
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cs2CO3-promoted defluorination and functionalization of α-CF3 carbonyl compounds in the presence of N-, O-, and/or S-nucleophiles†
Yue Wu,
Bingbing Zhang,
Yinying Zheng,
Yuheng Wang and
Xinsheng Lei *
*
School of Pharmacy, Fudan University, 826 Zhangheng Road, Pudong Zone, Shanghai 201203, China. E-mail: leixs@fudan.edu.cn; Fax: +86 21 5198 0128; Tel: +86 21 5198 0128
First published on 30th April 2018
Abstract
A simple, efficient, and mild method for defluorination and functionalization of 3,3,3-trifluoro carbonyl compounds has been developed. In the present method, Cs2CO3 can easily convert α-trifluoromethyl esters, amides, and ketones into β,β-S-, O- and/or N-substituted α,β-unsaturated carbonyl compounds in the presence of N-, O-, and S-nucleophiles with moderate to excellent yields, and furthermore, this transformation with α-trifluoromethyl ester and a series of 2-aminophenols can result in benzooxazoles in good yields.
Introduction
In the past six decades, hydrofluorocarbons have been widely used in pharmaceuticals, agrochemicals, materials, refrigeration, and air conditioning. Introduction of C–F bonds into pharmaceuticals or materials can modify the acidity, lipophilicity, conformation and metabolism of pharmaceuticals,1–3 improve the hydrophobic properties, chemical inertness, and elasticity of materials,4,5 and sometimes enhance their special water-/stain-resistant and non-sticky characteristics.6,7 This “fluorine effect” or “fluorine magic” stems from a very short bond length, low polarizability, being fairly inert and the strong inductive effects of the C–F bond.8,9Logically, chemical inertness of C–F bond will make hydrofluorocarbons resistent to biotransformation or biodegradation.10,11 Thus, continual and increasing use of hydrofluorocarbons in modern life has implications for the environment and human health that urgently require attention.12,13 In light of the environmental concerns associated with organofluorine compounds,14,15 the development of novel synthetic methods for the C–F cleavage and subsequent functionalization is highly required.16
3,3,3-Trifluoropropanoic acid derivatives are one of the very important building blocks to incorporate one CF3 group into organic molecules. The exploitation of their α-CF3 enolates as active nucleophiles for the introduction of a CF3 group have been achieved (Fig. 1, path a),17–21 but their defluorination and functionization are only scarcely explored,22 because their direct defluorinations usually require strong basic conditions23 and the resultant β,β-difluoro-α,β-unsaturated carbonyl compounds are prone to subsequent decomposition even at a low temperature (path b).24 Recently, defluorination and functionization of 3,3,3-trifluoropropanoic acid derivatives have been reported at −78 °C to prepare monofluoroalkenes via elimination and addition with organolithium or Grignard reagents as C-nucleophiles (path c).25 These results prompted us to examine defluorination and functionization of α-CF3 carbonyl compounds with other nucleophiles such as S-, O-, and N-nucleophiles. Herein, we described an efficient and mild defluorinated and functionized method of α-CF3 carbonyl compounds via a Cs2CO3-promoted elimination/addition in the presence of a series of N-, O-, S-nucleophiles.
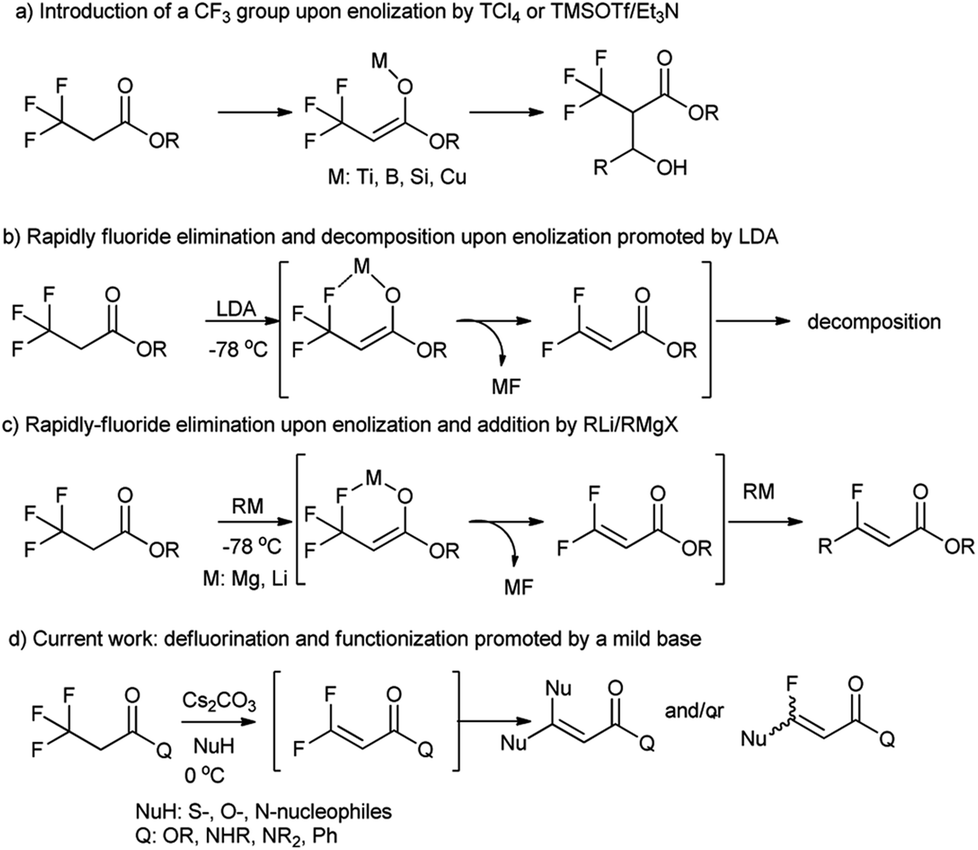 | ||
| Fig. 1 The enolization and defluorination of 3,3,3-trifluoropropanoic acid derivatives. | ||
Results and discussion
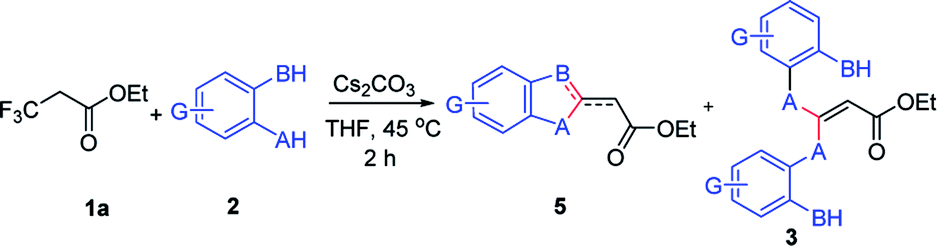
We initiated our work by studying the reaction of ethyl 3,3,3-trifluoropropanoate (1a) and 4-mercaptotoluene (2a) under the basic conditions. As shown in Table 1, in the presence of NaHMDS, the reaction was run at rt for 2 h, and expectedly, 2a was not consumed because of the almost quantitative recovery of 2a. When the reaction was run at 0 °C, only a small amount of defluorinated product could be isolated (entry 1, 37%), and its structure was subsequently determined as 3aa. Decreasing reaction temperature to −78 °C, only 3aa could be isolated in a higher yield (entry 2, 46%), but the partial-defluorinated product, such as 4aa, was still not isolated. Increasing the amount of 2a did not improve the reaction (entry 3, 47%), instead, increasing the amount of NaHMDS could increase the yield of 3aa up to 88%, albeit without 4aa. LDA had a similar performance to NaHMDS (entry 5, 64%). The above results implied that those bases might rapidly transform the ester into active β,β-difluoro-α,β-unsaturated carbonyl compounds, and then this unstable intermediate could be trapped by 2a to some extent while it was prone to decompose.
|
|||||
|---|---|---|---|---|---|
| Entry | 1a/2a/base (eq.) | Base | Temp. | Solvent | Yield (3aa)a,b |
| a Reaction conditions: to a solution of 1a (1.0 mmol), 2a in anhydrous THF (10 mL) was added one kind of base in one portion at the suitable temperature under Ar, and the reaction mixture was stirred for the indicated time.b The isolated yield based on 1a.c Not detected. | |||||
| 1 | 1.0/1.0/2.0 | NaHMDS | 0 °C | THF | 37% |
| 2 | 1.0/1.0/2.0 | NaHMDS | −78 °C | THF | 46% |
| 3 | 1.0/2.0/2.0 | NaHMDS | −78 °C | THF | 47% |
| 4 | 1.0/2.0/3.0 | NaHMDS | −78 °C | THF | 88% |
| 5 | 1.0/1.0/3.0 | LDA | −78 °C | THF | 64% |
| 6 | 1.0/2.0/3.0 | t-BuOK | 0 °C | THF | 54% |
| 7 | 1.0/2.0/3.0 | MeONa | 0 °C | THF | 56% |
| 8 | 1.0/2.0/3.0 | Na2CO3 | 0 °C | THF | NDc |
| 9 | 1.0/2.0/3.0 | K2CO3 | 0 °C | THF | 15% |
| 10 | 1.0/2.0/3.0 | AcONa | 0 °C | THF | NDc |
| 11 | 1.0/2.0/3.0 | NEt3 | 0 °C | THF | NDc |
| 12 | 1.0/2.0/2.0 | Cs2CO3 | 0 °C | THF | 96% |
| 13 | 1.0/2.0/2.0 | Cs2CO3 | 0 °C | DMF | 30% |
| 14 | 1.0/2.0/2.0 | Cs2CO3 | 0 °C | DMSO | 74% |
| 15 | 1.0/2.0/2.0 | Cs2CO3 | 0 °C | EtOH | NDc |
| 16 | 1.0/2.0/2.0 | Cs2CO3 | 25 °C | THF | 43% |
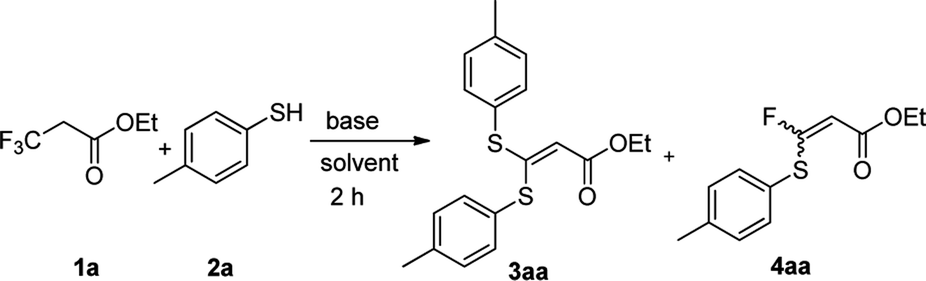
Based on the facts that only the trace partial-defluorinated product was observed and the major full-defluorinated product, as one kind of α-oxoketene-S,S-acetals, might be one potentially versatile three-carbon building blocks for the construction of various heterocyclic systems,26 we turned to optimize the full-defluorinated reaction by screening a variety of other bases for the reaction.
As shown in Table 1, t-BuOK gave a moderate yield (entry 6, 54%) at 0 °C, but a higher or lower temperature led to a poorer yield (at rt or −78 °C). Similarly, MeONa could give the almost same result (entry 7, 56%). The weaker bases, such as Na2CO3, AcONa and Et3N, could not initiate the reaction (entry 8, 10, 11) while K2CO3 just resulted in a very low yield (entry 9, 15%). To our delight, Cs2CO3 gave the almost quantitative yield and its amount could be reduced from 3.0 to 2.0 equivalents (entry 12, 98% vs. 96%). Subsequently, the solvent screening demonstrated that neither DMF nor EtOH was good to the reaction (entry 13, 15), and DMSO just gave a slightly decreased yield (entry 14, 74%). As a result, THF was chosen as the optimized solvent for the reaction, but therein, elevating the temperature seemed to have an adverse effect on the reaction (entry 16).
With the optimized condition in hand, we subsequently examined the nucleophile scope with a variety of mono-dentate S-, O-, and N-nucleophiles, and the results were depicted in Table 2. For the S-containing nucleophiles, aryl thiols generally gave their defluorinated products in good to excellent yields (3aa–3af, 46–96%), and the substituent electronic effect in the aryl ring may have little influence on the reaction, instead, the bulkier nucleophile seems to have a slight effect on the reaction (3ac, 46%). This reaction could successfully be expanded to the aliphatic thiol (3ag, 89%).
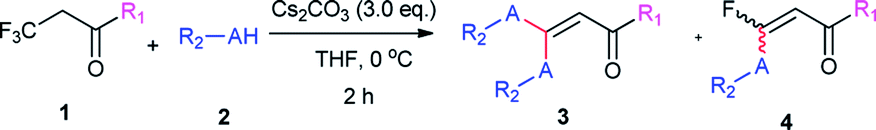
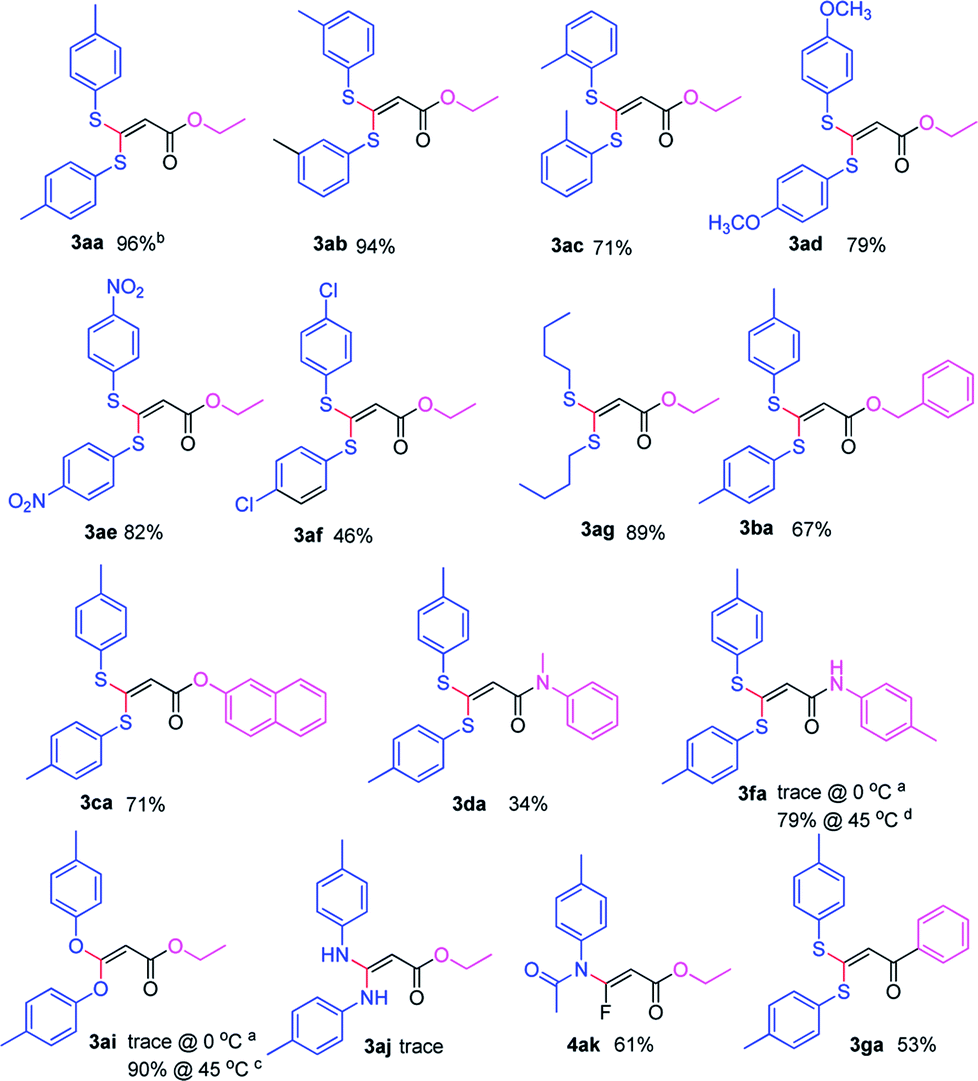
On the other hand, for the different 3,3,3-trifluoropropanoyl esters, the reaction also gave the similar results in the case of benzyl ester and β-naphthalenyl ester (3ba, 43%; 3ca, 71%). N,N-substituted 3,3,3-trifluoropropanamide was able to carry out this transformation with a lower yield (3da, 34%). In contrast, the N-monosubstituted amide only produced a trace amount of the desired product at 0 °C, but could afford the product at 45 °C in DMSO (3fa, 79%).
Furthermore, the O- and N-containing nucleophiles were tested in the reaction, for example, p-cresol successfully afforded the defluorinated product at a higher temperature (3ai, 90% at 45 °C). However, in terms of the N-containing nucleophile, p-toluidine did not afford the desired compound (3aj, trace) under Cs2CO3-, NaHMDS-, or t-BuOK-promoted conditions, possibly due to poor nucleophilicity of the amine or instability of 3aj. In contrast, the amide was able to produce the partial-defluorinated product (4ak, 61%) with Z-configuration. To our surprise, α-CF3 ketone was also proved flexible in the reaction, for example, 3,3,3-trifluoro-1-phenylpropan-1-one could react with 2a under the standard condition, and then gave 3ga in 53% yield.
Next, this defluorinative functionalization was switched from those mono-dentate nucleophiles to the bidentate nucleophiles, the results were depicted in Table 3. When one equivalent of catechol was used as one O,O-bidentate nucleophile, the reaction did not occur at 0 °C, but at a higher temperature (45 °C) the cyclized compound was smoothly produced (5am, 83%). Similarly, 2-mercaptophenol, one O,S-bidentate nucleophile, could smoothly afford the corresponding product (5an, 66%), and interestingly, the single crystal X-ray diffraction experiment demonstrated that the stereo configuration between H atom at the olefin and O atom at the heterocycle was syn-configuration,27 indicating there evidently existed bulkier repulse between H atom and S atom.
|
|---|
| a Reaction conditions: a solution of 1a (1.0 mmol), 2 (1.0 mmol) and Cs2CO3 (2.0 mmol)in anhydrous THF (10 mL) was stirred at 45 °C for 2 h under Ar. The isolated yield based on 1a. |
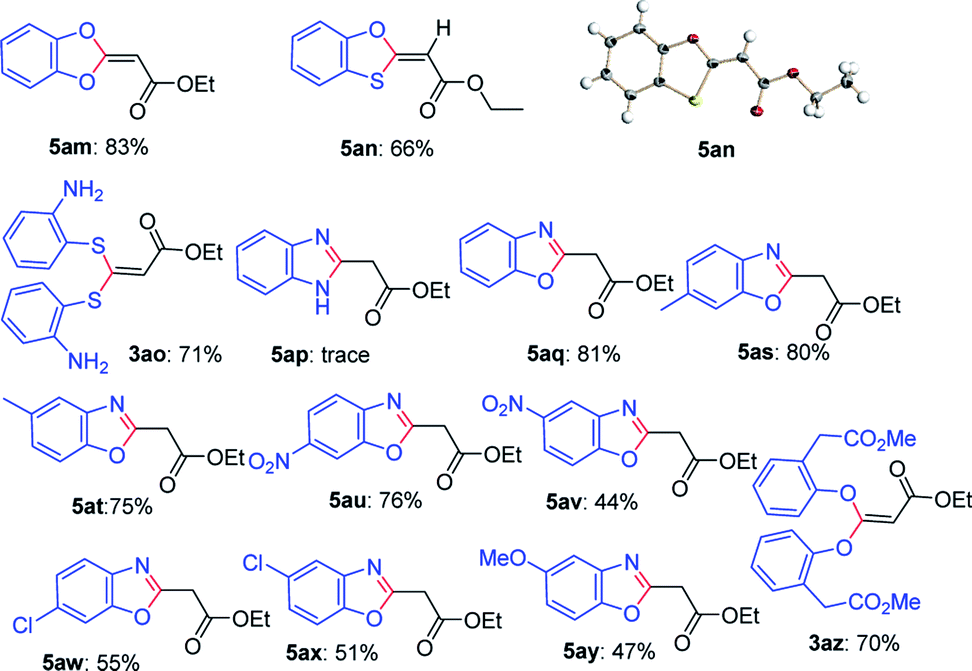 |
Unexpectedly, 2-aminothiophenol, as one N,S-bidentate nucleophile, did not give the desired product, instead, it resulted in the undesired product (3ao, 71%), possibly due to the weaker nucleophilicity of amine than that of thiol. Furthermore, the reaction using benzene-1,2-diamine as one N,N-bidentate nucleophile led to complicated products (5ap, trace).
Interestingly, 2-aminophenol, as one N,O-bidentate nucleophile, produced the benzooxazole derivative at 45 °C in a good yield (5aq, 81%) via defluorinative substitution, intramolecularly defluorinative substitution and aromatization. To our delight, a set of 2-aminophenols substituted at the aromatic ring could easily produce the corresponding ethyl 2-(benzo[d]oxazol-2-yl)acetates in moderate to good yields (5aq–5ay, 44–81%). However, when one C,O-bidentate nucleophile, such as methyl 2-(2-hydroxyphenyl)acetate, was used in the reaction, only O atom instead of C atom acted as the nucleophile (3az, 70%).
To understand the reaction mechanism, the control experiments were performed, as shown in Scheme 1. If the reaction was run in absence of Cs2CO3, the reaction did not occur (Scheme 1a). Meanwhile the trifluoropropanoylate ester was not decomposed by Cs2CO3 in the absence of nucleophile (Scheme 1b), although decomposition occurred with a strong base (such as NaHMDS). On the other hand, if methyl 3,3-difluoroacrylate was subjected to the same basic condition, the desired compound (3ha) could be produced (Scheme 1c), strongly indicating the 3,3-difluoroacrylate was the intermediate. Although we attempted to isolate the corresponding 3,3-difluoroacrylamide or monitor other fluoro organic compounds by 19F NMR in the case of 4ka, unfortunately we did not succeed (Scheme 1d).
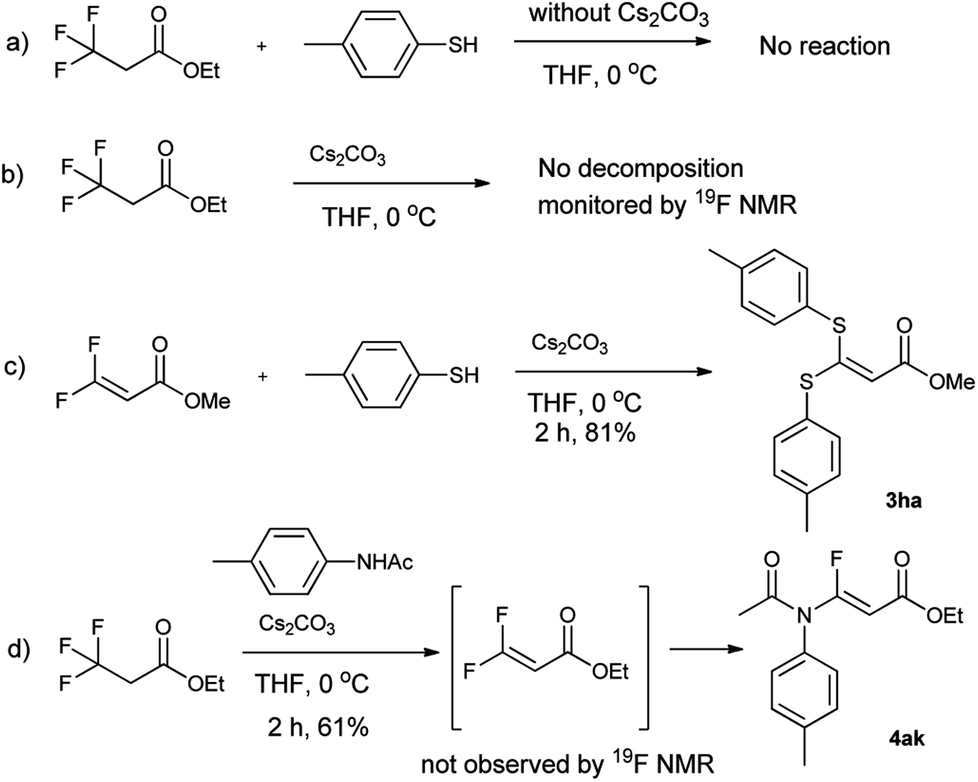 | ||
| Scheme 1 The control experiments. | ||
Together with the above results, we proposed a plausible mechanism (Scheme 2).22–24 At first, Cs2CO3 promoted the dehydrofluorinative elimination of trifluoropropanoylate ester in the presence of one suitable nucleophile, and the resultant 3,3-difluoroacrylate ester was rapidly added by the nucleophile, then the adduct continued to rapidly undergo elimination to afford α,β-unsaturated monofluorinated carbonyl compound, and in general, the further addition and dehydrofluorinative elimination continued to occur and finally resulted in the full-defluorinated product.
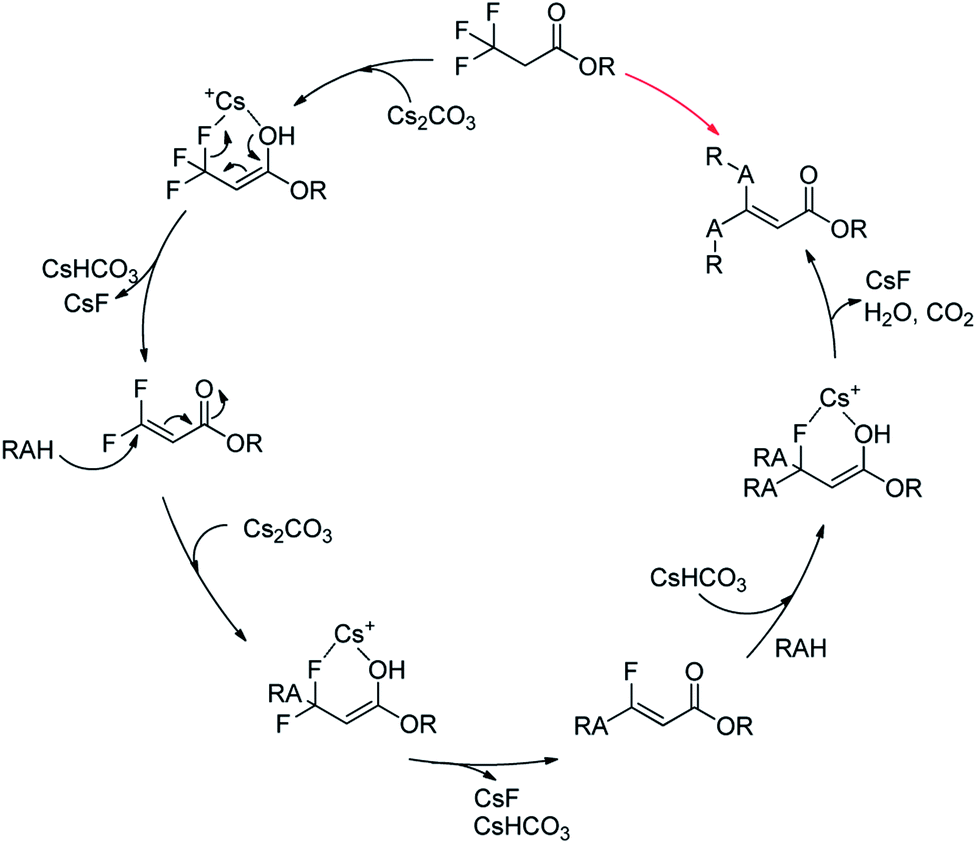 | ||
| Scheme 2 A plausible mechanism of the defluorination and functionization of ethyl 3,3,3-trifluoropropanoylate. | ||
Conclusions
In conclusion, we have developed an efficient defluorinated and functionized method of 3,3,3-trifluoropropanoic acid derivatives, and in this method, a mild base (Cs2CO3) enables to promote defluorination and functionalization of the β,β,β-trifluoro carbonyl compounds in the presence of a series of N-, O-, S-nucleophiles. By the current method, the α-trifluoromethyl carbonyl compounds easily afford β,β-N-, O-, and/or S-substituted α,β-unsaturated products in moderate to excellent yields, and α-trifluoromethyl ester can also be transformed into the corresponding benzooxazoles in good yields when 2-aminophenols were used as nucleophiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Natural Foundation of China (No. 21472024 and No. 21242008) for the financial support.Notes and references
- Y. Zhou, J. Wang, Z. N. Gu, S. N. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398 CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- Z. L. Chu and S. Seeger, Chem. Soc. Rev., 2014, 43, 2784 RSC.
- M. Pagliaro and R. Ciriminna, J. Mater. Chem., 2005, 15, 4981 RSC.
- I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef.
- W. Zhang, Chem. Rev., 2009, 109, 749 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed , and references therein.
- C. F. Ni and J. B. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC.
- D. O'Hagan and H. Deng, Chem. Rev., 2014, 115, 634 CrossRef PubMed.
- C. D. Murphy, Appl. Microbiol. Biotechnol., 2016, 100, 2617 CrossRef CAS PubMed.
- N. Khalil, A. Chen, M. Lee, S. A. Czerwinski, J. R. Ebert, J. C. DeWitt and K. Kannan, Environ. Health Perspect., 2016, 124, 81 CAS.
- H. Fromme, S. A. Tittlemier, W. Voelkel, M. Wilhelm and D. Twardella, Int. J. Hyg. Environ. Health, 2009, 212, 239 CrossRef CAS PubMed.
- K. Kato, L. Y. Wong, L. T. Jia, Z. Kuklenyik and A. M. Calafat, Environ. Sci. Technol., 2011, 45, 8037 CrossRef CAS PubMed.
- A. Rotander, A. Kärrman, B. van Bavel, A. Polder, F. Rigét, A. Auðunsson, G. Víkingsson, G. W. Gabrielsen, D. Blochg and M. Damh, Chemosphere, 2012, 86, 278 CrossRef CAS PubMed.
- O. Eisenstein, J. Milani, N. Robin and R. N. Perutz, Chem. Rev., 2017, 117, 8710 CrossRef CAS PubMed , and references therein.
- (a) A. Matsuzawa, H. Noda, N. Kumagai and M. Shibasaki, J. Org. Chem., 2017, 82, 8304 CrossRef CAS PubMed; (b) L. Brewitz, F. A. Arteaga, L. Yin, K. Alagiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2015, 137, 15929 CrossRef CAS PubMed; (c) P. V. Ramachandran, B. Otoo and P. B. Chanda, Tetrahedron Lett., 2015, 56, 3019 CrossRef CAS; (d) L. Yin, L. Brewitz, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2014, 136, 17958 CrossRef CAS PubMed; (e) J.-L. Chen, Z.-W. You and F.-L. Qing, J. Fluorine Chem., 2013, 155, 143 CrossRef CAS; (f) P. V. Ramachandran, G. Parthasarathy and P. D. Gagare, Tetrahedron Lett., 2011, 52, 6055 CrossRef; (g) P. V. Ramachandran, G. Parthasarathy and P. D. Gagare, Tetrahedron Lett., 2011, 52, 5359 CrossRef; (h) P. V. Ramachandran, G. Parthasarathy and P. D. Gagare, Org. Lett., 2010, 12, 4474 CrossRef CAS PubMed.
- K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817 CrossRef CAS PubMed , and references therein.
- (a) A.-L. Dreier, B. Beutel, C. Mück-Lichtenfeld, A. V. Matsnev, J. S. Thrasher and G. Haufe, J. Org. Chem., 2017, 82, 1638 CrossRef CAS PubMed; (b) A. Saito, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2017, 56, 5551 CrossRef CAS PubMed; (c) F. Grellepois, V. Kikelj, N. Coia and C. Portella, Eur. J. Org. Chem., 2012, 509 CrossRef CAS.
- (a) H.-W. Liang, W. Ding, K. Jiang, L. Shuai, Y. Yuan, Y. Wei and Y.-C. Chen, Org. Lett., 2015, 17, 2764 CrossRef CAS PubMed; (b) J. Xie, P. Xu, H. Li, Q. Xue, H. Jin, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 5672 RSC; (c) Y. Guo, X. Zhao, D. Zhang and S.-I. Murahashi, Angew. Chem., Int. Ed., 2009, 48, 2047 CrossRef CAS PubMed.
- (a) A. Gómez-Palomino, M. Pellicena, J. M. Romo, R. Solà, P. Romea, F. Urpí and M. Font-Bardia, Chem. – Eur. J., 2014, 20, 10153 CrossRef PubMed; (b) J. K. Augustine, A. Bombrun, B. Ramappa and C. Boodappa, Tetrahedron Lett., 2012, 53, 4422 CrossRef CAS; (c) E. Yoshioka, S. Kohtani and H. Miyabe, Angew. Chem., Int. Ed., 2011, 50, 6038 CrossRef PubMed; (d) Y. Liu, H. Lai, B. Rong, T. Zhou, J. Hong, C. Yuan, S. Zhao, X. Zhao, B. Jiang and Q. Fang, Adv. Synth. Catal., 2011, 353, 3161 CrossRef CAS.
- (a) H. Liu, S. Song, C.-Q. Wang, C. Feng and T.-P. Loh, ChemSusChem, 2017, 10, 58 CrossRef CAS PubMed; (b) K. N. Nandeesh, H. A. Swarup, N. C. Sandhya, C. D. Mohan, C. S. P. Kumar, M. N. Kumara, K. Mantelingu, S. Ananda and K. S. Rangappa, New J. Chem., 2016, 40, 2823 RSC.
- Y. Itoh, M. Yamanaka and K. Mikami, J. Am. Chem. Soc., 2004, 126, 13174 CrossRef CAS PubMed.
- (a) K. K. Nanda and B. W. Trotter, Tetrahedron Lett., 2008, 48, 5332 CrossRef; (b) T. Yokozawa, T. Nakai and N. Ishikawa, Tetrahedron Lett., 1984, 25, 3987 CrossRef CAS; (c) T. Yokozawa, T. Nakai and N. Ishikawa, Tetrahedron Lett., 1984, 25, 3991 CrossRef CAS.
- M. Lecea, A. Grassin, L. Ferreiro-Mederos, S. Choppin, A. Urbano, M. C. Carreno and F. Colobert, Eur. J. Org. Chem., 2013, 4486 CrossRef CAS.
- (a) D. Cheng, L. Wu, Z. Deng, X. Xu and J. Yan, Adv. Synth. Catal., 2017, 359, 4317 CrossRef CAS; (b) L. Zhu, H. Yu, Q. Guo, Q. Chen, Z. Xu and R. Wang, Org. Lett., 2015, 17, 1978 CrossRef CAS PubMed; (c) B. Liu, G. Zheng, X. Liu, C. Xu, J. Liu and M. Wang, Chem. Commun., 2013, 49, 2201 RSC; (d) G. K. Verma, R. K. Verma and M. S. Singh, RSC Adv., 2013, 3, 245 RSC, and references therein (e) G. K. Verma, R. K. Verma, G. Shukla, N. Anugula, A. Srivastava and M. S. Singh, Tetrahedron, 2013, 69, 6612 CrossRef CAS; (f) D. Liang, M. Wang, B. Bekturhun, B. Xiong and Q. Liu, Adv. Synth. Catal., 2010, 352, 1593 CrossRef CAS.
- Crystallographic data for compound 5an: CCDC 1815972.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1815972. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02353k |
| This journal is © The Royal Society of Chemistry 2018 |