
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEarly stage release control of an anticancer drug by drug-polymer miscibility in a hydrophobic fiber-based drug delivery system
Yue Yuana,
Kyoungju Choibc,
Seong-O Choi bc and
Jooyoun Kim*de
bc and
Jooyoun Kim*de
aDepartment of Textile Engineering, Chemistry and Science, North Carolina State University, Raleigh, NC 27695, USA
bDepartment of Anatomy and Physiology, Kansas State University, Manhattan, KS 66506, USA
cNanotechnology Innovation Center of Kansas State, Kansas State University, Manhattan, KS 66506, USA
dDepartment of Textiles, Merchandising and Fashion Design, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jkim256@snu.ac.kr
eResearch Institute of Human Ecology, Seoul National University, Seoul 08826, Republic of Korea
First published on 29th May 2018
Abstract
The drug release profiles of doxorubicin-loaded electrospun fiber mats were investigated with regard to drug-polymer miscibility, fiber wettability and degradability. Doxorubicin in hydrophilic form (Dox-HCl) and hydrophobic free base form (Dox-base) was employed as model drugs, and an aliphatic polyester, poly(lactic acid) (PLA), was used as a drug-carrier matrix. When hydrophilic Dox-HCl was directly mixed with PLA solution, drug molecules formed large aggregates on the fiber surface or in the fiber core, due to poor drug-polymer compatibility. Drug aggregates on the fiber surface contributed to the rapid initial release. The hydrophobic form of Dox-base was dispersed better with PLA matrix compared to Dox-HCl. When dimethyl sulfoxide (DMSO) was used as the solvent for Dox-HCl, the miscibility of drug in the polymer matrix was significantly improved, forming a quasi-monolithic solution scheme. The drug release from this monolithic matrix was slowest, and this slow release led to a lower toxicity to hepatocellular carcinoma. When an enzyme was used to promote PLA degradation, the release rates were closely correlated with degradation rates, demonstrating degradation was the dominant release mechanism. The possible drug release mechanisms were speculated based on the release kinetics. The results suggest that manipulation of drug-polymer miscibility and polymer degradability can be an effective means of designing drug release profiles.
Introduction
Cancer patients after surgical resection commonly receive postsurgical anticancer treatments to reduce the risk of recurrence. Due to the non-selective cytotoxicity of anticancer drugs, systemic treatments such as oral or intravenous administration may lead to severe side effects.1 With such concerns, targeted local drug delivery has been considered as an effective alternative for postsurgical treatment, which minimizes adverse side effects of systemic chemotherapy. As vehicles to carry therapeutic agents to target sites, fibrous materials have been studied extensively due to their advantages such as high specific surface area, porosity, and structural similarities to the extracellular matrix.2–6 Of the fiber-forming processes, electrospinning has been widely adopted as a versatile method to produce fibrous vehicles with fine-tuned morphology for effective delivery of therapeutic agents.1–3,5,7–20 For example, multi-axial electrospinning has been utilized to produce multi-compartmental fibers that allow multiple release stages or multi-drug delivery.5,10,18 This configuration has a potential application of treating patients who have developed resistance to specific drugs.To achieve the desired drug release profiles using polymeric carriers, polymers need to be strategically selected because the release rates are influenced by its degradability, wettability, diffusivity, etc.4,5,16,21,22 For degradable polymers, as their geometry changes during degradation, the release mechanism can be more complex than that of non-degradable drug-carriers.23 As a model anticancer drug, doxorubicin (Dox) has been studied in many researches1,24,25 due to its effectiveness for a wide range of cancers, including breast cancer, bladder cancer, lymphocytic leukemia and lymphoma. Dox exists either in a hydrophilic form, doxorubicin HCl (Dox-HCl) or in a hydrophobic form, doxorubicin free base (Dox-base),26 and their release profiles from polymeric carriers are influenced by the compatibility of drugs in a polymer/solvent system and by the structural characteristics of a drug carrier.24,27,28
While an earlier study29 partly demonstrated the effect of drug–polymer–solvent compatibility on drug release profiles, further analytical work is necessary to explain the phenomena. Of major interest of this study is to: (1) investigate the influence of drug–polymer miscibility on the drug release profiles from hydrophobic fiber mats; (2) analyze the drug localization in the polymeric matrix; and (3) examine the effectiveness of drug delivery by in vitro cytotoxicity assay. Drug-loaded fiber mats were prepared by electrospinning of solution precursors containing poly(lactic acid) (PLA) and either hydrophilic Dox (Dox-HCl) or hydrophobic Dox (Dox-base). The miscibility of polymer–drug–solvent system was altered by incorporating either hydrophilic or hydrophobic drugs directly in PLA solution or by utilizing a secondary solvent, dimethylsulfoxide (DMSO). The miscibility of drug and polymer was analyzed by observing the drug localization in polymer matrices. In vitro drug release profiles and release mechanisms were associated with drug–polymer miscibility, fiber wettability, and matrix degradation. Finally, the viability of hepatocellular carcinoma cell C3A with Dox-loaded fiber mats was examined. The findings of this study can be utilized as a simple selection guide of materials (drug, polymer, and solvents) in fabricating drug-loaded fiber mats, to attain desired drug release profiles from a hydrophobic fiber-based drug delivery system.
Materials and methods
Materials
Poly(lactic acid) (PLA) resin (Ingeo 4043D, 98% L-lactide, with weight average molecular weight of 111 kg mol−1 (ref. 30)) was purchased from NatureWorks (Minnetonka, MN, USA). Dimethyl sulfoxide (DMSO) and dimethylformamide (DMF) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Doxorubicin hydrochloride (99%, Dox-HCl) and doxorubicin free base (99%, Dox-base) were purchased from MedKoo Biosciences (Morrisville, NC, USA). Proteinase K (isolated from Tritirachium album) was purchased from Bio Basic (Amherst, NY, USA). Phosphate buffered saline (PBS, pH 7.4) and Tris–HCl buffer (pH 8.0) were purchased from Thermo Fisher (Waltham, MA, USA), and other chemicals were purchased from Fisher Scientific (Hampton, NH, USA) unless otherwise stated.Preparation of drug-loaded fiber webs
Prior to electrospinning of PLA mats (without drug), 10% (w/v) PLA solution was prepared in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of dichloromethane (DCM) and dimethyl formamide (DMF). For electrospinning (Spraybase®, Dublin, Ireland), a grounded aluminum collector was placed in front of a 22-gauge needle at a distance of 10 cm, and fibers were spun horizontally toward the collector rotating at 100 rpm. The feeding rate of a polymer solution was 1.5 mL h−1. The applied voltage was adjusted between 9.5–10.5 kV to produce non-beaded fibers.
:1 ratio of dichloromethane (DCM) and dimethyl formamide (DMF). For electrospinning (Spraybase®, Dublin, Ireland), a grounded aluminum collector was placed in front of a 22-gauge needle at a distance of 10 cm, and fibers were spun horizontally toward the collector rotating at 100 rpm. The feeding rate of a polymer solution was 1.5 mL h−1. The applied voltage was adjusted between 9.5–10.5 kV to produce non-beaded fibers.
For drug incorporation, either doxorubicin HCl (Dox-HCl) or free base (Dox-base) was added to the PLA polymer solution before electrospinning by different methods. The first method was mixing the drug particles directly into a 10% (w/v) PLA/DCM/DMF solution, stirring for 1 h at room temperature. The formulations for both PLA–HCl and PLA–base were: 0.02 g drug; 0.4 g PLA; 4 mL DCM/DMF (1:1 v/v). In another method, DMSO was used to dissolve Dox-HCl at 2% (w/v), and 1 mL of drug/DMSO solution was mixed with 4 mL of 10% (w/v) PLA solution. The formulations for PLA–HCl(DMSO) was: 0.02 g Dox-HCl; 1 mL DMSO; 0.4 g PLA; 4 mL DCM/DMF (1:1 v/v). The sample codes and descriptions are shown in Table 1.
| Code | Description |
|---|---|
| PLA | PLA web without drug |
| PLA-HCl | PLA web with Dox-HCl, drug was directly mixed in PLA/DCM/DMF solution |
| PLA-base | PLA web with Dox-base, drug was directly mixed in PLA/DCM/DMF solution |
| PLA-HCl(DMSO) | PLA web with Dox-HCl, drug was dissolved in DMSO, then mixed in PLA/DCM/DMF solution |
Characterization of drug-loaded fiber mats
Contact angle measurement
Static contact angles (CA) of 4 μL of distilled water on fibrous surfaces were measured by an optical tensiometer (Attension Theta, Biolin Scientific, Paramus, NJ, USA) at room temperature.31,32 All measurements were carried out at least five times from different locations of each sample.Differential scanning calorimetry (DSC)
Thermal properties of drug-loaded fibers were analyzed by DSC (Q20P, TA Instruments, New Castle, DE, USA). Approximately 3 mg samples of fiber webs were heated from 25 °C to 200 °C at a constant rate of 10 °C min−1 under a nitrogen atmosphere. The glass transition temperature (Tg), crystallization temperature (Tc) and melting temperature (Tm) were examined from the scanning. The degree of crystallinity was calculated by eqn (1), using a reference value H0 of 93 J g−1 for PLA.33
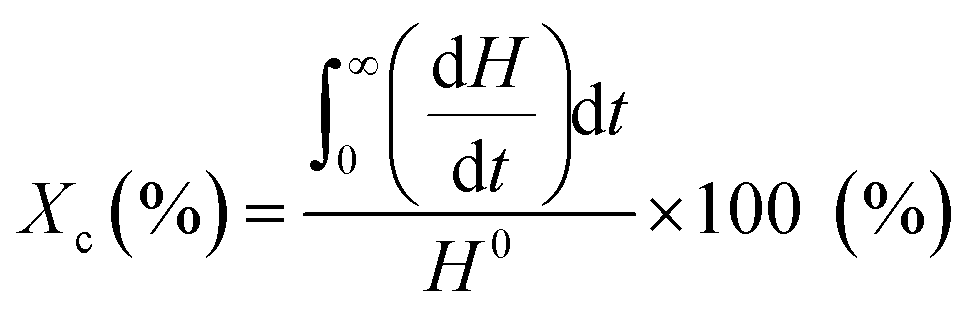 | (1) |
Calculation of drug loading content (LC) and loading efficiency (LE)
Drug loading content (LC%) was determined by the weight percentage of the drug that is actually contained in a fiber web (eqn (2)). For this measurement, drug-loaded fiber webs were completely dissolved in solvents (50% acetone/water for PLA), and the amount of drugs in the web was calculated from the fluorescence intensity of doxorubicin (at 600 nm emission) measured by a UV-visible micro plate reader (Synergy H1 Hybrid Multi-Mode Reader, BioTek, Winooski, VT, USA). Loading efficiency (LE%) of a drug-loaded fiber web was calculated by eqn (3).
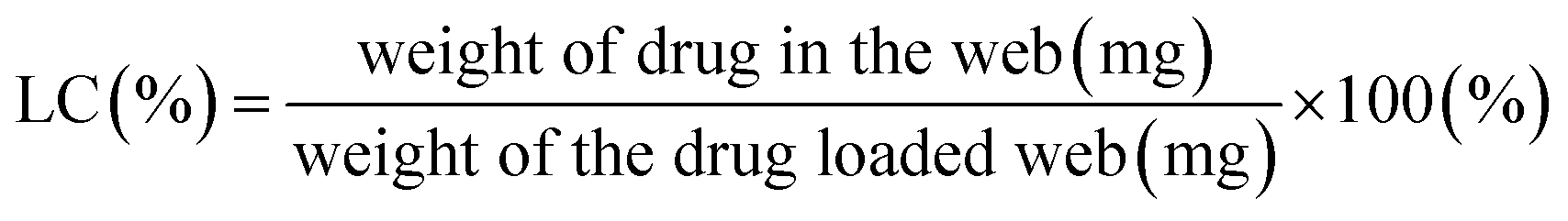 | (2) |
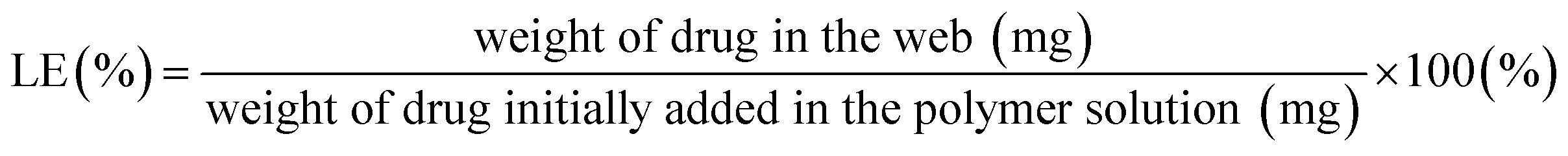 | (3) |
In vitro drug release analysis
Approximately 20 mg samples of the drug-loaded fiber mats were immersed in the vertical diffusion cell (PermeGear, Hellertown, PA, USA) with 3.5 mL of PBS (pH 7.4) and incubated at 37 °C under magnetic stirring at 100 rpm. At each sampling time, 3.5 mL of drug-released PBS medium was replaced with a fresh 3.5 mL PBS. The amount of drug released from the fiber webs at each sampling interval was quantified by comparing with a calibration curve of fluorescence intensities versus doxorubicin (Dox) concentrations. The cumulative percentage of released drug at each sampling point was calculated by eqn (4). All experiments were performed in triplicate and replicated at least two times.
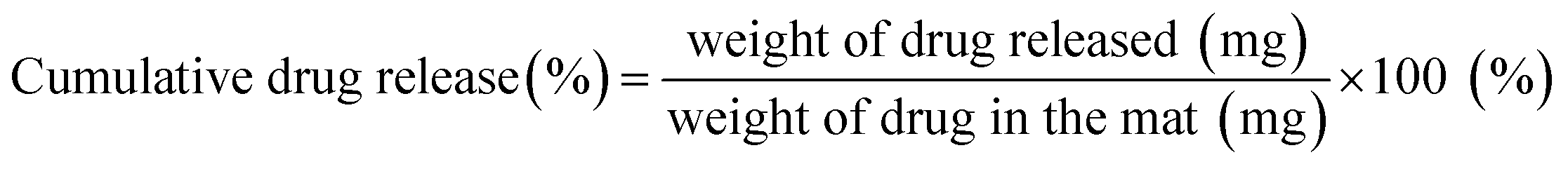 | (4) |
Accelerated degradation of PLA and release of doxorubicin (Dox)
PLA degradation was accelerated using proteinase K enzyme, and the Dox release profiles with PLA degradation was investigated. Enzymatic degradation was conducted in 20 mM Tris–HCl buffer solution (pH 8.0) with 0.5 mg mL−1 of proteinase K at 37 °C. Fiber samples weighing approximately 20 mg were cut and placed in a 3.5 mL Tris–HCl buffer solution. At each predetermined time, the samples were taken out from the buffer and dried at 45 °C for about 4.5 h, and the weight of the dried samples was measured. The enzyme solution was replaced at each measurement point to ensure the activity of proteinase K. The weight loss (%) of the drug-loaded fibers was calculated at each sampling time by eqn (5).
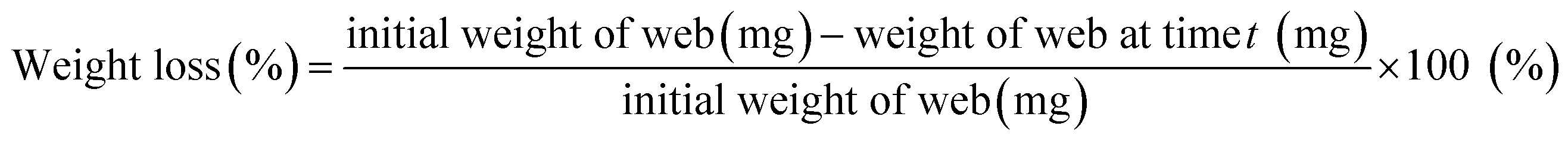 | (5) |
Cell culture
Hepatocellular carcinoma cell C3A were obtained from ATCC® (Manassas, VA). C3A cells were cultured in complete Eagle's Minimum Essential Medium (EMEM) supplemented with 10% fetal bovine serum; and expanded to approximately 80% confluence in T75 flask with medium changes every 4 days. After 0.25% (w/v) trypsin–0.53 mM ethylenediaminetetraacetic acid (EDTA) digestion, cells between passage 9 and 12 with an average viability of ∼90% were plated in the 24-well plates at 6.25 × 105 cells per well followed by an incubation at 37 °C in a humidified atmosphere of 95% air and 5% CO2. After 48 h incubation, cells were ready for dosing.Cytotoxicity of drug-loaded and drug-free PLA
The viability of C3A cells was determined using the alamarBlue® viability assay (Thermo Sci., Waltham, MA, USA) as previously described.34 Cells seeded in the 24-well plates were treated with Dox HCl-loaded PLA (42 μg mL−1 to 420 μg mL−1), Dox base-loaded PLA (126.7 μg mL−1 to 1266.7 μg mL−1), and Dox HCl(DMSO)-loaded PLA (30.7 μg mL−1 to 306.7 μg mL−1) as well as Dox-free PLA. At the end of 5 h and 24 h of incubation, 10% of alamarBlue® reagent in complete EMEM (v/v) was added to the cell culture and incubated for 3 h at 37 °C. Fluorescence (excitation 555/emission 585 nm) was quantified with the Synergy H1 hybrid multi-mode microplate reader. Controls assessed interaction between Dox and the active ingredient of alamarBlue® reagent, resazurin or a reduced product, resorufin. Resazurin (no cells) and culture medium (no cells) served as background controls. Fluorescence, proportional to cell viability, was normalized with respect to controls and expressed as percent viability.Results and discussion
Effect of drug-polymer miscibility on drug localization
The effect of drug-polymer miscibility on fiber morphology and drug distribution was observed in Fig. 1. When hydrophilic Dox-HCl was directly added to hydrophobic PLA polymer solution (PLA in DCM/DMF), the drug was neither dissolved nor stably dispersed in PLA solution and formed large drug aggregates (∼20 μm). The bright-field and fluorescence images in Fig. 1c and d clearly show Dox-HCl aggregates at the surface of fiber mats. Due to the immiscibility of Dox-HCl in the polymer, the PLA–HCl fiber mat appeared lighter in color with red speckles (Fig. 1b). PLA–base mat showed rather uniform dispersions of drug particles compared to PLA–HCl (Fig. 1c and d), due to better miscibility between hydrophobic Dox-base and hydrophobic PLA.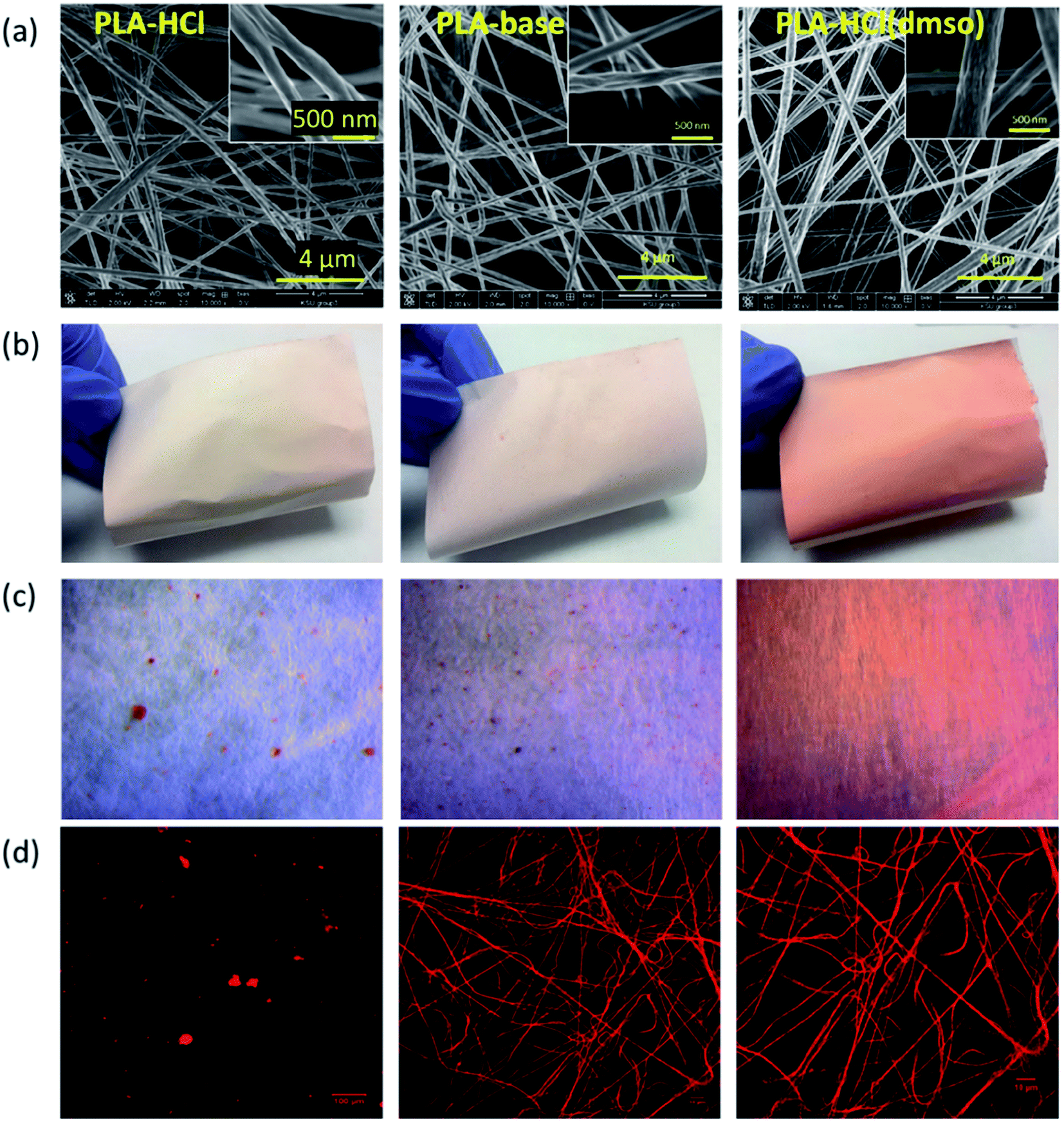 | ||
| Fig. 1 Photographic and microscopic images of doxorubicin-loaded fiber mats. (a) SEM images of drug-loaded PLA fibers; (b) fiber mats producing different colors depending on drug–polymer miscibility; (c) optical images displaying dissolution or aggregation of drug crystals in fiber webs; (d) fluorescence images by the fluoresced doxorubicin. | ||
When DMSO was used as a solvent for Dox-HCl, the drug was homogeneously distributed within fibers, indicated by more saturated color coming from doxorubicin red. While DMSO itself was not a good solvent for PLA, the use of DMSO as a solvent for drug in a PLA/DCM/DMF system helped the miscibility between Dox and PLA. Due to this enhanced miscibility of drug in the polymer matrix, there were little drug particles observable from the optical images of PLA–HCl(DMSO). From the fluorescence images (Fig. 1d), PLA–base and PLA–HCl(DMSO) showed uniform internalization of Dox in polymer matrices, which may be due to the enhanced dispersion or dissolution. Also, the red aggregates in PLA–HCl was identified to be doxorubicin crystals (not PLA polymer aggregates) by electron crystallography. The selected area diffraction pattern (SADP) of PLA–HCl sample in Fig. 2 clearly shows the crystalline phase of a Dox-HCl particle (region 1) and the amorphous phase of PLA fiber (region 2).
 | ||
| Fig. 2 Doxorubicin crystal aggregates in PLA–HCl mat observed by an optical microscope (a) and TEM (b). Dox particle is in crystalline phase (1) while PLA fiber was mostly amorphous (2). | ||
To further investigate the localization of doxorubicin in fibrous matrices, the cross-sections of Dox-loaded PLA fibers were observed by TEM (Fig. 3). Due to the difference of electron density between PLA and doxorubicin molecules, doxorubicin-rich region appeared darker than PLA. In Fig. 3a, the cross-section of PLA–HCl fibers exhibited obvious distinction between Dox-rich and PLA-rich regions. Due to the lack of miscibility between hydrophilic Dox-HCl and hydrophobic PLA, the cross-sectional area of the fiber was separated into two phases; Dox-rich region was observed in the core, surrounded by PLA-rich region. As the doxorubicin red was masked by PLA matrix, the PLA–HCl web appeared lighter in color in Fig. 1a. Taken together from Fig. 1 and 3, the immiscible Dox-HCl drug aggregates were localized in PLA matrix, partly in the core region and partly on the surface of fibers.
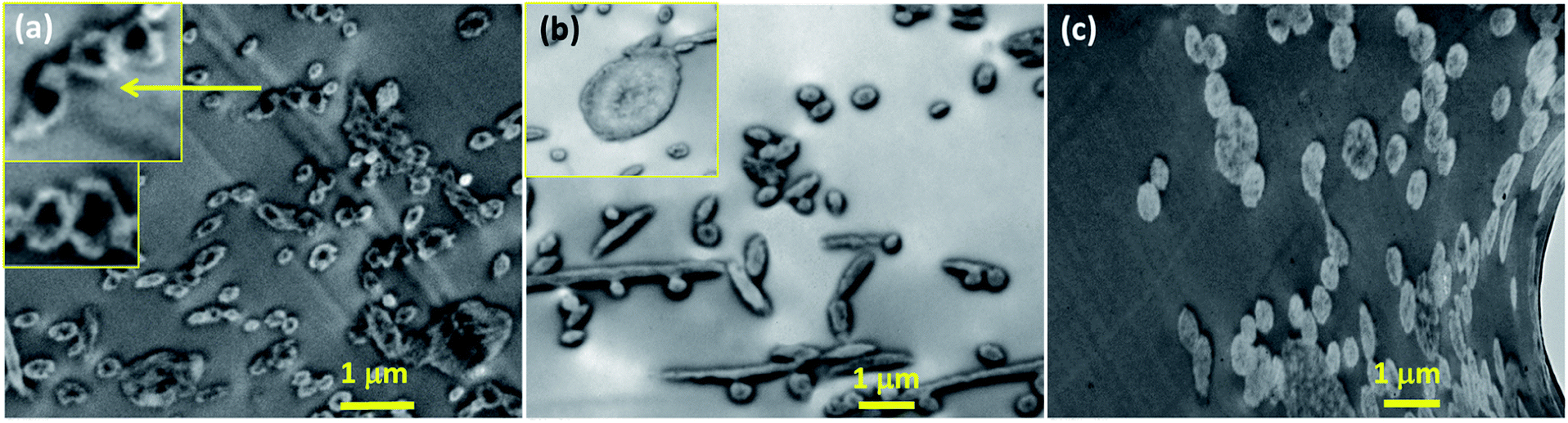 | ||
| Fig. 3 TEM cross-sectional images of Dox-loaded fibers. (a) PLA–HCl; (b) PLA–base; (c) PLA–HCl(DMSO). Note: the magnification of (a) is different, thus the length of 1 μm in (a) appears different. | ||
When DMSO was used as a solvent for Dox-HCl, the composition of drug–polymer matrix appeared homogeneous, and Dox-rich and polymer-rich regions were hardly separable, forming a monolithic-like compound (Fig. 3c). However, the PLA–base showed somewhat segregated Dox-rich phase in the fiber (Fig. 3b), though Dox-rich and PLA-rich regions were not as clearly separable as those in PLA–HCl. As shown in Fig. 1c, the miscibility of PLA–base was better than PLA–HCl but worse than PLA–HCl(DMSO). This intermediate miscibility of PLA–base among the tested is evidenced by the cross-sectional image (Fig. 3b) that shows partly localized drug molecules in PLA fibers. The results demonstrate that the use of DMSO as a solvent for the drug is beneficial for improving the drug–polymer miscibility and for forming homogeneous, monolithic-like drug-loaded mats.
As another parameter that could influence drug release profiles, wettability of drug-loaded fiber mats was examined. Previous study reported that the hydrophobicity of a drug-loaded fibers can delay the drug release by the impeded water permeation into the polymer matrix, and this hydrophobic characteristic was utilized to design delayed drug release profile.2,5,7,35 As a fibrous surface provides high specific surface area and porosity, the effect of wettability of fiber mats on drug release can be larger and complex than that of non-porous solid films.
The measured water contact angles (CA) of drug-loaded fibers are presented in Table 2. The CA of PLA web without drug was 160°. When the hydrophobic Dox-base was added to it, PLA–base maintained the same level of hydrophobicity. However, when the hydrophilic Dox-HCl was added, CA decreased to 139°. The CA measurement for PLA–HCl gave a large standard deviation because CA depended on the presence of Dox-HCl aggregates at the measured sites. Also, CA decreased to 148° when DMSO was added to help the dissolution of doxorubicin, yet the CA measurements of PLA–HCl(DMSO) produced a smaller standard deviation due to the homogeneous nature of the matrix.
| Sample | Fiber dia. (μm) | CA° | LC (wt%) | LE (wt%) | Morphology |
|---|---|---|---|---|---|
| PLA | 0.31 (±0.03) | 160° (±2.8) | NA | NA | Non-beaded, cylindrical fibers with grooves |
| PLA–HCl | 0.32 (±0.05) | 139° (±14.6) | 3.5 (±0.2) | 74 (±3.2) | Large Dox-HCl aggregates from the surface |
| PLA–base | 0.30 (±0.03) | 160° (±4.1) | 4.8 (±0.4) | 100 (±7.5) | Small Dox-base from the surface |
| PLA–HCl(DMSO) | 0.39 (±0.05) | 148° (±2.4) | 5.1 (±0.1) | 107 (±2.0) | No observable Dox-HCl particles |
The influence of drug–polymer miscibility on drug loading efficiency (LE) and loading content (LC) was examined. When producing electrospun mats, Dox was added to PLA by 4.8% (w/w). For PLA–base and PLA–HCl(DMSO), loading efficiency (LE) reached 100%, as the drug molecules were stably incorporated in the polymer matrix at 4.8% concentration. However, PLA–HCl had only 74% LE, producing the actual drug content of 3.5% (w/w), implicating that hydrophilic Dox-HCl was not stably blended in hydrophobic PLA. For PLA–HCl, precipitates of Dox-HCl particles were visually observed. Characteristics of drug-loaded fibers, including fiber diameters, water contact angle (CA), drug loading content (LC), loading efficiency (LE) and morphology of fibers, are summarized in Table 2.
The influence of drug addition to PLA on thermal properties and crystallinities were examined by DSC (Table 3). The temperatures for melting and crystallization were not changed by the addition of Dox to PLA. When DMSO was used as a solvent for Dox, the glass transition temperature (Tg) slightly decreased, probably due to the plasticization by the DMSO dissolved in PLA.36,37 When the drug was added to the polymer without DMSO, Tg was not affected; without DMSO, drug was not completely dissolved in the polymer matrix, which may result in no significant influence on Tg or intermolecular interactions. The addition of drug particles increased the crystallinity (%) because the drug crystals acted as a nucleating agent in the polymer, thereby facilitating crystal growth.38 The results show that the fiber mats had lower degree of crystallinity when DMSO was used as the drug solvent. Previous studies have demonstrated that the degree of crystallinity is influenced by the solvent evaporation rate.39,40 With the addition of DMSO (vapor pressure at 20 °C ∼55.6 Pa), the solvent evaporation rate of polymer solution would have been lowered (vapor pressure of DMF at 20 °C ∼360 Pa; vapor pressure of DCM at 20 °C ∼47091 Pa), adversely influencing the crystalline development. The results of this study indicate that use of DMSO helped the homogenous blending of Dox in PLA matrix, and the presence of DMSO in the polymer solution hindered intermolecular interactions, thereby decreasing Tg and crystallinity.
| Sample | PLA | PLA–HCl | PLA–base | PLA–HCl(DMSO) |
| Tg (°C) | 61.1 | 60.0 | 60.0 | 47.1 |
| Tc (°C) | 89.7 | 87.9 | 87.0 | 86.9 |
| Tm (°C) | 150.0 | 150.8 | 150.0 | 150.4 |
| Crystallinity (%) | 12.9 | 15.6 | 16.3 | 9.4 |
In vitro drug release profiles
Fig. 4 shows in vitro drug release profiles of the Dox-PLA fiber mats. Regardless of drug-polymer miscibility or wettability, all samples showed the rapid release of Dox within 5 h followed by the sustained release. However, drug release rates were distinct depending on drug hydrophilicity and the incorporation method to PLA matrix. PLA–HCl in which Dox-HCl formed crystal aggregates exhibited the fastest release during 5 h (50% of Dox-HCl was released). The rapid release of Dox-HCl in the earlier stage should be attributed to the dissolution of Dox-HCl aggregates exposed at the fiber surface. It is also possible that the dissolution of Dox-HCl particles on the surface created pores and voids in the mat, facilitating the access of release medium into the fibrous web. The reduced hydrophobicity (CA ∼ 139°) could be another contributor to the faster release. PLA–HCl exhibited extra 10–15% of release between 5 h and 24 h. The remaining Dox-HCl enveloped inside the core region of fibers (Fig. 3a), seemingly about 35% of total loaded drug, was not released until 6 days. The drugs in the core would be released when the polymer degradation or erosion occurs, and the release with degradation is discussed in the later section of this study.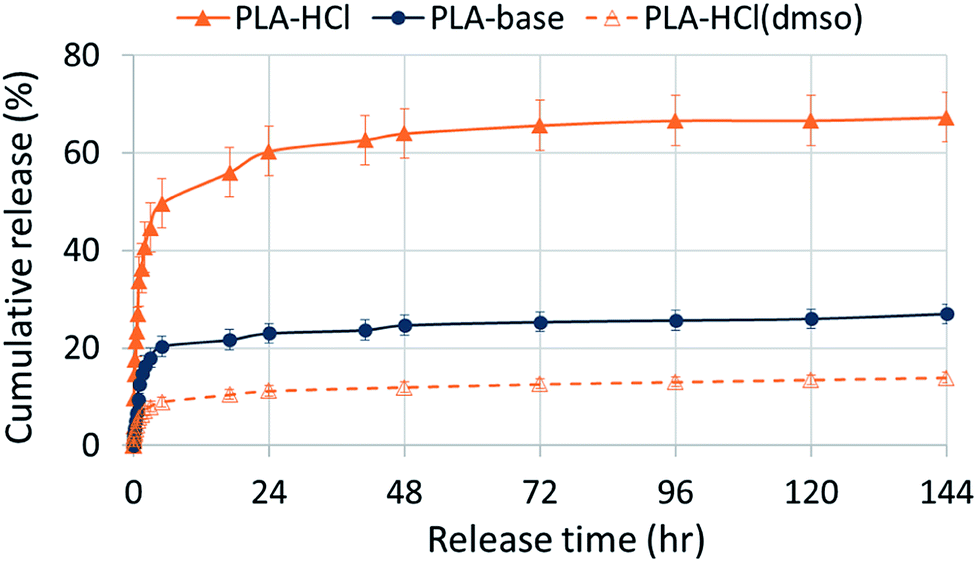 | ||
| Fig. 4 In vitro release profiles of doxorubicin from PLA fibrous carriers. | ||
PLA–base showed slower release rates than PLA–HCl. As the PLA–base improved the miscibility with the hydrophobic drug, the drug particles were dispersed rather uniformly, forming less aggregates compared to PLA–HCl. The initial 20% release during 5 h seemed to result from the dissolution of Dox-base particles dispersed on the fiber surface. The remaining drug embedded inside the polymer was then hardly released within 6 days of experiment. Generally, hydrophobic carriers such as PLA exhibit sustained release of internalized drugs by the impeded permeation of medium into the polymer matrix.
PLA–HCl(DMSO) exhibited the slowest release rate. The use of DMSO as a solvent for drugconsiderably improved the miscibility of Dox and PLA, leading to formation of homogeneous and monolithic-like matrix. As a monolithic compound, the embedded drug was hardly diffused out of the polymer matrix. About 10% of the loaded was released during the initial 5 h period. The dominant release mechanism for such a homogeneous matrix would be polymer degradation (or polymer erosion). It is noteworthy that increased wettability and decreased crystallinity of PLA–HCl(DMSO) had little influence on increasing release rates.
Drug release analysis with accelerated polymer degradation
The release profiles in Fig. 4 exhibited two different release stages. The faster release during the earlier stage seemed to be dominantly attributed to rapid dissolution of Dox deposited or embedded near the surfaces. The earlier release rate was the highest in PLA–HCl where large Dox aggregates were observed on the mat surface. For all samples, later stage of release was much slower compared to the first 5 h of release. As PLA is hardly swellable and its degradation takes a long period of time in buffer solutions (as long as 6 months),41 release mechanisms associated with degradation/erosion would occur very limitedly. Speculating that the later stage of drug release would be dominated by the polymer degradation, the release profile with accelerated degradation was further investigated.Fig. 5 shows the drug-loaded fibers that had gone through accelerated degradation by proteinase K for different time periods (1–16 h). The color of PLA–HCl and PLA–base changed as degradation proceeded, from their originally light color to doxorubicin red, due to staining of the mat as Dox was released. The weight loss (%) of the drug-loaded fiber is shown in Fig. 6a. The weight loss was faster for PLA–HCl and PLA–base than PLA–HCl(DMSO). The quick dissolution of drug particles/aggregates from those fibers would have created pores and voids in the fibers, and this might have facilitated the degradation process for PLA–HCl and PLA–base. The degradation rate of PLA–HCl(DMSO) was slower due to its homogeneous monolithic-like nature. From the DSC results, the crystallinity of PLA–HCl(DMSO) was slightly lower than that of the other two samples. However, the influence of crystallinity on degradation rates seems to be negligible in this study. It appears that there is a threshold crystallinity that influences the degradation, as Cai et al.42 explained. The study reported that the degradation of PLA can be decelerated when the degree of crystallinity is greater than 22%. As all the fibers in this study showed degree of crystallinities lower than this threshold, the influence of crystallinity on degradation rate was negligible.
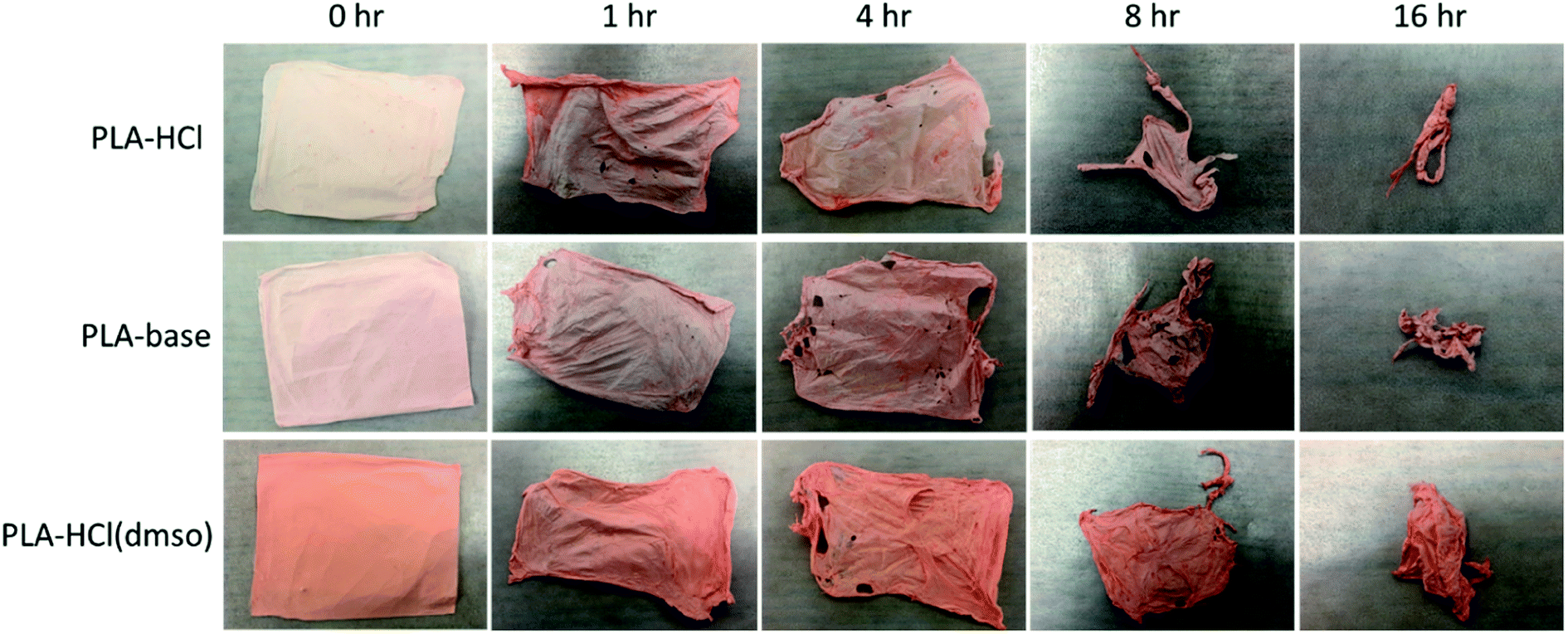 | ||
| Fig. 5 Accelerated degradation of drug-loaded mats in the presence of proteinase K. | ||
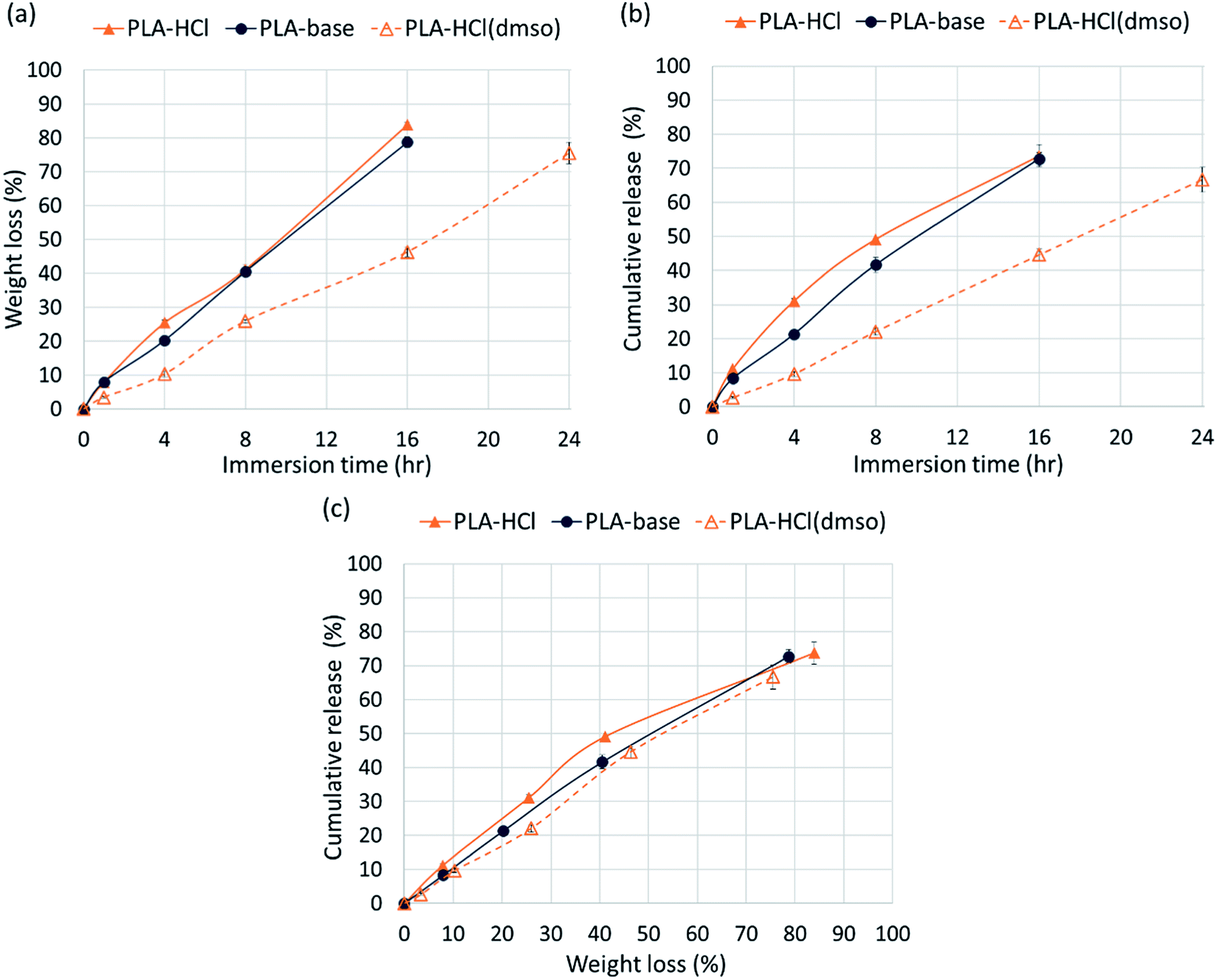 | ||
| Fig. 6 Drug release profiles with the accelerated degradation. (a) Weight loss (%) of fiber mats for 24 h; (b) cumulative drug release (%) of mats with time; (c) Dox release as a function of weight loss (%). | ||
From Fig. 6c, the release rates are linearly proportional to the degradation rates (weight loss), demonstrating that PLA degradation/erosion is the dominant drug release mechanism. This linear correlation between degradation (weight loss) and release was slightly deviant for PLA–HCl; the release was slightly higher than weight loss till 60% of degradation occurred. This result implicates that the quick dissolution of drug aggregates from PLA–HCl contributed additionally to the release mechanisms. Overall, the result from the accelerated degradation allows the prediction of release behavior when degradation is the dominant release mechanism.
Drug release kinetics
Drug-loaded fiber mats can exhibit several different release mechanisms, including immediate dissolution of drug molecules, permeation of fluid into mats followed by the diffusion of drug molecules and polymer degradation/erosion. To examine the release kinetics, the empirical power law shown in eqn (6)26,43,44 was applied to the results (Fig. 7).
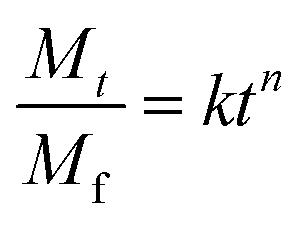 | (6) |
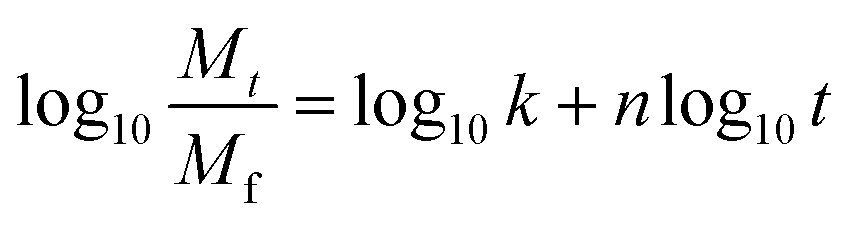 | (7) |
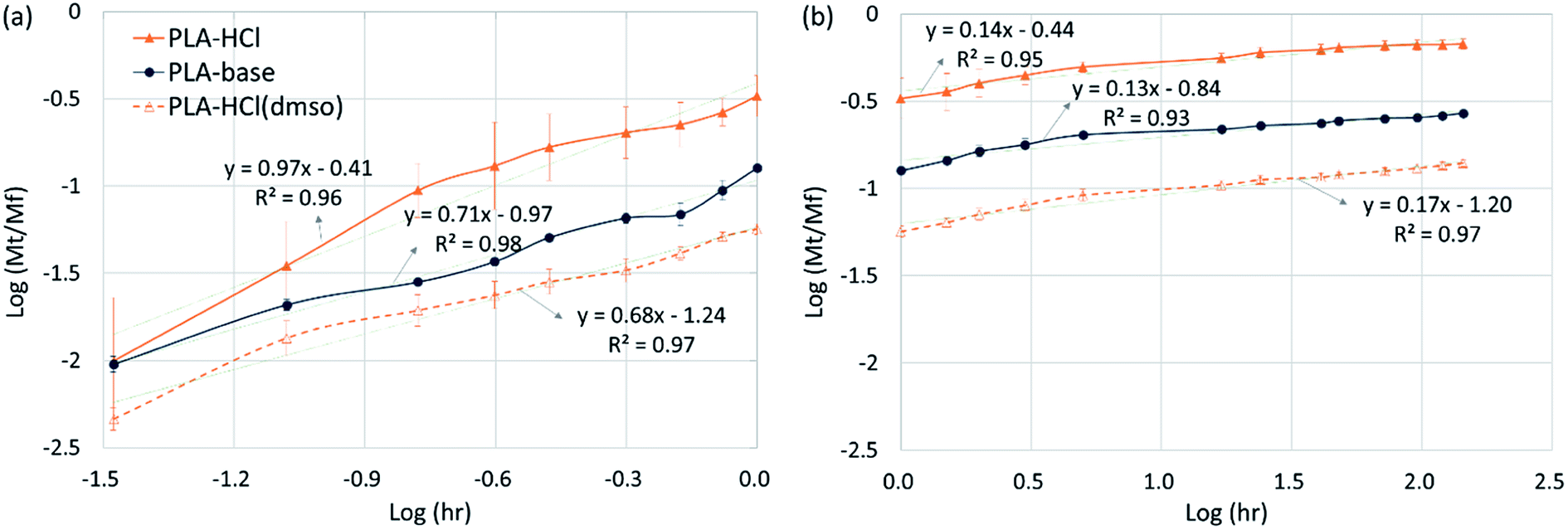 | ||
| Fig. 7 Drug release kinetics. (a) Initial stage release (0 to 1 h); (b) later stage release (1 h to 6 days). | ||
In eqn (7), the exponent ‘n’ has been used to explain the release mechanisms for different matrix geometries.26,43,44 For example, n for the Fickian release from slabs, cylinders and spheres are 0.50, 0.45 and 0.43, respectively, and those for zero-order release from non-swelling system is 1.26,44 The geometry of the drug delivery system in this study can be represented as cylinders that form a slab. For PLA–HCl in which the drugs were directly added without DMSO, drug molecules were not completely dissolved in the polymer solution, and the drug-rich region was separated from the polymer-rich region. For this fiber mat, drug particles and aggregates were localized in the core of fiber or at the surface of fiber mat, and this form of drug delivery system was assumed to be a ‘reservoir with deposits’.23 For PLA–base, drug particles were rather homogeneously mixed (either dissolved or dispersed) in the matrix like a ‘monolithic dispersion’.23 For PLA–HCl(DMSO), the drug molecules were homogeneously dissolved within the polymeric matrix like the ‘monolithic solution’.23 The release mechanisms would be affected by the schemes of drug delivery systems such as reservoir with deposits, monolithic dispersions and monolithic solution.
For all the fiber mats tested, there were two distinct stages of release kinetics. In Table 4, the slope n for two different release stages are presented (all R2 values were greater than 0.93). The very initial release period of 0–1 h (Fig. 7a) exhibited larger n with faster releases while the later period had similarly small n values (0.10–0.17). In the initial stage of release, both immediate dissolution of drug molecules and diffusion at the near surface would occur. The initial stage of exponent n was in the order of PLA–HCl > PLA–base > PLA–HCl(DMSO). The trend corresponded with the order of immiscibility between the drug and matrix.
| PLA–HCl | PLA–base | PLA–HCl(DMSO) | ||
|---|---|---|---|---|
| Scheme of drug delivery system23 | Slab reservoir with surface deposits | Slab with monolithic dispersions | Slab with monolithic solutions | |
| Initial stage release (0–1 h) | n | 0.97 | 0.71 | 0.68 |
| (R2) | (R2 = 0.96) | (R2 = 0.98) | (R2 = 0.97) | |
| Mech | Dissolution of drug particles near surfaces, diffusion | |||
| Later stage release (1 h to 6 d) | n | 0.14 | 0.13 | 0.17 |
| (R2) | (R2 = 0.95) | (R2 = 0.93) | (R2 = 0.97) | |
| Mech | Degradation/erosion, diffusion | |||
For monolithic solutions of PLA–HCl(DMSO), the exponent n was smaller compared to the monolith dispersions (PLA–base) and reservoir with deposits (PLA–HCl). As most of drug molecules were homogeneously distributed in the matrix, the initial rapid rate may result from the diffusion of drug molecules near the fiber surface. The n for PLA–HCl(DMSO) was 0.64, which represents the dominant mechanism of non-Fickian, anomalous diffusion.
The later release stage (1 h to 6 days) exhibited small n (0.13–0.17) regardless of the scheme of drug delivery systems. Small n for the later release appears to be associated with the slow degradation of PLA polymer. As PLA is rarely swellable, drug dispersions and solutes in the bulk matrix would be released mostly when PLA degrades. The low n demonstrates that PLA degradation within the test period occurred only limitedly. In the meanwhile, when the drug molecules near the surface are diffused or dissolved out, pores and voids could be created in the matrix, which could accelerate the degradation process by increasing the surface area exposed to the external aqueous environment. From the result of the accelerated degradation, the released amount of drug was linearly correlated with the degradation rate. This indicates that the negligible drug release in the later stage is attributed to slow degradation of PLA.
The possible drug release mechanisms for Dox-loaded PLA fiber mats are illustrated in Fig. 8. The results indicate that the drug release from the polymeric fibers is influenced by the drug–polymer miscibility, fiber wettability and degradability. As PLA is hardly swelling, drug diffusion through polymer matrix would be very limited. However, when drug molecules and particles are dispersed near the surface of polymer matrices or in the pore/voids of mats, diffusion mechanism could partially contribute to the overall release. PLA degradation/erosion would take months of process, and drug release by polymer degradation hardly occurred within a week of experiment. The monolithic solution of PLA–HCl(DMSO) exhibited sustained drug release as PLA degradation is the dominant release mechanism, and the degradation takes only for a prolonged time. When degradation was accelerated by the enzymatic activity, drug release was exactly correlated with the degradation rate, corroborating that the release by degradation/erosion was the dominant mechanism for PLA–base and PLA–HCl(DMSO). The results suggest that blending with hydrophilic or readily biodegradable polymer may significantly accelerate drug release rates.
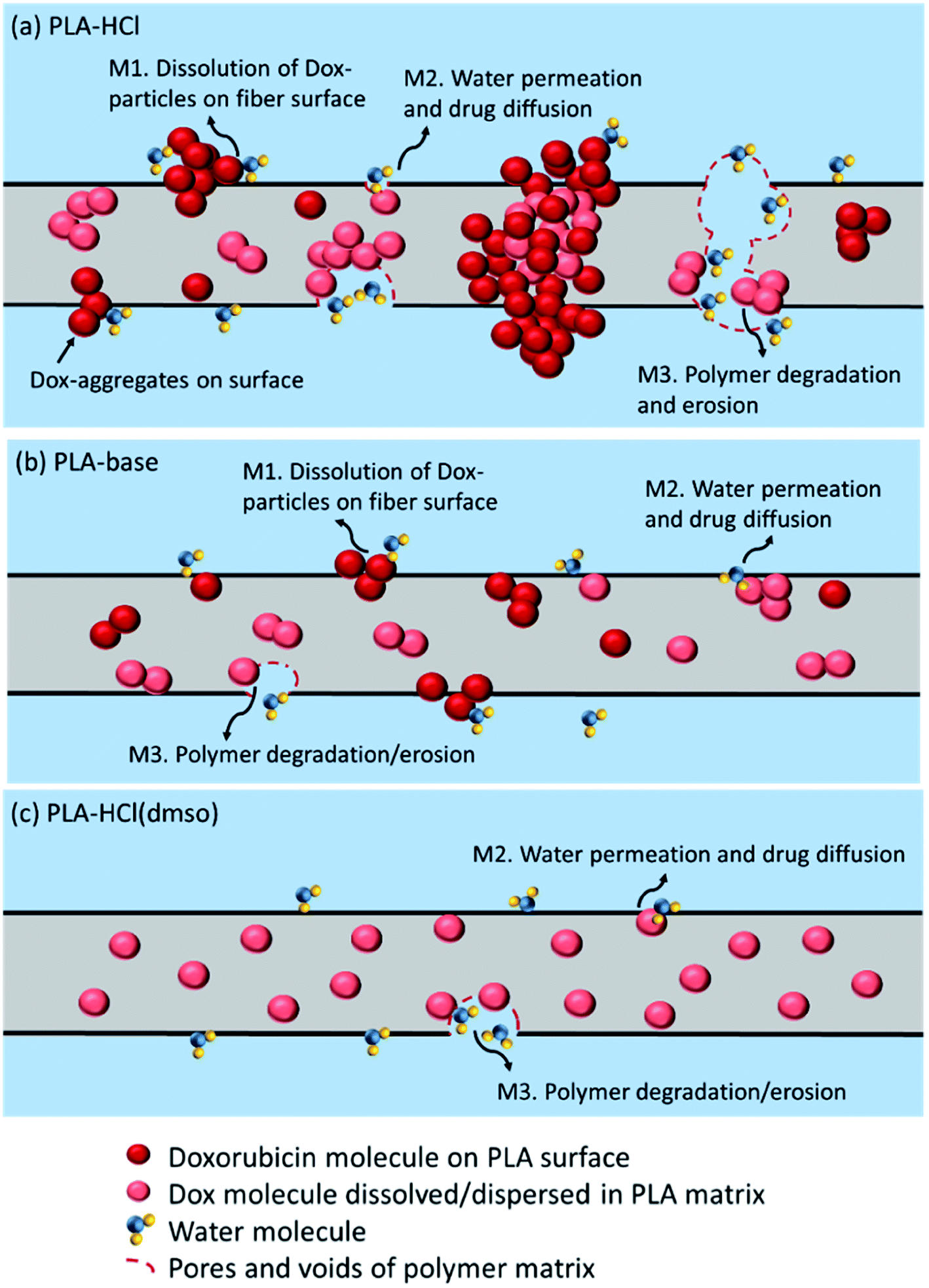 | ||
| Fig. 8 Illustration of doxorubicin drug release mechanisms from a PLA fiber matrix. (a) PLA–HCl; (b) PLA–base; (c) PLA–HCl(DMSO). M1, dissolution of drug molecules; M2, water permeation followed by drug diffusion; M3, polymer degradation followed by drug dissolution/diffusion. | ||
Cytotoxicity of Dox-loaded PLA
The cytotoxicity of all Dox-loaded PLA and Dox-free PLA was measured using the alamar blue assay. In Fig. 9, cell viability is shown as a function of Dox-amount (in fiber mats) that is present in mL of cell culture. All Dox-loaded PLA were toxic to C3A cells with corresponding concentrations from 184 μg mL−1 to 608 μg mL−1, while Dox-free PLA was not toxic. About 200 μg mL−1 of Dox incorporated in PLA–HCl and PLA–base substantially decreased the cell viability within the first 5 h (52% viability) and sustained up to 24 h (47.5–52% viability) compared to those of controls; 184 μg mL−1 of Dox in PLA–base showed 52% and 47.5% viability, respectively; 210 μg mL−1 of Dox in PLA–HCl had 52.3% and 52% viability; and 243 μg mL−1 of Dox in PLA–HCl(DMSO) produced 90% viability at both 5 h and 24 h. The lowest cytotoxicity of PLA–HCl(DMSO) is associated with the slowest release of Dox in PBS solution (Fig. 4), attributed to its monolithic solution-like scheme. For PLA–HCl and PLA–base, Dox molecules were not homogeneously blended with polymer matrix, and unblended particles were exposed to surface of fibers, producing substantial initial release that led to higher cytotoxicity.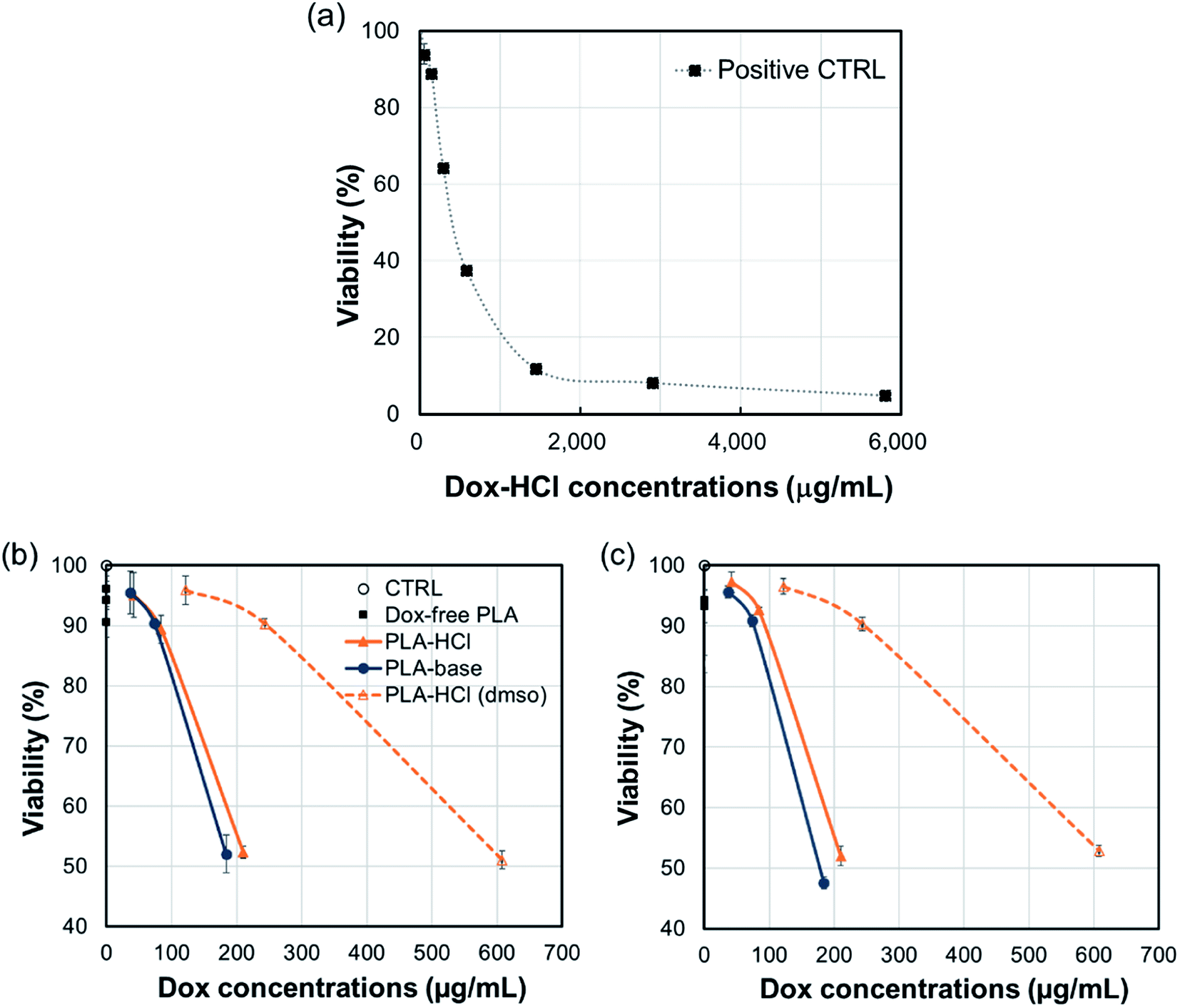 | ||
| Fig. 9 C3A viability for the positive control with Dox-HCl (a), Dox with PLA samples after 5 h incubation (b), and Dox with PLA samples after 24 h incubation (c). Data represent mean (n = 3) ± S.D. Positive CTRL, Dox-HCl with cells without PLA fiber mats; CTRL, only cells without drug or PLA fiber mats. LC50 approximately 413 μg mL−1. | ||
The result indicates that the release of doxorubicin occurs mostly within the first 5 h, which limits the application of this formulation. For practical application, the drug/polymer formulation should be adjusted to include higher drug contents or to facilitate polymer degradation.
Conclusions
Drug release profiles from PLA fibers were investigated with regard to drug–polymer miscibility, fiber wettability and degradability, employing doxorubicin in hydrophilic form (Dox-HCl) and hydrophobic form (Dox-base) as model drugs. Drug-loaded fiber mats were fabricated by electrospinning, with drug loading efficiency of about 74% for less compatible drug-polymer combination (PLA–HCl) and about 100% for all other mats. When hydrophilic Dox-HCl was directly mixed with the hydrophobic PLA solution, drug molecules aggregated either on the mat surface or inside the core region of fibers, due to poor drug–polymer compatibility. About 65% of initial drug release from PLA–HCl was attributed to quick dissolution of drug particles present near the surface. The hydrophobic Dox-base was better dispersed with PLA matrix forming monolithic dispersions. When DMSO was used as the solvent for Dox-HCl, the miscibility of the drug in the polymer matrix was significantly improved, forming a quasi-monolithic solution scheme. The drug release from this monolithic solution matrix was slowest as the immediate dissolution of unblended drug did not occur, and this slower release from PLA–HCl(DMSO) led to lower cytotoxicity to hepatocellular carcinoma cell C3A during 5 h and 24 h of incubation, compared to PLA–HCl and PLA–base. Drug incorporation methods affected Tg and degree of crystallinities of drug-loaded fibers, yet those properties hardly influenced the drug release rates associated with diffusion or degradation mechanism. When an enzyme was used to promote PLA degradation, the release rates were closely correlated with the degradation rates, demonstrating that degradation was the dominant release mechanism for PLA mats. For PLA–HCl, quick dissolution of the drug particles exposed on the surface contributed significantly to the fast initial release. The results demonstrated that the manipulation of drug–polymer miscibility and polymer degradability would be an effective means of modulating drug release profiles. Particularly, the results demonstrated that an improved miscibility of the hydrophilic drug in a hydrophobic PLA polymer can reduce the release rates. It is speculated that the release behavior of a drug from a hydrophilic polymer would be quite different; uniform blending of the drug in a hydrophilic polymer may lead to an increased release rate by the fast degradation in PBS solution. The future study validating the interplay of surface energy between drugs and polymers is recommended. Within the limited scope, the findings of this study can be applied as a facile design strategy for controlling the early stage release profiles in fiber-based drug delivery systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research Resettlement Fund for the new faculty of Seoul National University (350-20170057), and Johnson Cancer Research Center at Kansas State University. Authors appreciate the help of Dr Daniel L. Boyle in the Kansas State University Microscopy Facility for TEM analyses.References
- X. Xu, X. Chen, Z. Wang and X. Jing, Ultrafine PEG–PLA Fibers Loaded with Both Paclitaxel and Doxorubicin Hydrochloride and Their In Vitro Cytotoxicity, Eur. J. Pharm. Biopharm., 2009, 72, 18–25 CrossRef PubMed.
- J. A. Kaplan, et al., Prevention of Lung Cancer Recurrence Using Cisplatin-loaded Superhydrophobic Nanofiber Meshes, Biomaterials, 2016, 76, 273–281 CrossRef PubMed.
- F. Sharifi, et al., Fiber Based Approaches as Medicine Delivery Systems, ACS Biomater. Sci. Eng., 2016, 2, 1411–1431 CrossRef.
- I. Sebe, P. Szabó, B. Kállai-Szabó and R. Zelkó, Incorporating Small Molecules or Biologics into Nanofibers for Optimized Drug Release: A Review, Int. J. Pharm., 2015, 494, 516–530 CrossRef PubMed.
- S. Chou, D. Carson and K. A. Woodrow, Current Strategies for Sustaining Drug Release from Electrospun Nanofibers, J. Controlled Release, 2015, 220, 584–591 CrossRef PubMed.
- A. Balaji, et al., An Insight on Electrospun-Nanofibers-Inspired Modern Drug Delivery System in the Treatment of Deadly Cancers, RSC Adv., 2015, 5, 57984–58004 RSC.
- S. T. Yohe, Y. L. Colson and M. W. Grinstaff, Superhydrophobic Materials for Tunable Drug Release: Using Displacement of Air to Control Delivery Rates, J. Am. Chem. Soc., 2012, 134, 2016 CrossRef PubMed.
- J. Cui, L. Qiu, Y. Qiu, Q. Wang and Q. Wei, Co-electrospun Nanofibers of PVA-SbQ and Zein for Wound Healing, J. Appl. Polym. Sci., 2015, 132, 42565 Search PubMed.
- Q. Ding, et al., Preparation and Therapeutic Application of Docetaxel-loaded Poly(d,l-lactide) Nanofibers in Preventing Breast Cancer Recurrence, Drug Delivery, 2015, 1–9 Search PubMed.
- P. Dubey and P. Gopinath, Fabrication of Electrospun Poly(ethylene oxide)–Poly(capro lactone) Composite Nanofibers for Co-delivery of Niclosamide and Silver Nanoparticles Exhibits Enhanced Anti-cancer Effects In Vitro, J. Mater. Chem. B, 2016, 4, 726–742 RSC.
- X. Hu, et al., Electrospinning of Polymeric Nanofibers for Drug Delivery Applications, J. Controlled Release, 2014, 185, 12–21 CrossRef PubMed.
- M. Ignatova, I. Rashkov and N. Manolova, Drug-loaded Electrospun Materials in Wound-dressing Applications and in Local Cancer Treatment, Expert Opin. Drug Delivery, 2013, 10, 469–483 CrossRef PubMed.
- S. Jain, S. R. K. Meka and K. Chatterjee, Engineering a Piperine Eluting Nanofibrous Patch for Cancer Treatment, ACS Biomater. Sci. Eng., 2016, 2, 1376–1385 CrossRef.
- J. Lin, C. Li, Y. Zhao, J. Hu and L. Zhang, Co-electrospun Nanofibrous Membranes of Collagen and Zein for Wound Healing, ACS Appl. Mater. Interfaces, 2012, 4, 1050 Search PubMed.
- M. Chen, Y.-F. Li and F. Besenbacher, Electrospun Nanofibers-Mediated On-Demand Drug Release, Adv. Healthcare Mater., 2014, 3, 1721–1732 CrossRef PubMed.
- P. I. Morgado, A. Aguiar-Ricardo and I. J. Correia, Asymmetric Membranes as Ideal Wound Dressings: An Overview on Production Methods, Structure, Properties and Performance Relationship, J. Membr. Sci., 2015, 490, 139–151 CrossRef.
- M. Sampath, R. Lakra, P. Korrapati and B. Sengottuvelan, Curcumin Loaded Poly (lactic-co-glycolic) Acid Nanofiber for the Treatment of Carcinoma, Colloids Surf., B, 2014, 117, 128–134 CrossRef PubMed.
- J. Wei, J. Hu, M. Li, Y. Chen and Y. Chen, Multiple Drug-loaded Electrospun PLGA/gelatin Composite Nanofibers Encapsulated with Mesoporous ZnO Nanospheres for Potential Postsurgical Cancer Treatment, RSC Adv., 2014, 4, 28011–29019 RSC.
- J. Wu, et al., Poly (d,l-lactic acid) Electrospun Fibers with Tunable Surface Nanotopography for Modulating Drug Release Profiles, Mater. Lett., 2015, 161, 716–719 CrossRef.
- X. Zhou, et al., Electrospun Nanofibers Incorporating Self-decomposable Silica Nanoparticles as Carriers for Controlled Delivery of Anticancer Drug, RSC Adv., 2015, 5, 65897–66594 RSC.
- J. Liu, Y. Xiao and C. Allen, Polymer–Drug Compatibility: A guide to the Development of Delivery Systems for the Anticancer Agent, Ellipticine, J. Pharm. Sci., 2004, 93, 132–143 CrossRef PubMed.
- S. S. Shah, Y. Cha and C. G. Pitt, Poly (glycolic acid-co-dl-lactic acid): Diffusion or Degradation Controlled Drug Delivery?, J. Controlled Release, 1992, 18, 261–270 CrossRef.
- J. Siepmann and F. Siepmann, Modeling of Diffusion Controlled Drug Delivery. Modeling of Diffusion Controlled Drug Delivery, J. Controlled Release, 2012, 161, 351–362 CrossRef PubMed.
- J. Xu, Q. Zhao, Y. Jin and L. Qiu, High Loading of Hydrophilic/Hydrophobic Doxorubicin into Polyphosphazene Polymersome for Breast Cancer Therapy, Nanomedicine, 2014, 10, 349–358 CrossRef PubMed.
- K. Qiu, et al., Doxorubicin-loaded Electrospun Poly(l-lactic acid)/Mesoporous Silica Nanoparticles Composite Nanofibers for Potential Postsurgical Cancer Treatment, J. Mater. Chem. B, 2013, 1, 4601–4611 RSC.
- C. Sanson, et al., A Simple Method to Achieve High Doxorubicin Loading in Biodegradable Polymersomes, J. Controlled Release, 2010, 147, 428–435 CrossRef PubMed.
- D. Han and A. J. Steckl, Triaxial Electrospun Nanofiber Membranes for Controlled Dual Release of Functional Molecules, ACS Appl. Mater. Interfaces, 2013, 5, 8241 Search PubMed.
- Y. Z. Zhang, et al., Coaxial Electrospinning of (Fluorescein Isothiocyanate-Conjugated Bovine Serum Albumin)-Encapsulated Poly (ε-Caprolactone) Nanofibers for Sustained Release, Biomacromolecules, 2006, 7, 1049–1057 CrossRef PubMed.
- J. Zeng, et al., Influence of the Drug Compatibility with Polymer Solution on the Release Kinetics of Electrospun Fiber Formulation, J. Controlled Release, 2005, 105, 43–51 CrossRef PubMed.
- A. Kathuria, M. G. Abiad and R. Auras, Deterioration of Metal–Organic Framework Crystal Structure During Fabrication of Poly(l-lactic acid) Mixed-matrix Membranes, Polym. Int., 2013, 62, 1144–1151 CrossRef.
- Y. Yuan, M. P. Hays, P. R. Hardwidge and J. Kim, Surface Characteristics Influencing Bacterial Adhesion to Polymeric Substrates, RSC Adv., 2017, 7, 14254–14261 RSC.
- Y. Yuan, S. Choi and J. Kim, Analysis of Contact Area between Water and Irregular Fibrous Surface for Prediction of Wettability, RSC Adv., 2016, 6, 73313–73322 RSC.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Investigation of the Structure of Solution Grown Crystals of Lactide Copolymers by Means of Chemical Reactions, Kolloid-Zeitschrift und Zeitschrift für Polymere, 1973, 251, 980–990 CrossRef.
- K. Choi, J. E. Riviere and N. A. Monteiro-Riviere, Protein Corona Modulation of Hepatocyte Uptake and Molecular Mechanisms of Gold Nanoparticle Toxicity, Nanotoxicology, 2017, 11, 64–75 CrossRef PubMed.
- E. A. Ho, V. Vassileva, C. Allen and M. Piquette-Miller, In Vitro and In Vivo Characterization of a Novel Biocompatible Polymer–Lipid Implant System for the Sustained Delivery of Paclitaxel, J. Controlled Release, 2005, 104, 181–191 CrossRef PubMed.
- F. Siepmann, J. Siepmann and V. Le Brun, Drugs Acting as Plasticizers in Polymeric Systems: A Quantitative Treatment, J. Controlled Release, 2006, 115, 298–306 CrossRef PubMed.
- O. Martin and L. Avérous, Poly(lactic acid): Plasticization and Properties of Biodegradable Multiphase Systems, Polymer, 2001, 42, 6209–6219 CrossRef.
- S. C. Schmidt and M. A. Hillmyer, Polylactide Stereocomplex Crystallites as Nucleating Agents for Isotactic Polylactide, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 300–313 CrossRef.
- T. W. Chung, Y. Y. Huang, Y. L. Tsai and Y. Z. Liu, Effects of Solvent Evaporation Rate on the Properties of Protein-Loaded PLLA and PDLLA Microspheres Fabricated by Emulsion-Solvent Evaporation Process, J. Microencapsulation, 2002, 19, 463–471 CrossRef PubMed.
- J. Runt and P. B. Rim, Effect of Preparation Conditions on the Development of Crystallinity in Compatible Polymer Blends: Poly(styrene-co-acrylonitrile)/Poly(ε-caprolactone), Macromolecules, 1982, 15, 1018–1023 CrossRef.
- E. A. R. Duek, C. A. C. Zavaglia and W. D. Belangero, In Vitro Study of Poly(lactic acid) Pin Degradation, Polymer, 1999, 40, 6465–6473 CrossRef.
- H. Cai, V. Dave, R. A. Gross and S. P. McCarthy, Effects of Physical Aging, Crystallinity, and Orientation on the Enzymatic Degradation of Poly(lactic acid), J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2701–2708 CrossRef.
- Z. Miao, S. Cheng, X. Zhang and R. Zhuo, Study on Drug Release Behaviors of Poly-alpha,beta-[n-(2-hydroxyethyl)-L-aspartamide]-g-poly(epsilon-caprolactone) Nano- and Microparticles, Biomacromolecules, 2006, 7, 2020–2026 CrossRef PubMed.
- P. L. Ritger and N. A. A. Peppas, Simple Equation for Description of Solute Release I. Fickian and Non-Fickian Release from Non-swellable Devices in the Form of Slabs, Spheres, Cylinders or Discs, J. Controlled Release, 1987, 5, 23–36 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |