
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA first-principles study on Si24 as an anode material for rechargeable batteries†
Yu He ab,
Xia Luc and
Duck Young Kim*ad
ab,
Xia Luc and
Duck Young Kim*ad
aCenter for High Pressure Science and Technology Advanced Research, Shanghai 201203, China. E-mail: duckyoung.kim@hpstar.ac.cn
bKey Laboratory of High-Temperature and High-Pressure Study of the Earth's Interior, Institute of Geochemistry, Chinese Academy of Sciences, Guiyang, Guizhou 550081, China
cBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Engineering, Beijing 100029, China
dDivision of Advanced Nuclear Engineering, POSTECH, South Korea
First published on 4th June 2018
Abstract
Due to its intriguing geometry, possessing an open-channel structure, Si24 demonstrates potential for storing and/or transporting Li/Na ions in rechargeable batteries. In this work, first-principles calculations were employed to investigate the phase stability and Li/Na storage and transport properties of the Si24 anode to evaluate its electrochemical performance for batteries. The intercalation of Li and Na into the Si24 structure could deliver a capacity of 159 mA h g−1 (Li4Si24 and Na4Si24), and the average intercalation potentials were 0.17 V (vs. Li) and 0.34 V (vs. Na). Moreover, the volume change of Si24 upon intercalation proved very small (0.09% for Li, 2.81% for Na), indicating its “zero-strain” properties with stable cycling performance. Li+ and Na+ can diffuse along the channels inside the Si24 structure with barrier energies of 0.14 and 0.80 eV respectively, and the ionic conductivity of Li2.66Si24 was calculated to be as high as 1.03 × 10−1 S cm−1 at 300 K. Our calculations indicate that the fast Li-ionic conductivity properties make the Si24 structure a novel anode material for both lithium and sodium ion batteries.
Introduction
Rechargeable lithium ion batteries (LIBs) and sodium ion batteries (SIBs) have been proven to be efficient energy storage devices.1–5 However, with the wide use of electric vehicles and energy storage systems for renewable energy, the energy density, cycling stability, safety and cost issues of current rechargeable batteries cannot meet the increasingly high requirements. Accordingly, abundant, low-cost, nontoxic, stable and low-strain novel electrode materials must be developed for better batteries.As a great number of new materials have been synthesized at high pressure,6,7 novel materials for rechargeable batteries could also be found using high-pressure technology. Moreover, high-pressure induced phase transformations of several electrode materials, e.g., LixMPO4 (M = Fe and Co),8,9 LiMn2O4,10–12 Li15Si4,13 V2O5,14 and superoxide LiO2,15 have been reported. Some of these high-pressure polymorphs have revealed a higher Li+ ion diffusion coefficient than their normal-pressure phase and demonstrated potential as electrode materials for batteries.16 In addition to these high-pressure polymorphs of current electrode materials, many novel alkali compounds with an open-channel structure have been derived at high pressure.12,17–19 It has been proven that these channels likely provide a diffusion path for the alkali ions and lead to high ionic conductivities.12,18 Although these materials have potential application for batteries, related investigations are rare. Therefore, it is necessary to exploit their potential as electrode materials for LIBs and SIBs.
Recently, a novel Na–Si compound structure (Na4Si24), which has the same structure as Eu4Ga8Ge16 with open channels along the [100] direction, was found at a pressure above 8 GPa.19,20 In this structure, the inside channels are constructed from sp3-bonded Si atoms and filled with Na atoms. These Na atoms in the channels can be entirely removed by a thermal “degassing” process to form a new orthorhombic Si allotrope, namely Si24.20 Si24 with open channels for ionic storage and diffusion displays potential as an electrode material for LIBs or SIBs. Consequently, a thorough investigation of this material is warranted and its electrochemical performance as an electrode material should be evaluated. During the revision of this manuscript, Arrieta et al. published their theoretical research on NaxSi24 (1 ≤ x ≤ 4) for SIBs and believe it is a possible anode material. However, they did not consider its potential use for LIBs and Li+/Na+ conductivity properties.21 In this work we performed a systematic investigation of Si24 properties for use as an anode material for LIBs and SIBs with first-principles theoretical insight.
Computational details
First-principles calculations were executed using the Vienna Ab Initio Simulation Package (VASP) based on the density functional theory (DFT).22,23 The projected augmented wave (PAW) potentials24 were used to deal with the electronic exchange–correlation interaction along with the PBE-GGA functional.25 A plane wave representation for the wave function with a cutoff energy of 600 eV was applied. Geometry optimizations were performed using a conjugate gradient minimization until all the forces acting on the ions were less than 0.01 eV Å−1 per atom. A k-point mesh with a spacing of ca. 0.03 Å−1 was adopted. The Li+ and Na+ migration barrier energy was calculated by the CINEB method.26 A larger supercell of 3 × 1 × 1 containing 84 atoms for Li4Si24 and Na4Si24 was used for our simulation. For the large supercell adopted in the CINEB calculations, only the Γ point was adopted for k-point sampling to reduce the computational cost. The convergence check indicated that a denser k-mesh did not affect our conclusion.In molecular dynamic calculations, 3 × 1 × 1 supercells of Li2.66Si24 and Na2.66Si24 were employed. The energy cutoff was reduced to 400 eV and Brillouin zone sampling was performed at the Γ point. An equilibration step was first carried out in the canonical ensemble (constant N, V, T) using a Nosé thermostat.27 In general, the model system was thermally equilibrated for 2 ps first, followed by a MD run at different temperatures (400 to 1800 K) for 20 ps. The time average mean square displacements (MSD)28 of the different atoms were generated using the atomic configuration information from every finite MD time step, defined as:
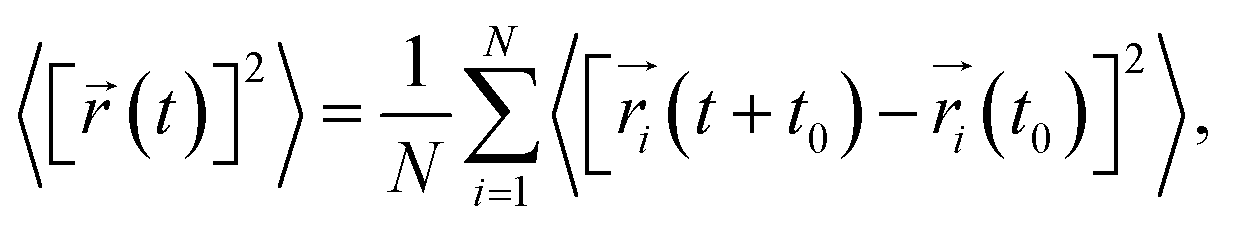 | (1) |
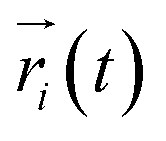 is the displacement of the ith Li or Na ion at time t, and N is the total number of Li or Na ions in the system. In practice, D is obtained by a linear fit to the time dependence of the average MSD. The obtained D at various temperatures can be fitted with the Arrhenius equation:
is the displacement of the ith Li or Na ion at time t, and N is the total number of Li or Na ions in the system. In practice, D is obtained by a linear fit to the time dependence of the average MSD. The obtained D at various temperatures can be fitted with the Arrhenius equation:
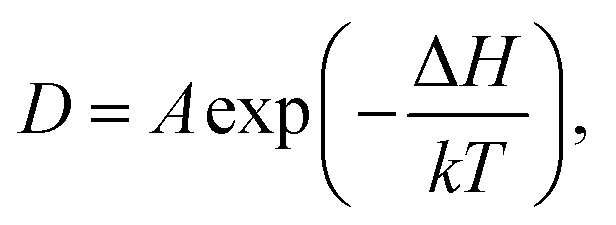 | (2) |
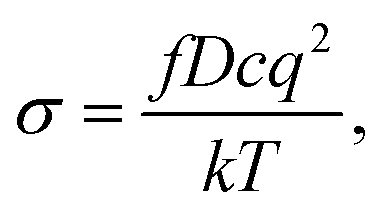 | (3) |
Results and discussion
Phase stability and intercalation potential
A unit cell of Si24 with four channels along the [100] direction is shown in Fig. 1a. It was reported that sodium atoms can be accommodated in the middle of the channels to form Na4Si24 (Fig. 1c), which was synthesized at high-pressure and is stable at ambient pressure and temperature.19,20 In our simulations, lithium atoms were placed in the same positions as sodium to construct the initial Li4Si24 structure (Fig. 1b) and then derive the final lithiation phase by structural relaxation. In this way, a total of 4 Li/Na atoms can be encapsulated in a Si24 unit cell, while delivering a theoretical capacity of 159 mA h g−1. In this work, the Li4Si24 and Na4Si24 phases are considered the fully lithiated and sodiated states of Si24 hereafter, respectively.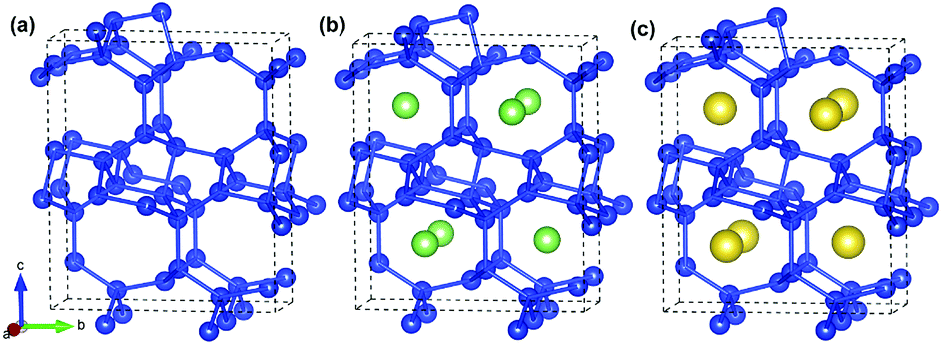 | ||
| Fig. 1 Crystal structures of (a) Si24, (b) Li4Si24 and (c) Na4Si24. Blue, green and yellow spheres represent the Si, Li and Na atoms, respectively. | ||
To evaluate the performance of Si24 as an electrode material for LIBs and SIBs, the phase stability of Si24 after Li/Na intercalation was investigated first. The stability of Si24 with different Li or Na contents is determined by the formation energy of the MxSi24 (M = Li and Na; 1 ≤ x ≤ 4) alloy, defined as:
| Eform = E(MxSi24) − E(Si24) − xE(M). | (4) |
The calculated formation energies of all the MxSi24 (M = Li and Na; 1 ≤ x ≤ 4) phase are negative and decrease with increasing Li/Na content, which means the Li and Na intercalation process is energetically favorable as shown in Fig. 2a. On the basis of the formation energies, the Li4Si24 and Na4Si24 phase can form during the charging process. For Na4Si24, it is synthesized at high pressure and is stable at ambient temperature and pressure. For Li4Si24, it might be synthesized by either chemical lithiation from Si24 or an ion exchange method from Na4Si24. These stabilized Li or Na rich materials could demonstrate promising applications for compensating Li/Na loss during the formation of a solid electrolyte interphase (SEI) or pair with cathode materials without a Li/Na component such as V2O5 and some organic cathode materials, which are much cheaper than Li/Na rich cathode materials.29–32
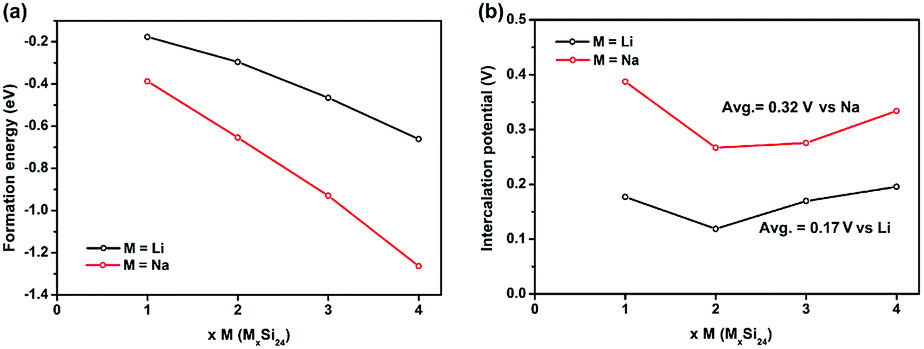 | ||
| Fig. 2 (a) Formation energies and (b) intercalation potentials of LixSi24 and NaxSi24 (1 ≤ x ≤ 4) upon Li/Na insertion into the Si24 structure. | ||
The average Li/Na intercalation potential is a critical parameter for electrode materials, which is related to the difference in the Gibbs free energy between the charged and discharged states, and can be determined by the following equation:
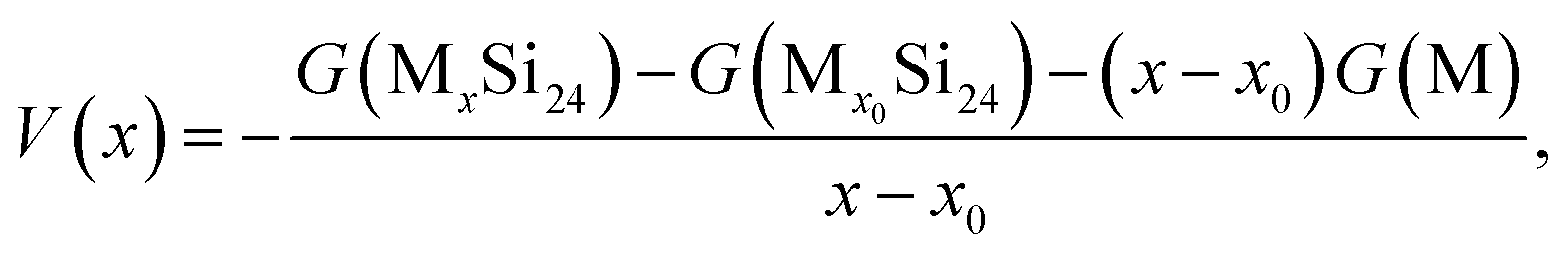 | (5) |
Volume change
It is known that significant volume expansion happens during the lithiation process of conventional silicon-based anode materials, leading to a poor cycling performance (Table S1†).33,34 However, our calculation results demonstrate that the volume changes of Si24 during Li/Na intercalation is very small. As shown in Fig. 3, the volume expansion of Si24 after Li intercalation is only 0.09%, which is negligible compared with the volume expansion of commercialized graphite (∼10%) and cubic silicon (∼300%) anode materials. The volume change after Na intercalation is larger because of the larger ionic radius of Na. However, the 2.81% volume change is still quite small compared with Li4Ti5O12 (12.5%) and other metal or metalloid anode materials (>200%) for SIBs.35–38 Due to the very small volume changes of Si24 during Li/Na intercalation, the good cycling stability of Si24 could be expected when used as anode material for both LIBs and SIBs.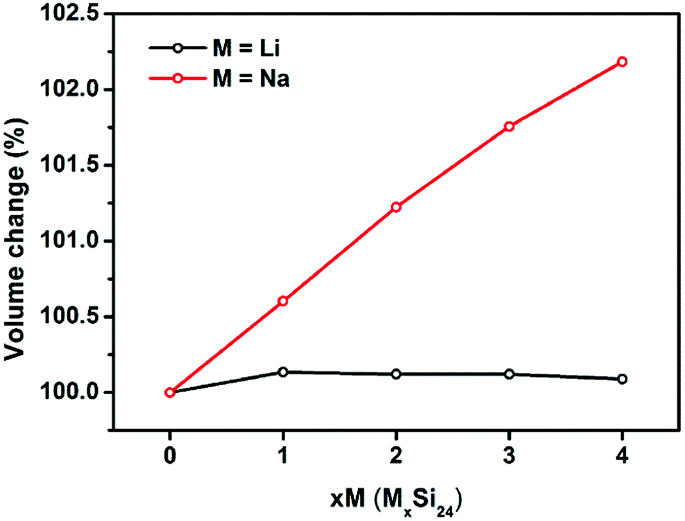 | ||
| Fig. 3 Normalized volume changes of Si24 upon Li/Na intercalation. | ||
Next the cell parameters were examined after Li/Na intercalation. As shown in Table 1, the cell parameter a is elongated while b and c are compressed during Li/Na intercalation. The increase of a is 4.16% for Li and 9.35% for Na, while the decrease of b and c is 0.74% and 1.73% for Li and 2.42% and 4.40% for Na. These parameter changes show limited influence on the ionic preferential diffusion along the b-axis and/or c-axis in the Si24 structure.
| Compound | a (Å) | b (Å) | c (Å) | Volume (Å3) |
|---|---|---|---|---|
| Si24 | 3.85 | 10.75 | 12.74 | 527.28 |
| Li4Si24 | 4.01 | 10.67 | 12.52 | 528.11 |
| Na4Si24 | 4.21 | 10.49 | 12.18 | 537.90 |
Electronic structure
The electronic conductivity is very important in electrode materials. To evaluate the electron conductivity change after Li/Na intercalation in Si24, the band structures of Si24, LixSi24 and NaxSi24 (1 ≤ x ≤ 4) were calculated and compared in Fig. S1 and S2,† and the band gaps (direct and indirect) are shown in Table 2. From our calculations, Si24 with an indirect band gap of 0.44 eV exhibits a semiconductor-like property. When Li/Na atoms intercalate, the band gaps decrease significantly to 0.18 eV for Li4Si24 and 0 eV for Na4Si24, which suggests that the electronic conductivity can be greatly enhanced after Li/Na intercalation. The improvement of electronic conductivity by Li/Na doping will certainly benefit the electron transport property of the electrode and improve its rate performance during the charge and discharge process.| MxSi24 | Li (eV) | Na (eV) | ||
|---|---|---|---|---|
| Indirect | Direct | Indirect | Direct | |
| 0 | 0.44 | 0.54 | 0.44 | 0.54 |
| 1 | 0.29 | 0.46 | 0.14 | 0.28 |
| 2 | 0.29 | 0.29 | 0.15 | 0.28 |
| 3 | 0.23 | 0.34 | 0.16 | 0.32 |
| 4 | 0.18 | 0.23 | 0 | 0 |
Li/Na ion transport
In addition to electronic conductivity, the Li/Na ion transport properties of electrode materials are also essential to the battery performances such as power density, over potential, and rate performance. Therefore, thoroughly understanding the Li/Na ion transport properties of an electrode material is necessary. Using the CINEB method, we calculated the barrier energies of Li+ and Na+ migration in Si24. There are two possible ion diffusion paths in Si24 as shown in Fig. 4a. Path 1 is along the channel direction (a-axis), and path 2 is between the channels along the b-axis. The calculated barrier energies of Li+ and Na+ along path 1 are 0.14 and 0.80 eV, respectively (Fig. 4b). Along path 2, the barrier energies are 0.87 eV for Li+ and 2.98 eV for Na+ (Fig. 4c). It is obvious that path 1 (a-axis) is the most favorable migration path with dramatically lower energy barriers for both Li+ and Na+ ionic migration. Accordingly, from an activation energy perspective, the diffusion of Li+ in Si24 framework can mainly proceed along the a-axis with a plausible fast ionic diffusion kinetic for such a low energy barrier. While for the Na ion, the migration barrier energy of 0.80 eV is a little bit higher than normal electrode material. This can be a drawback of Si24 as an anode for SIBs.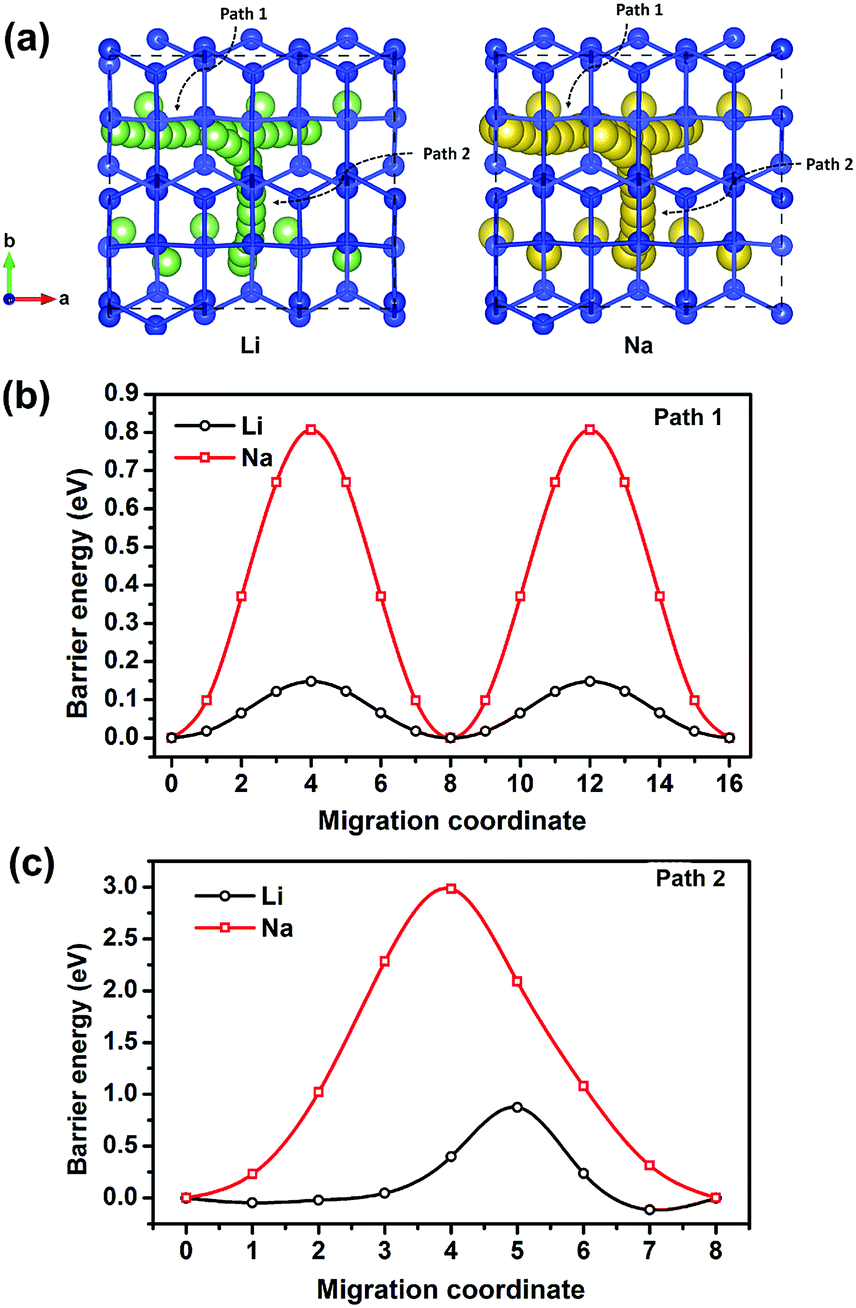 | ||
| Fig. 4 (a) Possible migration paths for Li+ and Na+ in Si24 and the calculated barrier energy of Li+ and Na+ along (b) path 1 and (c) path 2. Normalized volume changes of Si24 upon Li/Na intercalation. | ||
The diffusion properties and ionic conductivity of Li+/Na+ in Si24 were subsequently investigated by a first-principles molecular dynamic (FPMD) method at high temperature. For Li4Si24 and Na4Si24, no measurable net MSD results for Li+/Na+ diffusion were seen up to the utmost temperature before melting (Fig. S3†). This result signals that the ideal crystal M4Si24 (M = Li and Na) does not provide enough space for Li+/Na+ diffusion because all the possible positions in the channels are occupied by Li/Na atoms. However, in reality, for electrode materials, ion diffusion happens before the fully lithiated/sodiated state. It means the ionic conductivity of M4Si24 (M = Li and Na) with Li/Na vacancies could be considered in electrode material. To assess this case, 4 Li/Na vacancies were created in a 3 × 1 × 1 supercell corresponding to M2.66Si24 (M = Li and Na), with a total of 80 atoms in the supercell. In these partially lithiated/sodiated states, Li/Na ions diffusion was observed at the temperature before melting, as shown in Fig. 5a and b. The one-dimensional Li+/Na+ transport property was also confirmed by the simulated trajectories (Fig. 5a and b) and MSD (Fig. S4†). At high temperature, the diffusion effect becomes pronounced with the trajectory of the Li/Na ions distributing inside all the channels, indicating potentially high ionic conductivity. With an increase in the simulated temperature, the MSD of the Si ions also starts to increase with time (Fig. S5†), which signals the melting of the entire crystal. Based on our simulation results, Li2.66Si24 and Na2.66Si24 start to melt at temperatures above 1000 and 1500 K, respectively.
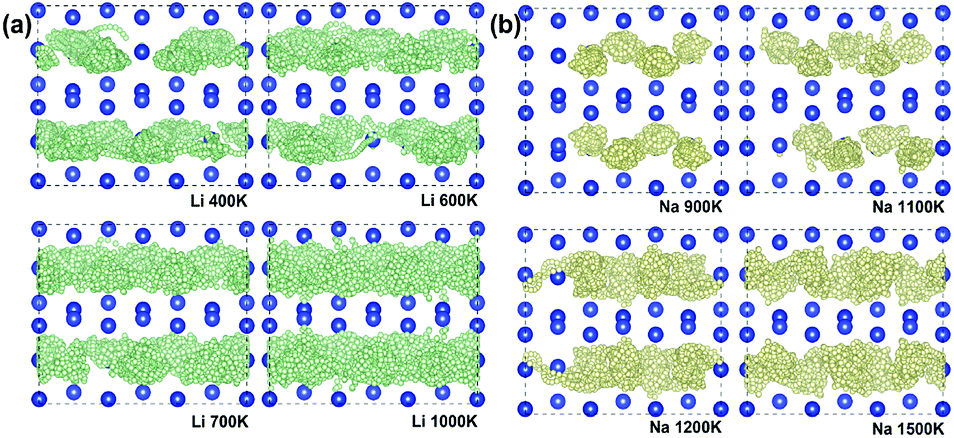 | ||
| Fig. 5 Trajectories of (a) Li+ (small green bullets) at 400, 600, 700, and 1000 K and (b) Na+ (small yellow bullets) at 900, 1100, 1200, and 1500 K for 20 ps viewed from (001) plane. | ||
The diffusion coefficients of Li/Na ions can be deduced from the simulated MSD over time at different temperatures. The convergence of the diffusion coefficients is achieved with approximately 20 ps of MD simulations because of the relatively fast Li+/Na+ diffusion in this material (Fig. S6†). The diffusion coefficients are fitted through the Arrhenius equation at temperatures varying from 400 to 1000 K for Li and 900 to 1500 K for Na, respectively (Fig. 6), from which the migration enthalpies of 0.031 eV for Li+ and 0.239 eV for Na+ were derived. Using the Nernst–Einstein equation, we calculated the Li/Na ions conductivity in Li2.66Si24 and Na2.66Si24 and plotted it in Fig. 7. The extrapolated Li+ and Na+ conductivities at 300 K are 1.03 × 10−1 and 1.26 × 10−8 S cm−1. Due to the very low activation enthalpy for Li+ migration in Si24, the Li+ conductivity is quite high at room temperature. The fast Li+ migration path along the channels of Si24 makes it a superionic conductor at room temperature, which is favorable for use as an anode material for LIBs that can be charged and discharged at a very high rate. On the contrary, the Na+ conductivity changes a lot with temperature and exhibits high ionic conductivity at higher temperatures. The ionic conductivity of Na+ in Si24 is a little bit low at room temperature, although it can be a Na+ superionic conductor at high temperature. It is worth to note that Si24 with a preferential orientation along b-axis and/or c-axis could be used to decrease the Na+ diffusion distance and promote the rate performance of the electrode.
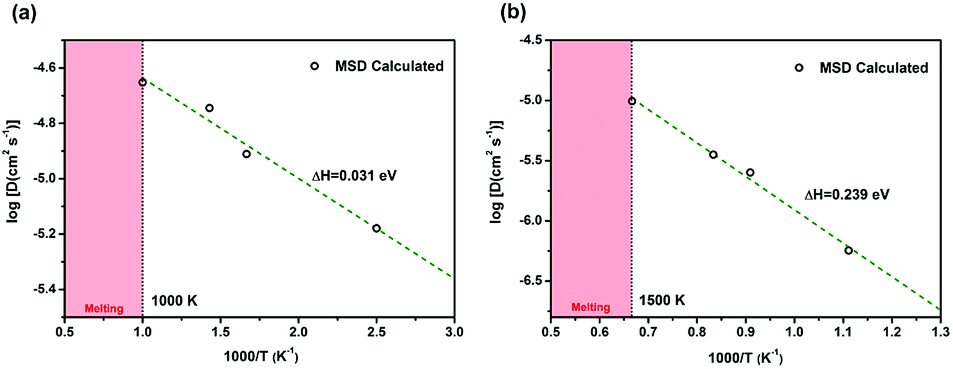 | ||
| Fig. 6 The diffusion coefficients of (a) Li+ and (b) Na+. The linear fitted results are shown by the green dashed line. The crystal structures begin melting at 1000 K for Li2.66Si24 and 1500 K for Na2.66Si24 (red areas). | ||
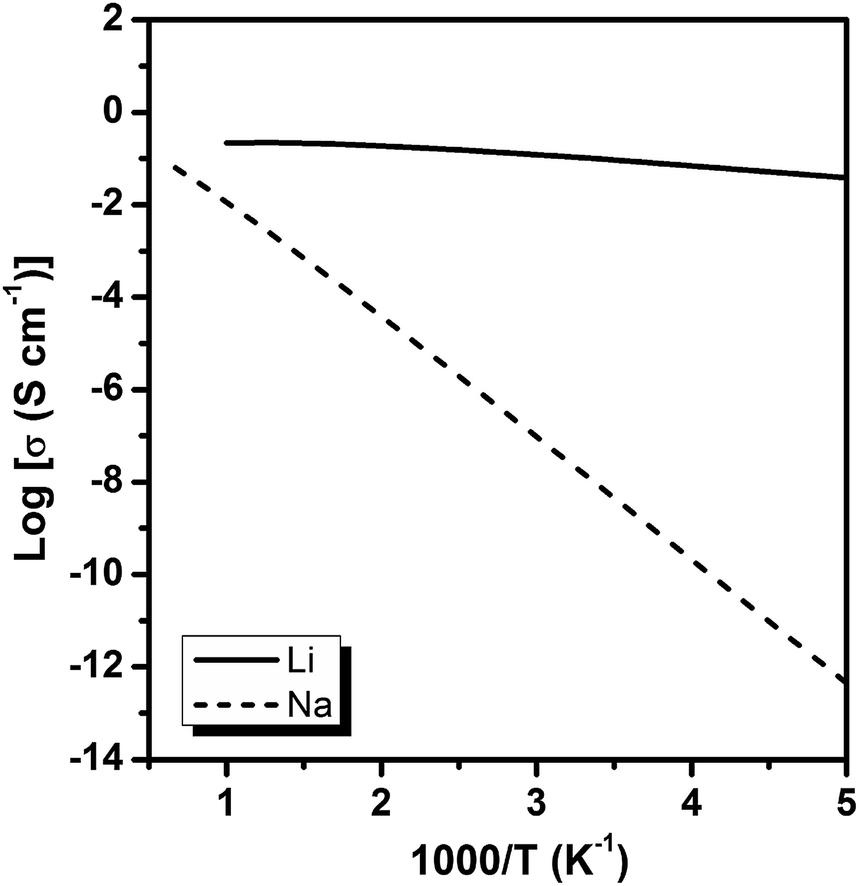 | ||
| Fig. 7 The calculated Li+ and Na+ conductivity in Li2.66Si24 and Na2.66Si24. | ||
Conclusions
Si24 with an open-channel structure was thoroughly investigated by a first-principles method to evaluate its performance as an anode for LIBs and SIBs. Various properties of Si24, including its phase stability, theoretical capacity, intercalation potentials, volume changes, electronic structures, and ionic transport property were calculated. Based on the calculation results, the advantages of Si24 as an anode material for lithium/sodium ion batteries are summarized as follows:(i) Li/Na can be stored in the channels of Si24 to form Li4Si24/Na4Si24, which is stable and energetically favorable.
(ii) The theoretical capacity of Si24 is 159 mA h g−1, and the average intercalation potentials of LixSi24 and NaxSi24 are 0.17 V (vs. Li) and 0.34 V (vs. Na), respectively. The flat and low intercalation potentials can benefit its energy density.
(iii) Si24 shows little volume change during Li/Na intercalation, which is favorable for cycling stability.
(iv) The electronic conductivity of Si24 can be improved by Li/Na intercalation. This property can lead to a high-rate performance.
(v) The ionic conductivity of Li2.66Si24 is very high, reaching 1.03 × 10−1 S cm−1 at 300 K, whereas the ionic conductivity of Na2.66Si24 is a little bit low at room temperature. However, this difficulty can be overcome by using a nanoscale material or a material with a preferential orientation along the b-axis and/or c-axis.
Due to the advantages described above, Si24 has great capability for use as an anode material in both LIBs and SIBs. In addition, M4Si24 (M = Li and Na) can also be used as lithium or sodium-rich anode materials to reduce the cost of batteries.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the support of the National Natural Science Foundation of China (Grant No. 51302259, 11774015 and 11704019), and the Youth Innovation Promotion Association of CAS. DYK acknowledges the support by NRF (NRF-2017R1D1A1B03031913). Prof. X. Lu thanks the funding supports for State Key Laboratory of Organic-Inorganic Composites (oic-201701011).Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef
.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef PubMed
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef
- Z. Jian, Y.-S. Hu, X. Ji and W. Chen, Adv. Mater., 2017, 29, 1601925 CrossRef PubMed
- J. V. Badding, Annu. Rev. Mater. Sci., 1998, 28, 631–658 CrossRef
- A. San-Miguel, Chem. Soc. Rev., 2006, 35, 876–889 RSC
- O. Garcia-Moreno, M. Alvarez-Vega, J. Garcia-Jaca, J. M. GallardoAmores, M. L. Sanjuan and U. Amador, Chem. Mater., 2001, 13, 1570–1576 CrossRef
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores and U. Amador, Electrochem. Solid-State Lett., 2005, 8, A564–A569 CrossRef
- P. Piszora, W. Nowicki and J. Darul, J. Mater. Chem., 2008, 18, 2447–2452 RSC
- K. Yamaura, Q. Z. Huang, L. Q. Zhang, K. Takada, Y. Baba, T. Nagai, Y. Matsui, K. Kosuda and E. Takayama-Muromachi, J. Am. Chem. Soc., 2006, 128, 9448–9456 CrossRef PubMed
- L. Chen and F. Mizuno, Chem. Mater., 2013, 25, 3062–3071 CrossRef
- h. Zeng, Q. Zeng, N. Liu, A. R. Oganov, Q. Zeng, Y. Cui and W. L. Mao, Adv. Energy Mater., 2015, 5, 1500214 CrossRef
- M. E. Arroyoy de Dompablo, J. M. Gallardo-Amores, U. Amador and E. Moran, Electrochem. Commun., 2007, 9, 1305–1310 CrossRef
- W. Yang, D. Y. Kim, L. Yang, N. Li, L. Tang, K. Amine and H.-K. Mao, Adv. Sci., 2017, 4, 1600453 CrossRef PubMed
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jonesc and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238 Search PubMed
- L. G. Liu, Geophys. Res. Lett., 1977, 4, 183–186 CrossRef
- Y. He, Y. Sun, X. Lu, J. Gao, H. Li and H. Li, Geophys. Res. Lett., 2016, 43, 6228–6233 CrossRef
- O. O. Kurakevych, T. A. Strobel, D. Y. Kim, T. Muramatsu and V. V. Struzhkin, Cryst. Growth Des., 2012, 13, 303–307 Search PubMed
- D. Y. Kim, S. Stefanoski, O. O. Kurakevych and T. A. Strobel, Nat. Mater., 2015, 14, 169–173 CrossRef PubMed
- U. Arrieta, N. A. Katcho, O. Arcelus and J. Carrasco, Sci. Rep., 2017, 7, 5350 CrossRef PubMed
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef
- P. E. Blochl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef
- J. Nose, J. Chem. Phys., 1984, 81, 511 CrossRef
- Y. Mo, S. P. Ong and G. Ceder, Chem. Mater., 2012, 24, 15 CrossRef
- X. Zhao, X. Zhang, D. Wu, H. Zhang, F. Ding and Z. Zhou, J. Mater. Chem. A, 2016, 4, 16606–16611 Search PubMed
- S. Wang, S. Li, Y. Sun, X. Feng and C. Chen, Energy Environ. Sci., 2011, 4, 2854–2857 Search PubMed
- K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165, 870–873 CrossRef
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef
- Y. He, X. Yu, Y. Wang, H. Li and X. Huang, Adv. Mater., 2011, 23, 4938–4941 CrossRef PubMed
- Y. He, X. Yu, G. Li, R. Wang, H. Li, Y. Wang, H. Gao and X. Huang, J. Power Sources, 2012, 216, 131–138 CrossRef
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed
- Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045 CrossRef PubMed
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897 CrossRef PubMed
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01829d |
| This journal is © The Royal Society of Chemistry 2018 |