
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid detection of staphylococcal enterotoxin B in milk samples based on fluorescence hybridization chain reaction amplification
Yanyang Xu *a,
Bingyang Huoa,
Xuan Sunb,
Baoan Ningc,
Yuan Pengc,
Jialei Baic and
Zhixian Gao*c
*a,
Bingyang Huoa,
Xuan Sunb,
Baoan Ningc,
Yuan Pengc,
Jialei Baic and
Zhixian Gao*c
aCollege of Food Science and Engineering, Jilin University, Changchun 130022, P. R. China. E-mail: hbyaijbx@163.com
bHuazhong Agricultural University, College of Life Science and Technology, Wuhan 430070, P. R. China
cTianjin Key Laboratory of Risk Assessment and Control Technology for Environment and Food Safety, Institute of Environmental and Operational Medicine, Academy of Military Medical Science, Academy of Military Science, Tianjin 300050, P. R. China
First published on 30th April 2018
Abstract
A rapid, simple, and sensitive method has been developed to detect staphylococcal enterotoxin B (SEB). To establish the hybridization chain reaction-based aptasensor, we described the new probes of two hairpins (H1 and H2), which were first designed based on the partial complementary sequence of the SEB aptamer (cDNA). The H1 labeled with a fluorophore and a quencher can act as a molecular fluorescence “switch”. Hence, in the presence of SEB, the aptamer binds SEB, while the unbound cDNA triggers HCR to carry out the cyclic hybridization of H1 and H2 so as to turn “ON” the fluorescence through forming long nicked DNA. By using this new strategy, SEB can be sensitively detected within the range of 3.13 ng mL−1 to 100 ng mL−1 with a detection limit of 0.33 ng mL−1 (S/N = 3). Furthermore, the developed method could facilitate the detection of SEB effectively in milk samples.
1. Introduction
Food borne diseases have always been a major threat to human health and are considered as a widespread public health concern worldwide. Contamination of foods is mainly caused by bacteria, toxins, and chemical agents, leading to nausea, vomiting, and abdominal cramping.1 As the second most common cause of food poisoning,2 Staphylococcus aureus produces various enterotoxins (SEs) throughout the transition from the exponential to the stationary phase.3 Among these SEs, staphylococcal enterotoxin B (SEB) is a heat-stable and enzymatic hydrolysis resistance toxin that can incapacitate people for up to two weeks with as low as 0.4 ng kg−1.4 Additionally, SEB is also considered to be a bioterrorism threat agent both in the UK and in the US.5 Thus, the simple and rapid detection of SEB has received significant interest.At present, numerous immunoassay methods including ELISA,6 electrochemical,7,8 chemiluminescence,9,10 immuno-polymerase chain reaction (PCR),11 POCT,12 piezoelectric crystal immunosensor,13 magnetoelastic immunosensor,14 lateral flow assay,15 and LC-LC/MS16 have been established to detect SEB. The methods described above manifest low sensitivity and high specificity based on the antigen–antibody interactions. However, these methods are difficult to promote because of their multiple assay steps and additional exogenous reagents.
Aptamers from specific oligonucleotide sequences, have emerged as a novel molecular recognition element. These sequences are obtained by an exponential enrichment process (SELEX)17 and have the ability to recognize the targeted molecules. High purity, low-cost preparation, and easy modification with functional groups,18 render aptamers as potential candidates for replacing antibodies. One of the most comprehensive “aptamer–target” detection strategies is called “structure-switching signaling”.19 Among these detection methods, the sensitivity of the traditional aptasensor fluorescence method is largely influenced by the affinity of aptamers.
For solving this problem and further improving the detection sensitivity, a series of enzyme-free nucleic acid amplification techniques have been gradually developed and received considerable attention. Compared with enzyme amplification techniques, including polymerase chain reaction,20 ligase chain reaction,21 loop-mediated isothermal amplification,22,23 helicase-dependent amplification,24 and rolling circle amplification,25 hybridization chain reaction (HCR)26 provides an isothermal, simple signaling amplification method among the enzyme-free approaches. Based on toehold-mediated strand displacement and introducing of an initiator single-stranded DNA,27 HCR can be achieved by two kinetically trapped nucleic acid hairpin molecules, which can form DNA nanostructures with long repeating units.
To date, this signal amplification has been applied in various biosensor platforms, including color,28,29 fluorescence,7,30–32 chemiluminescence,33–35 and electrochemical.8,36–39 Mostly traditional HCR-based aptasensor need to wash unbound probes and long incubation period. For example, Song et al.40 developed a HCR-based aptasensor, where hairpins controllable assembly only the conformation of PDGF-BB aptamer. Then, the fluorescent signal appeared by using SA-QDs. However, it needs repeated washing steps and a relatively long time to accomplish the experiment.
To solve this problem, a simple, enzyme-free, and ingenious detection of SEB aptasensor platform is proposed. With the introduction of HCR amplification as a new strategy for aptamer-based molecular switching, this method offers the advantage of the design of an intelligent hairpin “switch” based on the fluorescent resonance energy transfer (FRET). This signal “switch” mainly depends on the excellent signal transduction of molecular beacons (MBs). In our design, the 6-FAM and BHQ-1 are linked covalently at the end of H1's arm. The stem of H1 leads to the close proximity of the fluorescence and quencher which result in the lowest fluorescence intensity. This competitive aptasensor for sensitive detecting SEB is constructed combing with HCR amplification. In the presence of SEB, the SEB–aptamer complex is formed, and the unbound cDNA (denoted as initiator) triggers two designed hairpin DNA probes (H1 and H2) successive hybridization chain reaction. The H1 unfolds the stem of hairpin structure and separates the fluorophore (6-FAM) and the quencher (BHQ-1), thus performing the FRET for signal transduction and allowing the fluorophore of 6-FAM to fluoresce. The introduction of this HCR amplification technique is expected to provide a rapid, low-cost, and sensitive aptasensor fluorescence strategy.
2. Materials and methods
2.1 Materials
Staphylococcal enterotoxin B (SEB, 1 mg mL−1), staphylococcal enterotoxin A (SEA, 1 mg mL−1), bovine serum albumin (BSA), ovalbumin (OVA), ricin toxin (RT, 1 mg mL−1), the liquid milk used for the analysis was purchased from a local store. The hybridization buffer containing 20 mM of Tris–HCl, 400 mM of NaCl, and 18.2 MΩ cm−1 NANOpure water (Millipore purified) was used in all experiments. A 50 bp DNA ladder, a 200 bp ladder, and a D2000 marker were purchased from Takara (Dalian, China), while agarose (G-10, BIOWEST) and Genegreen were purchased from Tiangen Co., Ltd. (Beijing, China). All DNA sequences were synthesized and subjected to HPLC purification by Sangon Co., Ltd. The sequences of the synthetic DNA are shown in Table 1.| Assay | Oligonucleotides | Sequence (5′–3′)a |
|---|---|---|
| a The sequence of SEB aptamer was reported by Jeffrey A DeGrasse.41 H1 was marked with 6-FAM on the 5′ side and BHQ-1 in the underlined ‘T’ base. SEB aptamer1 was labeled with 6-FAM in the underlined ‘T’ base and cDNA1 was labeled with BHQ-1 on the 3′ side. | ||
| HCR | SEB aptamer | GGTATTGAGGGTCGCATCCACTGGTCGTTGTTGTCTGTTGTCTGTTATGTTGTTTCGTGATGGCTCTAACTCTCCTCT |
| cDNA (H0) | GACAACAACGACCAGCTTATCCCA | |
| H1 | AACGACCAGCTTATCCCACCTTAATGGGATAAGCTGGTCGT![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) GTTGTC GTTGTC |
|
| H2 | TTAAGGTGGGATAAGCTGGTCGTTGACAACAACGACCAGCTTATCCCA | |
| Traditional method | SEB aptamer1 | GGTATTGAGGGTCGCATCCACTGGTCGTTGTTGTCGTTGTCTGTTATGTTGTTTCGTGATGGCTCTAACTCTCCTCT |
| cDNA1 | GACAACAACGACCAGCTTATCCCA | |
2.2 Instruments
Fluorescence intensity was measured with F97pro device (Lengguang Tech.). FAM was excited at 488 nm within the recorded range from 510 nm to 600 nm. All excitation and emission slits were set to 10 nm. The following apparatus were used. FE28-Standard pH meter (Mettler Toledo), IKA rotating shaker (MS3), Digital Heating Shaking Drybath (TMS-200, Hangzhou, China), and ImageQuant350 Gel imaging system (GE Healthcare).2.3 Gel electrophoresis
Hairpin 1 (H1) and hairpin 2 (H2) samples were heated to 95 °C for 10 min and gradient cooled within 2 h to 25 °C by using a PCR instrument. Different concentrations of initiator DNA reacted with 3.3 μM of H1 and H2 in the hybridization buffer at 37 °C for 2 h. The 2% agarose gels was prepared with 1× TAE buffer (40 mM Tris-acetate, 1 mM EDTA, and 20 mM acetic acid). Also, the aptamer-HCR system was characterized through agarose gel. Subsequently, the gel was run at 110 V for 120 min and visualized under UV light through the ImageQuant system.2.4 HCR-based aptasensor fluorescence measurement for SEB detection
Before use, H1 and H2 should be heated to 95 °C for 10 min and then gradient cooled within 2 h to 25 °C by using a PCR instrument. Then, 20 μL of SEB aptamer (1 μM) was incubated with different concentrations of SEB and incubated at 37 °C for 30 min. Next, 30 μL of partial complementary strand (1 μM) was added to the mixture and reacted at 37 °C for 30 min. Subsequently, 20 μL of H1/H2 (2.5 μM) was added into the hybridization buffer to reach a final volume of 250 μL. The signal of HCR amplification was detected after incubation at 37 °C for 45 min through a fluorescence spectrophotometer. Each concentration was measured for five times using the F97pro device.2.5 Aptamer-based fluorescent assay for SEB detection
Before use, the SEB aptamer was heated to 95 °C for 5 min and placed in an ice water bath for 10 min quickly. Then, 20 μL volumes of SEB aptamer solution (1 μM) were mixed with 100 μL of SEB standard solutions at different concentrations. Each mixture was incubated at 37 °C for 30 min. Afterward, 20 μL of cDNA solution (1 μM) was added into the mixture to hybridize with the remaining SEB aptamer. The final volume was added to the buffer to make up 250 μL. Each concentration was measured for five times using the F97pro device.2.6 SEB detection in milk sample
The performance of the SEB in the competitive HCR-based aptasensor was assessed in milk, which is commonly implicated in SEB foodborne illnesses. The samples were prepared and spiked with known concentrations of SEB. Before testing, the samples were centrifuged at 5000 rpm for 10 min, and the substances were transferred into a fresh 1.5 mL tube after removing the fat. Different concentrations of SEB were then added to the prepared samples (100 μL) for 2 h at 25 °C. Standard addition and recovery experiments were performed on each sample for five times.3. Results and discussion
3.1 Design of principle HCR-based aptasensor fluorescent sensing strategy
The principle of the SEB detection based competitive HCR signal amplification strategy is shown in Scheme 1. The proposed HCR-based aptasensor mainly composed of SEB aptamer, partial complementary strand (cDNA), the 6-FAM and BHQ-1 labeled hairpin 1, and hairpin 2. As described in the Scheme 1, the cDNA was designed to be paired with SEB aptamer partly. In the absence of SEB, the cDNA would hybridize with the aptamer closely through complementary pairing. On the contrary, a certain concentration of SEB can be determined by employing aptamer–SEB interaction. Once the aptamer/target is formed, the region of essential nucleotides of aptamer is occupied, and cDNA can not interact with aptamer through hydrogen bonding. Then the cDNA (H0) acts as the initiator for the signal amplification. The linear HCR system contained two hairpin substrates (H1 and H2) and a single strand DNA (H0). Two hairpin probes (H1 and H2) were designed based on the toehold strand displacement with the aid of NUPACK and the Mfold software. First, the additional 6 nt (a*) sticky end of H1 and its adjacent stems (b*) are paired with H0. Then, the loop of 6 nt (c) and the stem of 18 nt (b) of H1 would hybridize with H2 and displace the stem of H2. The exposed strand of H2 is identical to that of H0 in sequence. Finally, the H1 and H2 would bind with each other continuously and generate a long chain of H0–(H1–H2)n double helix complex. H1 is labeled with 6-FAM at the end of its 5′ arm, which is opposite to the BHQ-1 quencher in the hairpin conformation. The fluorophore and quencher adjacent to each other can quench the fluorescence of the 6-FAM through resonance energy transfer.41 In the absence of SEB, the SEB aptamer binds with cDNA which can not trigger the HCR, and the molecular beacons (H1) are switched OFF. On the contrary, because the affinity of SEB/aptamer is higher than that of aptamer/cDNA. And the H1 is switched on (the fluorophore and quencher are separated) and release fluorescence signal. Thus, different concentrations of H0 can trigger corresponding fluorescence signal intensities, which could be used for the indirect determination of SEB concentration. | ||
| Scheme 1 Competitive HCR-based aptasensor for SEB detection. | ||
3.2 Characterization of HCR system
Agarose gel (2%) was used to characterize and verify the HCR amplification and aptamer HCR system. As shown in Fig. 1A, lanes 2 to 3 show the base number of 1 μM H1 and H2, respectively. In the absence of H0, there is only one clear (lane 9) stripe, which was due to the absence of HCR, existed in the channel. However, upon the addition of H0 with different concentrations, lanes 4 to 8 present a clear increasing average molecular weight trend opposite the concentration of the H0. Fig. 1B further validates the potential quantitative sensing. Given that H1 is labeled with 6-FAM and BHQ-1 in the corresponding position, the fluorescence is quenched because of fluorescence resonance energy transfer (FRET). H0 concentration exhibits a good linear relationship with fluorescence intensity (R2 = 0.9813), with increasing H0 concentration translates to high fluorescence intensity. This linear HCR kinetics can be applied to signal amplification for the quantitative detection of SEB in this experiment. Fig. 2 characterized the SEB aptamer HCR system. Lane 2 shows the traditional HCR amplification after adding H0. Lane 3 shows the aptamer HCR system (SEB aptamer + H0 + H1 + H2) without SEB, and only two stripes appeared (one stripe shows the base number of SEB aptamer, and the other represents the mixture of H1 and H2) because no HCR happened. However, when adding SEB, an increase at the base number stripes illustrate the HCR amplification can be triggered (lane 4). Also, lane 5 demonstrates the SEB aptamer can not trigger the HCR amplification, which indicates that this possesses good specificity.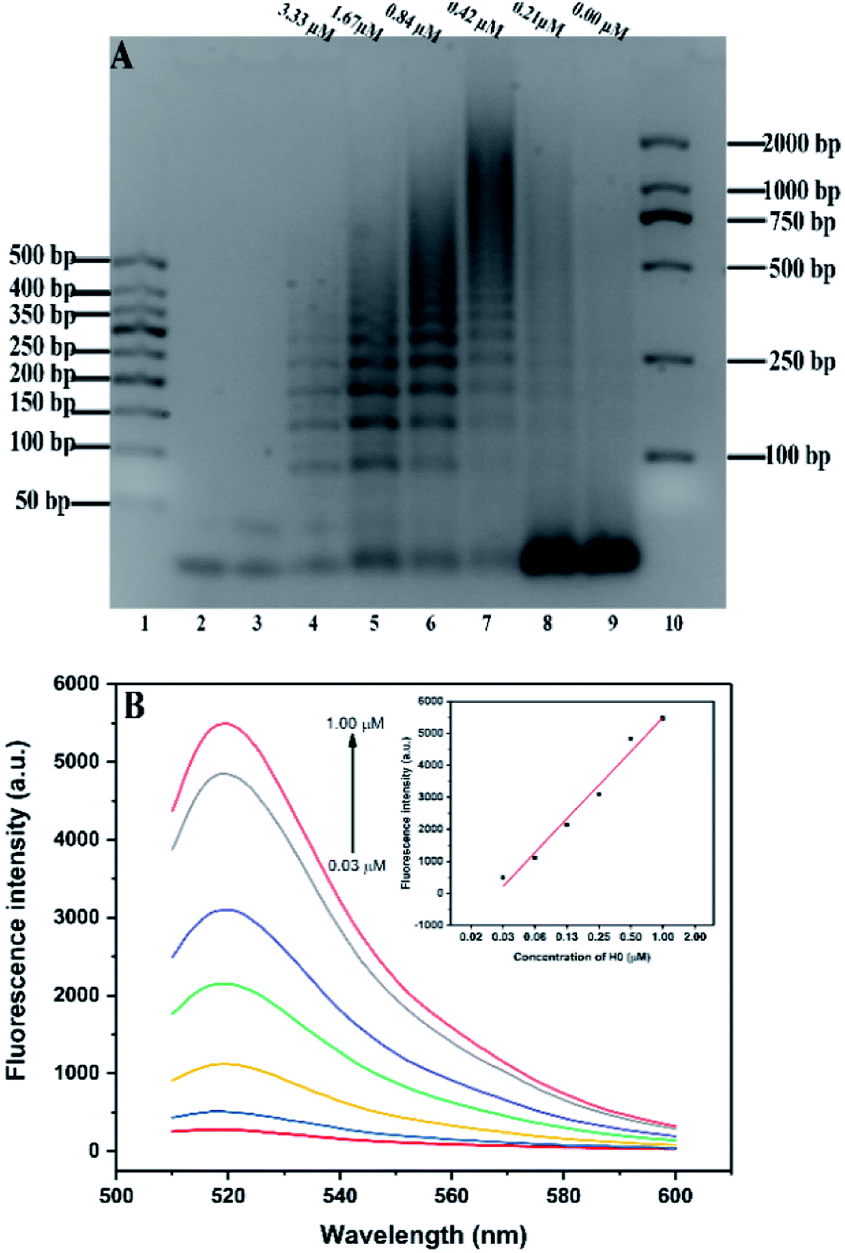 | ||
| Fig. 1 (A) Agarose gel (2%) analysis of different concentrations of H0 on HCR amplification. Lanes 1 and 10: two kinds of DNA markers. Lanes 2 and 3: 1 μM H1 and H2, separately; lane 4–9: six different concentrations of H0 (3.33, 1.67, 0.84, 0.42, 0.21, and 0.00 μM) in a 3.3 μM mixture of H1 and H2. (B) HCR kinetics verification through fluorescence intensity. | ||
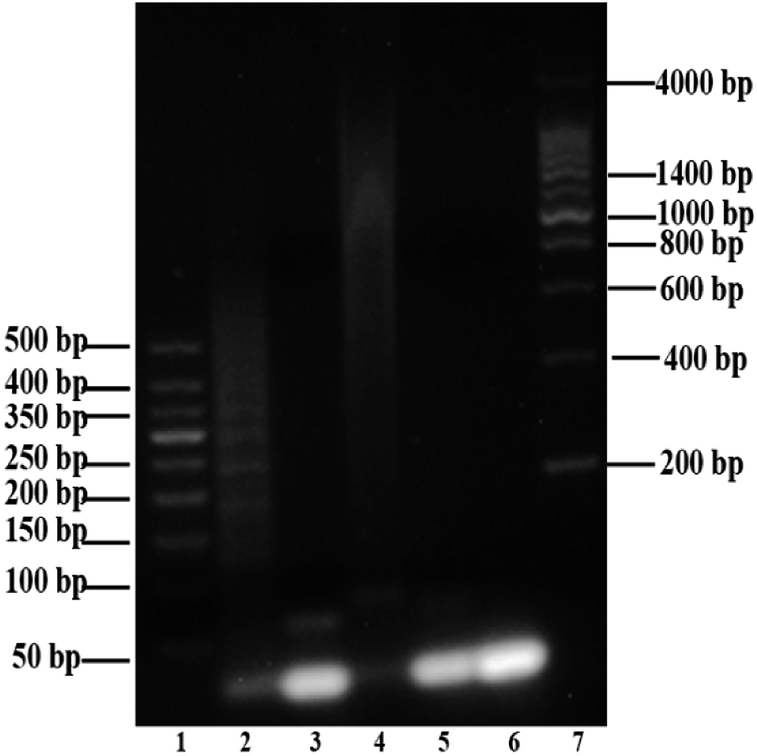 | ||
| Fig. 2 Agarose gel (2%) analysis of SEB aptamer HCR system. Lanes 1 and 7: two kinds of DNA markers; lane 2: addition of simple initiator trigger HCR (H0 + H1 + H2); lane 3: negative control HCR experiment in the absence of SEB (SEB aptamer + H0 + H1 + H2); lane 4: SEB recognition through HCR amplification (SEB aptamer + H0 + H1 + H2); lane 5: non-specific amplification verification (SEB aptamer + H1 + H2). Lane 6: mixture of H1 and H2. | ||
3.3 Optimization of the experimental conditions
Several conditions were optimized in order to achieve the sensitivity of competitive HCR-based aptasensor. The fluorescence values were normalized using the fluorescence value in the group to the highest fluorescence value. Fig. 3A presents the optimal addition volume of H1/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). It was found that the relative fluorescence intensity increased with the increasing volume of H1/H2 (1:1). A plateau was reached at the volume of 25 μL. When the added hairpins continued to increase, the high concentration of H1/H2 led to the steric hindrance,42 which decreased the fluorescence intensity. However, considering the cost requirements, 20 μL of H1/H2 was selected in this experiment.
:1). It was found that the relative fluorescence intensity increased with the increasing volume of H1/H2 (1:1). A plateau was reached at the volume of 25 μL. When the added hairpins continued to increase, the high concentration of H1/H2 led to the steric hindrance,42 which decreased the fluorescence intensity. However, considering the cost requirements, 20 μL of H1/H2 was selected in this experiment.
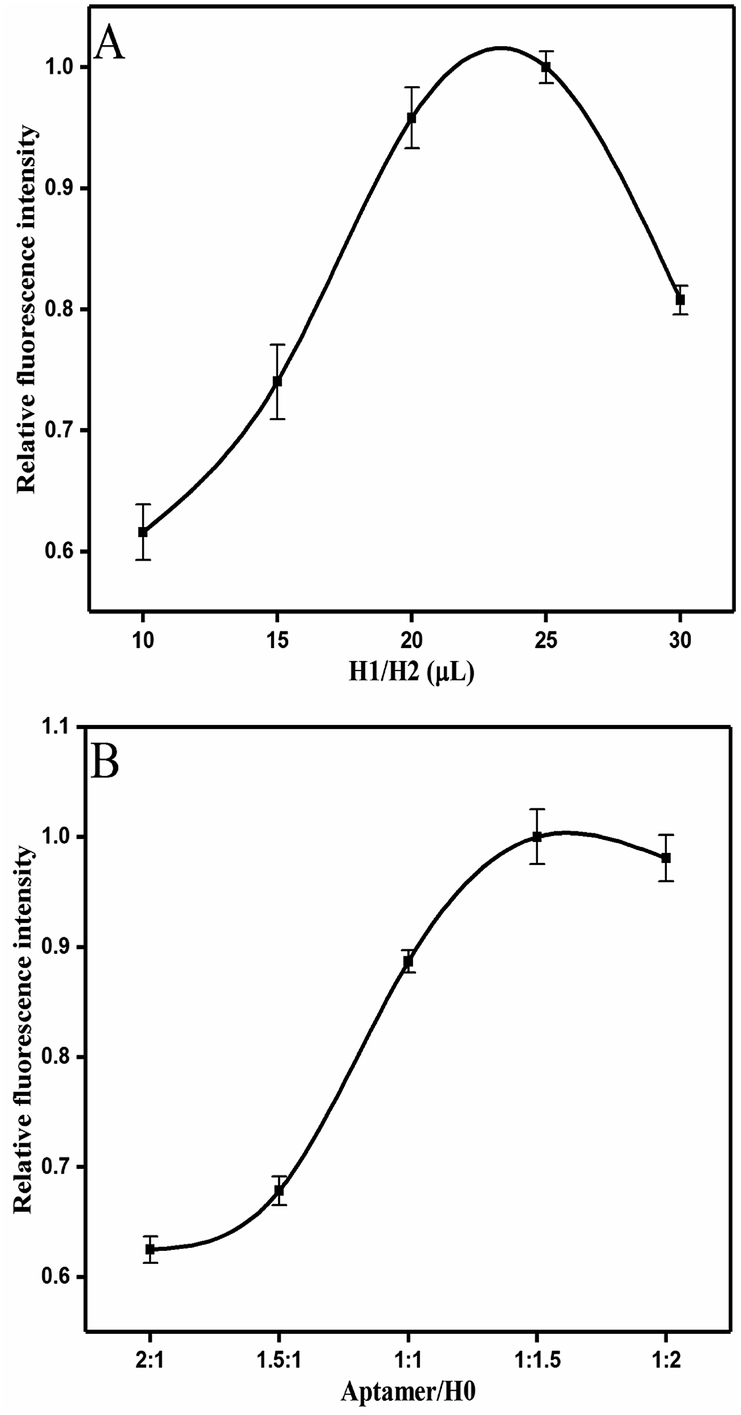 | ||
| Fig. 3 Optimizations of experiment conditions: (A) added volume of H1/H2 (1:1). (B) The ratio of aptamer/H0. Error bars were calculated from three measurement replicates. | ||
The results on the effect of the molar ratio of aptamer on H0 is shown in Fig. 3B. The figure clearly reveals that the relative fluorescence intensity increases along with the increase of molar ratio (aptamer/H0) from 2:1 to 1:2 because the concentration of H0 relatively improves. Thus, this outcome triggers increased hybridization chain reaction and opening of more hairpin stems. Meanwhile, when the molar ratio of aptamer/H0 increases to 1:1.5, the fluorescence intensity stabilizes. Hence, the molar ratio of aptamer to H0 was set to 1:1.5. Fig. 4A shows that the maximal response occurred at pH 7, whereas lower and higher pH values decreased fluorescence intensity. This result may be partly attributed to the structure changes in the aptamer, resulting in its functional binding sites.43 Another explanation is that low pH leads to tensile surface stress, whereas high pH decreases the steric interaction.44
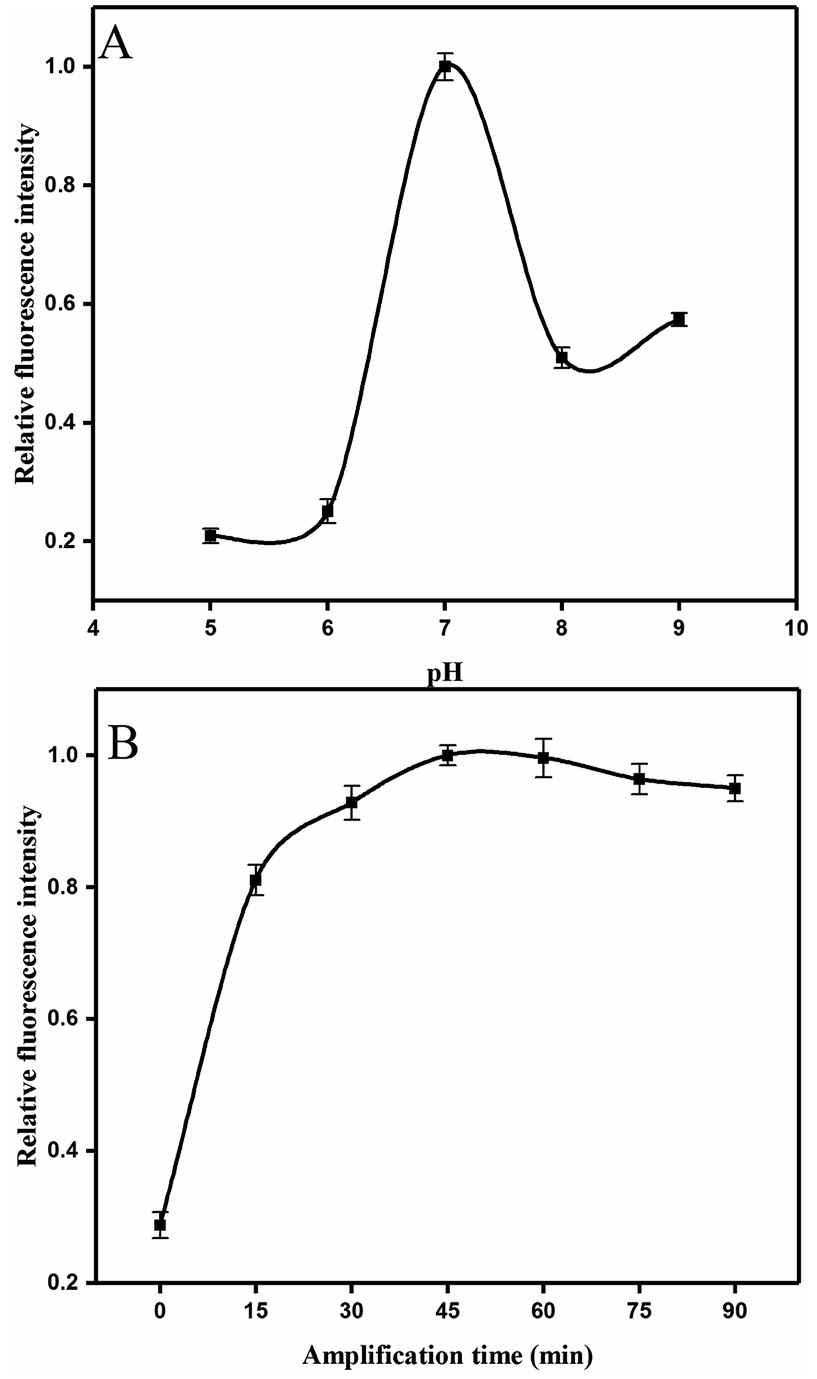 | ||
| Fig. 4 Optimizations of experiment conditions: (A) the pH of reaction system. (B) Evaluation of time of the linear HCR. Error bars were calculated from three measurement replicates. | ||
In addition, the time of HCR amplification was studied. Intensity increased evidently in the first 30 min (Fig. 4B), and this finding was consistent with those in previous studies.45When the HCR is initiated, numerous H0 units bind with the H1 and displaces the original stem of H1. Consequently, the 6-FAM and BHQ-1 labeled on the H1 were segregated and the fluorescence intensity was recovered. Fluorescence intensity was apparently saturated at the reaction time of 45 min. Thus, 45 min was selected for this HCR amplification.
3.4 Performance of the HCR-based sensor
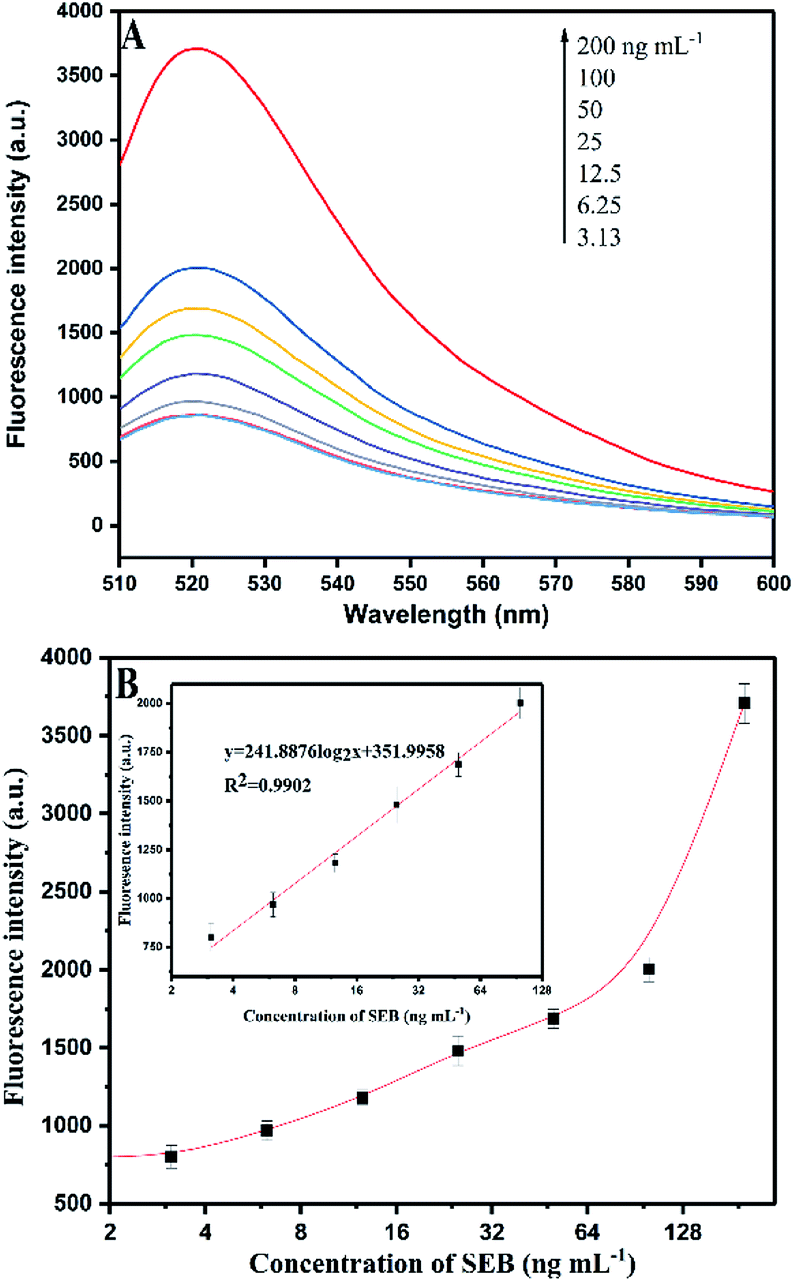 | ||
| Fig. 5 (A) Fluorescence signal responses of the HCR-based aptasensor with different concentrations of SEB. (B) Calibration curves for SEB detection using HCR signal amplification. | ||
Fig. 5B shows that a linear relation of HCR-based amplification assay is constructed between the fluorescence intensity and the logarithm concentration of SEB (3.13–100 ng mL−1). The linear equation is y = 241.8876log2x + 351.9958 (R2 = 0.9902), where y is the fluorescence intensity and x is the SEB concentration. The detection limit is 0.33 ng mL−1 (S/N = 3). Fig. 6A shows the traditional aptamer-based method for detection of SEB, the linear equation is y = 142.453log2x + 1732.1943 (R2 = 0.9860), where y is the fluorescence intensity and x is the logarithm concentration of SEB (31.25–1000 ng mL−1). The detection limit is 6.94 ng mL−1 (S/N = 3). This result indicates that the HCR-based aptasensor can achieve a 21-fold higher sensitivity than traditional aptamer-based fluorescence detection. Also, this proposed HCR-based aptasensor is simple as compared to ELISA (Table 3) that requires repeated washing steps and long incubation time. At the same time, the aptamer-based methods have the advantage of designing “molecule switch”, which can effectively avoid the number of steps (i.e., elution steps).
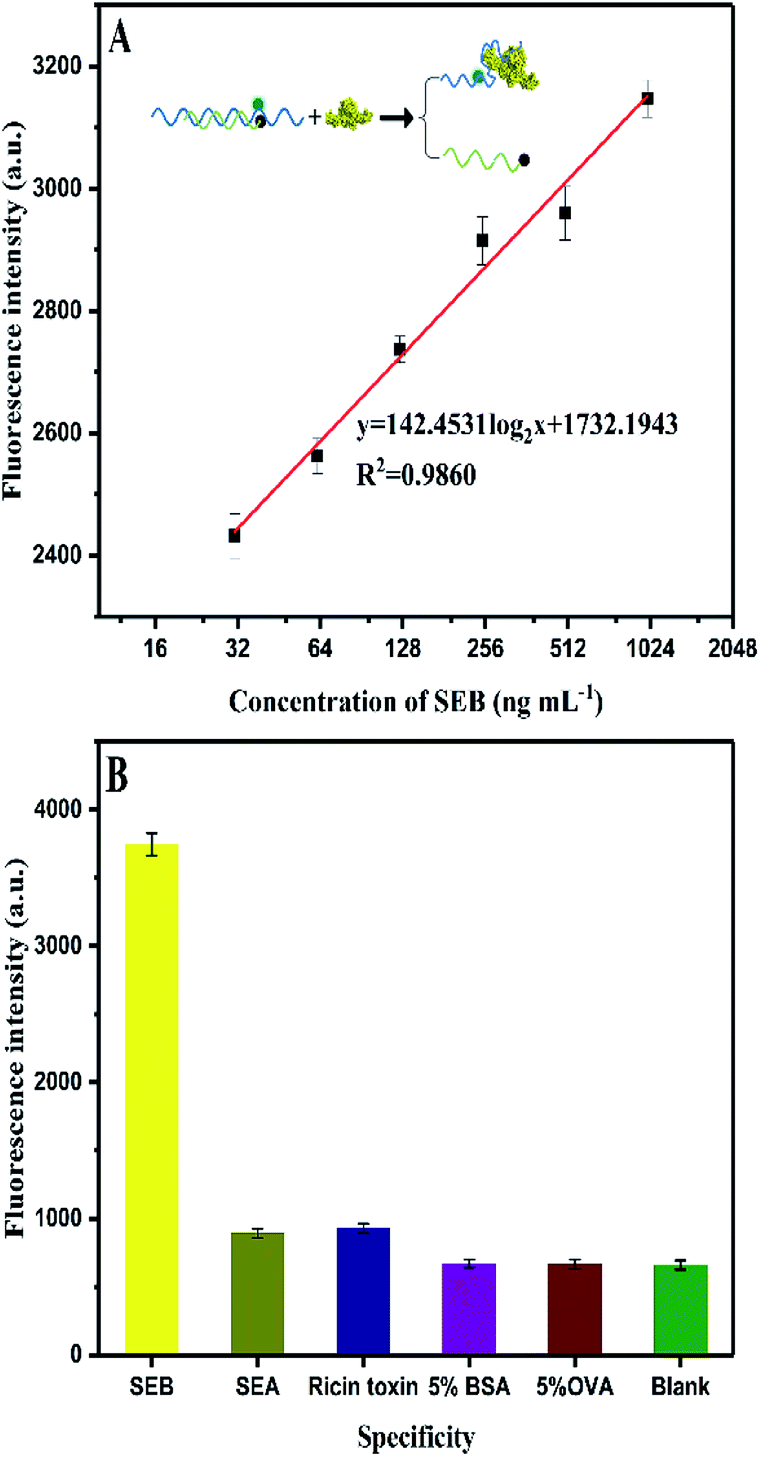 | ||
| Fig. 6 (A) Aptamer-based fluorescence assay for SEB detection. (B) Specificity for aptamer-based assay for SEB (200 ng mL−1) detection against other analogues. | ||
| Sample | Found (ng mL−1) | Added (ng mL−1) | Detection (ng mL−1) | Recovery ratio (%) |
|---|---|---|---|---|
| a Nd: not detected. | ||||
| No. 1 | Nd | 10 | 9.62 | 96.15 ± 3.23 |
| 20 | 22.54 | 112.68 ± 2.94 | ||
| 80 | 70.86 | 88.58 ± 1.70 | ||
| No. 2 | 15.61 | 10 | 26.32 | 107.09 ± 2.30 |
| 20 | 33.17 | 87.83 ± 2.91 | ||
| 80 | 99.56 | 104.94 ± 2.43 | ||
| No. | Method | LOD (ng mL−1) | Number of steps | Time of analysis (min) | Ref. |
|---|---|---|---|---|---|
| 1 | ELISA | 0.1 | 8 | 240 | 46 |
| 2 | BIA-MS | 1 | 5 | — | 47 |
| 3 | Piezoelectric (PZ) | 250 | 6 | 120 | 13 |
| 4 | SPR | 5 | 5 | 140 | 48 |
| 5 | Magnetoelastic | 0.5 | 9 | 300 | 49 |
| 6 | Electroluminescence | 3.9 | 6 | 240 | 50 |
| 7 | Flow-based microarray | 4 | 6 | 15 | 51 |
| 8 | Hydrogel-based protein-microarray | 1 | 4 | >150 | 52 |
| 9 | The proposed assay | 0.33 | 4 | 105 |
3.5 SEB detection in milk sample by the proposed method by HCR-based amplification
In this assay, two SEB-spiked randomly samples with a concentration of 10, 20, and 80 ng mL−1 concentrations were investigated using the HCR-based aptasensor scheme. In particular, the proposed method was as follows: 100 μL of samples were incubated with 20 μL of SEB aptamer (1 μM) at 37 °C for 30 min. Then, 30 μL of partial complementary strand (1 μM) was added to the mixture and reacted at 37 °C for 30 min. Subsequently, 20 μL of H1/H2 (2.5 μM) were reacted in the hybridization buffer at 37 °C for 45 min. Table 2 indicates the recoveries were between 87.83% ± 2.91% and 112.68% ± 2.94%, which represent the accuracy of the proposed method.4. Conclusion
In summary, a simple, rapid, and enzyme-free aptasensor combined with a HCR-based isothermal amplification strategy was developed. In our design, we attempted to introduce both H1 and H2, which are similar to the molecular beacon design into the SEB detection. However, the fluorescence intensity showed no significant increase which may result from the location relative to each group. Therefore, only fluorescent and quenching groups were labeled for H1 in our experiment, whereas H2 did not make any modification. This binding force of the aptamer–SEB target conjugates competes with cDNA (H0), triggering the subsequent HCR and thus producing a strong signal response. The LOD of the proposed method was 21-fold lower than that of traditional aptasensor fluorescence method, and the total detection was less than 2 h. Moreover, this assay demonstrates the promising application in the detection of other targets. However, this linear HCR does not substantially improve the sensitivity to a great extent (Table 3), and a portion of the fluorescence is quenched due to the amplification of HCR, resulting in a certain decrease in fluorescence intensity. Therefore, in our future research, HCR should be combined with different signal output means to further improve the sensitivity is necessary in our next step of research.Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of the manuscript.Acknowledgements
This work was supported by 2017YFC1200903 National Natural Science Foundation of China (21477162, 81502847, 81602896, AWS15J006).Notes and references
- M. A. Argudin, M. C. Mendoza and M. R. Rodicio, Toxins, 2010, 2, 1751–1773 CrossRef CAS PubMed.
- M. C. Enright, N. P. Day, C. E. Davies, S. J. Peacock and B. G. Spratt, J. Clin. Microbiol., 2000, 38, 1008–1015 CAS.
- S. Derzelle, F. Dilasser, M. Duquenne and V. Deperrois, Food Microbiol., 2009, 26, 896–904 CrossRef CAS PubMed.
- S. G. Ler, F. K. Lee and P. Gopalakrishnakone, J. Chromatogr. A, 2006, 1133, 1–12 CrossRef CAS PubMed.
- C. D. Lindsay and G. D. Griffiths, Hum. Exp. Toxicol., 2013, 32, 606–619 CAS.
- K. E. Sapsford, J. Francis, S. Sun, Y. Kostov and A. Rasooly, Anal. Bioanal. Chem., 2009, 394, 499–505 CrossRef CAS PubMed.
- A. Sharma, V. K. Rao, D. V. Kamboj, R. Gaur, S. Upadhyay and M. Shaik, Biotechnol Rep., 2015, 6, 129–136 CrossRef PubMed.
- M. P. Chatrathi, J. Wang and G. E. Collins, Biosens. Bioelectron., 2007, 22, 2932–2938 CrossRef CAS PubMed.
- B. Zhang, B. Liu, D. Tang, R. Niessner, G. Chen and D. Knopp, Anal. Chem., 2012, 84, 5392–5399 CrossRef CAS PubMed.
- M. Yang, Y. Kostov, H. A. Bruck and A. Rasooly, Int. J. Food Microbiol., 2009, 133, 265–271 CrossRef CAS PubMed.
- L. Panneerseelan and P. M. Muriana, J. Food Prot., 2009, 72, 2538–2546 CrossRef CAS PubMed.
- M. Yang, S. Sun, Y. Kostov and A. Rasooly, Anal. Biochem., 2011, 416, 74–81 CrossRef CAS PubMed.
- H.-C. Lin and W.-C. Tsai, Biosens. Bioelectron., 2003, 18, 1479–1483 CrossRef CAS PubMed.
- C. Ruan, K. Zeng, O. K. Varghese and C. A. Grimes, Biosens. Bioelectron., 2004, 20, 585–591 CrossRef CAS PubMed.
- S. Rong-Hwa, T. Shiao-Shek, C. Der-Jiang and H. Yao-Wen, Food Chem., 2010, 118, 462–466 CrossRef CAS.
- I. Sospedra, R. Marín, J. Mañes and J. M. Soriano, Food Chem., 2012, 133, 163–166 CrossRef CAS.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- M. Blind and M. Blank, Mol. Ther.--Nucleic Acids, 2015, 4, e223 CrossRef PubMed.
- R. Nutiu and Y. Li, Chem.–Eur. J., 2004, 10, 1868–1876 CrossRef CAS PubMed.
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis and H. A. Erlich, Science, 1988, 239, 487–491 CrossRef CAS PubMed.
- F. Barany, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 189–193 CrossRef CAS.
- T. Notomi, Jpn. J. Clin. Med., 2007, 65, 957–961 Search PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- M. Vincent, Y. Xu and H. Kong, EMBO Rep., 2004, 5, 795–800 CrossRef CAS PubMed.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- S. S. Lu, T. Hu, S. Wang, J. Sun and X. R. Yang, et al., ACS Appl. Mater. Interfaces, 2016, 9, 167–175 Search PubMed.
- P. Liu, X. Yang, S. Sun, Q. Wang, K. Wang, J. Huang, J. Liu and L. He, Anal. Chem., 2013, 85, 7689–7695 CrossRef CAS PubMed.
- Z. Li, X. Miao, Z. Cheng and P. Wang, Sens. Actuators, B, 2017, 243, 731–737 CrossRef CAS.
- S. Wu, N. Duan, X. Ma, Y. Xia, H. Wang and Z. Wang, Anal. Chim. Acta, 2013, 782, 59–66 CrossRef CAS PubMed.
- S. Niu, Y. Jiang and S. Zhang, Chem. Commun., 2010, 46, 3089–3091 RSC.
- L. Chen, Z. Zhang, X. Zhang, A. Fu, P. Xue and R. Yan, Food Chem., 2012, 135, 208–212 CrossRef CAS.
- X. Wang, C. Lau, M. Kai and J. Lu, Analyst, 2013, 138, 2691–2697 RSC.
- H. Wang, L. Peng, Y. Q. Chai and R. Yuan, Anal. Chem., 2017, 89, 11076–11082 CrossRef CAS PubMed.
- J. Zhuang, L. Fu, M. Xu, H. Yang, G. Chen and D. Tang, Anal. Chim. Acta, 2013, 783, 17–23 CrossRef CAS PubMed.
- M. Liu, J. Xu, F. Yang, Y. Gu, H. Chen, Y. Wang and F. Li, Anal. Bioanal. Chem., 2017, 409, 6821–6829 CrossRef CAS PubMed.
- J. Han, Y. Zhuo, Y. Chai, Y. Yu, N. Liao and R. Yuan, Anal. Chim. Acta, 2013, 790, 24–30 CrossRef CAS PubMed.
- J. Zhao, C. Chen, L. Zhang, J. Jiang and R. Yu, Biosens. Bioelectron., 2012, 36, 129–134 CrossRef CAS PubMed.
- W. Song, K. Zhu and Z. Cao, et al., Analyst, 2012, 137, 1396–1401 RSC.
- J. A. DeGrasse, PLoS One, 2012, 7, e33410 CAS.
- S. Tyagi, S. A. Marras and F. R. Kramer, Nat. Biotechnol., 2000, 18, 1191–1196 CrossRef CAS PubMed.
- Q. Guo, J. J. Han, S. Shan, D. F. Liu, S. S. Wu, Y. H. Xiong and W. H. Lai, Biosens. Bioelectron., 2016, 86, 990–995 CrossRef CAS PubMed.
- T. Hianik, V. Ostatná, M. Sonlajtnerova and I. Grman, Bioelectrochemistry, 2007, 70, 127–133 CrossRef CAS PubMed.
- J. Zhang, H. P. Lang, G. Yoshikawa and C. Gerber, Langmuir, 2012, 28, 6494–6501 CrossRef CAS PubMed.
- M. A. Poli, V. R. Rivera and D. Neal, Toxicon, 2002, 40, 1723–1726 CrossRef CAS PubMed.
- D. Nedelkov, A. Rasooly and R. W. Nelson, Int. J. Food Microbiol., 2000, 60, 1–13 CrossRef CAS PubMed.
- J. Homola, J. Dostalek, S. F. Chen, R. Avraham, S. Y. Jiang and S. S. Yee, Int. J. Food Microbiol., 2002, 75, 61–69 CrossRef CAS PubMed.
- C. Ruan, K. Zeng, O. K. Varghese and C. A. Grimes, Biosens. Bioelectron., 2004, 20, 585–591 CrossRef CAS PubMed.
- K. E. Sapsford, J. Francis, S. Sun, K. Yordan and R. Avraham, Anal. Bioanal. Chem., 2009, 394, 499–505 CrossRef CAS PubMed.
- J. B. Delehanty and F. S. Ligler, Anal. Chem., 2002, 74, 5681–5687 CrossRef CAS PubMed.
- P. T. Charles, C. R. Taitt, E. R. Goldman, J. G. Rangasammy and D. A. Stenger, Langmuir, 2004, 20, 270–272 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |