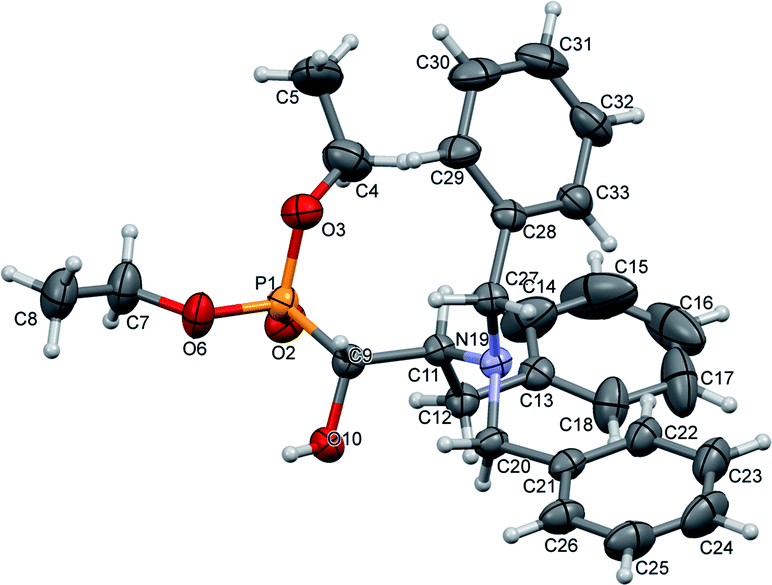 | ||
| Fig. 2 A perspective view of 10a, showing the numbering scheme. Ellipsoids were drawn at the 30% probability level; hydrogen atoms are represented by spheres of arbitrary radii. | ||
At the same time, the addition performed on (2S)-prolinal and (4S)-serial derivatives yielded (1R,2S)16a–17a, and (R,4S)19a–21a as major diastereoisomers, confirmed by NOESY experiments or X-ray diffraction analysis. The diastereoselectivity of the Pudovik reaction in the case of 20a,b–21a,b was analogous to that obtained by Wróblewski et al.28
Considering only a few examples of ring opening reactions of trifluoromethylated N-unactivated aziridines with nitrogen nucleophiles in the literature,29 we tried to open the ring of aziridin-2-ylphosphonates 6a,b with BnNH2 under acidic conditions, in the presence of Sc(OTf)3, Yb(OTf)3, Bi(OTf)3, PBu3, B(C6F5)3, BiCl3, TiCl4 in different solvents, but all attempts failed. Only unreacted 6a,b were observed in the reaction mixtures. Then, we decided to carry out the reaction under basic conditions in the presence of K2CO3 and acetonitrile as a solvent. As a result, the phosphate 22 (δ = −5.01 in 31P NMR) was formed (Scheme 2).
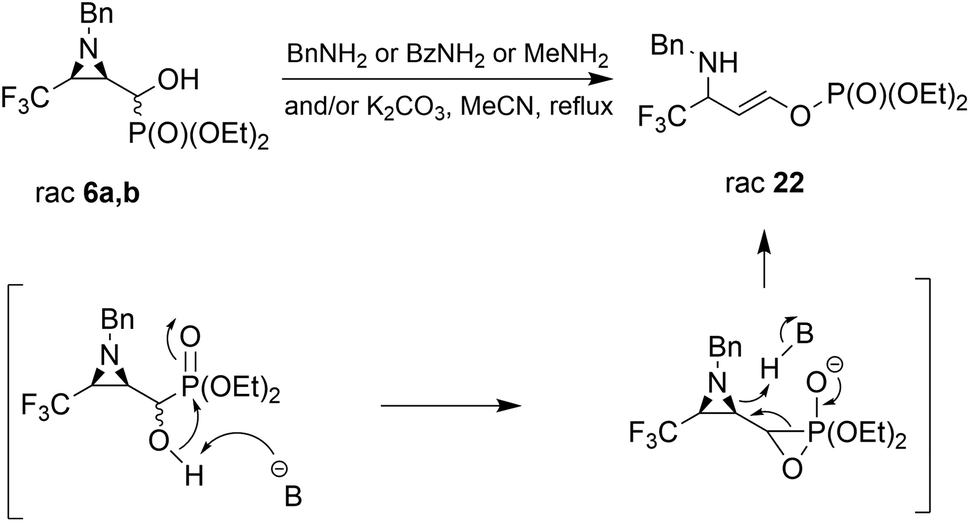 | ||
| Scheme 2 Reactions of 6a,b with different nitrogen nucleophiles under K2CO3 conditions. | ||
During the experiment, we observed the formation of diethyl phosphite (δ = 7.32 in 31P NMR) and an aldehyde 1a (δ = 9.30 in 1H NMR), which vanished at the end of the reaction. Apparently, besides the rearrangement of the α-hydroxyphosphonate, due to proton extraction from the hydroxyl group by base the aldehyde was formed with concomitant phosphonate elimination, supporting the retro-Abramov reaction mechanism proposed by Gancarz.13d Furthermore, the presence of the electron-withdrawing CF3 moiety in the aziridine ring allowed the α-hydroxyphosphonate intramolecular rearrangement with subsequent aziridine ring opening to phosphate 22. The phosphonate/phosphate conversion was already studied in the case of the fluorene molecule when stronger amines were used by Gancarz et al.30
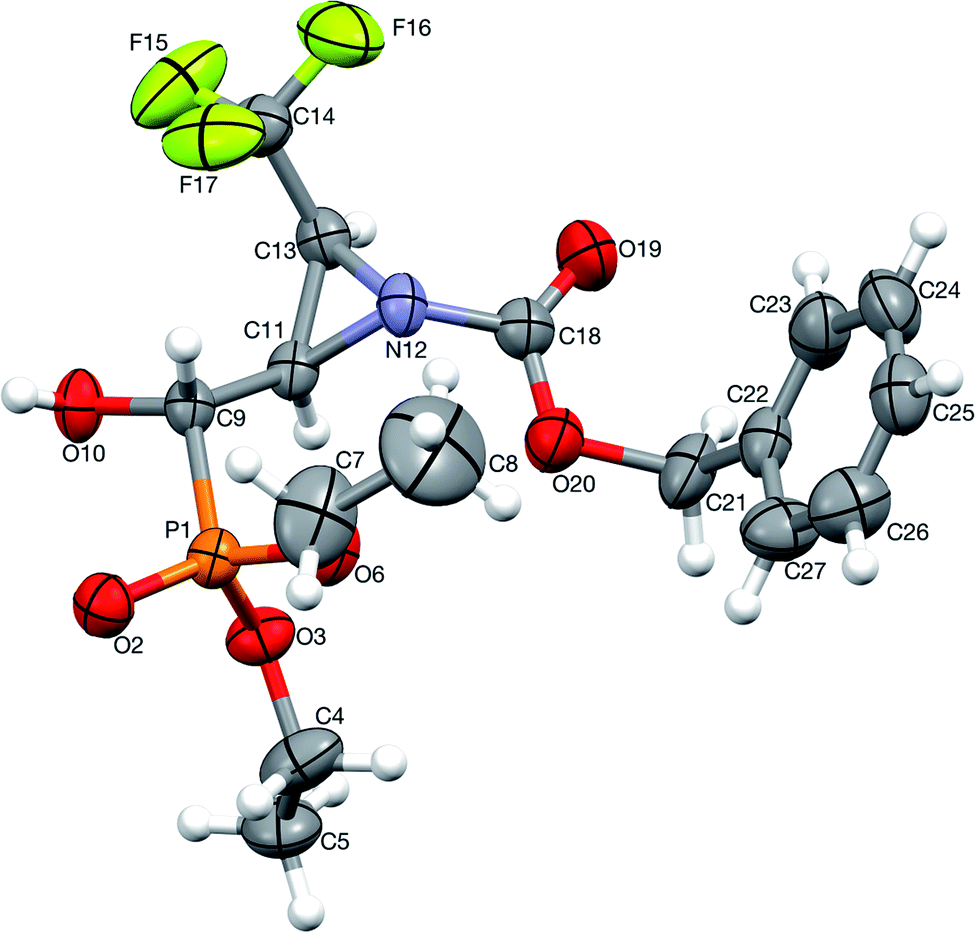
Moreover, the application of MeNH2 or BzNH2 (instead of BnNH2), under the same conditions, as well as using K2CO3 without nitrogen nucleophiles, gave phosphate 22 (Scheme 2). Surprisingly, when para-toluenesulfonamide (TsNH2) was used along with K2CO3, aminophosphonates 23a,b were obtained as the sole products. The structure and stereochemistry of compound 23a was determined by X-ray diffraction analysis and indicated the rac(1S,2R,3S)-23a configuration, analogous to 6a,26 obtained by Pudovik addition (Fig. 3).
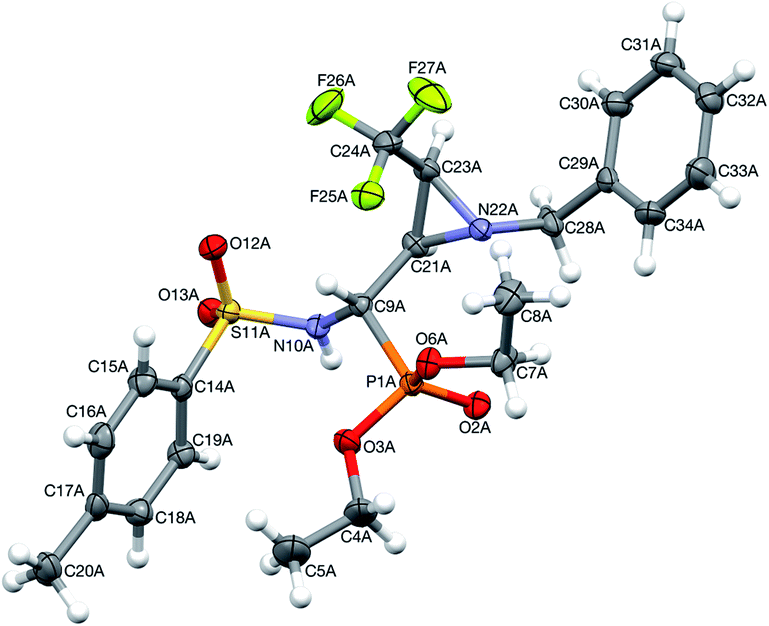 | ||
| Fig. 3 A perspective view of 23a, showing the numbering scheme. Ellipsoids were drawn at the 50% probability level and hydrogen atoms are represented by spheres of arbitrary radii. | ||
The obtained results were contrary to known methods leading to N-tosylamide derivatives. Usually, the hydroxyl group reacts with sulfonamides under acidic conditions,17 or under basic conditions the substitution of leaving groups such as O-mesyl is applied.15a On the other hand, a similar transformation of hydroxyphosphonates to aminophosphonates with amines in a basic environment was reported by Gancarz.20 Likewise, the application of Lewis bases such as CaCl2 with aniline was announced by Kaboudin et al.31 The results concerning the applied reaction conditions in the case of compounds 6a,b with TsNH2 are presented below (Table 2).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | d.r. | Base (eq.) | Solvent | Temp. [°C] | Product | Yielda [%] | d.r.b |
| a 19F NMR and/or 31P NMR yield.b Crude reaction mixture (19F NMR and/or 31P NMR). | |||||||
| 1 | 20:1 |
K2CO3 (1.2 eq.) | MeCN | Reflux | 23a,b | 74 | 6:1 |
| 2 | 1:1 |
K2CO3 (1.2 eq.) | MeCN | Reflux | 23a,b | 87 | 6:1 |
| 3 | 1:1 |
K2CO3 (5 eq.) | MeCN | Reflux | 23a,b | 73 | 9:1 |
| 4 | 20:1 |
K2CO3 (12 eq.) | MeCN | Reflux | 23a,b | 80 | 9:1 |
| 5 | 1:1 |
K2CO3 (12 eq.) | MeCN | Reflux | 23a,b | 96 | 9:1 |
| 6 | 1:1 |
K2CO3 (1.2 eq.) | DMF | 100 | Decomp. | — | — |
| 7 | 1:1 |
K2CO3 (1.2 eq.) | THF | Reflux | n.r. | — | — |
| 8 | 1:1 |
K2CO3 (1.2 eq.) | EtOH | Reflux | 23a,b | 60 | 7:1 |
| 9 | 1:1 |
None | MeCN | Reflux | n.r. | — | — |
| 10 | 1:1 |
TEA (1.2 eq.) | MeCN | Reflux | n.r. | — | — |
| 11 | 1:1 |
TEA (1.2 eq.) | EtOH | Reflux | n.r. | — | — |
| 12 | 1:1 |
NaHCO3 (1.2 eq.) | MeCN | Reflux | 1a | 20 | — |
| 13 | 1:1 |
NaHCO3 (12 eq.) | MeCN | Reflux | 1a | 20 | — |
| 14 | 1:1 |
NaH (1.2 eq.) | THF | Reflux | 23a,b | 18 | 8:1 |
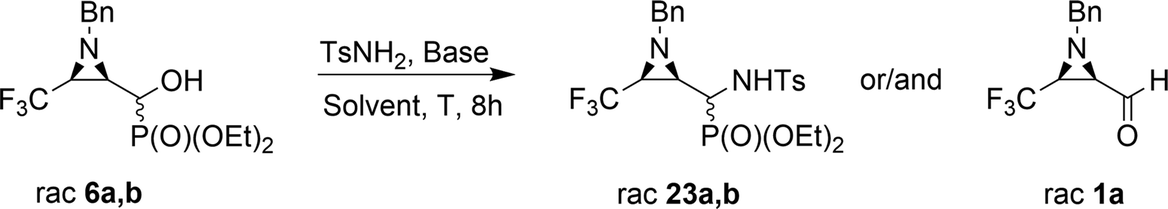
The presented experiments indicate that the best conditions leading to 23a,b involved 1.2 eq. (5 eq.) of K2CO3 and MeCN as a solvent, while the reaction mixture was refluxed for 8 h. When we monitored this reaction at lower temperatures (40 °C → 60 °C), only signals of substrates were detected (19F, 31P NMR). Besides, increasing the amount of K2CO3 from 1.2 eq. to 12 eq. led to slightly better yields and higher diastereoselectivity (Table 2, entry 2, 5). The application of THF as a solvent gave no reaction, while reaction in DMF led to the decomposition of the starting material to a number of undefined products. Moreover, reaction in EtOH, contrary to results reported by Gancarz,13d decreased the reaction yield (60% 19F, 31P NMR). On the other hand, the reaction without base failed. Surprisingly, the employment of other bases led to distinct results. In the presence of TEA, there were no reactions in MeCN, neither in EtOH. When NaHCO3 was used, mainly unreacted substrates were observed, together with diethyl phosphite and aldehyde 1a (NMR) but with lower yield, likewise in the case of reactions with BnNH2, MeNH2 and BzNH2. The last examined base, NaH (in anhydrous THF), allowed α-aminophosphonates 23a,b, but with very poor yields (Table 2, entry 14). Additionally, we decided to examine the reaction of 1a with TsNH2 in detail using 19F NMR monitoring. Similar to studies reported by Keglevich et al.,5b the appropriate N-tosylaldimine 1a′ as a transient species was detected during the reaction in a crude mixture but in very low concentration (δ = −78.16 ppm in 19F NMR, which corresponded to the chemical shifts of β-CF3–imines in the literature ref. 32). Subsequent addition of diethyl phosphite and K2CO3 (1.2 eq.) to the reaction led to 23a,b but in poor yields (30% in 31P NMR, with accompanying dominance of the diethyl phosphite signal). Finally, we decided to monitor the reactions of 6a,b with TsNH2 and K2CO3 (1.2 eq.). In the employed basic conditions, the elimination of the hydroxyl group gave rise to aldimine 1a′, which most probably occurred by the E1cB mechanism involving the participation of the sulfonimidate anion; we observed the same signal of imine 1a′ in the 19F NMR during the reaction. This signal was completely suppressed at the end of the reaction. Subsequent nucleophilic addition of dialkyl phosphite to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of imine 1a′ (Pudovik reaction) gave 23a,b (Scheme 3). These observations support the imine path of the Kabachnik–Fields reaction (KFR), proving that the imine is the most rational intermediate in the synthesis of α-aminophosphonates.
N bond of imine 1a′ (Pudovik reaction) gave 23a,b (Scheme 3). These observations support the imine path of the Kabachnik–Fields reaction (KFR), proving that the imine is the most rational intermediate in the synthesis of α-aminophosphonates.
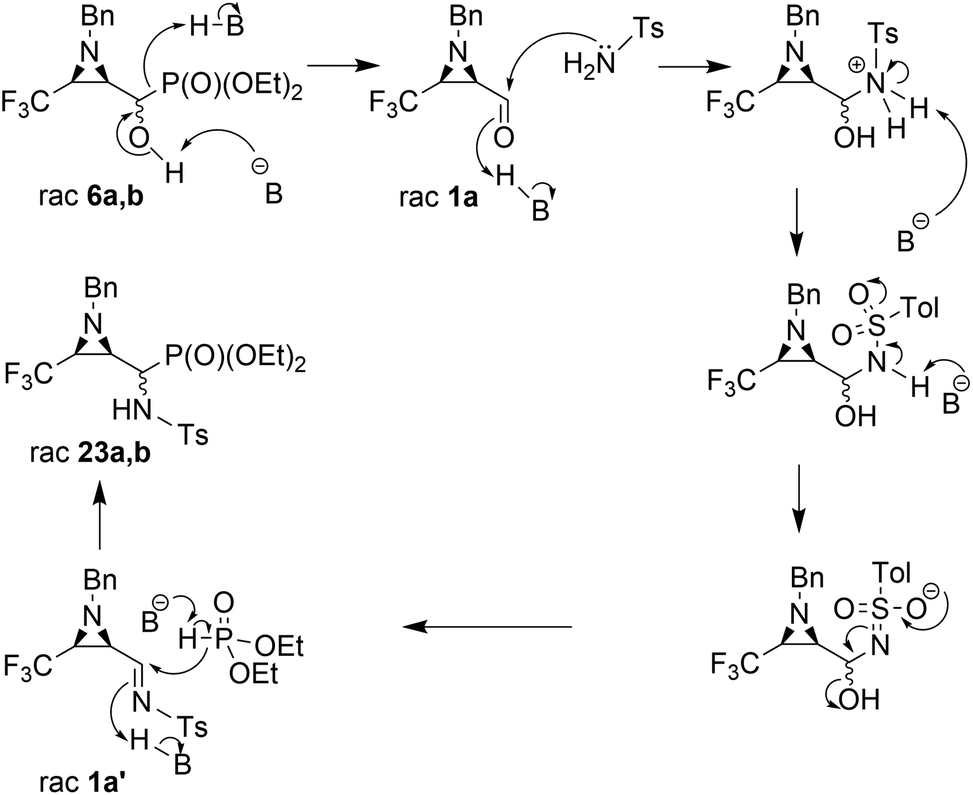 | ||
| Scheme 3 Proposed reaction mechanism of 6a,b with TsNH2 under K2CO3 conditions. | ||
The optimized results prompted us to examine the scope of this particular transformation in the reactions of aziridines 6a,b–9a,b as well as the amino acid origin of α-hydroxyphosphonates 10a,b–12a,b to N-tosylamide phosphonates 23a,b–37a,b. Initially, we used 12 eq. of K2CO3 as a base, but decreasing yields in some cases led us to choose 1.2 eq. as a standard base concentration (Table 3).
|
||||||
|---|---|---|---|---|---|---|
| Substrate | d.r. | Product | PG | Yielda [%] | d.r.b | |
| a Isolated yield.b Crude reaction mixture (19F NMR and/or 31P NMR).c After additional 10 hours of heating.d Configuration (2S).e Configuration (2R). | ||||||
| 6a,b | 1:1 |
 |
23a,b | Bn | 87 | 6:1 |
| 7a,b | 1:1 |
24a,b | H | 80 | 6:1 |
|
| 8a,b | 1:99 |
25a,b | Boc | — | — | |
| 9a,b | 3:1 |
 |
38 | Cbz | 41(74)c | — |
| 10a,b | 19:1 |
 |
26a,b | Bn2 | 81 | 99:1 |
| 11a,b | 1.9:1 |
27a,b | Boc | 78 | 2.5:1 |
|
| 12a,b | 2.7:1 |
 |
28a,b | Cbz | 40 | 2.4:1 |
| 39 | 11 | 99:1 |
||||
| 13a,b | 9:1 |
 |
29a,b | Bn2 | — | — |
| 14a,b | 5.4:1 |
30a,b | Boc | 51 | 3.6:1 |
|
| 15a,b | 1.7:1 |
31a,b | Cbz | 42 | 5.2:1 |
|
| 16a,bd | 1.3:1 |
 |
32a,b | Bn | — | — |
| 17a,bd | 4:1 |
33a,bd | Boc | 59 | 4.4:1 |
|
| 18a,be | 3.3:1 |
34a,be | Cbz | 75 | 3.7:1 |
|
| 19a,b | 2.6:1 |
 |
35a,b | Bn | — | — |
| 20a,b | 99:1 |
36a,b | Boc | 72 | 12:1 |
|
| 21a,b | 13:1 |
37a,b | Cbz | 75 | 12:1 |
|
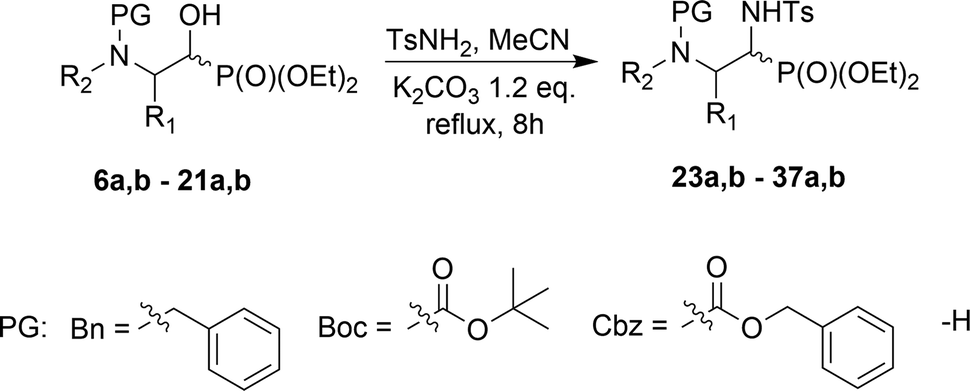
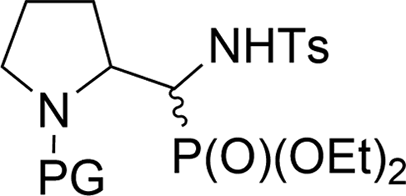
It is noteworthy that the stereoselectivity as well as the stereochemistry of the major diastereoisomers of 23a,b–37a,b were always analogous to the ratio and configurations of those obtained in the Pudovik reaction, the major isomers of 6a,b–21a,b; slightly different results were reported by Dimukhametov.33 Thus, higher stereoselectivity was obtained in the case of the application of chiral imines in the Pudovik reaction, compared to the use of chiral amines in the three-component Kabachnik–Fields reaction. The employment of two diastereoisomers of 6a,b (1:1, NMR ratio) under standard conditions led to the corresponding 23a,b with very good yield. On the contrary, in the case of 12a,b and 15a,b, the reactions proceeded with moderate yields. The stereochemistry of 24a,b, 26a,b–31a,b/33a,b, 37a,b was confirmed by NMR as well as NOESY experimental analysis. Moreover, the steric hindrance in 8b, between the N-Boc substituent and the phosphonate moiety (confirmed by interactions between protons on 1D ROESY experiments) caused no access to the hydroxyl group by the base (K2CO3) and subsequently, only the substrate 8b was observed in the reaction mixture. On the other hand, the very good diastereoselectivity of the addition, yielding 36a,b and 37a,b, can be explained by the Felkin–Ahn model as well as additional interactions substantiated the addition of the dialkyl phosphite on the CN unit of N-tosylimine 5a′ derived from the appropriate aldehyde 5a. According to this, a H-bond was formed between the P(O)H moiety of the phosphite and the nitrogen atom of the pyrrolidine, arranging the five-membered transition state (Scheme 4). It seems probable that the actual mechanism is dependent on the components of the reaction since the reaction of carbohydrate derived α-hydroxyphosphonates (e.g. two epimeric carbohydrates, 5C-phosphonate with L-ido- or D-gluco-configurations)34 with TsNH2 failed, presumably due to a lack of nitrogen heteroatoms in the analogous neighborhood of the reaction center.
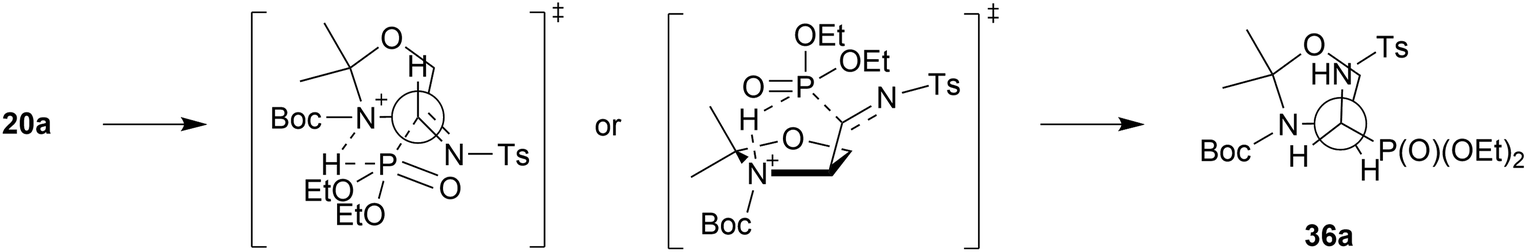 | ||
| Scheme 4 The Felkin–Ahn model of the addition of diethyl phosphite to α-hydroxyphosphonate 20a. | ||
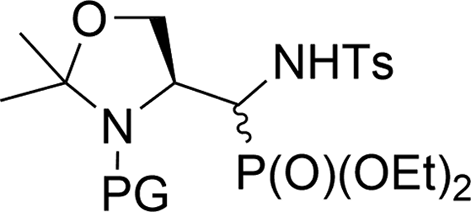
Additionally, the X-ray crystal structure determinations in the case of 34a and 36a (Fig. 4) were performed. Interestingly, both compounds existed as a racemic mixture rac(1S,2R)-34a and rac(R,4S)-36a in the studied crystals. Apparently, during the reaction of 20a,b (K2CO3, TsNH2) yielding 36a,b, besides the N-tosylimine formation via aldehyde 5b, the competitive enolization took place, followed by proton addition from both sides of the enol double bond leading to racemization at C4. The phenomenon of partial racemization during aldehyde formation was already reported in the case of serine derivatives.35
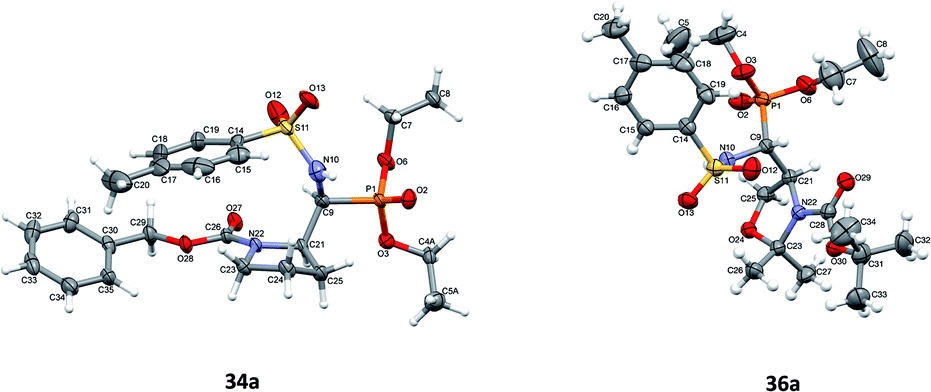 | ||
| Fig. 4 A perspective view of 34a and 36a showing the numbering scheme. Ellipsoids were drawn at the 50% probability level and hydrogen atoms are represented by spheres of arbitrary radii. Only one of the alternative conformations of the C4–C5 ethyl group is shown. | ||
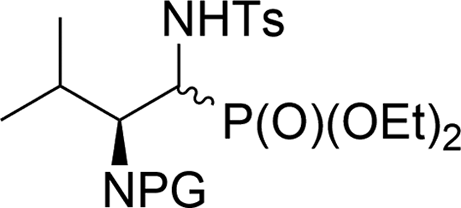
In the case of the reactions of 13a,b, 16a,b and 19a,b with TsNH2 in the presence of K2CO3, the retro-Abramov reactions took place. Thus, in the reaction mixture only diethyl phosphite and the appropriate aldehydes 3a–5a were detected (monitored by NMR). Additionally, in the case of reactions of 10a,b and 20a,b [(R,S)/(S,S) 19:1 and 99:1 ratios, respectively] with K2CO3 (1.2 eq.) as well as with K2CO3 and amines (MeNH2, BnNH2) or benzamide (BzNH2), partial racemization at carbon α regarding phosphonate moieties occurred, leading to 10a,b and 20a,b [(R,S)/(S,S) in 1.5:1 ratio and 3.6:1 ratio, respectively]. These results confirmed the formation of aldehydes due to base treatment (K2CO3) during the analyzed reactions. Similar observations were reported by Wróblewski et al.36 Thus, treatment of the single diastereomeric 1,2-oxaphospholane derivative with sodium methoxide led to the retro-Abramov reaction followed by phosphite addition and cyclization, yielding the corresponding mixture of diastereoisomers. On the other hand, these observations were in contradiction to the results of the analogous reactions of 6a,b, which led almost exclusively to phosphate 22. Gancarz et al. explained the distinction between the retro-Abramov reaction yielding aldehyde and the intramolecular rearrangement towards phosphate, based on kinetic and NMR studies.30 Their observations were based on the reactions between various α-hydroxyphosphonates and amines, assuming that the differentiation of these two routes was dependent on the electronic effect of the substituents. They concluded that the retro-Abramov reaction is preferred when the electron-donating substituents appear in the α-hydroxyphosphonates. In our case, the presence of the strongly electron-withdrawing CF3 group in 6a,b facilitated the intramolecular rearrangement over the retro-Abramov reaction. In this particular reaction, the in situ formed alkoxide ion substituted the phosphorus atom, leading to the formation of the three-membered cyclic intermediate. Subsequent electron pair transfer led to phosphate while stabilization of the partial negative charge on the α-carbon atom followed by aziridine ring opening finally gave the vinyl phosphate 22.37
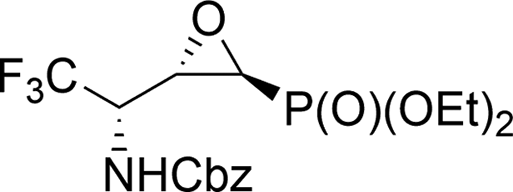
On the other hand, the reactions of 9a,b (1:1.2, d.r.) with TsNH2 (1.2 eq. K2CO3, MeCN, reflux) after 8 h of heating, gave only one diastereoisomer of epoxide 38 (41%), while the remaining diastereoisomer 9b was present in the reaction mixture. In the 31P NMR spectrum, the signal of epoxide 38 was shifted distinctly upfield compared to the aziridinyl substrates 9a,b and α-sulfonamide derivatives 23a,b–24a,b (δP = 15.89 vs. approx. 20 ppm). Furthermore, additional heating of the remaining reaction mixture for 10 h under the same reaction conditions led to 38 (74% after isolation), due to the total consumption of starting materials 9a,b. The reaction was monitored by 19F and 31P NMR. Apparently, treatment of α-hydroxyphosphonate 9a with base led to aziridine ring opening by the attack of a previously formed alkoxide anion on an adjacent nitrogen atom (anti to alkoxide ion) of the Cbz moiety. Furthermore, due to proton abstraction followed by aldehyde 1c (PG = Cbz) formation, the racemization, such as in case of 5b, via enol took place. Subsequent phosphite addition to the CO bond of aldehyde 1c led to 9a,b as an equilibrating mixture of diastereoisomers, where only one diastereoisomer 9a reacted with base to give 38 (Scheme 5). The structure and stereochemistry of 38 as rac(1S,2S,3S) were confirmed by NMR analysis and X-ray crystal structure determination (Fig. 5).
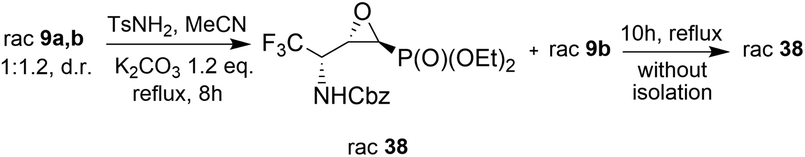 | ||
| Scheme 5 Reaction of N-Cbz protected aziridines 9a,b under K2CO3 conditions. | ||
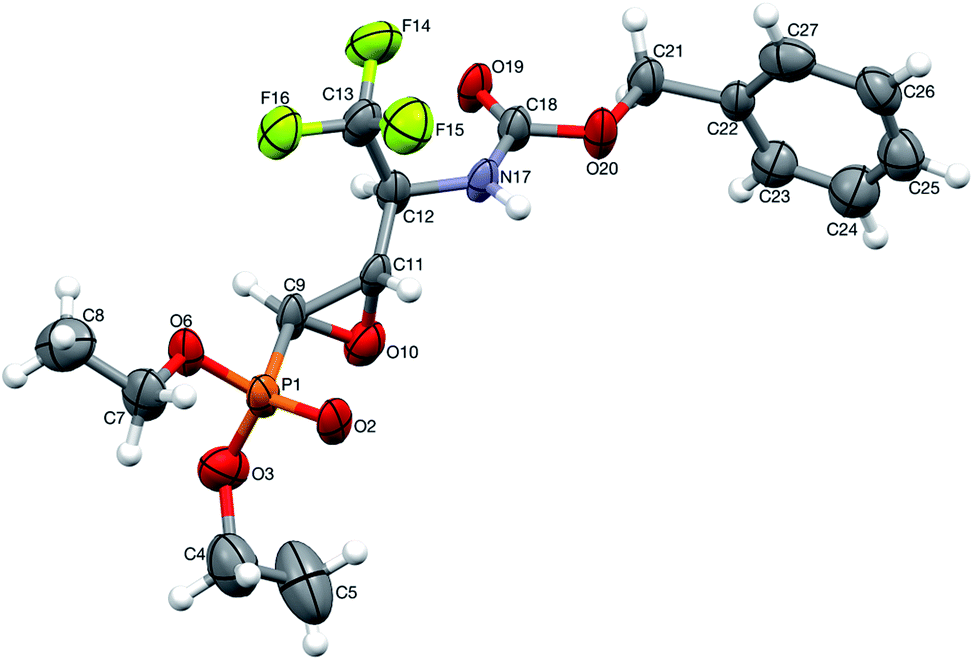 | ||
| Fig. 5 A perspective view of 38 showing the numbering scheme. Ellipsoids were drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
This aza-Payne rearrangement of non-fluorinated N-Boc38 protected aziridinemethanols, as well as Ts,38,39 Mts40 and Ms38 protected aziridinemethanols, was previously reported. To the best of our knowledge, there are limited numbers of publications reporting the synthesis of trifluoromethylated epoxide phosphonates41 that could be biologically promising derivatives of non-fluorinated epoxide phosphonates possessing antibiotic activity.42 On the other hand, epoxide 38 can easily provide trifluoromethylated hydroxyphosphonates, whose biological activities were already evaluated.43
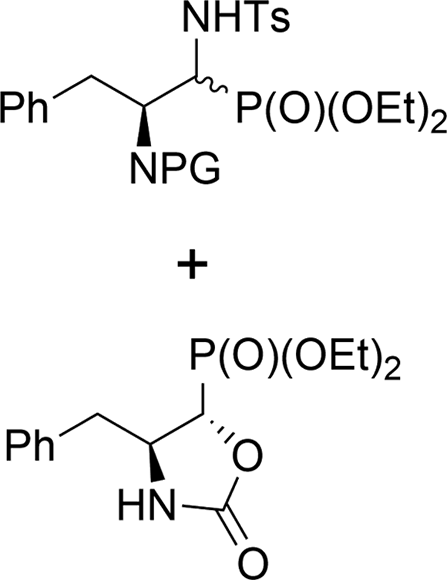
When phenylalanine N-Cbz protected derivatives 12a,b (2.7:1, d.r.) were subjected to the reaction with TsNH2 under the same conditions, the α-(N-tosyl)aminophosphonates 28a,b were obtained as major products, together with compound 39 (Scheme 6). Apparently, the alkoxide ion formed from α-hydroxyphosphonate attacked the carbonyl carbon atom of the Cbz moiety, instead of the adjacent carbon, as was in the case of 9b where the formation of fused three and five membered rings was excluded. Subsequent leaving of the benzyloxide ion led to the formation of the oxazolidin-2-one function in 39. A similar displacement of the N-amide group leading to the corresponding oxazolidin-2-one was reported by Patel et al.11f Based on the detailed analysis of 1H NMR data we were able to assign the stereochemistry of compound 39. Thus, diagnostic signals appeared at 4.45 (dd, J = 6.1, 0.7 Hz, CHP) and 4.33–4.25 ppm (m, CHN) in 1H NMR, which corresponded to the (1S,2S) diastereoisomer of 39.44 These data indicated that during the reaction of Cbz protected phenylalanine derivatives 12a,b under basic conditions with TsNH2, only the (1S,2S)-12b diastereoisomer reacted towards (1S,2S)-39.
 | ||
| Scheme 6 Reaction of N-Cbz protected phenylalanine derivatives 12a,b under K2CO3 conditions. | ||
Fig. 1–5 show the perspective views of the molecules. Of all six compounds, only 10a crystallized in the chiral P41212 space group as a single enantiomer (1R,2S). All other compounds crystallized in the centrosymmetric space group, which means that both enantiomers were present in the crystals. This difference was also visible in the supramolecular motifs created by hydrogen bonds in the crystal structures. In 10a the O–H⋯O hydrogen bonds connect molecules into infinite chains (Fig. 1 and Table 2 in ESI†), expanding along the z-direction (molecules related by fourfold, right-hand screw axis). In all other crystal structures, the well-defined, directional O–H⋯O or N–H⋯O (Fig. 2 and Table 2 in ESI†) hydrogen bonds made centrosymmetric dimers, arranged by two different enantiomers. In compound 23a, each of the symmetry-independent molecules made the dimer with its own symmetry-related mate (i.e. A–A and B–B). It is possible that the relative ease of making centrosymmetric dimers is one of the reasons that all these compounds crystallized as racemates.
Conclusions
In summary, our results demonstrate the synthesis of a wide range of α-hydroxyphosphonates that subsequently underwent reactions with nitrogen nucleophiles. Only para-toluenesulfonamide provided access to fluorinated3k,45 and non-fluorinated α-aminophosphonates,46 an important group of compounds that are mimics of the naturally occurring α-amino acids,47 and could be explored as versatile substrates in the synthesis of biologically active species. Further deprotection of the α-amino group could be considered in the design of important phosphonated building blocks employed in the synthesis of useful compounds such as peptide analogues. According to the establishments concerning the reaction mechanism, this study gives further proof that the Kabachnik–Fields reaction occurs via the imine intermediate, which immediately undergoes the addition of dialkyl phosphite towards N-tosyl-α-aminophosphonates.Experimental section
General methods
1H NMR, 13C NMR, 19F NMR and 31P NMR spectral measurements were performed on Bruker ASCEND 400 (400 MHz), Bruker ASCEND 600 (600 MHz) spectrometers. All 2D and 1D selective NMR spectra were recorded on the Bruker ASCEND 600 (600 MHz) spectrometer. Chemical shifts of 1H NMR were expressed in parts per million downfield from tetramethylsilane (TMS) as an internal standard (δ = 0) in CDCl3 or using the residual solvent peak in the case of CD3CN (δ = 1.96). Chemical shifts of 13C NMR were expressed in parts per million downfield and upfield from CDCl3 as an internal standard (δ = 77.0). Chemical shifts of 19F NMR were expressed in parts per million upfield from CFCl3 as an internal standard (δ = 0) in CDCl3. Chemical shifts of 31P NMR were expressed in parts per million in CDCl3 and CD3CN. All d.r. ratios were evaluated on the basis of 19F NMR or/and 31P NMR in the crude reaction mixture. High-resolution mass spectra were recorded by electron spray (MS-ESI) techniques using a QToF Impact HD Bruker spectrometer. The melting points were measured on a Boetius apparatus and were uncorrected. Reagent grade chemicals were used. Solvents were dried by refluxing with sodium metal-benzophenone·(THF), CaH2·(CH2Cl2, CH3CN) and NaH·(Et2O), then distilled under an argon atmosphere. Absolute ethanol was stored under argon and over molecular sieves (3 Å). All moisture sensitive reactions were carried out under an argon atmosphere using oven-dried glassware. Reactions at temperatures below 0 °C were performed using a cooling bath (liquid N2/n-hexane or liquid N2/i-PrOH). TLC was performed on Merck Kieselgel 60-F254 with EtOAc/n-hexane and MeOH/CHCl3 as developing systems, and products were detected by inspection under UV light (254 nm) and with a solution of potassium permanganate. Merck Kieselgel 60 (0.063–0.200 μm), Merck Kieselgel 60 (0.040–0.063 μm), Merck Kieselgel 60 (0.015–0.004 μm), were used for column chromatography. X-ray diffraction data were collected by the ω-scan technique on a Rigaku four-circle Xcalibur (Eos detector) diffractometer with graphite-monochromatized MoKα radiation (λ = 0.71073 Å): for 9a, 10a and 36a at room temperature, for 23a and 38 at 130(1) K, and for 34a at 100(1) K. The data were corrected for Lorentz-polarization and absorption effects.48 Accurate unit-cell parameters were determined by a least-squares fit of 7906 (9a), 4321 (10a), 6595 (23a), 3219 (34a), 7252 (36a) and 1078 (38) reflections of highest intensity, chosen from the whole experiment. The structures were solved with SHELXT49 and refined with the full-matrix least-squares procedure on F2 by SHELXL-2014/7.49 All non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in the calculated positions and refined as the ‘riding model’ with the isotropic displacement parameters set at 1.2 (1.5 for methyl groups) times the Ueq. value for the appropriate non-hydrogen atoms. In 9a, 34a, 36a and 38, the lengths of terminal C–C bonds in the ethyl groups C4–C5 and C7–C8 were constrained to the typical values, due to the significant shortening resulting from large thermal motion; additionally, in 9a, 34a and 38 weak constraints were applied to the selected anisotropic displacement parameters. Relevant crystal data are listed in Table 1 (see ESI†), together with refinement details. In structure 34a one of the ethyl groups was disordered over two alternative conformations; an s.o.f. of 0.5 was assigned to both positions.Crystallographic data for the structural analysis was deposited with the Cambridge Crystallographic Data Centre, no. CCDC – 1568454 (9a), CCDC – 1569663 (10a), CCDC – 1568455 (23a), CCDC – 1568456 (34a), CCDC – 1568457 (36a) and CCDC – 1568458 (38).
Procedure for the synthesis of aldehydes 1–5
All aldehydes were prepared from the corresponding alcohols. Compounds 1a,26 2–3,27b 4a,50 4c,51 5a,52 5b53 were prepared as described. The NMR data for 2–3,54 4b,27b 5c55 were in good agreement.O), 135.66, 128.77, 128.23, 128.14 (4 × s, Ph), 123.46 (q, J = 274.7 Hz, CF3), 62.37 (s, CH2Ph), 47.39 (s, CHCHCF3), 44.47 (q, J = 40.8 Hz, CHCF3). 19F NMR (376 MHz, CDCl3) δ = −66.26 (dd, J = 6.2, 2.7 Hz).Procedure for the addition of diethyl phosphite to aldehyde 1a
Compounds 6a,b26 were prepared as described. The NMR data for 6a were in good agreement.Procedure for N-deprotection of 6a,b
To a round-bottom flask with aziridinyl phosphonates 6a,b (1:1, d.r.) (1.68 mmol, 616 mg) dissolved in ethanol (10 mL), a catalytic amount of palladium hydroxide was added. The flask was then connected by three-way valve to a vacuum pump and a gasbag filled with gaseous hydrogen. Hydrogen was then introduced inside the flask at 0 °C and vigorously stirred. This cycle was repeated 10 times and the reaction mixture was stirred overnight at room temperature. The catalyst was then filtered out and crude products 7a,b were isolated using column chromatography (chloroform/methanol 99:1, v/v).
Procedure for the introduction of the Boc protecting group into 7a,b
Aziridinyl phosphonates 7a,b (1:1, d.r.) (0.72 mmol, 200 mg, 1 eq.) were dissolved in acetonitrile (5 mL) under argon at 0 °C. DMAP (0.87 mmol, 106 mg, 1.2 eq.) was added and the reaction mixture was stirred for 15 min. Next, di-tert-butyl dicarbonate (99%, 1.08 mmol, 238 mg, 1.5 eq.) was introduced and the reaction mixture was stirred overnight at room temperature. The reaction mixture was then diluted with water (10 mL), extracted with CH2Cl2 (3 × 10 mL) and the layers were separated. The organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product 8b was isolated using column chromatography (n-hexane/ethyl acetate 10:90, v/v → ethyl acetate/methanol 99:1, v/v).
:1 r.r. that could be isolated). Major rotamer: 1H NMR (600 MHz, CDCl3) δ = 5.03 (t, J = 9.6 Hz, 1H, CHP), 4.30–4.15 (m, 4H, 2 × OCH2CH3), 2.97–2.84 (m, 2H, CHCF3, CHCHCF3), 1.53 (s, 9H, C(CH3)3), 1.36 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 152.13 (s, CO), 124.08 (q, J = 274.5 Hz, CF3), 83.54 (s, C(CH3)3), 69.87 (d, J = 169.3 Hz, CHP), 63.43 (d, J = 6.8 Hz, OCH2CH3), 62.95 (d, J = 6.7 Hz, OCH2CH3), 35.02 (q, J = 41.4 Hz, CHCF3), 34.27 (d, J = 8.9 Hz, CHCHCF3), 27.68 (s, C(CH3)3), 16.36 (d, J = 5.9 Hz, OCH2CH3), 16.23 (d, J = 5.8 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −66.94 (d, J = 6.6 Hz). 19F{/1H} NMR (565 MHz, CDCl3) δ = −66.94 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = 16.47 (s). HRMS (ESI) calcd for C13H23F3NO6PNa ([M + Na]+): 400.1112, found: 400.1119. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ = 4.88 (t, J = 9.5 Hz, 1H, CHP), 4.32–4.18 (m, 4H, 2 × OCH2CH3), 3.00–2.70 (m, 2H, CHCF3, CHCHCF3), 1.51 (s, 9H, C(CH3)3), 1.38 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 151.60 (s, CO), 124.13 (q, J = 275.5 Hz, CF3), 83.43 (s, C(CH3)3), 69.42 (d, J = 164.1 Hz, CHP), 63.32 (d, J = 7.1 Hz, OCH2CH3), 63.26 (d, J = 6.8 Hz, OCH2CH3), 33.58 (dq, J = 39.7, 11.2 Hz, CHCF3), 32.66 (d, J = 2.6 Hz, CHCHCF3), 27.54 (s, C(CH3)3), 16.43 (d, J = 5.8 Hz, OCH2CH3), 16.35 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −66.95 (br s). 19F{/1H} NMR (376 MHz, CDCl3) δ = −66.96 (s). 31P{/1H} NMR (162 MHz, CDCl3) δ = 17.64 (s). HRMS (ESI) calcd for C13H23F3NO6PK ([M + K]+): 416.0852, found: 416.0846.Procedure for the introduction of the Cbz protecting group into 7a,b
Aziridinyl phosphonates 7a,b (1:1, d.r.) (0.72 mmol, 200 mg, 1 eq.) were dissolved in CH2Cl2 (5 mL) under argon at 0 °C. NaHCO3 (0.87 mmol, 67 mg, 1.2 eq.) was added and the reaction mixture was stirred for 15 min. Benzyl chloroformate (95%, 1.08 mmol, 194 mg, 162 μL, 1.5 eq.) was introduced and the reaction mixture was stirred overnight at room temperature. Next, the reaction mixture was diluted with water (10 mL) then extracted with CH2Cl2 (3 × 10 mL). The organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude products 9a,b were isolated using column chromatography (n-hexane/ethyl acetate 10:90, v/v → ethyl acetate/methanol 99:1, v/v).
O), 134.92, 128.61, 128.44, 128.16 (4 × s, Ph), 122.98 (q, J = 275.0 Hz, CF3), 69.06 (s, CH2Ph), 65.22 (d, J = 161.6 Hz, CHP), 63.42 (d, J = 7.3 Hz, OCH2CH3), 63.26 (d, J = 7.0 Hz, OCH2CH3), 40.88 (d, J = 4.8 Hz, CHCHCF3), 38.71 (dq, J = 40.6, 12.4 Hz, CHCF3), 16.34 (d, J = 5.5 Hz, OCH2CH3), 16.28 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −67.26 (br s). 19F{/1H} NMR (565 MHz, CDCl3) δ = −67.27 (d, J = 2.2 Hz). 31P{/1H} NMR (243 MHz, CDCl3) δ = 20.68 (s). HRMS (ESI) calcd for C16H21F3NO6PNa ([M + Na]+): 434.0956, found: 434.0952.O), 134.84, 128.52, 128.35, 128.06 (4 × s, Ph), 122.91 (q, J = 275.1 Hz, CF3), 68.95 (s, CH2Ph), 65.09 (dd, J = 161.7, 1.2 Hz, CHP), 63.32 (d, J = 7.3 Hz, OCH2CH3), 63.15 (d, J = 7.0 Hz, OCH2CH3), 40.82 (d, J = 5.2 Hz, CHCHCF3), 38.61 (dq, J = 40.5, 12.6 Hz, CHCF3), 16.25 (d, J = 5.7 Hz, OCH2CH3), 16.19 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −67.18 (d, J = 6.0 Hz). 19F{/1H} NMR (565 MHz, CDCl3) δ = −67.18 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = 19.15 (s). HRMS (ESI) calcd for C16H21F3NO6PNa ([M + Na]+): 434.0956, found: 434.0935.Procedure for the addition of diethyl phosphite to aldehydes 2–5
Compounds 10–15 were prepared according to the previously reported procedure.27b The NMR data for 10,27b 13,27b 17,56 2028 were in good agreement.Procedure A. To the respective aldehydes, diethyl phosphite (1 eq.) and TEA (0.2 eq.) were added and the reaction mixture was kept at 50 °C for 1 day (monitored by TLC or NMR). The reaction mixture was then diluted with water (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with aqueous sodium bicarbonate then brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude products were isolated using column chromatography (chloroform/methanol 99:1, v/v → chloroform/methanol 95:5).
Procedure B. Analogous treatment of aldehyde 4a and diethyl phosphite (1 eq.) and i-Pr2EtN (1.1 eq.) in CH2Cl2, (reflux) for 1 day gave crude products 16a,b, which were isolated using column chromatography (chloroform/methanol 99:1, v/v → chloroform/methanol 95:5).
O), 138.07, 129.37, 128.25, 126.22 (4 × s, Ph), 78.95 (s, C(CH3)3), 67.63 (d, J = 163.0 Hz, CHP), 62.96 (d, J = 7.1 Hz, OCH2CH3), 62.43 (d, J = 7.3 Hz, OCH2CH3), 52.89 (s, CHCH2Ph), 38.12 (s, CH2Ph), 28.26 (s, C(CH3)3), 16.42–16.27 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 23.25 (s). HRMS (ESI) calcd for C18H30NO6PNa ([M + Na]+): 410.1708, found: 410.1701.O), 137.84, 136.66, 129.43, 128.43, 128.39, 128.34, 127.88, 126.44 (8 × s, Ph), 67.52 (d, J = 162.7 Hz, CHP), 66.43 (s, OCH2Ph), 63.15 (d, J = 7.0 Hz, OCH2CH3), 62.64 (d, J = 7.2 Hz, OCH2CH3), 53.51 (s, CHCH2Ph), 38.03 (s, CH2Ph), 16.28 (d, J = 4.5 Hz, OCH2CH3), 16.24 (d, J = 5.7 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 23.06 (s). HRMS (ESI) calcd for C21H28NO6PNa ([M + Na]+): 444.1552, found: 444.1543.O), 79.14 (s, C(CH3)3), 68.32 (d, J = 162.7 Hz, CHP), 63.02 (d, J = 7.0 Hz, OCH2CH3), 62.55 (d, J = 7.6 Hz, OCH2CH3), 56.76 (s, CHCH(CH3)2), 29.89 (d, J = 11.9 Hz, CH(CH3)2), 28.38 (s, C(CH3)3), 19.67 (s, CH3), 19.19 (s, CH3), 16.48 (d, J = 4.6 Hz, OCH2CH3), 16.45 (d, J = 4.2 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 23.47 (s). HRMS (ESI) calcd for C14H31NO6P ([M + H]+): 340.1889, found: 340.1882.O), 136.79, 128.35, 127.99, 127.96 (4 × s, Ph), 67.84 (d, J = 163.3 Hz, CHP), 66.51 (s, CH2Ph), 63.19 (d, J = 7.3 Hz, OCH2CH3), 62.57 (d, J = 7.4 Hz, OCH2CH3), 56.90 (s, CHCH(CH3)2), 30.39 (d, J = 12.3 Hz, CH(CH3)2), 19.54 (s, CH3), 19.21 (s, CH3), 16.40 (d, J = 5.4 Hz, OCH2CH3), 16.30 (d, J = 6.1 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 23.23 (s). HRMS (ESI) calcd for C17H29NO6P ([M + H]+): 374.1732, found: 374.1736.:1, r.r.). Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.39–7.29 (m, 5H, Ph), 5.18 (d, J = 12.7 Hz, 1H, CHHPh), 5.12 (d, J = 12.6 Hz, 1H, CHHPh), 4.35 (br d, J = 5.6 Hz, 1H, CHP), 4.26–4.14 (m, 5H, 2 × OCH2CH3, CHCHP), 3.64 (ddd, J = 11.3, 7.9, 4.0 Hz, 1H, NCHH), 3.46–3.41 (m, 1H, NCHH), 2.35–2.25 (m, 1H, CHHCH), 2.13–2.04 (m, 1H, CHHCH), 2.04–1.96 (m, 1H, NCH2CHH), 1.77–1.70 (m, 1H, NCH2CHH), 1.35 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.29 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 156.60 (s, CO), 136.46, 128.49, 128.06, 127.88 (4 × s, Ph), 70.16 (d, J = 156.0 Hz, CHP), 67.19 (s, CH2Ph), 62.68 (d, J = 7.2 Hz, OCH2CH3), 62.53 (d, J = 6.8 Hz, OCH2CH3), 60.74 (d, J = 5.4 Hz, CHCHP), 47.58 (s, NCH2), 27.51 (s, CH2CH), 24.54 (s, NCH2CH2), 16.51–16.44 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.90 (s). Minor rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.39–7.29 (m, 5H, Ph), 5.16 (d, J = 12.7 Hz, 1H, CHHPh), 5.11 (d, J = 12.6 Hz, 1H, CHHPh), 4.45 (d, J = 11.1 Hz, 1H, CHP), 4.15–4.05 (m, 5H, 2 × OCH2CH3, CHCHP), 3.61–3.57 (m, 1H, NCHH), 3.46–3.41 (m, 1H, NCHH), 2.35–2.25 (m, 1H, CHHCH), 2.13–2.04 (m, 1H, CHHCH), 2.04–1.96 (m, 1H, NCH2CHH), 1.77–1.70 (m, 1H, NCH2CHH), 1.29 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 154.70 (s, CO), 136.46, 128.49, 128.06, 127.88 (4 × s, Ph), 70.08 (d, J = 156.4 Hz, CHP), 67.19 (br s, CH2Ph), 62.68 (d, J = 7.2 Hz, OCH2CH3), 62.53 (d, J = 6.8 Hz, OCH2CH3), 58.16 (d, J = 10.0 Hz, CHCHP), 47.58 (s, NCH2), 26.53 (s, CH2CH), 24.34 (s, NCH2CH2), 16.51–16.44 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 22.47 (s). HRMS (ESI) calcd for C17H27NO6P ([M + H]+): 372.1571, found: 372.1579.:1, r.r.). Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.39–7.32 (m, 5H, Ph), 5.21 (br s, 1H, OH), 5.17 (d, J = 12.0 Hz, 1H, CHHPh), 5.15 (d, J = 12.0 Hz, 1H, CHHPh), 4.32 (ddd, J = 10.1, 6.9, 3.7 Hz, 1H, CHCHP), 4.28–4.14 (m, 4H, 2 × OCH2CH3), 3.83 (dd, J = 9.4, 4.6 Hz, 1H, CHP), 3.53 (br q, J = 7.9 Hz, 1H, NCHH), 3.43 (ddd, J = 11.0, 7.5, 4.7 Hz, 1H, NCHH), 2.26–2.21 (m, 1H, CHHCH), 2.08–2.03 (m, 1H, CHHCH), 1.96–1.90 (m, 1H, NCH2CHH), 1.90–1.85 (m, 1H, NCH2CHH), 1.37 (br t, J = 6.6 Hz, 6H, 2 × OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 158.65 (s, CO), 136.14, 128.54, 128.20, 127.96 (4 × s, Ph), 72.68 (d, J = 159.4 Hz, CHP), 67.77 (s, CH2Ph), 63.14 (d, J = 7.3 Hz, OCH2CH3), 62.55 (d, J = 7.0 Hz, OCH2CH3), 59.88 (d, J = 8.9 Hz, CHCHP), 46.96 (s, NCH2), 28.27 (s, CH2CH), 24.10 (s, NCH2CH2), 16.51 (d, J = 6.1 Hz, OCH2CH3), 16.45 (d, J = 5.7 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.22 (s). Minor rotamer: 31P{/1H} NMR (162 MHz, CDCl3) δ = 22.08 (s).:1, r.r.). Major rotamer: 1H NMR (600 MHz, CD3CN) δ = 7.45–7.36 (m, 5H, Ph), 5.17–5.11 (m, 2H, CH2Ph), 4.36 (dd, J = 12.4, 7.2 Hz, 1H, CHP), 4.32 (br dd, J = 9.2, 2.6 Hz, 1H, OCHH), 4.26–4.21 (m, 1H, CHCHP), 4.15–4.10 (m, 2H, OCH2CH3), 4.08–3.96 (m, 3H, OCHH, OCH2CH3), 1.59 (s, 3H, C(CH3)2), 1.50 (s, 3H, C(CH3)2), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CD3CN) δ = 153.54 (s, CO), 138.28, 129.96, 129.45, 129.30 (4 × s, Ph), 95.44 (s, C(CH3)2), 68.03 (d, J = 159.9 Hz, CHP), 67.78 (s, CH2Ph), 64.80 (s, OCH2), 64.02 (d, J = 7.0 Hz, OCH2CH3), 63.56 (d, J = 6.9 Hz, OCH2CH3), 59.04 (d, J = 13.6 Hz, CHCHP), 26.33 (s, C(CH3)2), 24.16 (s, C(CH3)2), 17.27 (d, J = 5.4 Hz, OCH2CH3), 17.25 (d, J = 5.8 Hz, OCH2CH3). 31P{/1H} (243 MHz, CD3CN) δ = 21.72 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = 21.84 (s). Minor rotamer: 1H NMR (600 MHz, CD3CN) δ = 7.45–7.36 (m, 5H, Ph), 5.20 (d, J = 12.5 Hz, 1H, CHHPh), 5.17–5.11 (m, 1H, CHHPh), 4.52 (dd, J = 11.4, 7.1 Hz, 1H, CHP), 4.28 (br dd, J = 8.3, 3.3 Hz, 1H, OCHH), 4.26–4.21 (m, 1H, CHCHP), 4.15–4.10 (m, 1H, OCHH), 4.08–3.96 (m, 4H, 2 × OCH2CH3), 1.54 (s, 3H, C(CH3)2), 1.47 (s, 3H, C(CH3)2), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CD3CN) δ = 154.32 (s, CO), 138.28, 129.96, 129.45, 129.30 (4 × s, Ph), 95.12 (s, C(CH3)2), 68.12 (s, CH2Ph), 66.70 (d, J = 159.9 Hz, CHP), 64.33 (s, OCH2), 64.16 (d, J = 7.1 Hz, OCH2CH3), 63.56 (d, J = 6.9 Hz, OCH2CH3), 60.17 (d, J = 12.7 Hz, CHCHP), 26.99 (s, C(CH3)2), 25.94 (s, C(CH3)2), 17.27 (d, J = 5.4 Hz, OCH2CH3), 17.20 (d, J = 5.8 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CD3CN) δ = 22.04 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = 22.13 (s). HRMS (ESI) calcd for C18H28NO7PNa ([M + Na]+): 424.1496, found: 424.1495.Procedure for reactions of 6a,b, 10a,b and 20a,b with nitrogen nucleophiles (BnNH2, MeNH2, BzNH2) and/or with K2CO3
To a solution of benzylamine or methylamine (HCl salt) or benzamide (1.2 eq.) in acetonitrile (5 mL), potassium carbonate (1.2 eq. or 2.2 eq. in the case of methylamine HCl) was added. After 30 min, the α-hydroxyphosphonates (1 eq.) were added to the stirred solution. The reaction mixture was refluxed for 8 h. When the reaction was completed, water was added (10 mL) followed by extraction with a few portions of CH2Cl2. The organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude products were isolated using column chromatography (chloroform/methanol or n-hexane/ethyl acetate → ethyl acetate/methanol).CH), 5.33 (ddd, J = 12.1, 9.0, 0.8 Hz, 1H, OCHCH), 4.23–4.15 (m, 4H, 2 × OCH2CH3), 3.93 (d, J = 13.4 Hz, 1H, CHHPh), 3.79 (d, J = 13.4 Hz, 1H, CHHPh), 3.53 (m, 1H, CHCF3), 1.38 (t, J = 6.8 Hz, 3H, OCH2CH3), 1.37 (t, J = 6.7 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 141.86 (d, J = 5.2 Hz, OCHCH), 138.71, 128.62, 128.13, 127.45 (4 × s, Ph), 125.25 (q, J = 281.7 Hz, CF3), 109.3 (d, J = 10.5, 1.8 Hz, OCHCH), 64.74 (d, J = 6.0 Hz, 2 × OCH2CH3), 50.56 (s, CH2Ph), 16.07 (d, J = 6.8 Hz, OCH2CH3), 16.06 (d, J = 6.5 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −75.84 (d, J = 6.9 Hz). 19F{/1H} NMR (565 MHz, CDCl3) δ = −75.84 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = −5.02 (s). HRMS (ESI) calcd for C15H22F3NO4P ([M + H]+): 368.1239, found: 368.1236.Note 1: An analogous reaction of 6a,b with K2CO3 (1.2 eq.) in acetonitrile (reflux, 8 h) according to the above procedure gave 22.
Note 2: Reaction of 10a,b (19:1, d.r.) with K2CO3 (1.2 eq.) or K2CO3 (1.2 eq.) and BzNH2 (1.2 eq.) or with K2CO3 (1.2 eq.) and BnNH2 (1.2 eq.) in acetonitrile (reflux, 8 h) according to the above procedure gave a partially racemized mixture of 10a,b (1.5:1, d.r.).
Note 3: Reaction of 20a,b (99:1, d.r.) with K2CO3 (1.2 eq.) or K2CO3 (1.2 eq.) and BzNH2 (1.2 eq.) or with K2CO3 (1.2 eq.) and BnNH2 (1.2 eq.) in acetonitrile (reflux, 8 h) according to the above procedure gave a partially racemized mixture of 20a,b (3.6:1, d.r.).12
Procedure for the reactions of 6a,b–21a,b with TsNH2
To a solution of para-toluenesulfonamide (1.2 eq.) in acetonitrile (5 mL), potassium carbonate (1.2 eq.) was added. After 30 min, the α-hydroxyphosphonates 6a,b–21a,b (1 eq.) were added to the stirred reaction mixture. The solution was refluxed for 8 hours. When the reaction was completed, the reaction mixture was diluted with H2O (10 mL) and extracted with a few portions of CH2Cl2. The organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude products 23a,b–39 were isolated using column chromatography (chloroform/methanol or n-hexane/ethyl acetate → ethyl acetate/methanol).O), 143.40, 138.02, 137.54, 129.53, 129.36, 128.35, 127.17, 126.46 (8 × s, Ar, Ph), 79.58 (s, C(CH3)3), 63.15–62.87 (m, 2× OCH2CH3), 53.37 (d, J = 153.7 Hz, CHP), 52.25 (s, CHCH2Ph), 37.94 (s, CH2Ph), 28.20 (s, C(CH3)3), 21.49 (s, ArCH3), 16.35 (d, J = 5.7 Hz, OCH2CH3), 16.24 (d, J = 5.8 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.41 (s). HRMS (ESI) calcd for C25H37N2O7PSNa ([M + Na]+): 563.1957, found: 563.1954.O), 143.44, 137.17, 137.04, 129.76, 129.08, 128.50, 127.25, 126.63 (8 × s, Ar, Ph), 79.95 (s, C(CH3)3), 64.08 (d, J = 7.4 Hz, OCH2CH3), 62.82 (d, J = 6.7 Hz, OCH2CH3), 53.39 (d, J = 159.1 Hz, CHP), 51.92 (s, CHCH2Ph), 38.07 (s, CH2Ph), 28.27 (s, C(CH3)3), 21.55 (s, ArCH3), 16.43 (d, J = 5.6 Hz, OCH2CH3), 16.30 (d, J = 6.1 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 20.34 (s). HRMS (ESI) calcd for C25H37N2O7PSNa ([M + Na]+): 563.1957, found: 563.1964.O), 143.56, 139.10, 137.99, 137.23, 136.36, 129.69, 129.63, 129.23, 128.41, 127.88, 127.14, 126.46 (12 × s, Ar, Ph), 66.67 (s, OCH2Ph), 63.16 (d, J = 6.7 Hz, OCH2CH3), 63.12 (d, J = 7.0 Hz, OCH2CH3), 53.20 (d, J = 152.8 Hz, CHP), 52.73 (s, CHCH2Ph), 37.66 (s, CH2Ph), 21.51 (s, ArCH3), 16.32 (d, J = 5.6 Hz, OCH2CH3), 16.20 (d, J = 5.9 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 20.97 (s). HRMS (ESI) calcd for C28H36N2O7PS ([M + H]+): 575.1981, found: 575.1980.O), 143.23, 138.29, 129.38, 127.09 (4 × s, Ar), 79.58 (s, C(CH3)3), 62.87 (d, J = 7.1 Hz, 2 × OCH2CH3), 56.03 (d, J = 5.9 Hz, CHCH(CH3)2), 52.09 (d, J = 155.7 Hz, CHP), 29.21 (d, J = 7.4 Hz, CH(CH3)2), 28.30 (s, C(CH3)3), 21.48 (s, ArCH3), 20.38 (s, CH3), 17.45 (s, CH3), 16.32 (d, J = 5.7 Hz, OCH2CH3), 16.23 (d, J = 5.7 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 22.29 (s). HRMS (ESI) calcd for C21H37N2O7PSNa ([M + Na]+): 515.1957, found: 515.1952.O), 143.29, 138.26, 136.43, 129.41, 128.45, 128.03, 127.98, 127.02 (8 × s, Ar, Ph), 66.89 (s, OCH2Ph), 62.95 (d, J = 7.5 Hz, OCH2CH3), 62.93 (d, J = 6.9 Hz, OCH2CH3), 56.62 (d, J = 5.9 Hz, CHCH(CH3)2), 52.06 (d, J = 155.4 Hz, CHP), 29.12 (d, J = 6.9 Hz, CH(CH3)2), 21.47 (s, ArCH3), 20.39 (s, CCH3), 17.45 (s, CCH3), 16.28 (d, J = 6.0 Hz, OCH2CH3), 16.21 (d, J = 6.1 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.79 (s). HRMS (ESI) calcd for C24H36N2O7PS ([M + H]+): 527.1981, found: 527.1978.:1, r.r.), mp. 119–121 °C. Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.76 (d, J = 8.1 Hz, 2H, Ar), 7.27 (d, J = 8.1 Hz, 2H, Ar), 4.30 (dd, J = 21.4, 0.9 Hz, 1H, CHP), 4.18–4.05 (m, 4H, 2 × OCH2CH3), 4.04–3.91 (m, 1H, CHCHP), 3.13–2.96 (m, 2H, NCH2), 2.41 (s, 3H, ArCH3), 2.10–1.98 (m, 1H, CHHCH), 1.98–1.81 (m, 2H, NCH2CHH, CHHCH), 1.64–1.58 (m, 1H, NCH2CHH), 1.48 (s, 9H, C(CH3)3), 1.33 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.31 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 155.34 (s, CO), 142.95, 138.36, 129.44, 127.18 (4 × s, Ar), 79.66 (s, C(CH3)3), 62.74 (d, J = 6.8 Hz, 2 × OCH2CH3), 58.26 (d, J = 8.1 Hz, CHCHP), 52.25 (d, J = 150.5 Hz, CHP), 47.10 (s, NCH2), 28.47 (s, C(CH3)3), 27.99 (s, CH2CH), 24.38 (s, NCH2CH2), 21.47 (s, ArCH3), 16.52–16.22 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.03 (s). Minor rotamer: 1H NMR (400 MHz, CDCl3): δ = 7.68 (d, J = 8.1 Hz, 2H, Ar), 7.27 (d, J = 8.1 Hz, 2H, Ar), 4.60 (dd, J = 22.9, 1.4 Hz, 1H, CHP), 4.04–3.91 (m, 4H, 2 × OCH2CH3), 3.93–3.86 (m, 1H, CHCHP), 3.86–3.79 (m, 1H, NCHH), 3.36–3.25 (m, 1H, NCHH), 2.41 (br s, 3H, ArCH3), 2.23–2.10 (m, 1H, CHHCH), 1.98–1.81 (m, 2H, NCH2CHH, CHHCH), 1.64–1.58 (m, 1H, NCH2CHH), 1.55 (s, 9H, C(CH3)3), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 153.67 (s, CO), 142.86, 138.91, 129.53, 126.58 (4 × s, Ar), 80.48 (s, C(CH3)3), 63.02 (d, J = 7.4 Hz, 2 × OCH2CH3), 57.03 (d, J = 14.0 Hz, CHCHP), 52.17 (d, J = 147.0 Hz, CHP), 46.01 (s, NCH2), 28.45 (s, C(CH3)3), 27.31 (s, CH2CH), 23.92 (s, NCH2CH2), 21.47 (s, ArCH3), 16.52–16.22 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.54 (s). HRMS (ESI) calcd for C21H36N2O7PS ([M + H]+): 491.1975, found: 491.1986.:1, r.r.). Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.73 (d, J = 8.1 Hz, 2H, Ar), 7.27 (d, J = 8.1 Hz, 2H, Ar), 4.18–4.05 (m, 5H, 2 × OCH2CH3, CHP), 3.55 (dd, J = 14.6, 10.4 Hz, 1H, CHCHP), 3.36–3.25 (m, 1H, NCHH), 2.79–2.71 (m, 1H, NCHH), 2.41 (s, 3H, ArCH3), 2.23–2.10 (m, 1H, CHHCH), 1.81–1.74 (m, 1H, NCH2CHH), 1.64–1.58 (m, 2H, CHHCH, NCH2CHH), 1.46 (s, 9H, C(CH3)3), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 157.30 (s, CO), 142.90, 138.45, 129.37, 127.07 (4 × s, Ar), 80.05 (s, C(CH3)3), 62.50 (d, J = 6.8 Hz, 2 × OCH2CH3), 56.62 (d, J = 11.1 Hz, CHCHP), 55.13 (d, J = 156.8 Hz, CHP), 46.55 (s, NCH2), 29.47 (s, CH2CH), 28.41 (s, C(CH3)3), 23.38 (s, NCH2CH2), 21.47 (s, ArCH3), 16.52–16.22 (m, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.27 (s). Minor rotamer: 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.40 (s).:1, r.r.), mp. 136–138 °C. Major rotamer: 1H NMR (600 MHz, CDCl3) δ = 7.74 (d, J = 8.3 Hz, 2H, Ar), 7.42–7.35 (m, 5H, Ph), 7.22 (d, J = 8.3 Hz, 2H, Ar), 6.35 (br s, 1H, NH), 5.16 (d, J = 12.4 Hz, 1H, CHHPh), 5.12 (d, J = 12.5 Hz, 1H, CHHPh), 4.36 (dd, J = 22.1, 9.8 Hz, 1H, CHP), 4.17–4.09 (m, 2H, OCH2CH3), 4.07–4.00 (m, 2H, OCH2CH3), 3.97–3.91 (m, 1H, CHCHP), 3.43 (ddd, J = 12.0, 7.8, 4.7 Hz, 1H, NCHH), 3.15–3.11 (m, 1H, NCHH), 2.36 (br s, 3H, ArCH3), 2.13–2.07 (m, 1H, CHHCH), 2.01–1.94 (m, 1H, CHHCH), 1.92–1.88 (m, 1H, NCH2CHH), 1.66–1.59 (m, 1H, NCH2CHH), 1.30 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.21 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 155.61 (s, CO), 143.05, 138.3, 136.63, 129.48, 128.45, 127.96, 127.83, 127.04 (8 × s, Ar, Ph), 66.93 (s, CH2Ph), 63.17 (d, J = 7.3 Hz, OCH2CH3), 62.89 (d, J = 7.0 Hz, OCH2CH3), 58.73 (d, J = 8.8 Hz, CHCHP), 51.94 (d, J = 150.4 Hz, CHP), 46.89 (s, NCH2), 27.87 (s, CH2CH), 24.45 (s, NCH2CH2), 21.44 (s, ArCH3), 16.30 (d, J = 5.9 Hz, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 20.49 (s). Minor rotamer: 1H NMR (600 MHz, CDCl3) δ = 7.57 (d, J = 8.0 Hz, 2H, Ar), 7.42–7.35 (m, 5H, Ph), 7.22 (d, J = 8.0 Hz, 2H, Ar), 5.55 (dd, J = 9.7, 6.9 Hz, 1H, NH), 5.23 (d, J = 12.0 Hz, 1H, CHHPh), 5.12 (d, J = 12.0 Hz, 1H, CHHPh), 4.49 (ddd, J = 22.5, 9.7, 2.0 Hz, 1H, CHP), 4.24–4.20 (m, 2H, OCH2CH3), 4.00–3.91 (m, 2H, OCH2CH3), 3.92–3.87 (m, 1H, CHCHP), 3.27 (dt, J = 9.8, 6.6 Hz, 1H, NCHH), 2.61 (dt, J = 10.7, 6.8 Hz, 1H, NCHH), 2.41 (s, 3H, ArCH3), 2.28–2.20 (m, 1H, CHHCH), 2.00–1.94 (m, 1H, CHHCH), 1.92–1.88 (m, 1H, NCH2CHH), 1.66–1.59 (m, 1H, NCH2CHH), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.19 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 154.15 (s, CO), 143.00, 138.56, 136.16, 129.48, 128.53, 127.96, 127.83, 126.65 (8 × s, Ar, Ph), 67.43 (s, CH2Ph), 63.17 (d, J = 7.3 Hz, OCH2CH3), 62.89 (d, J = 7.0 Hz, OCH2CH3), 57.13 (d, J = 13.7 Hz, CHCHP), 51.71 (d, J = 146.6 Hz, CHP), 46.40 (s, NCH2), 27.60 (s, CH2CH), 23.94 (s, NCH2CH2), 21.50 (s, ArCH3), 16.48 (d, J = 5.6 Hz, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 20.71 (s). HRMS (ESI) calcd for C24H34N2O7PS ([M + H]+): 525.1819, found: 525.1834.:1, r.r.). Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.67 (d, J = 8.2 Hz, 2H, Ar), 7.42–7.35 (m, 5H, Ph), 7.16 (d, J = 8.0 Hz, 2H, Ar), 6.22 (d, J = 8.3 Hz, 1H, NH), 5.09 (d, J = 12.4 Hz, 1H, CHHPh), 5.02 (d, J = 12.4 Hz, 1H, CHHPh), 4.29–4.25 (m, 3H, CHCHP, OCH2CH3), 4.07–4.00 (m, 2H, OCH2CH3), 3.66–3.60 (m, 1H, CHP), 3.15–3.11 (m, 1H, NCHH), 2.72 (ddd, J = 10.3, 7.1, 3.1 Hz, 1H, NCHH), 2.31 (s, 3H, ArCH3), 2.13–2.07 (m, 1H, CHHCH), 2.01–1.94 (m, 1H, CHHCH), 1.84–1.81 (m, 1H, NCH2CHH), 1.74–1.70 (m, 1H, NCH2CHH), 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.19 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 157.43 (s, CO), 143.05 (br s), 138.36, 136.40, 129.52, 128.57, 127.83, 126.14, 126.69 (8 × s, Ar, Ph), 67.38 (s, CH2Ph), 63.48 (d, J = 7.5 Hz, OCH2CH3), 62.63 (d, J = 6.9 Hz, OCH2CH3), 57.26 (d, J = 11.1 Hz, CHCHP), 55.15 (d, J = 156.3 Hz, CHP), 46.40 (s, NCH2), 29.20 (s, CH2CH), 23.37 (s, NCH2CH2), 21.38 (s, ArCH3), 16.21 (d, J = 6.1 Hz, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 20.49 (s). Minor rotamer: 31P{/1H} NMR (162 MHz, CDCl3) δ = 20.88 (s).:1, r.r.), mp. 152–153 °C. Major rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.78 (d, J = 8.1 Hz, 2H, Ar), 7.29 (d, J = 8.6 Hz, 2H, Ar), 5.39 (dd, J = 10.0, 4.2 Hz, 1H, NH), 4.53–4.45 (m, 1H, CHP), 4.28–4.22 (m, 2H, CHCHP, OCHH), 4.07–4.00 (m, 2H, OCHH, OCHHCH3), 3.96–3.90 (m, 1H, OCHHCH3), 3.90–3.86 (m, 1H, OCHHCH3), 3.62–3.56 (m, 1H, OCHHCH3), 2.42 (s, 3H, ArCH3), 1.67 (s, 3H, C(CH3)2), 1.56 (s, 9H, C(CH3)3), 1.47 (s, 3H, C(CH3)2), 1.25–1.21 (m, 3H, OCH2CH3), 1.10 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 152.57 (s, CO), 143.06, 138.47, 129.33, 127.35 (4 × s, Ar), 94.30 (s, C(CH3)2), 80.80 (s, C(CH3)3), 63.83 (s, OCH2), 62.66 (d, J = 7.1 Hz, OCH2CH3), 62.56 (d, J = 7.3 Hz, OCH2CH3), 57.28 (d, J = 13.0 Hz, CHCHP), 49.80 (d, J = 151.6 Hz, CHP), 28.45 (s, C(CH3)3), 26.10 (s, C(CH3)2), 24.48 (s, C(CH3)2), 21.46 (s, ArCH3), 16.29 (d, J = 5.6 Hz, OCH2CH3), 16.18 (d, J = 5.5 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.34 (s). Minor rotamer: 1H NMR (400 MHz, CDCl3) δ = 7.76 (d, J = 8.4 Hz, 2H, Ar), 7.29 (d, J = 8.6 Hz, 2H, Ar), 5.08 (dd, J = 9.2, 3.4 Hz, 1H, NH), 4.53–4.45 (m, 1H, CHP), 4.28–4.22 (m, 2H, CHCHP, OCHH), 4.07–4.00 (m, 4H, OCHH, OCHHCH3, OCH2CH3), 3.90–3.86 (m, 1H, OCHHCH3), 2.42 (s, 3H, ArCH3), 1.58 (s, 9H, C(CH3)3), 1.44 (s, 3H, C(CH3)2), 1.39 (s, 3H, C(CH3)2), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.25–1.21 (m, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 151.43 (s, CO), 143.27, 138.84, 129.44, 126.98 (4 × s, Ar), 94.61 (s, C(CH3)2), 80.66 (s, C(CH3)3), 64.06 (s, OCH2), 62.97 (d, J = 7.3 Hz, OCH2CH3), 62.45 (d, J = 7.0 Hz, OCH2CH3), 56.36 (d, J = 14.3 Hz, CHCHP), 51.59 (d, J = 152.1 Hz, CHP), 28.45 (s, C(CH3)3), 24.99 (s, C(CH3)2), 22.51 (s, C(CH3)2), 21.46 (s, ArCH3), 16.35 (d, J = 5.6 Hz, OCH2CH3), 16.31 (d, J = 6.0 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz, CDCl3) δ = 21.19 (s). HRMS (ESI) calcd for C22H38N2O8PS ([M + H]+): 521.2081, found: 521.2090.:1, r.r.). Major rotamer: 1H NMR (600 MHz, CDCl3) δ = 7.60 (d, J = 8.3 Hz, 2H, Ar), 7.50–7.47 (m, 2H, Ph), 7.41–7.36 (m, 3H, Ph), 7.22 (d, J = 8.0 Hz, 2H, Ar), 5.47 (dd, J = 9.6, 4.1 Hz, 1H, NH), 5.26 (d, J = 12.0 Hz, 1H, CHHPh), 5.12 (d, J = 12.0 Hz, 1H, CHHPh), 4.36 (ddd, J = 19.2, 9.5, 2.6 Hz, 1H, CHP), 4.28–4.22 (m, 2H, OCHH, CHCHP), 4.07–4.02 (m, 2H, OCHH, OCHHCH3), 3.98–3.95 (m, 1H, OCHHCH3), 3.90–3.85 (m, 1H, OCHHCH3), 3.80–3.75 (m, 1H, OCHHCH3), 2.40 (s, 3H, ArCH3), 1.56 (s, 3H, C(CH3)2), 1.49 (s, 3H, C(CH3)2), 1.09 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.10 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 152.03 (s, CO), 143.33, 138.51, 136.29, 128.67, 128.31, 128.08, 127.41, 127.14 (8 × s, Ar, Ph), 95.19 (s, C(CH3)2), 67.39 (s, CH2Ph), 64.37 (s, OCH2), 63.02 (d, J = 7.7 Hz, OCH2CH3), 62.70 (d, J = 6.9 Hz, OCH2CH3), 56.41 (d, J = 14.6 Hz, CHCHP), 50.77 (d, J = 150.2 Hz, CHP), 25.06 (s, C(CH3)2), 23.03 (s, C(CH3)2), 21.57 (s, ArCH3), 16.32 (d, J = 5.9 Hz, OCH2CH3), 16.31 (d, J = 5.9 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 20.56. Minor rotamer: 1H NMR (600 MHz, CDCl3) δ = 7.77 (d, J = 8.3 Hz, 2H, Ar), 7.41–7.34 (m, 5H, Ph), 7.24 (d, J = 8.1 Hz, 2H, Ph), 5.65 (dd, J = 9.7, 4.6 Hz, 1H, NH), 5.23 (d, J = 12.3 Hz, 1H, CHHPh), 5.17 (d, J = 12.3 Hz, 1H, CHHPh), 4.58 (ddd, J = 19.7, 9.6, 2.5 Hz, 1H, CHP), 4.28–4.22 (m, 2H, OCHH, CHCHP), 4.07–4.02 (m, 1H, OCHH), 3.98–3.95 (m, 1H, OCHHCH3), 3.75–3.62 (m, 3H, OCH2CH3, OCHHCH3), 2.38 (s, 3H, ArCH3), 1.54 (s, 3H, C(CH3)2), 1.42 (s, 3H, C(CH3)2), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.14 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 153.42 (s, CO), 143.29, 138.42, 136.14, 128.67, 128.23, 128.08, 127.41, 127.14 (8 × s, Ar, Ph), 94.71 (s, C(CH3)2), 67.61 (s, CH2Ph), 64.03 (s, OCH2), 63.10 (d, J = 7.3 Hz, OCH2CH3), 62.94 (d, J = 6.9 Hz, OCH2CH3), 57.79 (d, J = 13.5 Hz, CHCHP), 49.63 (d, J = 151.2 Hz, CHP), 25.97 (s, C(CH3)2), 24.81 (s, C(CH3)2), 21.55 (s, ArCH3), 16.36 (d, J = 5.8 Hz, OCH2CH3), 16.23 (d, J = 5.8 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 20.89 (s). HRMS (ESI) calcd for C25H36N2O8PS ([M + H]+): 555.1924, found: 555.1919.O), 135.34, 128.68, 128.59, 128.25 (4 × s, Ph), 124.04 (q, J = 282.4 Hz, CF3), 68.04 (s, CH2Ph), 63.59 (d, J = 6.2 Hz, OCH2CH3), 63.29 (d, J = 6.3 Hz, OCH2CH3), 51.72 (d, J = 1.6 Hz, CHCHCF3), 51.24 (q, J = 31.5 Hz, CHCF3), 45.81 (d, J = 204.2 Hz, CHP), 16.45 (d, J = 5.4 Hz, OCH2CH3), 16.43 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −75.49 (d, J = 7.8 Hz). 19F{/1H} NMR (565 MHz, CDCl3) δ = −75.49 (s). 31P{/1H} NMR (243 MHz, CDCl3) δ = 15.89 (s). HRMS (ESI) calcd for C16H21F3NO6P ([M + Na]+): 434.0956, found: 434.0955.O), 135.46, 129.16, 129.13, 127.52 (4 × s, Ar), 74.98 (d, J = 171.9 Hz, CHP), 64.03 (d, J = 6.7 Hz, OCH2CH3), 63.54 (d, J = 6.8 Hz, OCH2CH3), 54.90 (s, CHCH2Ph), 42.22 (d, J = 8.9 Hz, CH2Ph), 16.49 (d, J = 4.4 Hz, OCH2CH3), 16.46 (d, J = 4.4 Hz, OCH2CH3). 31P{/1H} NMR (243 MHz, CDCl3) δ = 15.91 (s). HRMS (ESI) calcd for C14H21NO5P ([M + H]+): 314.1157, found: 314.1155.Conflicts of interest
There are no conflicts to declare.References
- M. Dryjanski and R. F. Pratt, Biochemistry, 1995, 34, 3569–3575 CrossRef CAS PubMed.
- G. Pochetti, E. Gavuzzo, C. Campestre, M. Agamennone, P. Tortorella, V. Consalvi, C. Gallina, O. Hiller, H. Tschesche, P. A. Tucker and F. Mazza, J. Med. Chem., 2006, 49, 923–931 CrossRef CAS PubMed.
- (a) P. J. Roberts, G. A. Foster, N. A. Sharif and J. F. Collins, Brain Res., 1982, 238, 475–479 CrossRef CAS PubMed; (b) P. Kafarski, B. Lejczak, P. Mastalerz, D. Dus and C. Radzikowski, J. Med. Chem., 1985, 28, 1555–1558 CrossRef CAS PubMed; (c) B. Lejczak, P. Kafarski and J. Zygmunt, Biochemistry, 1989, 28, 3549–3555 CrossRef CAS PubMed; (d) S. De Lombaert, M. D. Erion, J. Tan, L. Blanchard, L. El-Chehabi, R. D. Ghai, Y. Sakane, C. Berry and A. J. Trapani, J. Med. Chem., 1994, 37, 498–511 CrossRef CAS PubMed; (e) J. Oleksyszyn and J. C. Powers, Methods Enzymol., 1994, 244, 423–441 CAS; (f) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley-Interscience, New York, 2000 Search PubMed; (g) P. Kafarski and B. Lejczak, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 301–312 CrossRef CAS PubMed; (h) S. R. Walker and E. J. Parker, Bioorg. Med. Chem. Lett., 2006, 16, 2951–2954 CrossRef CAS PubMed; (i) F. Orsini, G. Sello and M. Sisti, Curr. Med. Chem., 2010, 17, 264–289 CrossRef CAS PubMed; (j) Z. H. Kudzin, M. H. Kudzin, J. Drabowicz and C. V. Stevens, Curr. Org. Chem., 2011, 15, 2015–2071 CrossRef CAS; (k) K. V. Turcheniuk, V. P. Kukhar, G.-V. Röschenthaler, J. L. Aceña, V. A. Soloshonok and A. E. Sorochinsky, RSC Adv., 2013, 3, 6693–6716 RSC.
- (a) M. I. Kabachnik and T. Y. Medved, Dokl. Akad. Nauk SSSR, 1952, 83, 689–692 CAS; (b) E. K. Fields, J. Am. Chem. Soc., 1952, 74, 1528–1531 CrossRef CAS.
- (a) R. A. Cherkasov and V. I. Galkin, Russ. Chem. Rev., 1998, 67, 857–882 CrossRef; (b) G. Keglevich and E. Bálint, Molecules, 2012, 17, 12821–12835 CrossRef CAS PubMed.
- H.-J. Ha and G.-S. Nam, Synth. Commun., 1992, 22, 1143–1148 CrossRef CAS.
- V. S. Abramov, Zh. Obshch. Khim., 1952, 22, 647–652 CAS.
- H. Gröger and B. Hammer, Chem.–Eur. J., 2000, 6, 943–948 CrossRef.
- (a) P. G. Baraldi, M. Guarneri, F. Moroder, G. P. Pollini and D. Simoni, Synthesis, 1982, 653–655 CrossRef CAS; (b) T. Minami and J. Motoyoshiya, Synthesis, 1992, 333–349 CrossRef CAS; (c) T. Yokomatsu and S. Shibuya, Tetrahedron: Asymmetry, 1992, 3, 377–378 CrossRef CAS; (d) T. Gajda and M. Matusiak, Synthesis, 1992, 367–368 CrossRef CAS; (e) E. Öhler and S. Kotzinger, Synthesis, 1993, 497–502 CrossRef; (f) J. J. Kiddle and J. H. Babbler, J. Org. Chem., 1993, 58, 3572–3574 CrossRef CAS; (g) S. Berté-Verrando, F. Nief, C. Patois and P. Savignac, J. Chem. Soc., Perkin Trans. 1, 1995, 2045–2048 RSC.
- (a) H. Fleisch, Endocr. Rev., 1998, 19, 80–100 CrossRef CAS PubMed; (b) J. Neyts and E. De Clercq, Antimicrob. Agents Chemother., 1997, 41, 2754–2756 CAS; (c) R. Snoeck, A. Holý, C. Dewolf-Peeters, J. Van Den Oord, E. De Clercq and G. Andrei, Antimicrob. Agents Chemother., 2002, 46, 3356–3361 CrossRef CAS PubMed; (d) M. V. Lee, E. M. Fong, F. R. Singer and R. S. Guenette, Cancer Res., 2001, 61, 2602–2608 CAS.
- (a) D. V. Patel, K. Rielly-Gauvin and D. E. Ryono, Tetrahedron Lett., 1990, 31, 5591–5594 CrossRef CAS; (b) J. F. Dellaria Jr, R. G. Maki, H. H. Stein, J. Cohen, D. Whittern, K. Marsh, D. J. Hoffman, J. J. Plattner and T. J. Perun, J. Med. Chem., 1990, 33, 534–542 CrossRef CAS; (c) T. R. Burke Jr, Z.-H. Li, J. B. Bolen and V. E. Marquez, J. Med. Chem., 1991, 34, 1577–1581 CrossRef CAS; (d) B. Stowasser, K. H. Budt, L. Jian-Qi, A. Peyman and D. Ruppert, Tetrahedron Lett., 1992, 33, 6625–6628 CrossRef CAS; (e) J. A. Sikorski, M. J. Miller, D. S. Braccolino, D. G. Cleary, S. D. Corey, J. L. Font, K. J. Gruys, C. Y. Han, K.-C. Lin, P. D. Pansegrau, J. E. Ream, D. Schnur, A. Shah and M. C. Walker, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 76, 115–118 CrossRef; (f) D. V. Patel, K. Rielly-Gauvin, D. E. Ryono, C. A. Free, W. L. Rogers, S. A. Smith, J. M. DeForrest, R. S. Oehl and E. W. Petrillo Jr, J. Med. Chem., 1995, 38, 4557–4569 CrossRef CAS PubMed; (g) M. Tao, R. Bihovsky, G. J. Wells and J. P. Mallamo, J. Med. Chem., 1998, 41, 3912–3916 CrossRef CAS PubMed.
- (a) O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS; (b) T. Gajda, Tetrahedron: Asymmetry, 1994, 5, 1965–1972 CrossRef CAS; (c) T. Yokomatsu, Y. Yoshida and S. Shibuya, J. Org. Chem., 1994, 59, 7930–7933 CrossRef CAS; (d) D. L. Hughes, The Mitsunobu Reaction in Organic Reactions, ed. L. A. Paquette et al., John Wiley & Sons, Inc., New York, 2004, vol. 42, pp. 335–656 Search PubMed; (e) D. Skropeta, R. Schwörer and R. R. Schmidt, Bioorg. Med. Chem. Lett., 2003, 13, 3351–3354 CrossRef CAS PubMed.
- (a) X. Creary, C. C. Geiger and K. Hilton, J. Am. Chem. Soc., 1983, 105, 2851–2858 CrossRef CAS; (b) X. Creary and T. L. Underiner, J. Org. Chem., 1985, 50, 2165–2170 CrossRef CAS; (c) T. Hanaya, A. Miyoshi, A. Noguchi, H. Kawamoto, M. Armour, A. M. Hogg and H. Yamamoto, Bull. Chem. Soc. Jpn., 1990, 63, 3590–3594 CrossRef CAS; (d) R. Gancarz, I. Gancarz and U. Walkowiak, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 104, 45–52 CrossRef CAS.
- J. Bird, R. C. De Mello, G. P. Harper, D. J. Hunter, E. H. Karran, R. E. Markwell, A. J. Miles-Williams, S. S. Rahman and R. W. Ward, J. Med. Chem., 1994, 37, 158–169 CrossRef CAS PubMed.
- (a) A. A. Thomas and K. B. Sharpless, J. Org. Chem., 1999, 64, 8379–8385 CrossRef CAS PubMed; (b) M. Otmar, L. Poláková, M. Masojídková and A. Holý, Collect. Czech. Chem. Commun., 2001, 66, 507–516 CrossRef CAS; (c) C. Pousset and M. Larchevêque, Tetrahedron Lett., 2002, 43, 5257–5260 CrossRef CAS; (d) D. G. Piotrowska and A. E. Wróblewski, Tetrahedron, 2003, 59, 8405–8410 CrossRef CAS; (e) D. G. Piotrowska and A. E. Wróblewski, Tetrahedron, 2009, 65, 4310–4315 CrossRef CAS; (f) A. E. Wróblewski and J. Drozd, Tetrahedron: Asymmetry, 2011, 22, 200–206 CrossRef.
- X. Wang, Y. Cai, J. Chen and F. Verpoort, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1268–1273 CrossRef CAS.
- (a) J. A. Mikroyannidis and A. K. Tsolis, J. Heterocycl. Chem., 1982, 19, 1179–1183 CrossRef CAS; (b) G. Pallikonda and M. Chakravarty, RSC Adv., 2013, 3, 20503–20511 RSC.
- N. Z. Kiss, Z. Rádai, Z. Mucsi and G. Keglevich, Heteroat. Chem., 2016, 27, 260–268 CrossRef CAS.
- J.-C. Monbaliu and J. Marchand-Brynaert, Tetrahedron Lett., 2008, 49, 1839–1842 CrossRef CAS.
- (a) M. I. Kabachnik and T. Medved, Izv. Akad. Nauk, Ser. Khim., 1953, 1126–1128 CAS; (b) M. I. Kabachnik and T. Medved, Izv. Akad. Nauk, Ser. Khim., 1954, 1024–1026 CAS.
- K. A. Petrov, V. A. Chauzov and T. S. Erokhina, Zh. Obshch. Khim., 1975, 45, 737–748 CAS.
- V. I. Krutikov, A. I. Lavrentiev and E. W. Sukhanowskaya, Zh. Obshch. Khim., 1991, 61, 1321–1325 CAS.
- V. I. Galkin, E. R. Zvereva, A. A. Sobanov, I. V. Galkina and R. A. Cherkasov, Zh. Obshch. Khim., 1993, 63, 2224–2227 CAS.
- I. V. Galkina, A. A. Sobanov, V. I. Galkin and R. A. Cherkasov, Russ. J. Gen. Chem., 1998, 68, 1398–1401 CAS.
- (a) N. S. Zefirov and E. D. Matveeva, ARKIVOC, 2008,(i), 1–17 CAS; (b) R. Gancarz, Tetrahedron, 1995, 51, 10627–10632 CrossRef CAS.
- T. Cytlak, M. Saweliew, M. Kubicki and H. Koroniak, Org. Biomol. Chem., 2015, 13, 10050–10059 CAS.
- (a) M. T. Reetz, Chem. Rev., 1999, 99, 1121–1162 CrossRef CAS PubMed; (b) M. Kaźmierczak and H. Koroniak, J. Fluorine Chem., 2012, 139, 23–27 CrossRef.
- A. E. Wróblewski and K. B. Balcerzak, Tetrahedron: Asymmetry, 2001, 12, 427–431 CrossRef.
- T. Katagiri, M. Takahashi, Y. Fujiwara, H. Ihara and K. Uneyama, J. Org. Chem., 1999, 64, 7323–7329 CrossRef CAS.
- R. Gancarz, I. Gancarz and A. Deron, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 161, 61–69 CrossRef CAS.
- B. Kaboudin and H. Zahedi, Chem. Lett., 2008, 37, 540–541 CrossRef CAS.
- (a) S. Fustero, E. Salavert, B. Pina, C. Ramírez de Arellano and A. Asensio, Tetrahedron, 2001, 57, 6475–6486 CrossRef CAS; (b) J. A. Barten, E. Lork and G.-V. Röschenthaler, J. Fluorine Chem., 2004, 125, 1039–1049 CrossRef CAS; (c) Z.-J. Liu, Y.-Q. Mei and J.-T. Liu, Tetrahedron, 2007, 63, 855–860 CrossRef CAS.
- M. N. Dimukhametov, E. V. Bayandina, E. Y. Davydova, A. T. Gubaidullin, I. A. Litvinov and V. A. Alfonsov, Mendeleev Commun., 2003, 13, 150–151 CrossRef.
- M. Rapp, P. Mrowiec and H. Koroniak, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 745–751 CrossRef CAS.
- A. Dondini and D. Perrone, Synthesis, 1997, 527–529 CrossRef.
- A. E. Wróblewski and W. T. Konieczko, Monatsh. Chem., 1984, 115, 785–791 CrossRef.
- (a) V. S. Abramov, Y. A. Bochkova and A. D. Polyakova, Zh. Obshch. Khim., 1953, 23, 1013–1016 CAS; (b) J. Li and P. Beak, J. Am. Chem. Soc., 1992, 114, 9206–9207 CrossRef CAS.
- F. Xichun, Q. Guofu, L. Shucai, T. Hanbing, W. Lamei and H. Xianming, Tetrahedron: Asymmetry, 2006, 17, 1394–1401 CrossRef.
- T. Ibuka, K. Nakai, H. Habashita, Y. Hotta, A. Otaka, H. Tamamura, N. Fujii, N. Mimura, Y. Miwa, Y. Chounan and Y. Yamamoto, J. Org. Chem., 1995, 60, 2044–2058 CrossRef CAS.
- H. Tamamura, T. Kato, A. Otaka and N. Fujii, Org. Biomol. Chem., 2003, 1, 2468–2473 CAS.
- (a) Y. Yamauchi, T. Kawate, T. Katagiri and K. Uneyama, Tetrahedron, 2003, 59, 9839–9847 CrossRef CAS; (b) I. D. Titanyuk, I. P. Beletskaya, A. S. Peregudov and S. N. Osipov, J. Fluorine Chem., 2007, 128, 723–728 CrossRef CAS.
- N. Katayama, S. Tsubotani, Y. Nozaki, S. Harada and H. Ono, J. Antibiot., 1990, 43, 238–246 CrossRef CAS PubMed.
- Y. Xu, M. Tanaka, H. Arai, J. Aoki and G. D. Prestwich, Bioorg. Med. Chem. Lett., 2004, 14, 5323–5328 CrossRef CAS PubMed.
- T. Yokomatsu, T. Yamagishi and S. Shibuya, Tetrahedron: Asymmetry, 1993, 4, 1401–1404 CrossRef CAS.
- T. Cytlak, M. Kaźmierczak, M. Skibińska and H. Koroniak, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 602–620 CrossRef CAS.
- F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899–932 CrossRef CAS PubMed.
- M. Ordonez, J. L. Viveros-Ceballos, C. Cativiela and A. Arizpe, Curr. Org. Synth., 2012, 9, 310–341 CrossRef CAS.
- CrysAlis PRO (Version 1.171.38.41), Rigaku Oxford Diffraction, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- K. Thai, L. Wang, T. Dudding, F. Bilodeau and M. Gravel, Org. Lett., 2010, 12, 5708–5711 CrossRef CAS PubMed.
- (a) Y. St-Denis and T. H. Chan, J. Org. Chem., 1992, 57, 3078–3085 CrossRef CAS; (b) C. Mazzini, L. Sambri, H. Regeling, B. Zwanenburg and G. J. F. Chittenden, J. Chem. Soc., Perkin Trans. 1, 1997, 3351–3356 RSC.
- E. Richmond, K. B. Ling, N. Duguet, L. B. Manton, N. Çelebi-Ölçüm, Y.-H. Lam, S. Alsancak, A. M. Z. Slawin, K. N. Houk and A. D. Smith, Org. Biomol. Chem., 2015, 13, 1807–1817 CAS.
- (a) P. Garner, Tetrahedron Lett., 1984, 25, 5855–5858 CrossRef CAS; (b) P. Garner and J. M. Park, J. Org. Chem., 1987, 52, 2361–2364 CrossRef CAS.
- (a) J. Wu, S. Gao, G. Liao, H. Lin and A. Nie, Synth. Commun., 2012, 42, 2907–2916 CrossRef CAS; (b) C. A. Flentge, J. T. Randolph, P. P. Huang, L. L. Klein, K. C. Marsh, J. E. Harlan and D. J. Kempf, Bioorg. Med. Chem. Lett., 2009, 19, 5444–5448 CrossRef CAS PubMed; (c) S. Mirilashvili, N. Chasid-Rubinstein and A. Albeck, Eur. J. Org. Chem., 2010, 4671–4686 CrossRef CAS; (d) G. Berger, M. Gelbcke, E. Cauët, M. Luhmer, J. Nève and F. Dufrasne, Tetrahedron Lett., 2013, 54, 545–548 CrossRef CAS; (e) S. Kitahata, T. Chiba, T. Yoshida, M. Ri, S. Iida, A. Matsuda and S. Ichikawa, Org. Lett., 2016, 18, 2312–2315 CrossRef CAS PubMed.
- (a) P. L. Beaulieu and P. W. Schiller, Tetrahedron Lett., 1988, 29, 2019–2022 CrossRef CAS; (b) N. I. Martin, J. J. Woodward, M. B. Winter and M. A. Marletta, Bioorg. Med. Chem. Lett., 2009, 19, 1758–1762 CrossRef CAS PubMed.
- O. O. Kolodyazhnaya, A. O. Kolodyazhnaya and O. I. Kolodyazhnyi, Russ. J. Gen. Chem., 2014, 84, 169–170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compounds characterization, 1H, 13C, 31P and 2D NMR spectra of compounds and crystallographic data. CCDC 1568454–1568458 and 1569663. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra01656a |
| This journal is © The Royal Society of Chemistry 2018 |