
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore@shell structured Co–CoO@NC nanoparticles supported on nitrogen doped carbon with high catalytic activity for oxygen reduction reaction†
Zihao Zhena,
Zhongqing Jiang bc,
Xiaoning Tianc,
Lingshan Zhoua,
Binglu Denga,
Bohong Chena and
Zhong-Jie Jiang*a
bc,
Xiaoning Tianc,
Lingshan Zhoua,
Binglu Denga,
Bohong Chena and
Zhong-Jie Jiang*a
aGuangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Research Institute, College of Environment and Energy, South China University of Technology, Guangzhou 510006, Guangdong, P. R. China. E-mail: Zhongjiejiang1978@hotmail.com; eszjiang@scut.edu.cn
bDepartment of Physics, Key Laboratory of ATMMT Ministry of Education, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China
cSchool of Materials and Chemical Engineering, Ningbo University of Technology, Ningbo 315211, Zhejiang, P. R. China
First published on 18th April 2018
Abstract
A composite with a hierarchical structure consisting of nitrogen doped carbon nanosheets with the deposition of nitrogen doped carbon coated Co–CoO nanoparticles (Co–CoO@NC/NC) has been synthesized by a simple procedure involving the drying of the reaction mixture containing Co(NO3)2, glucose, and urea and its subsequent calcination. The drying step is found to be necessary to obtain a sample with small and uniformly sized Co–CoO nanoparticles. The calcination temperature has a great effect on the catalytic activity of the final product. Specifically, the sample prepared at the calcination temperature of 800 °C shows better catalytic activity of the oxygen reduction reaction (ORR). Urea in the reaction mixture is crucial to obtain the sample with the uniformly sized Co–CoO nanoparticles and also plays an important role in improving the catalytic activity of the Co–CoO@NC/NC. Additionally, there exists a strong electronic interaction between the Co–CoO nanoparticles and the NC. Most interestingly, the Co–CoO@NC/NC is highly efficient for the ORR and can deliver an ORR onset potential of 0.961 V vs. RHE and a half-wave potential of 0.868 V vs. RHE. Both the onset and half-wave potentials are higher than those of most catalysts reported previously and even close to those of the commercial Pt/C (the ORR onset and half-wave potential of the Pt/C are 0.962 and 0.861 V vs. RHE, respectively). This, together with its high stability, strongly suggests that the Co–CoO@NC/NC could be used as an efficient catalyst for the ORR.
Introduction
The ever-increasing global demands in sustainable energy have prompted worldwide efforts in developing alternative energy conversion and storage devices, such as fuel cells, and metal air batteries.1,2 Exploiting efficient electrocatalysts that can enhance the kinetics of the oxygen reduction reaction (ORR) has therefore attracted a great deal of attention, since the sluggishness of the ORR has greatly limited practical applications of these devices.3,4 Currently, the best catalysts reported for the ORR are Pt and its alloys, such as Pt/Ni, Pt/Co, and Pt/Cu, etc.5–8 These catalysts, however, suffer from severe problems of low stability, poor tolerance to poisoning, scarcity in earth, and high cost, which have largely hindered their applications as the electrocatalysts for the ORR in fuel cells and metal air batteries.9,10 Developing alternative catalysts that can boost the ORR kinetics and simultaneously meet the requirements of low cost and high stability is therefore highly desirable.In response to reduce the cost and improve the catalytic activities for the ORR, efforts on developing ORR catalysts have been mainly focused on noble metal-free or even metal-free materials.11,12 Transition metal based catalysts are of particular interest due to their advantages of superior catalytic activity and earth abundance.13,14 As reported previously, transition metals (TMs) and their oxides (TMOs) can exhibit excellent catalytic activities for the ORR when they are synthesized properly.15–18 The main problem associated with the practical applications of the TMs and TMOs, however, is their poor stability in the complicated electrochemical environments.19,20 Strategies to address this issue are therefore needed to promote the applications of the TMs and TMOs as the ORR electrocatalysts. Encapsulating with the carbon shell is a promising way to improve the stability of the TMs and TMOs.16,21–23 As demonstrated recently, the encapsulation cannot only protect the encapsulated TMs and TMO cores from degradation through avoiding their direct contact with the electrolyte solutions, but also improve the catalytic activities of the TMs and TMOs since the ultrathin carbon shells can greatly promote penetration of electrons from the TM and TMO cores to the carbon surface, participating in the catalytic reactions.24,25 In particular, when the carbon shell is doped with heteroatoms, the synergistic interaction between heteroatom doped carbon shell and the TM/TMO core can increase the electron density on the carbon shell, further improving their capability for the catalytic applications.24,26,27
Metallic cobalt (Co) and its oxide (CoO) are typical TM based materials, both of which have been demonstrated to be usable as the catalysts for the ORR.28,29 The catalytic activities of the single component Co or CoO material, however, remain lower in comparison to the Pt-based catalysts, although they might be enhanced through the integration of Co or CoO with carbonaceous materials.20,30 Recently, the composite materials consisting of two or more components that are individually active for the ORR are receiving growing attention, since the synergistic between multiple components might generate more fascinating properties for the catalytic applications.2,12,31–36 For example, the Fe–Fe2O3 particles deposited N-doped graphene shows enhanced catalytic activity for the ORR.12 The Ni–NiO nanoparticles on PDDA-modified graphene are highly active for oxygen reduction reaction.37 In this sense, the combination of Co with CoO is therefore expectable to produce a composite particle with higher catalytic activity for the ORR.
In this work, we investigate the ORR catalytic activity of nitrogen doped carbon coated Co–CoO nanoparticles (Co–CoO@NC). Specifically, a composite material with a hierarchical structure consisting of the nitrogen doped carbon nanosheets with the deposition of the Co–CoO@NC (Co–CoO@NC/NC) has been synthesized through a simple calcination of a mixture containing Co(NO3)2, glucose, and urea. The effects of the drying step and the calcination temperature on the catalytic activity of the final product has been studied. It shows that the drying step is indispensable in obtaining the sample with the small and uniformly sized Co–CoO nanoparticles. The sample prepared at the calcination temperature of 800 °C shows better catalytic activity for the ORR, since at this temperature glucose and urea are well carbonized and the obtained product consists of small and uniform sizes of the Co–CoO nanoparticles (NPs) and a high content of the doping nitrogen in the carbon. Urea in the reaction mixture is found to play an important role in the formation of the Co–CoO@NC/NC with higher catalytic activity. It does not only provide a nitrogen source for the NC, but also facilitate the formation of the Co–CoO nanoparticles with small and uniform sizes. Most notably, there exists a strong interaction between the Co–CoO nanoparticles and the NC. When used as the catalyst, the Co–CoO@NC/NC at the calcination temperature of 800 °C (Co–CoO@NC/NC-800) could exhibit an ORR onset potential of 0.961 V vs. RHE and a half-wave potential of 0.868 V vs. RHE. These values are higher than the onset and half-wave potentials of the single component Co and CoO based catalysts and even close to those of the commercial Pt/C, strongly suggesting that the Co–CoO@NC/NC is an efficient catalyst for the ORR. Up to now, although a significant amount of the catalysts has been reported to be catalytically active for the ORR, those with activities higher than or even close to the commercial Pt/C has less been reported. The work presented here is therefore of great interest since it develops a new and high efficient catalyst using a simple synthetic method and would be also helpful to design other catalysts.
Experimental
Chemicals and reagents
Urea (99%) and glucose (99%) were obtained from Guangzhou Chemical Reagent Co. Ltd. Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99%) was obtained from Tianjin Kermel Chemical Reagent Co. Ltd. Potassium hydroxide (KOH, 85%), methanol and isopropanol were obtained from Tianjin chemical reagent Co. Ltd. Nafion (5.0 wt%) was purchased from DuPont Company. All the chemicals were used as received without further purification. Deionized (DI) water with a resistance of ∼18 MΩ cm−1 was used in all the reactions.Material synthesis
In a typical synthesis, 50.0 mg of glucose and 1.0 g of urea were dissolved with 40 mL of DI water. The obtain mixture was stirred vigorously for 30 min, followed by addition of 0.6 mmol of Co(NO3)2. The obtained pink homogenous solution was heated at 80 °C under stirring to remove water. After vaporization, the obtained solid was loaded on a crucible, then thoroughly dried at 200 °C for 2 h, and finally calcined at 700, 800 or 900 °C for 3 h in a tube furnace under the N2 flow. The products obtained from the calcination temperature of 700, 800 and 900 °C were named as Co–CoO@NC/NC-700, Co–CoO@NC/NC-800, and Co–CoO@NC/NC-900, respectively. The same synthetic procedure was performed for the synthesis of the Co–CoO@C/C-800 and the NC in the absence of urea or Co(NO3)2, respectively, while keeping other parameters constant.Physical characterizations
X-ray diffraction patterns (XRD) of the samples were recorded on a Bruker D8 X-ray diffractometer with the Cu Kα radiation and were analyzed using Bruker EVA and Bruker TopAs 4.2 software. Scanning electron microscopy (SEM) was performed on Merlin with an operation voltage of 20.0 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) measurements were performed on a JEM-2100F with an accelerating voltage of 200 kV. Raman spectra were collected on a LabRAM HR Evolution system using an Ar laser with excitation wavelength of 514.5 nm. The chemical compositions of the samples were determined by X-ray photoelectron spectroscopy (XPS) performed on a PHI X-tool (Japan) with an Al Kα X-ray radiation (1486 eV). The Brunauer–Emmett–Teller (BET) surface areas and pore size distributions of the samples were determined by the N2 adsorption/desorption isotherms recorded at 77 K using Micromeritics ASAP 2020.Electrochemical measurement
A standard three-electrode cell was used for the electrochemical measurements using an electrochemical workstation (CHI 760D, China). Platinum wire and a saturated calomel electrode served as the counter and the reference electrodes, respectively. The working electrode was prepared by dropping drops of the catalyst ink onto the glassy carbon electrode and then naturally dried at room temperature. The catalyst ink with a catalysts concentration of 2 mg mL−1 was prepared by dispersing the catalyst and 10 μL of Nafion (5 wt%) into a mixture of water and isopropanol with a volume ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.0, followed by ultrasonication to obtain a homogeneous solution. The loading of the catalyst on the working electrode was well controlled to ∼0.1 mg cm−2. The working electrode with the Pt/C (20 wt% Pt on the carbon, Johnson Matthey) used for comparison was prepared in a similar manner. The cyclic voltammograms (CVs) were recorded at ambient temperature in an 0.1 M KOH solution. Before the measurements, the solution was aerated with N2 or O2 for at least 30 min. The N2 or O2 flow was maintained above the solution to keep the N2 or O2 saturation during the electrochemical measurements. The linear sweep voltammograms (LSVs) obtained from the rotating disk electrode (RDE) at the different rotating rates were collected in the O2 saturated 0.1 M KOH at a rate of 5 mV s−1 from 0.3 V to −0.8 V (vs. SCE). The kinetic analysis was performed using the Koutecky–Levich (K–L) equations shown below:
:3.0, followed by ultrasonication to obtain a homogeneous solution. The loading of the catalyst on the working electrode was well controlled to ∼0.1 mg cm−2. The working electrode with the Pt/C (20 wt% Pt on the carbon, Johnson Matthey) used for comparison was prepared in a similar manner. The cyclic voltammograms (CVs) were recorded at ambient temperature in an 0.1 M KOH solution. Before the measurements, the solution was aerated with N2 or O2 for at least 30 min. The N2 or O2 flow was maintained above the solution to keep the N2 or O2 saturation during the electrochemical measurements. The linear sweep voltammograms (LSVs) obtained from the rotating disk electrode (RDE) at the different rotating rates were collected in the O2 saturated 0.1 M KOH at a rate of 5 mV s−1 from 0.3 V to −0.8 V (vs. SCE). The kinetic analysis was performed using the Koutecky–Levich (K–L) equations shown below:
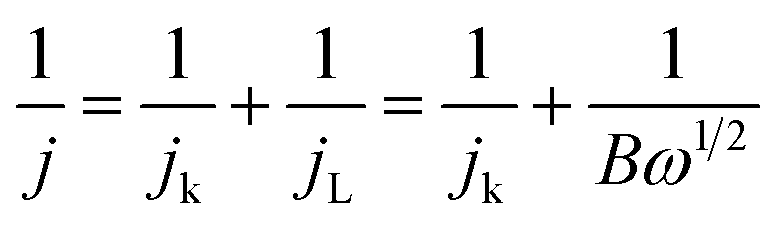 | (1) |
| B = 0.62nFCOD2/3v−1/6 | (2) |
500C mol−1), n transferred electron number, D the O2 diffusion coefficient of the electrolyte (D = 1.9 × 10−5 cm−2 s−1), v the kinetic viscosity (v = 0.01 cm2 s−1).
The rotating ring-disk electrode (RRDE) technique was also employed to analyze the ORR properties of the catalysts. The electron transfer number and the amount of HO2− generated during ORR from RRDE test were determined through the equations below:
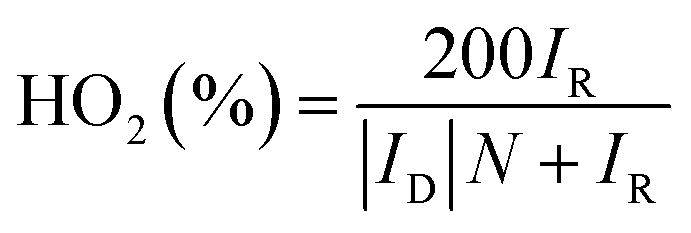 | (3) |
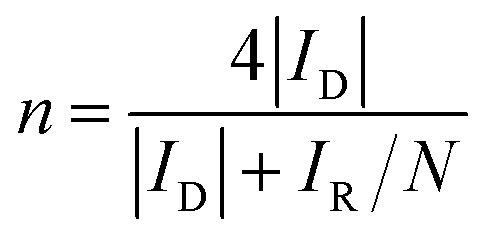 | (4) |
Results and discussion
Fig. 1a shows a typical SEM image of the Co–CoO@NC/NC-800 synthesized by a procedure involving the drying of the reaction mixture of Co(NO3)2, glucose and urea at 200 °C and its subsequent calcination at the high temperature of 800 °C. It reveals that the Co–CoO@NC/NC-800 has a morphology resembling to the crumpled papers with the deposition of the small sized particles. This is also verifiable by its TEM image in Fig. 1b, which shows a structure consisting of the sheet-like carbon material and uniformly sized nanoparticles. The average size of the nanoparticles is ∼14.8 nm, as estimated by the particle size distribution histogram in Fig. 1c. The high-resolution TEM image of the Co–CoO@NC/NC-800 in Fig. 1d indicates that the nanoparticles are composed of mixed phases of Co and CoO. As shown in Fig. 1d, the lattice fringes corresponding to the (111) and (200) planes of the face-centered cubic (fcc) structure cobalt and the (220) plane of the rock salt cubic structure CoO could be identified. The presence of the mixed phases of Co and CoO suggests that a fraction of Co2+ has been reduced by glucose and urea or their calcination products during the calcination process, while the residual has changed into CoO through the decomposition of Co(NO3)2 or through its reaction with oxygen from the decomposed glucose and urea. Most interestingly, each Co/CoO nanoparticle is surrounded by a crystallized shell of 1.2 nm (Fig. 1d). The d-spacing measured from the lattice fringes of the shell is 0.335, well consistent with the interlayer spacing of the (002) plane of the graphitic carbon. This suggests that the Co/CoO nanoparticles are coated with the carbon shells. These carbon shells could be attributed to the carbonized product of glucose and urea formed during the high-temperature calcination processes.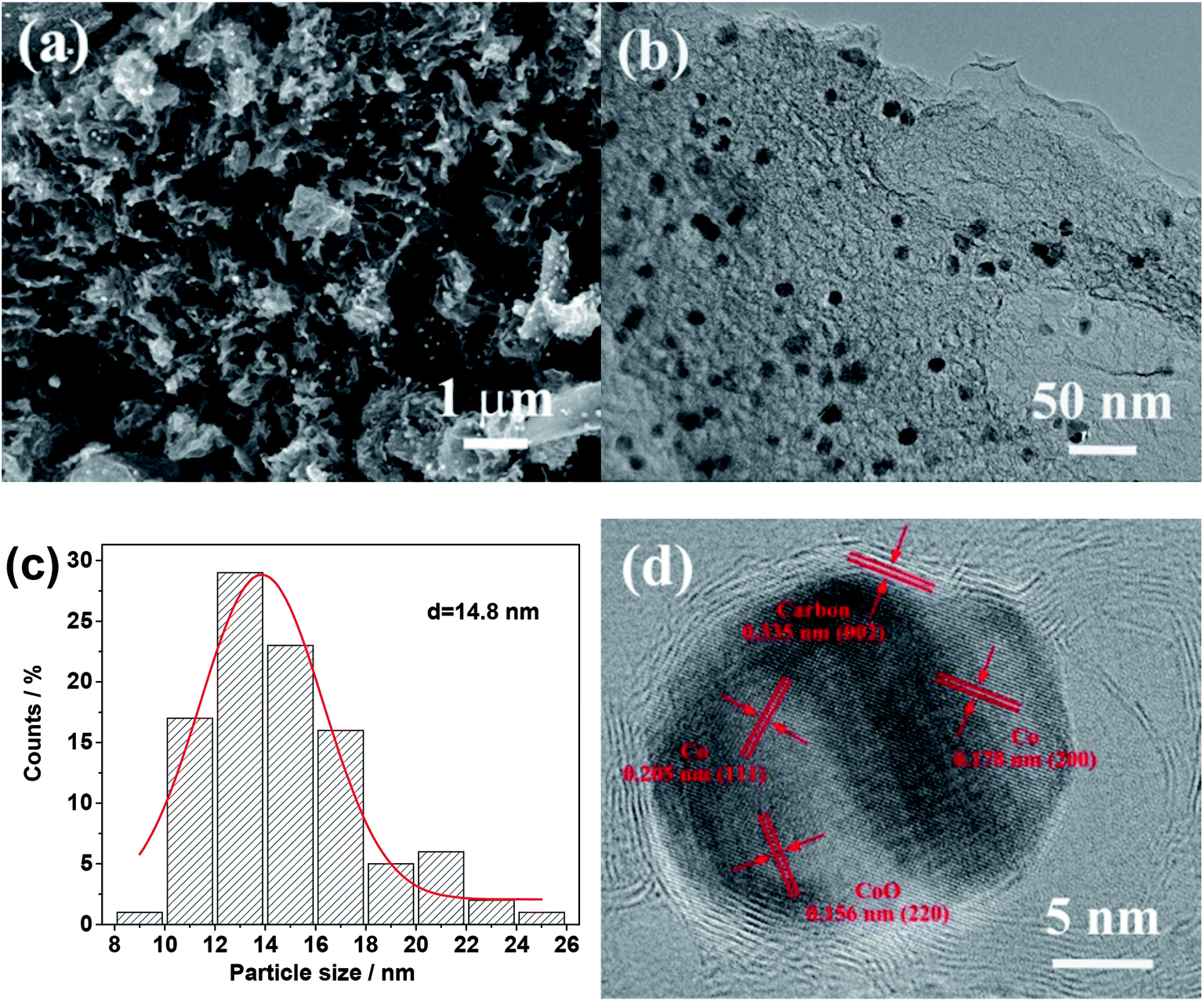 | ||
| Fig. 1 . (a) SEM and (b) TEM images of the Co–CoO@NC/NC-800. (c) Particle size distribution histogram of the Co–CoO NPs in the Co–CoO@NC/NC-800. (d) High resolution TEM image of the Co–CoO@NC/NC-800. | ||
The X-ray diffraction (XRD) pattern of the Co–CoO@NC/NC-800 is given in Fig. 2a. It shows three distinct peaks at 2θ = 44.2°, 51.6° and 75.8°, corresponding to the (111), (200), and (220) reflections of the fcc structure metallic Co (PDF no. 15-0806), and two weak peaks 2θ = 36.6 and 42.3°, corresponding to the (111) and (200) reflections of the rock salt cubic structure CoO (PDF no. 48-1719). This further demonstrates the co-existence of Co and CoO in the Co–CoO@NC/NC-800. The reason that the Co–CoO@NC/NC-800 exhibits low diffraction peaks corresponding to CoO could be attributed to its relatively low content in comparison to that of the metallic cobalt (as demonstrated by the XPS results shown below) and the low X-ray scattering factor of cobalt oxide. Besides these peaks, the XRD pattern of the Co–CoO@NC/NC-800 also exhibits a broad peak at 2θ = 25.9°, which could be assigned to the (002) reflection of the layer-by-layer aligned graphitic nanosheets, suggesting the presence of the graphitic carbon in the Co–CoO@NC/NC-800. This could be further corroborated by the Raman spectrum of the Co–CoO@NC/NC-800 in Fig. 2b, which shows pronounced peaks at 1350 cm−1 and 1580 cm−1, assignable to the D band (arising from the disorder carbon) and G band (associated with tangential stretching mode of highly order graphitic carbon) of the graphitic structure. Worthnoting is that the Raman spectrum of the Co–CoO@NC/NC-800 also exhibits peaks at 473, 517, and 679 cm−1, assignable to active vibrational modes Eg, F2g, and A1g of the cubic structure CoO, respectively.38,39 This further supports the presence of CoO in the Co–CoO@NC/NC-800.
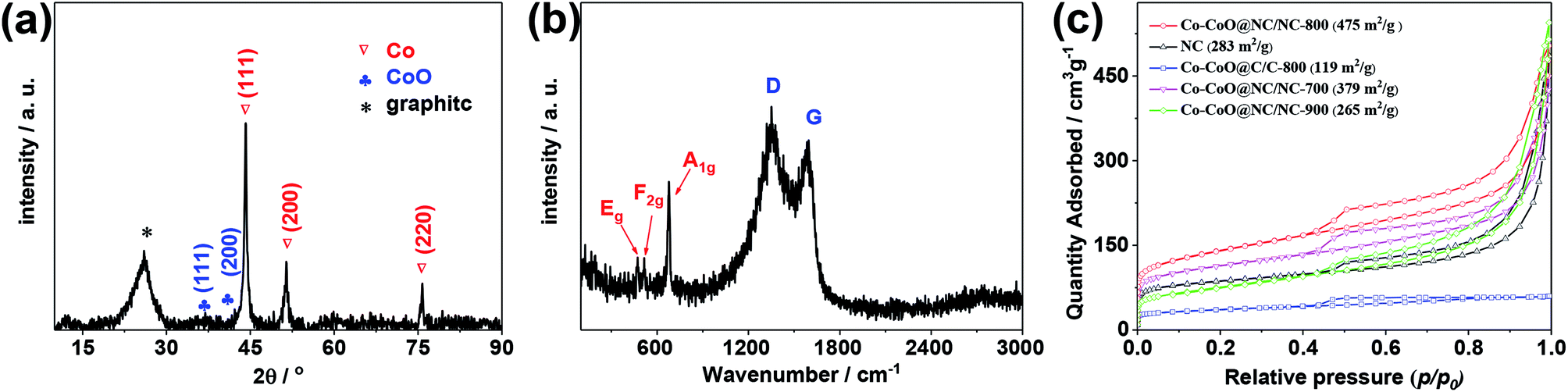 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra of the Co–CoO@NC/NC-800. (c) N2 adsorption/desorption isotherms of the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, the Co–CoO@NC/NC-900. | ||
The XPS measurements were carried out to gain information on the elemental composition of the Co–CoO@NC/NC-800. Fig. 3a shows the XPS survey spectrum of the Co–CoO@NC/NC-800, which indicates that the Co–CoO@NC/NC-800 is composed of Co, O, N, and C. The deconvolution of the Co 2p spectrum in Fig. 3b clearly demonstrates the presence of both Co(0) and Co(II) in the Co–CoO@NC/NC-800. As shown in Fig. 3b, the peaks corresponding to Co 2p3/2 and 2p1/2 of Co(II) could be observed at 780.5 eV and 796.5 eV, respectively, while the peak corresponding to Co 2p3/2 and 2p1/2 of Co(0) is observable at 778.8 eV and 794.1 eV, respectively. Based on the deconvoluted spectrum shown in Fig. 3b, the relative atomic ratio of Co(0):Co(II) is estimated to ∼3:2. It implies that Co in the Co–CoO@NC/NC-800 mainly exists in the metallic form. Fig. 3c displays the deconvoluted C 1s spectrum of the Co–CoO@NC/NC-800, which shows four components at binding energies of 284.5, 285.3, 286.3, and 288.2 eV, corresponding to sp2 C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/C–O, C–O–C/C–N, and O–CO, respectively. The domination of the peak corresponding to sp2 C suggests the carbon in the Co–CoO@NC/NC-800 is well graphitized. This is in line with the result shown by the Raman spectrum (Fig. 2b), which exhibits pronounced peaks corresponding to the D and G bands of the graphitic structure. The existence of the peaks corresponding to CN and C–N in the deconvoluted C 1s spectrum indicates that carbon in the Co–CoO@NC/NC-800 is doped with nitrogen. This could be also verified by the deconvoluted N 1s spectrum in Fig. 3d, which shows four peaks at binding energies of 397.9, 399.6, 400.7, and 402.3 eV, assignable to pyridinic, pyrrolic, graphitic, and oxidized nitrogen, respectively. This is similar to the NC reported previously.40,41 Since the Co–CoO@NC/NC-800 was synthesized from a mixture, in which the reactants were well dispersed, we believe that both the carbon shell of the Co–CoO nanoparticles and the carbon sheet observed in the SEM and TEM images of the Co–CoO@NC/NC-800 (Fig. 1a and b) are doped with nitrogen. The elemental composition of the Co–CoO@NC/NC-800 could be further demonstrated by its elemental mapping image in Fig. S1,† which clearly shows the uniform distribution of Co, O, N, and C. Based on the XPS results and the TGA curve of the Co–CoO@NC/NC-800 in Fig. S2,† the weight percentage of Co–CoO in the Co–CoO@NC/NC-800 is estimated to be 28.7% (the detailed calculation is given in the ESI†).
N/C–O, C–O–C/C–N, and O–CO, respectively. The domination of the peak corresponding to sp2 C suggests the carbon in the Co–CoO@NC/NC-800 is well graphitized. This is in line with the result shown by the Raman spectrum (Fig. 2b), which exhibits pronounced peaks corresponding to the D and G bands of the graphitic structure. The existence of the peaks corresponding to CN and C–N in the deconvoluted C 1s spectrum indicates that carbon in the Co–CoO@NC/NC-800 is doped with nitrogen. This could be also verified by the deconvoluted N 1s spectrum in Fig. 3d, which shows four peaks at binding energies of 397.9, 399.6, 400.7, and 402.3 eV, assignable to pyridinic, pyrrolic, graphitic, and oxidized nitrogen, respectively. This is similar to the NC reported previously.40,41 Since the Co–CoO@NC/NC-800 was synthesized from a mixture, in which the reactants were well dispersed, we believe that both the carbon shell of the Co–CoO nanoparticles and the carbon sheet observed in the SEM and TEM images of the Co–CoO@NC/NC-800 (Fig. 1a and b) are doped with nitrogen. The elemental composition of the Co–CoO@NC/NC-800 could be further demonstrated by its elemental mapping image in Fig. S1,† which clearly shows the uniform distribution of Co, O, N, and C. Based on the XPS results and the TGA curve of the Co–CoO@NC/NC-800 in Fig. S2,† the weight percentage of Co–CoO in the Co–CoO@NC/NC-800 is estimated to be 28.7% (the detailed calculation is given in the ESI†).
 | ||
| Fig. 3 (a) XPS survey spectra of the Co–CoO@NC/NC-800 and the NC. (b) High resolution Co 2p spectra of the Co–CoO@NC/NC-800 and the Co–CoO@C/C-800. (c) High resolution C 1s spectra of the Co–CoO@NC/NC-800 and the NC. (d) High resolution N 1s spectra of the Co–CoO@NC/NC-800 and the NC. | ||
The results shown above make it clear that the Co–CoO@NC/NC-800 has a structure consisting of the sheet-like NC with the deposition of the NC coated Co–CoO nanoparticles. To understand the role of urea in the formation of the Co–CoO@NC/NC-800, the same synthetic procedure was performed in the absence of urea while keeping other parameters constant. It shows the formation of the Co–CoO@C/C-800 consisting of largely sized Co–CoO particles with a big inhomogeneity in the size distribution (the TEM image, XPS spectra, XRD diffraction pattern, and Raman spectrum of the Co–CoO@C/C-800 is given in Fig. S3a, S4, S5, and S6,† respectively). This suggests that urea in the reaction mixture does not simply serve as the nitrogen source for the doping of carbon, but plays an important role in promoting the formation the Co–CoO@NC/NC-800 with uniformly nanosized Co–CoO particles. It is imaginable that during the high temperature calcinations the explosive pyrolysis of urea would facilitate the doping of nitrogen into graphitic structure of carbon with different forms, such as pyridinic, pyrrolic, graphitic, and oxidized nitrogen. Due to the high electronegativity of nitrogen, the doping sites in the forms of pyridinic, pyrrolic, and graphitic nitrogen are negatively charged,42,43 making them capable of adsorbing the Co2+ ions through the coordination and electrostatic interactions. This would prevent the aggregation of the Co2+ ions during its transformation into Co–CoO nanoparticles, facilitating the formation of the Co–CoO nanoparticles with a uniform and small size.
The calcination temperature in the synthetic procedure is found to have a profound effect on the final product. As demonstrated by the TEM image of the Co–CoO@NC/NC-700 in Fig. S3b,† the low temperature calcination would lead to the formation of smaller Co–CoO nanoparticles with a more uniform size. The XPS spectrum in Fig. S4 and Table S1† show that the Co–CoO@NC/NC-700 contains relatively higher atomic percentages of oxygen and nitrogen, suggesting that the urea and glucose are not well carbonized at the low calcination temperature. On the other hand, when the reaction mixture was calcined at the high temperature of 900 °C, the formation of the Co–CoO nanoparticles with the relatively larger size and big size inhomogeneity is observed, as indicated by the TEM image of the Co–CoO@NC/NC-900 in Fig. S3c.† This indicates that the mild reaction condition at the lower temperature is favorable for the formation of the Co–CoO nanoparticles with small and uniform sizes, while at the higher calcination temperature, the improved uncontrollability of the decomposition of Co(NO3)2, glucose, and urea due to the increase of the pyrolysis rates would promote the formation of large Co–CoO nanoparticles with a big inhomogeneity in the size distribution (the XPS spectrum, XRD diffraction pattern, and Raman spectra of the Co–CoO@NC/NC-900 is given in Fig. S4, S5, and S6,† respectively). Table S1† shows that the Co–CoO@NC/NC-900 contains relatively lower atomic percentages of oxygen and nitrogen, suggesting that the high calcination temperature would decrease the contents of oxygen and nitrogen containing groups in the final product. This is well consistent with the results reported previously,44,45 which showed that the high calcination temperature would lead to the significant decomposition of nitrogen and oxygen containing groups in the NC. Additionally, we also found that the drying step in the synthetic procedure is necessary to obtain the Co–CoO@NC/NC-800 with uniformly sized Co–CoO nanoparticles. As shown in Fig. S3d,† when the reaction mixture was directly calcined at 800 °C, the significant aggregation of the Co–CoO nanoparticles is observed in the final product. This might be attributed to the highly corrosive nature of water vapor at the high temperature. Its presence in the reaction mixture would greatly accelerate the pyrolysis of Co(NO3)3, glucose, and urea, leading to uncontrollable aggregation and coalescence of the Co–CoO nanoparticles during their nucleation and growth.
The textural property of the Co–CoO@NC/NC-800 was investigated by the N2 adsorption/desorption isotherms. As shown in Fig. 2c, the N2 adsorption/desorption isotherms of the Co–CoO@NC/NC-800 exhibit a rapid uptake at p/p0 < 0.04 followed by a H3-type hysteresis in the mesopore region. This indicates the co-existence of both the micropores and macropores in the sample. Additionally, the big uptake of N2 at p/p0 > 0.9 in the adsorption/desorption isotherms also demonstrates the existence of macropores in the Co–CoO@NC/NC-800. Base on the N2 adsorption/desorption isotherms in Fig. 2c, the specific surface area of the Co–CoO@NC/NC-800 estimated using the BET method is 475 m2 g−1. The presence of urea in the reaction precursor is found to have a significant effect on the BET specific surface area of the sample. As shown in Fig. 2c, the Co–CoO@C/C exhibits significantly lower BET specific surface area than that of the Co–CoO@NC/NC-800. This might be due to the reason that the thermal decomposition of urea during the calcination would produce a large amount of gases, such as CO2, NH3, and other gaseous products,46,47 which are helpful to the formation of the porous structure in the Co–CoO@NC/NC-800. Most interestingly, although the Co–CoO@NC/NC-700 and the Co–CoO@NC/NC-900 exhibit the N2 adsorption/desorption isotherms comparable to those of the Co–CoO@NC/NC-800, their BET specific surface areas are much lower than that of the Co–CoO@NC/NC-800. This suggests that the calcination temperature also has a great effect on the BET specific surface area of the sample. As demonstrated above, the calcination temperature influences the average size of Co–CoO nanoparticles and the doping content of nitrogen in the NC, and it also affects the pyrolysis rate of Co(NO3)2, glucose, and urea. These might be the reasons leading to the variations of the BET specific surface areas of the samples with the calcination temperature. The high specific BET surface area is helpful for the application of the Co–CoO@NC/NC-800 as the electrocatalyst for the ORR, since it allows for its good accessibility to the electrolyte.
The catalytic activity of the Co–CoO@NC/NC-800 for the ORR is first assessed by its CV curves in the N2 or O2 saturated 0.1 M KOH solution. As shown in Fig. 4a, the CV curve of the Co–CoO@NC/NC-800 in the N2 saturated 0.1 M KOH solution shows featureless voltammetric currents, while that in the O2 saturated 0.1 M KOH solution shows a well-defined cathodic peak corresponding to the reduction of oxygen. This clearly demonstrates the catalytic activity of the Co–CoO@NC/NC-800 toward the ORR. Fig. 4b displays the LSV of the Co–CoO@NC/NC-800 for the ORR in the O2 saturated 0.1 M KOH solution. It clearly shows that the Co–CoO@NC/NC-800 can exhibit an ORR onset potential of 0.961 V vs. RHE and half-wave potential of 0.868 V vs. RHE. Most interestingly, the ORR onset and half-wave potentials of the Co–CoO@NC/NC-800 are very close to those of the Pt/C (the ORR onset and half-wave potential of the Pt/C are 0.962 and 0.861 V vs. RHE, respectively, which are close to the reported values48,49). This clearly suggests that the Co–CoO@NC/NC-800 is an efficient electrocatalyst for the ORR. Comparison in Fig. 4d shows that the Co–CoO@NC/NC-800 can even exhibit the ORR half-wave potential higher than those of most catalysts reported previously (Table S2† shows a more detailed comparison of the ORR onset and half-wave potentials), strongly suggesting that the Co–CoO@NC/NC-800 is one of the best ORR catalysts among those reported (Table 1).
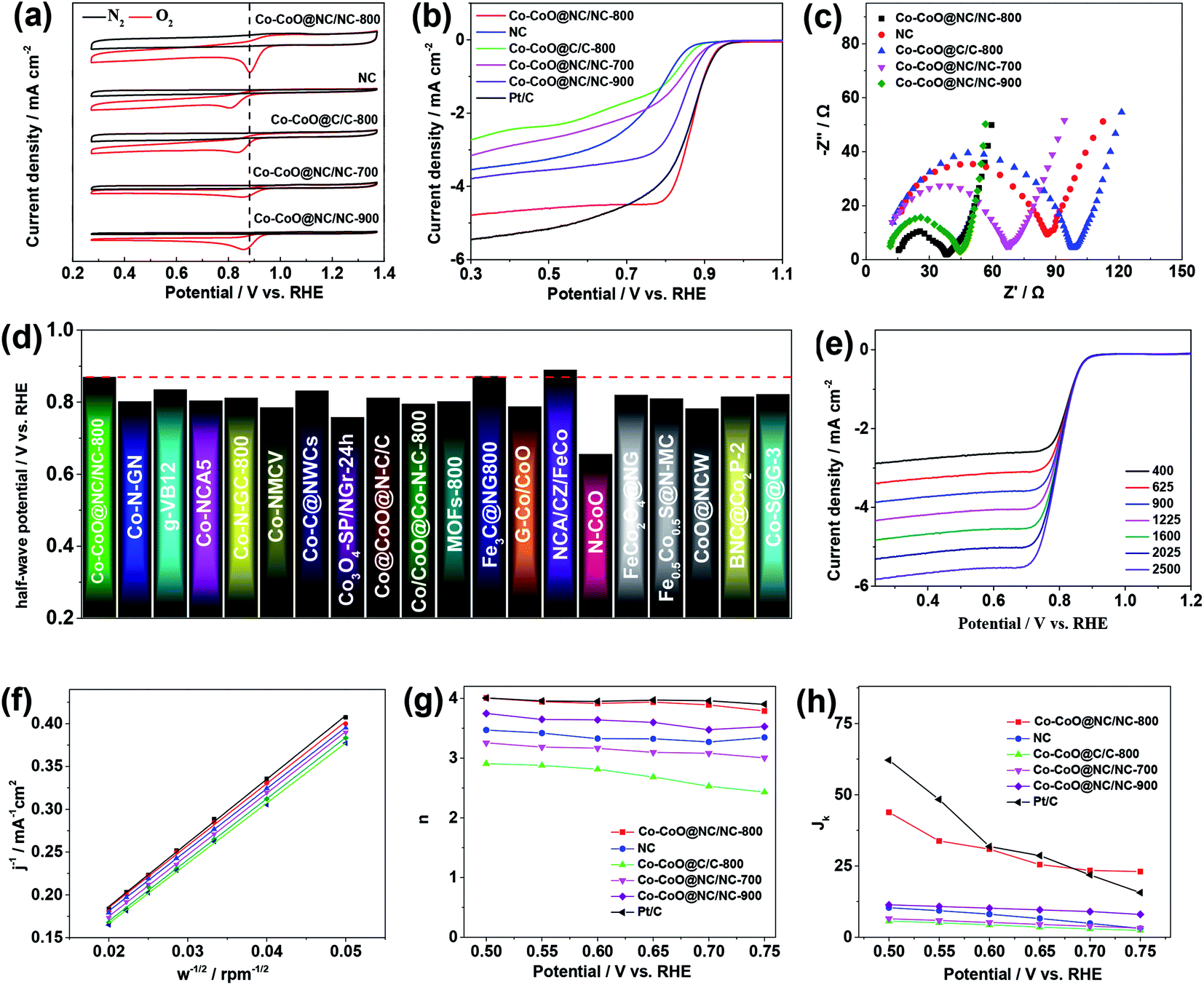 | ||
| Fig. 4 (a) CV curves of the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and the Co–CoO@NC/NC-900 in O2 or N2 saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 (b) LSVs for the ORR by the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, the Co–CoO@NC/NC-900, and the Pt/C in an O2-saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 (c) EIS spectra of the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and the Co–CoO@NC/NC-900. (d) Comparison of the ORR half-wave potential of the Co–CoO@NC/NC-800 with those of the catalysts reported. (e) LSVs for the ORR by the Co–CoO@NC/NC-800 in an O2-saturated 0.1 M KOH solution at various rotation rates at a scan rate of 5 mV s−1 (f) K–L plots of the ORR by the Co–CoO@NC/NC-800. (g) Electron transfer number and (h) kinetic current density of the ORR by the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, the Co–CoO@NC/NC-900 and the Pt/C. | ||
| Catalyst | Onset potential/V vs. RHEa | Half-wave potential/V vs. RHE |
|---|---|---|
| a The ORR onset potential is defined at which the current density reaches to 0.1 mA cm−2. | ||
| Co–CoO@NC/NC-800 | 0.961 | 0.868 |
| NC | 0.889 | 0.777 |
| Co–CoO@C/C-800 | 0.899 | 0.817 |
| Co–CoO@NC/NC-700 | 0.921 | 0.841 |
| Co–CoO@NC/NC-900 | 0.920 | 0.842 |
| Pt/C | 0.961 | 0.862 |
To understand the origin of the high ORR activity of the Co–CoO@NC/NC-800, the LSVs of the NC (synthesized from a same synthetic procedure while in the absence of Co(NO3)2), the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and the Co–CoO@NC/NC-900 in the O2 saturated 0.1 M KOH solutions were also recorded. Fig. 4a and b shows that although the NC is electrochemically active for the ORR, its activity is much lower than that of the Co–CoO@NC/NC-800. This clearly indicates a great importance of the Co–CoO nanoparticles in the high catalytic activity of the Co–CoO@NC/NC-800. The nitrogen doped structure of the carbon is also found to be important in improving its catalytic activity of the Co–CoO@NC/NC-800 for the ORR. As shown in Fig. 4a and b, the Co–CoO@C/C-800 exhibits significantly lower catalytic activity for the ORR than that of the Co–CoO@NC/NC-800. Previous work has demonstrated that the NC could be also highly active for the ORR, when its composition is well controlled.50–53 We therefore assume that the high catalytic activity of the Co–CoO@NC/NC-800 is a result of a synergistic effect between two electroactive catalysts of the Co–CoO nanoparticles and the NC. Worthnoting is that the catalytic activity of the sample is also effected by the calcination temperature. As shown in Fig. 4a and b, the catalytic activity of the Co–CoO@NC/NC-800 is higher than those of the Co–CoO@NC/NC-700 and Co–CoO@NC/NC-900. This might be due to the fact that the Co–CoO@NC/NC-800 synthesized at the calcination temperature of 800 °C has small and uniformly sized Co–CoO nanoparticles and exhibits a higher BET specific surface area, which expose more active sites and increase the accessibility of the Co–CoO@NC/NC-800 to the ORR. Additionally, the Co–CoO@NC/NC-800 has a well carbonized structure with the doping of an appropriate amount of nitrogen, which increases the electrical conductivity and hydrophilicity of the Co–CoO@NC/NC-800. Fig. 4c shows the electrochemical impedance spectra (EISs) of the Co–CoO@NC/NC-800, the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and the Co–CoO@NC/NC-900, which clearly demonstrate that the Co–CoO@NC/NC-800 has higher electrical conductivity.
The work reported previously has suggest that the deposition of transition metal based catalysts onto the surface of carbon would impose a strong electronic coupling between two materials, which could also improve the catalytic activities of the composite materials.48 The nitrogen doping could promote the electronic coupling between transition metal based catalysts and carbon.39,48 It is therefore expectable that the strong electronic coupling between the Co–CoO nanoparticles and the NC is an additional reason leading to the high catalytic activity of the Co–CoO@NC/NC-800 for the ORR. To demonstrate the presence of the electronic coupling between the Co–CoO nanoparticles and the NC, the XPS spectra of the NC were carefully analyzed. The XPS survey spectra in Fig. 3a shows the absence of the peak corresponding to Co in the NC. Most notably, although the high-resolution N 1s spectrum of the NC exhibits the components similar to that of the Co–CoO@NC/NC-800, their peak positions are slightly shifted to the lower binding energies, as shown in Fig. 3d. This clearly corroborates the presence of the strong electronic coupling between the Co–CoO nanoparticles and the NC with a possible electron transfer from the Co–CoO nanoparticles to the NC. Indeed, the strong electronic coupling between the Co–CoO nanoparticles and the NC could also be demonstrated by the C 1s spectra in Fig. 3c, which shows the shifts of the peak positions of CN/C–O, and C–O–C/C–N in the NC to lower binding energies in comparison to those in the Co–CoO@NC/NC-800, and by Co 2p spectra, which shows the shifts of the peaks of Co(0) and Co(II) in the Co–CoO@NC/NC-800 to higher binding energies in comparison to those in the Co–CoO@C/C-800. Additionally, Fig. 3d also shows a difference between the N 1s spectra profiles of the NC and the Co–CoO@NC/NC-800. This might be due to the fact that the growth of the Co–CoO nanoparticles on the nitrogen doping sites makes some of the nitrogen atoms undetectable by the XPS spectroscopy due to the low penetrability of X-ray photoelectrons, or the presence of the Co precursor in the reaction mixture changes the relative percentages of the different nitrogen components in the Co–CoO@NC/NC-800.
Previous work have demonstrated that the presence of metal–N bond in the metal and nitrogen coexisting system is responsible for the high catalytic activity for the ORR.54,55 In the work presented here, however, the metal–N bonds are not detected in both the XPS spectra of Co 2p and N 1s, which clearly rules out the presence of the metal–N bond in the Co–CoO@NC/NC-800 and its contribution to the higher catalytic activity of the Co–CoO@NC/NC-800. On the basis of this and the results shown above, we therefore attribute the following aspects to the main reasons responsible for the high catalytic activity of the Co–CoO@NC/NC-800: (1) strong electronic interaction between the Co–CoO nanoparticles and the NC, (2) high specific surface area of the Co–CoO@NC/NC-800, (3) small and uniformly sized Co–CoO nanoparticles, (4) good electric conductivity of the Co–CoO@NC/NC-800 due to its well carbonized structure, (5) good hydrophilicity of the Co–CoO@NC/NC-800 due to the presence of the NC.30,56 Additionally, the interaction between Co and CoO could be an additional reason leading to the high catalytic activity of the Co–CoO@NC/NC-800. As reported previously, the composites consisting of TMs and their oxides exhibit greatly enhanced catalytic activities in comparison to their single component counterparts.36,57,58
To gain insight into the reaction kinetics and mechanism of the ORR by the Co–CoO@NC/NC-800, its LSVs at the different rotating rates were recorded. Fig. 4e shows an improvement of the measured current density with increase of the rotating rate. Analysis of the LSVs using the K–L equation shows a linear relationship between the inverse of the current density (j−1) and the inverse of the root of the rotating rate (ω−1/2) at the different potentials (Fig. 4f). This indicates that the ORR by the Co–CoO@NC/NC-800 follows a first order reaction kinetics with respect to the concentration of dissolved oxygen. According to the slopes of the K–L plots, the electron transfer number (n) of the ORR can be extracted. Fig. 4g shows that the electron transfer number of the ORR by the Co–CoO@NC/NC-800 is always higher than 3.8 and close to that of the Pt/C over the potential range covered in this work, demonstrating that the ORR by the Co–CoO@NC/NC-800 proceeds mainly via a favorable four-electron pathway. This is different from the ORR by the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and Co–CoO@NC/NC-900 (the K-L plots of the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and Co–CoO@NC/NC-900 are given in Fig. S7†), in which a two-electron pathway plays an increasing role in the reduction of oxygen. As shown in Fig. 4g, the electron transfer numbers of the ORR by the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and Co–CoO@NC/NC-900 are much lower than 4.0. Fig. 4h presents the ORR kinetic current density (jk) of the catalysts. It shows that the Co–CoO@NC/NC-800 exhibits higher ORR kinetic current density than the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and Co–CoO@NC/NC-900, further demonstrating that the Co–CoO@NC/NC-800 is a more efficient catalyst for the ORR than the NC, the Co–CoO@C/C-800, the Co–CoO@NC/NC-700, and Co–CoO@NC/NC-900.
The rotating ring-disk electrode (RRDE) measurement was perform to further investigate the ORR by the Co–CoO@NC/NC-800. Fig. 5a shows the LSV of the ORR by the Co–CoO@NC/NC-800 obtained from the disk electrode of the RRDE shows a profile similar to that directly obtained from the rotating disk electrode (Fig. 4b). Most interestingly, the ORR by the Co–CoO@NC/NC-800 shows a comparable ring current corresponding to hydrogen peroxide ions (H2O−) to that by the Pt/C. Fig. 5b reveals that the H2O− yield of the ORR by the Co–CoO@NC/NC-800 is below 5% with an electron transfer number higher than 3.9 over the potential range from 0.2 to 0.8 V. The H2O− yield and electron transfer number of the ORR by the Co–CoO@NC/NC-800 are very close to those by the Pt/C, as shown Fig. 5b. Most interestingly, the H2O− yield of the ORR by the Co–CoO@NC/NC-800 is lower than those of most catalysts reported previously, while its electron transfer number is higher than those of the ORR by most catalysts reported previously.36,59,60 This further demonstrates that the Co–CoO@NC/NC-800 has a catalytic activity close to that of the Pt/C.
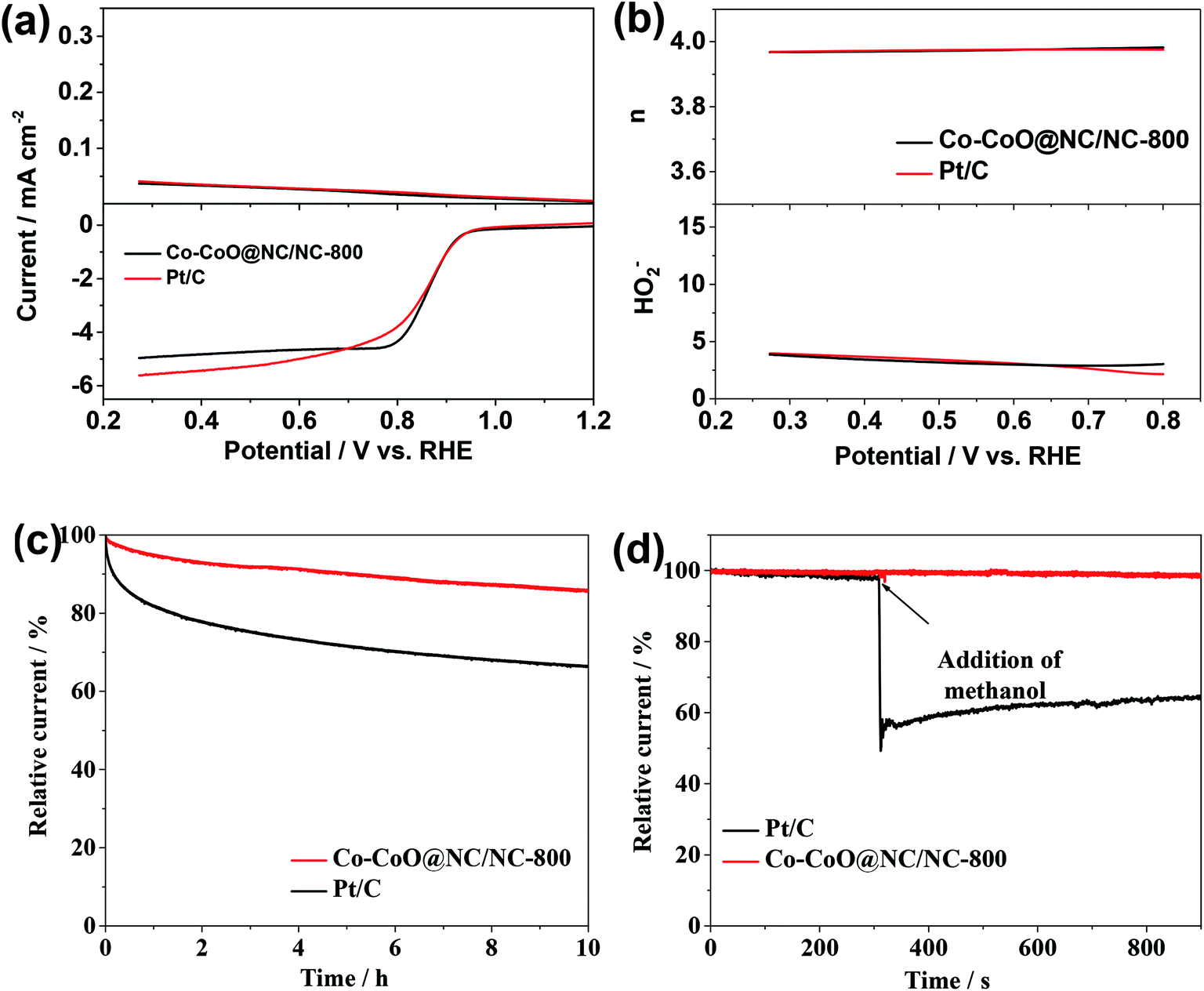 | ||
| Fig. 5 (a) LSV curves of the Co–CoO@NC/NC-800 and the Pt/C obtained from the RRDE measurements at a rotation rate of 1600 rpm in the O2 saturated 0.1 M KOH solution. (b) HO2− yields (top) and electron transfer number (n, bottom) of the Co–CoO@NC/NC-800 and the Pt/C obtained from the RRDE measurements. (c) Chronoamperometric curves of the Co–CoO@NC/NC-800 and the Pt/C at 0.67 V vs. RHE in the O2-saturated 0.1 mol L−1 KOH solution. (d) Chronoamperometric curves of the Co–CoO@NC/NC-800 and the Pt/C electrodes with addition of 0.3 M methanol. The arrow indicates the addition of methanol. | ||
In view that the Co–CoO@NC/NC-800 is highly active for the ORR with great potential for the replacement of the commercial Pt/C, its stability and durability towards the methanol crossover were tested. The current–time (i–t) chronoamperometric curve in Fig. 5c shows that the current density of the ORR by the Pt/C decreases rapidly with increase of the operating time. This could be attributed to the low stability of the Pt/C in the alkaline environment, which undergoes a fast degradation of the Pt/C over the time due to the dissolution and aggregation of the Pt nanoparticles and their surface oxidation.61,62 The introduction of methanol causes a sharp decrease in its ORR current density due to the poisoning of the ORR active sites by the oxidized products of methanol (Fig. 5d).62,63 The stability of the Co–CoO@NC/NC-800 and its durability toward the methanol crossover are much higher than those of the Pt/C. As shown in Fig. 5c and d, the current density of the ORR by the Co–CoO@NC/NC-800 decreases much slowly in comparison to that by the Pt/C, and the introduction of methanol has a negligible effect on its ORR current density. These results strongly suggest that the specific coating structure of the Co–CoO nanoparticles by the NC shell in the Co–CoO@NC/NC-800 could effectively protect the Co–CoO nanoparticles from the degradation and poisoning of the ORR active sites by the oxidized products of methanol.
Conclusions
In summary, the Co–CoO@NC/NC-800, which consists of the NC nanosheets with deposition of the NC coated Co–CoO nanoparticles, has been synthesized through a simple calcination of the dried reaction mixture of Co(NO3)2, glucose, and urea. The Co–CoO@NC/NC-800 is highly active for the ORR and can deliver an onset potential of 0.961 V vs. RHE and a half-wave potential of 0.868 V vs. RHE. The onset and the half-wave potentials of the Co–CoO@NC/NC-800 are very close to those of the Pt/C. This, in conjunction with its higher stability and better durability towards the methanol crossover, strongly suggests that the Co–CoO@NC/NC-800 is a promising ORR catalyst for practical uses. The work presented here does not simply introduce a new and simple method for the synthesis of the ORR catalysts of high efficiency, and the same method might be also applicable to design and synthesize other catalysts for various applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported from the Chinese National Natural Science Foundation (No. 11474101 and U1532139), the “Outstanding Talent and Team Plans Program” of South China University of Technology, the Guangdong Provincial Natural Science Foundation (No. 2017A030313092), the Guangdong Innovative and Entrepreneurial Research Team Program (No. 2014ZT05N200), and the Ningbo Natural Science Foundation (No. 2017A610059).References
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CAS.
- L. Li, C. Liu, G. He, D. Fan and A. Manthiram, Energy Environ. Sci., 2015, 8, 3274–3282 CAS.
- K. Mohanraju and L. Cindrella, RSC Adv., 2014, 4, 11939–11947 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850–11859 CrossRef CAS PubMed.
- G. Hu, E. Gracia-Espino, R. Sandström, T. Sharifi, S. Cheng, H. Shen, C. Wang, S. Guo, G. Yang and T. Wågberg, Catal. Sci. Technol., 2016, 6, 1393–1401 CAS.
- J. Ding, X. Zhu, L. Bu, J. Yao, J. Guo, S. Guo and X. Huang, Chem. Commun., 2015, 51, 9722–9725 RSC.
- Y. Zhao, J. Liu, Y. Zhao and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 19298–19306 RSC.
- D. U. Lee, M. G. Park, H. W. Park, M. H. Seo, X. Wang and Z. Chen, ChemSusChem, 2015, 8, 3129–3138 CrossRef CAS PubMed.
- P. Sennu, M. Christy, V. Aravindan, Y.-G. Lee, K. S. Nahm and Y.-S. Lee, Chem. Mater., 2015, 27, 5726–5735 CrossRef CAS.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- H. J. Choi, N. A. Kumar and J. B. Baek, Nanoscale, 2015, 7, 6991–6998 RSC.
- J. E. Kim, J. Lim, G. Y. Lee, S. H. Choi, U. N. Maiti, W. J. Lee, H. J. Lee and S. O. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 1571–1577 CAS.
- M. Shen, C. Ruan, Y. Chen, C. Jiang, K. Ai and L. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 1207–1218 CAS.
- Y. Zhan, M. Lu, S. Yang, C. Xu, Z. Liu and J. Y. Lee, ChemCatChem, 2016, 8, 372–379 CrossRef CAS.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- M. Sun, H. Liu, Y. Liu, J. Qu and J. Li, Nanoscale, 2015, 7, 1250–1269 RSC.
- J. Xu, Q. Yu, C. Wu and L. Guan, J. Mater. Chem. A, 2015, 3, 21647–21654 CAS.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- H. Yu, Y. Li, X. Li, L. Fan and S. Yang, Chem.–Eur. J., 2014, 20, 3457–3462 CrossRef CAS PubMed.
- M. Li, X. Bo, Y. Zhang, C. Han, A. Nsabimana and L. Guo, J. Mater. Chem. A, 2014, 2, 11672–11682 CAS.
- Z. Wang, S. Xiao, Z. Zhu, X. Long, X. Zheng, X. Lu and S. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 4048–4055 CAS.
- Y. Li, F. Cheng, J. Zhang, Z. Chen, Q. Xu and S. Guo, Small, 2016, 12, 2839–2845 CrossRef CAS PubMed.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., 2015, 127, 2128–2132 CrossRef.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- Y. Wang, X. Ding, F. Wang, J. Li, S. Song and H. Zhang, Chem. Sci., 2016, 7, 4284–4290 RSC.
- M. Zhuang, X. Ou, Y. Dou, L. Zhang, Q. Zhang, R. Wu, Y. Ding, M. Shao and Z. Luo, Nano Lett., 2016, 16, 4691–4698 CrossRef CAS PubMed.
- T. Kottakkat and M. Bron, ChemElectroChem, 2014, 1, 2163–2171 CrossRef CAS.
- K. Kreek, A. Sarapuu, L. Samolberg, U. Joost, V. Mikli, M. Koel and K. Tammeveski, ChemElectroChem, 2015, 2, 2079–2088 CrossRef CAS.
- C. Han, X. Bo, Y. Zhang, M. Li, A. Nsabimana and L. Guo, Nanoscale, 2015, 7, 5607–5611 RSC.
- X. Zhang, R. Liu, Y. Zang, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2016, 52, 5946–5949 RSC.
- H. Zhong, Y. Luo, S. He, P. Tang, D. Li, N. Alonso-Vante and Y. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 2541–2549 CAS.
- K. Liu, X. Huang, H. Wang, F. Li, Y. Tang, J. Li and M. Shao, ACS Appl. Mater. Interfaces, 2016, 8, 34422–34430 CAS.
- A. B. Yousaf, M. Imran, N. Uwitonze, A. Zeb, S. J. Zaidi, T. M. Ansari, G. Yasmeen and S. Manzoor, J. Phys. Chem. C, 2017, 121, 2069–2079 Search PubMed.
- Y. Cheng, S. Dou, M. Saunders, J. Zhang, J. Pan, S. Wang and S. P. Jiang, J. Mater. Chem. A, 2016, 4, 13881–13889 CAS.
- Z. Wu, J. Wang, L. Han, R. Lin, H. Liu, H. L. Xin and D. Wang, Nanoscale, 2016, 8, 4681–4687 RSC.
- T.-Y. Yung, L.-Y. Huang, T.-Y. Chan, K.-S. Wang, T.-Y. Liu, P.-T. Chen, C.-Y. Chao and L.-K. Liu, Nanoscale Res. Lett., 2014, 9, 444 CrossRef PubMed.
- X. Dong, H. Xu, X. Wang and Y. Huang, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- Z. J. Jiang and Z. Jiang, Sci. Rep., 2016, 6, 27081 CrossRef CAS PubMed.
- S. Wang, J. Zhang, P. Shang, Y. Li, Z. Chen and Q. Xu, Chem. Commun., 2014, 50, 12091–12094 RSC.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS.
- C. Ma, X. Shao and D. Cao, J. Mater. Chem. A, 2012, 22, 8911–8915 RSC.
- Y.-X. Yu, Phys. Chem. Chem. Phys., 2013, 16819–16827 RSC.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
- X. Zhang, D. Yu, Y. Zhang, W. Guo, X. Ma and X. He, RSC Adv., 2016, 6, 104183–104192 RSC.
- S. Cao, H. Chen, F. Jiang and X. Wang, Appl. Catal., B, 2018, 224, 222–229 CrossRef CAS.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- Y. Liang, H. Wang, P. Diao, W. Chang, G. Hong, Y. Li, M. Gong, L. Xie, J. Zhou, J. Wang, T. Z. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2012, 134, 15849–15857 CrossRef CAS PubMed.
- D. Gao, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- Z. Jiang, Z.-j. Jiang, X. Tian and W. Chen, J. Mater. Chem. A, 2014, 2, 441–450 CAS.
- Z.-J. Jiang and Z. Jiang, J. Mater. Chem. A, 2014, 2, 14071–14081 CAS.
- C. Li, Z. Han, Y. Yu, Y. Zhang, B. Dong, A. Kong and Y. Shan, RSC Adv., 2016, 6, 15167–15174 RSC.
- Y. Jiang, Y. Lu, X. Wang, Y. Bao, W. Chen and L. Niu, Nanoscale, 2014, 6, 15066–15072 RSC.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 CAS.
- V. M. Dhavale, S. K. Singh, A. Nadeem, S. S. Gaikwad and S. Kurungot, Nanoscale, 2015, 7, 20117–20125 RSC.
- X. X. Liu, J. B. Zang, L. Chen, L. B. Chen, X. Chen, P. Wu, S. Y. Zhou and Y. H. Wang, J. Mater. Chem. A, 2017, 5, 5865–5872 CAS.
- C. Han, X. Bo, Y. Zhang, M. Li, A. Wang and L. Guo, Chem. Commun., 2015, 51, 15015–15018 RSC.
- G. L. Tian, M. Q. Zhao, D. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- H. Uchida, K. Izumi and M. Watanabe, J. Phys. Chem. B, 2006, 110, 21924–21930 CrossRef CAS PubMed.
- F. Tian and A. B. Anderson, J. Phys. Chem. C, 2008, 112, 18566–18571 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Element mapping; TGA curve; TEM images; XPS survey spectra; XRD patterns; Raman spectra; LSVs and K-L plots; elemental composition; performance comparison. See DOI: 10.1039/c8ra01680a |
| This journal is © The Royal Society of Chemistry 2018 |