
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA short and efficient total synthesis of the bromotyrosine-derived alkaloid psammaplysene A†
Jingjing Xu ab,
Kai Wanga and
Jinlong Wu*a
ab,
Kai Wanga and
Jinlong Wu*a
aLaboratory of Asymmetric Catalysis, Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: wyz@zju.edu.cn
bDepartment of Chemistry, Hangzhou Medical College, Hangzhou 310053, China
First published on 12th April 2018
Abstract
Psammaplysene A, an inhibitor of FOXO1a-mediated nuclear export, has been synthesized by a concise and improved route from tyrosine-derived acid and amine fragments which were easily constructed using commercially available p-hydroxybenzaldehyde and tyramine as starting material, respectively. The strategy provides an efficient access of psammaplysene analogues that can be explored for potential pharmaceutical or biological activities.
Introduction
PTEN, a tumor suppressor gene, plays an important role in blocking cell cycle progression, inducing apoptosis, and negatively regulating PI3-K/AKT signaling pathway.1–3 Thus, loss-of-function mutation in PTEN can result in an unbalance of stimulatory and inhibitory signals and have been observed in a large number of cancers at high frequency.4–6 Since discovering small molecules that could directly compensate for PTEN loss-of-function mutation has been quite difficult, attention has turned to identify small molecules modulating downstream targets of such mutation, thus to indirectly compensate for PTEN mutation. FOXO1a, a member of the Forkhead family of transcription factors, is a downstream target of PTEN and can negatively regulate cell survival and cell cycle progression.7,8 As a result of loss of PTEN phosphatase activity, FOXO1a was translocated from the nucleus to cytoplasm, leading to a disturbance of regulation of cell cycle progression.9 When a high-throughput screen was performed to identify small molecules able to localize FOXO1a in nucleus, psammaplysene A (1) was identified as one of the most potent modulators (IC50 = 5 μM), along with the closely related psammaplysene B (2) as the somewhat less active one (IC50 = 20 μM).10 The exact target of psammaplysene A in the PI3-K/PTEN/AKT signaling pathway is not known, but small alterations of the basic structure, the removal of a methyl group to give psammaplysene B (2) for example, significantly diminish activity. In addition to inhibition of FOXO1a nuclear export, psammaplysene A was also found to have neuroprotective activity by binding to heterogeneous nuclear ribonucleoprotein K (HNRNPK), a RNA-binding protein, to protect against neuronal death.11Psammaplysene A (1) and psammaplysene B (2), isolated from Indian Ocean marine sponge Psammaplysilla sp., are bromotyrosine alkaloids10,12 formed by combining two bromotyrosine-derived subunits through amide bonds. Due to the limited supply of material from marine from sponge collections, Clardy and co-workers have developed an efficient synthesis of psammaplysene A and B by amidation between fragments 3 and 4 (Fig. 1).13,14 Both fragments were synthesized from the common starting material, p-iodophenol. The acid fragments 3 was prepared with the α,β-unsaturated carbonyl moiety assembled via Heck reaction, while the amine fragments 4 was synthesized by forming 2-(N,N-dimethylamino)ethyl moiety via Sonogashira reaction with trimethylsilyl acetylene followed by desilylative bromination and aminolysis. Although their synthetic method is very efficient with four steps for fragment 3 and six steps for fragment 4, the synthesis of psammaplysene A using Pd-catalyzed cross-coupling reaction twice is harsh reaction conditions, occurrence of several side products, and difficulty in recovery and reusability of the catalysts. In addition, some of the catalysts employed are expensive. To improve such shortcoming, we proposed a new efficient route to concisely synthesize psammaplysene A without the use of Pd catalyst.
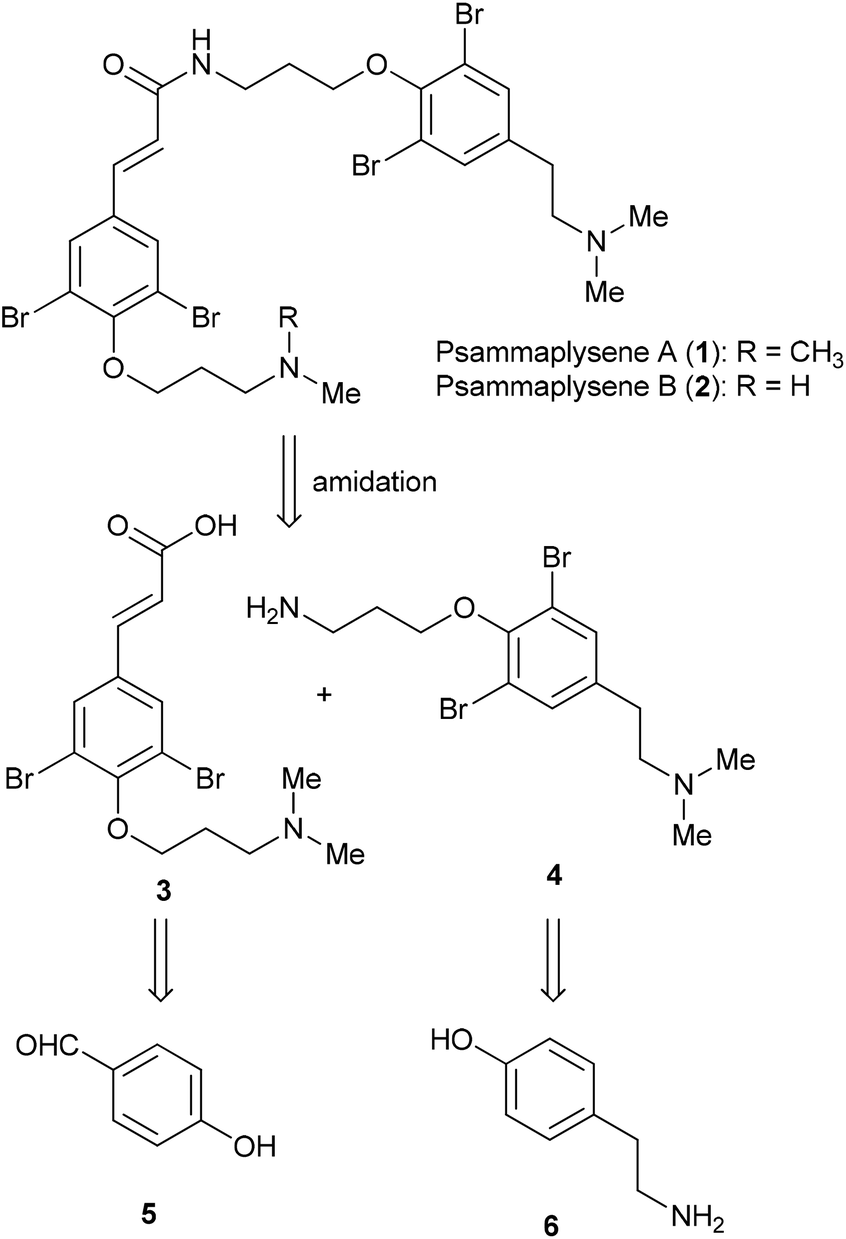 | ||
| Fig. 1 Structures of psammaplysene A (1) and B (2) and main retrosynthetic disconnection. | ||
Results and discussion
The retrosynthetic analysis of psammaplysene A is outlined in Scheme 1, providing the acid and amine fragments, 3 and 4. These two fragments possess a dibromophenol ring with the oxygen appended to an 3-aminopropyl group and are distinguishable from each other by the substituent at para position of the phenol ring, i.e. the α,β-unsaturated carbonyl moiety in 3 vs. the 2-(N,N-dimethylamino)ethyl moiety in 4. The acid building block 3 could be prepared from commercially available p-hydroxybenzaldehyde 5 in a simple manner. On the other hand, the amide building block 4 could be readily accessible from tyramine 6. | ||
| Scheme 1 Synthesis of the acid fragment (3). | ||
The synthesis of 3 began with p-hydroxybenzaldehyde 5 via ortho-bromination by using Br2 with NaOAc in HOAc to furnish dibromoaldehyde 7 in 94% yield.15,16 We initially explored a route involving Wittig olefination of 7 with the ylide Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me prior to O-alkylation; however, no desired olefination product was observed in reaction mixture. Hence, we turned to direct O-alkylation of 7 with N,N-dimethyl-3-chloropropylamine; however, low conversions were observed under various conditions,17 likely due to the poor nucleophilicity of dibromophenol with an electron-withdrawing formyl group at para position. Fortunately, compound 7, after being protected as glycol acetal, was readily converted into 8 via O-alkylation with N,N-dimethyl-3-chloropropylamine in the presence of Cs2CO3 and catalytic amount of NaI at 80 °C. Subsequent cleavage of the glycol acetal under acid condition gave the aldehyde 9 (97%), which then underwent Knoevenagel condensation18 with malonic acid in the presence of triethylamine (TEA) and a catalytic amount of piperidine in toluene at reflux to furnish the acid building block 3 (80%).
CHCO2Me prior to O-alkylation; however, no desired olefination product was observed in reaction mixture. Hence, we turned to direct O-alkylation of 7 with N,N-dimethyl-3-chloropropylamine; however, low conversions were observed under various conditions,17 likely due to the poor nucleophilicity of dibromophenol with an electron-withdrawing formyl group at para position. Fortunately, compound 7, after being protected as glycol acetal, was readily converted into 8 via O-alkylation with N,N-dimethyl-3-chloropropylamine in the presence of Cs2CO3 and catalytic amount of NaI at 80 °C. Subsequent cleavage of the glycol acetal under acid condition gave the aldehyde 9 (97%), which then underwent Knoevenagel condensation18 with malonic acid in the presence of triethylamine (TEA) and a catalytic amount of piperidine in toluene at reflux to furnish the acid building block 3 (80%).
Scheme 2 depicts the synthesis of the amide building block 4 in four steps starting from tyramine 6 in an approach similar to the literature precedent.19,20 Reductive methylation of tyramine 6 with aqueous formaldehyde was performed under catalytic hydrogenation conditions (10% Pd/C) to provide dimethyltyramine 10 in 98% yield. Bromination of 10 with Br2 in the presence of KBr afforded compound 11 in 81% yield.21 Subsequent O-alkylation of 11 with Boc-protected 3-bromopropylamine gave 12 (93%), which was converted into the dihydrochloride salt of amine 4 upon treatment with 4 M HCl in methanol.
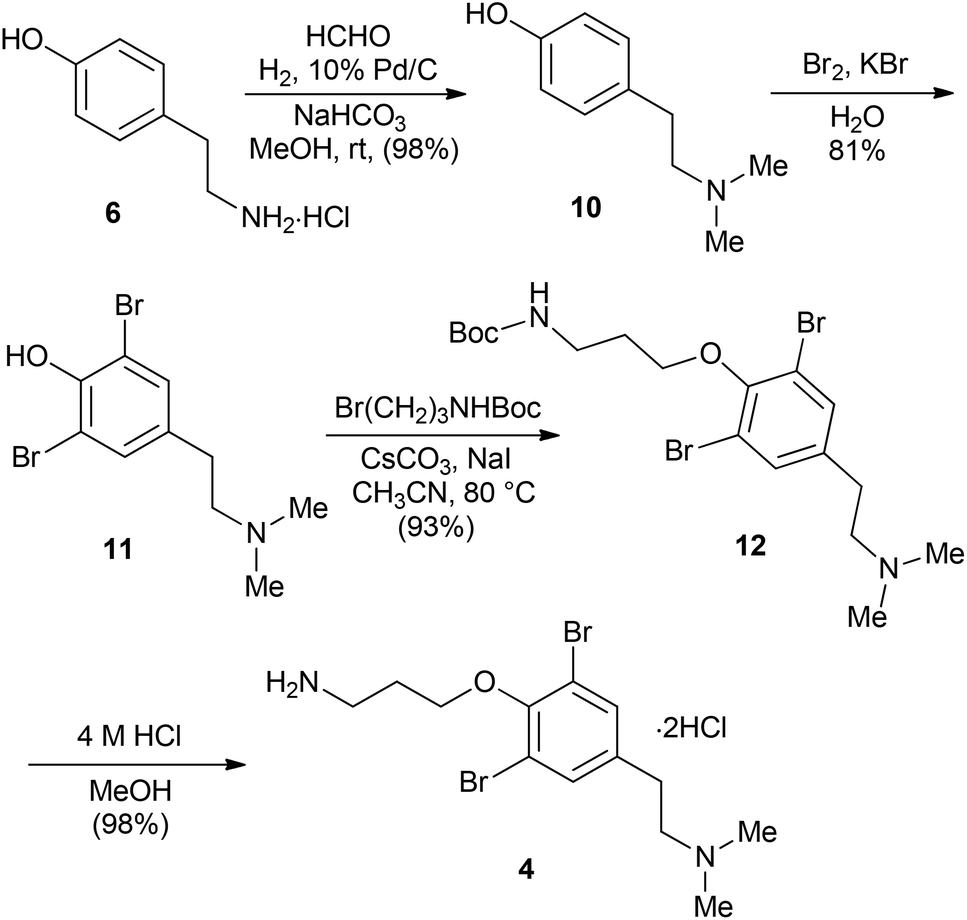 | ||
| Scheme 2 Synthesis of the amide fragment (4). | ||
Finally, the amidation between 3 and 4 were performed by using DIC with a catalytic amount of DMAP in the presence of TEA in DCM, leading to the formation of psammaplysene A (Scheme 3). The spectroscopic data of our synthetic psammaplysene A were identical to those reported for the natural product (1H NMR, 13C NMR, and HRMS).10,13
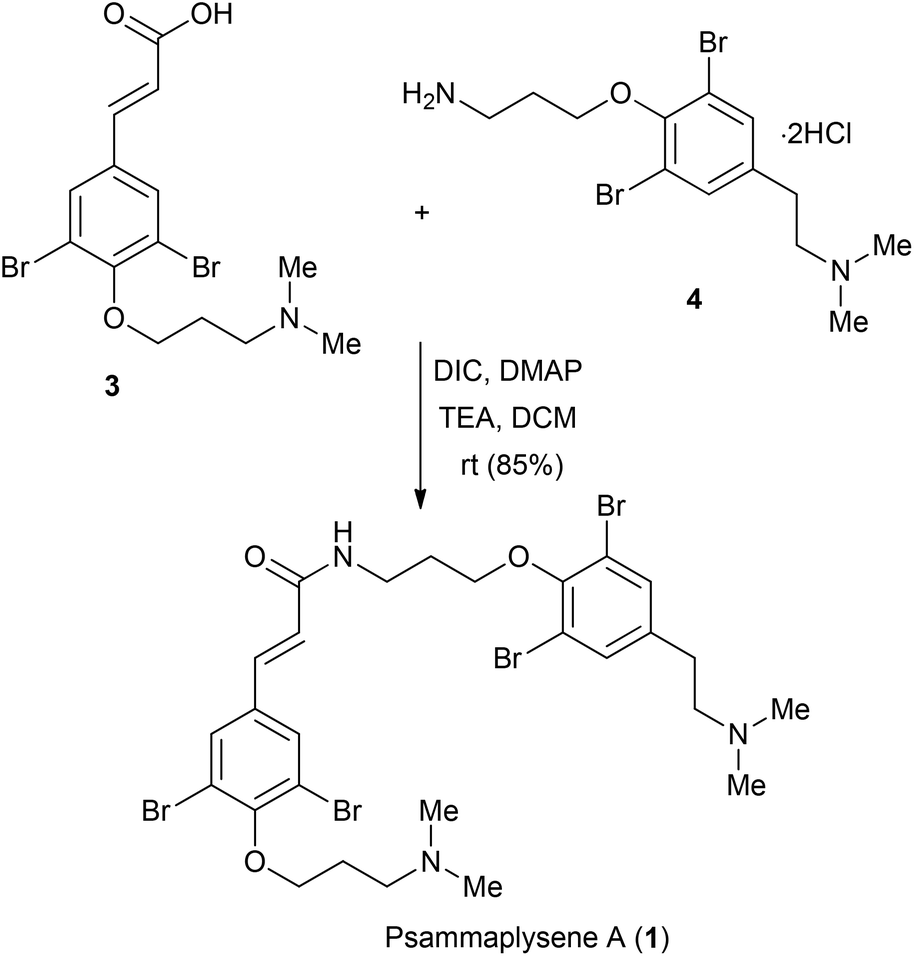 | ||
| Scheme 3 Synthesis of psammaplysene A (1). | ||
Conclusions
In summary, we have established an improved and efficient synthesis of psammaplysene A in 50% overall yield from p-hydroxybenzaldehyde 5 via Knoevenagel condensation and O-alkylation as key steps. The two major fragments 3 and 4 were concisely assembled from commercially available p-hydroxybenzaldehyde 5 in five steps (59%) and tyramine 6 in four steps (71%), respectively. Our approach is concise and might be applied in the synthesis of psammaplysene analogues on gram scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by research grants from Hangzhou Medical College Foundation (No. 2015B03) and Natural Science Foundation of Zhejiang Province (No. Y404302). The authors also thank Dr Saurav Bera for his assistance.Notes and references
- B. T. Hennessy, D. L. Smith, P. T. Ram, Y. Lu and G. B. Mills, Nat. Rev. Drug Discovery, 2005, 4, 988–1004 CrossRef CAS PubMed.
- L. Simpson and R. Parsons, Exp. Cell Res., 2001, 264, 29–41 CrossRef CAS PubMed.
- K. M. Yamada and M. Araki, J. Cell Sci., 2001, 114, 2375–2382 CAS.
- S. I. Wang, J. Puc, J. Li, J. N. Bruce, P. Cairns, D. Sidransky and R. Parsons, Cancer Res., 1997, 57, 4183–4186 CAS.
- F. Vazquez and W. R. Sellers, Biochim. Biophys. Acta, 2000, 1470, M21–M35 CAS.
- K. Kondo, M. Yao, K. Kobayashi, S. Ota, M. Yoshida, S. Kaneko, M. Baba, N. Sakai, T. Kishida, S. Kawakami, H. Uemura, Y. Nagashima, Y. Nakatani and M. Hosaka, Int. J. Cancer, 2001, 91, 219–224 CrossRef CAS PubMed.
- D. Accili and K. C. Arden, Cell, 2004, 117, 421–426 CrossRef CAS PubMed.
- H. Huang and D. J. Tindall, Future Oncol., 2006, 2, 83–89 CrossRef CAS PubMed.
- T. R. Kau, F. Schroeder, S. Ramaswamy, C. L. Wojciechowski, J. J. Zhao, T. M. Roberts, J. Clardy, W. R. Sellers and P. A. Silver, Cancer Cell, 2003, 4, 463–476 CrossRef CAS PubMed.
- F. C. Schroeder, T. R. Kau, P. A. Silver and J. Clardy, J. Nat. Prod., 2005, 68, 574–576 CrossRef CAS PubMed.
- M. Boccitto, N. Lee, S. Sakamoto, L. A. Spruce, H. Handa, J. Clardy, S. H. Seeholzer and R. G. Kalb, Mar. Drugs, 2017, 15, 246–257 CrossRef PubMed.
- G. W. Gribble, The Alkaloids: Chemistry and Biology, ed. H. J. Knölker, Academic Press, 2012, vol. 71, pp. 1–165 Search PubMed.
- S. N. Georgiades and J. Clardy, Org. Lett., 2005, 7, 4091–4094 CrossRef CAS PubMed.
- S. N. Georgiades and J. Clardy, Org. Lett., 2006, 8, 4251–4254 CrossRef CAS PubMed.
- G. Zhu, F. Yang, R. Balachandran, P. Höök, R. B. Vallee, D. P. Curran and B. W. Day, J. Med. Chem., 2006, 49, 2063–2076 CrossRef CAS PubMed.
- H. Gan, Y. Huang, W. Feng, W. Zhu and K. Guo, J. Chem. Res., 2015, 39, 336–339 CrossRef CAS.
- Various conditions were examined: NaH and CsCO3 as base, and THF, DMF and MeCN as solvents at different temperatures.
- H. S. Pawar, A. S. Wagh and A. M. Lali, New J. Chem., 2016, 40, 4962–4968 RSC.
- D. M. Swanson, S. J. Wilson, J. D. Boggs, W. Xiao, R. Apodaca, A. J. Barbier, T. W. Lovenberg and N. I. Carruthers, Bioorg. Med. Chem. Lett., 2006, 16, 897–900 CrossRef CAS PubMed.
- H. Kigoshi, K. Kanematsu, K. Yokota and D. Uemura, Tetrahedron, 2000, 56, 9063–9070 CrossRef CAS.
- E. García-Egido, J. Paz, B. Iglesias and L. Muñoz, Org. Biomol. Chem., 2009, 7, 3991–3999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and copies of 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c8ra02052c |
| This journal is © The Royal Society of Chemistry 2018 |