
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence20(S)-Ginsenoside Rg2 attenuates myocardial ischemia/reperfusion injury by reducing oxidative stress and inflammation: role of SIRT1
Wenwen Fu†
a,
Huali Xu†a,
Xiaofeng Yua,
Chen Lyua,
Yuan Tiana,
Minyu Guoa,
Jiao Sun*b and
Dayun Sui *a
*a
aDepartment of Pharmacology, School of Pharmaceutical Sciences, Jilin University, Changchun 130021, China. E-mail: suidy@jlu.edu.cn
bSchool of Nursing, Jilin University, Changchun, 130021, China. E-mail: sunjiao@jlu.edu.cn
First published on 2nd July 2018
Abstract
Previously we demonstrated that 20(S)-ginsenoside Rg2 protects cardiomyocytes from H2O2-induced injury by inhibiting reactive oxygen species (ROS) production, increasing intracellular levels of antioxidants and attenuating apoptosis. We explored the protective effect of 20(S)-ginsenoside Rg2 on myocardial ischemia/reperfusion (MI/R) injury and to clarify its potential mechanism of action. Rats were exposed to 20(S)-ginsenoside Rg2 in the presence/absence of the silent information regulator SIRT(1) inhibitor EX527 and then subjected to MI/R. 20(S)-Ginsenoside Rg2 conferred a cardioprotective effect by improving post-ischemic cardiac function, decreasing infarct size, reducing the apoptotic index, diminishing expression of creatine kinase-MB, aspartate aminotransferase and lactate dehydrogenase in serum, upregulating expression of SIRT1, B-cell lymphoma-2, procaspase-3 and procaspase-9, and downregulating expression of Bax and acetyl (Ac)-p53. Pretreatment with 20(S)-ginsenoside Rg2 also resulted in reduced myocardial superoxide generation, gp91phox expression, malondialdehyde content, cardiac pro-inflammatory markers and increased myocardial activities of superoxide dismutase, catalase and glutathione peroxidase. These results suggested that MI/R-induced oxidative stress and inflammation were attenuated significantly by 20(S)-ginsenoside Rg2. However, these protective effects were blocked by EX527, indicating that SIRT1 signaling may be involved in the pharmacological action of 20(S)-ginsenoside Rg2. Our results demonstrated that 20(S)-ginsenoside Rg2 attenuates MI/R injury by reducing oxidative stress and inflammatory responses via SIRT1 signaling.
Introduction
Myocardial infarction is a major cause of mortality worldwide. Although early reperfusion therapy is necessary for myocardial salvage, reperfusion triggers “myocardial ischemia/reperfusion” (MI/R). MI/R is characterized by cardiomyocyte apoptosis, cardiac hypofunction, arrhythmia and other disorders,1,2 which exacerbate myocardial damage.3Several factors contribute to the development of MI/R injury: cellular calcium loading; “no reflow” due to cell swelling; impaired vascular relaxation; and formation of “white cell plugs”. The enhanced oxidative stress observed after reperfusion, however, is believed to be the most likely cause.4 Oxidative stress leads to reactive oxygen species (ROS) production, increases in malondialdehyde (MDA) content, and subsequent cytotoxic injury.4,5 These processes play key parts in the progression of ischemic heart disease, and can contribute to cardiomyocyte apoptosis.5,6 Subsequently, ROS trigger leukocyte chemotaxis and cause inflammation, further aggravating severe cardiac damage.4,7 Increasing evidence suggests that ROS accumulation in mitochondria can lead to apoptotic cell death.8 ROS may also have direct effects on the structure and function of cells, which can lead to myocardial remodeling and might develop into heart failure.9,10 Therefore, protecting cardiomyocytes from ROS damage could be a rational method for ameliorating MI/R injury.
Ginsenosides are the major active components of ginseng. The latter has a range of pharmacologic and therapeutic uses, including vasorelaxation as well as anti-neoplastic, anti-diabetes mellitus, anti-inflammatory and anti-oxidative effects.11–16 20(S)-Ginsenoside Rg2 is a major active protopanaxatriol-type compound present in the roots and stem leaves of ginseng (Fig. 1). Recent studies have shown that 20(S)-ginsenoside Rg2 can block calcium channels and displays activity against free radicals.17 Our recent study demonstrated that 20(S)-ginsenoside Rg2 protects cardiomyocytes from H2O2-induced injury by increasing the intracellular levels of antioxidants, reducing ROS production, and subsequent apoptosis.18 However, few studies have explored the anti-oxidative and anti-inflammatory effects of 20(S)-ginsenoside Rg2 in MI/R injury.
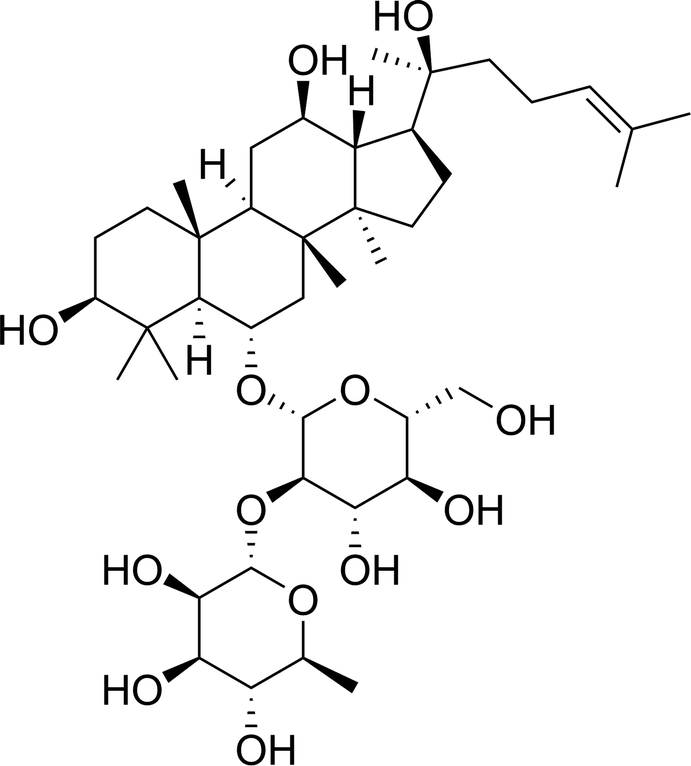 | ||
| Fig. 1 Chemical structure of 20(S)-ginsenoside Rg2. | ||
Silent information regulator (SIRT)1 is a type of histone deacetylase, the activity of which is dependent on nicotinamide adenine dinucleotide (NAD+). SIRT1 regulates the acetylation of p53, thereby inhibiting cardiomyocyte apoptosis.19 Studies have shown that activation of SIRT1 signaling protects against MI/R injury.20,21 It has been demonstrated that SIRT1 expression is reduced after MI/R-induced injury, and that activation of SIRT1 signaling with curcumin, melatonin or berberine confers protection against MI/R-induced injury by suppressing oxidative stress.22–24 Our recent study showed that 20(S)-ginsenoside Rg2 can protect H9c2 cells against H2O2-induced injury through its anti-oxidative and anti-apoptotic actions.18 Conversely, ginsenosides Rb1, Rc, Rh3, 20(S)-ginsenoside Rg3 and extract of red ginseng can activate SIRT1.25–30 In particular, 20(S)-ginsenoside Rg2 and some of its metabolites have high potential as SIRT1 activators, such as resveratrol.31 EX527 is a highly specific inhibitor of SIRT1 which suppresses NAD-dependent deacetylation in various cell types and conditions.32,33 Presumably, EX527 can be used to identify if SIRT1 can mediate the cardioprotective effect of 20(S)-ginsenoside Rg2, and whether it relates to oxidative stress and inflammation through inhibition of the SIRT1 activation triggered by 20(S)-ginsenoside Rg2.
We wished to ascertain if 20(S)-ginsenoside Rg2 confers anti-oxidative and anti-inflammatory effects in MI/R, and to investigate the role of SIRT1 signaling in the cardioprotective effect of 20(S)-ginsenoside Rg2.
Materials and methods
Chemicals and reagents
20(S)-Ginsenoside Rg2 was provided by Professor Yifa Zhou (School of Life Sciences, Northeast Normal University, Changchun, China). High-performance liquid chromatography showed that the purity of 20(S)-ginsenoside Rg2 was >98%. It was dissolved in 0.5% sodium carboxymethyl cellulose (CMC-Na) to give the final concentrations.EX527 was purchased from Selleck Chemicals (Houston, TX, USA). Creatine kinase-MB (CK-MB), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), MDA, catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GSH-PX), interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay using an in situ cell death detection kit was obtained from Roche Molecular Biochemicals (Mannheim, Germany). A quantitative polymerase chain reaction (qPCR) kit was obtained from Beijing TransGen Biotech (Beijing, China). A bicinchoninic acid (BCA) protein assay kit was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Antibodies against SIRT1, acetyl (Ac)-p53, gp91phox, procaspase-3, procaspase-9, B-cell leukemia (Bcl)-2 and Bax were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody against β-actin was obtained from ZSGB-BIO (Beijing, China). Goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from Beijing Dingduo Changsheng Biotechnology (Beijing, China).
Animals
Animals were treated according to the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health (NIH), Bethesda, MD, USA) and the Committee for the Care and Use of Laboratory Animals of Jilin University (Changchun, China). The study protocol was approved by the Ethics Committee of Jilin University.Female Sprague-Dawley rats (230–250 g) were provided by the Experimental Animal Center of Jilin University. All animals were allowed free access to food and water, and maintained at 22–24 °C under a 12 h light–dark cycle.
Experimental protocol
Experimental model of MI/R injury
Sprague-Dawley rats were anesthetized with 3% pentobarbital sodium. Myocardial ischemia was induced by exteriorizing the heart through a left-thoracic incision, placing a 6-0 silk suture and making a slipknot around the left anterior descending coronary artery. After 30 min of ischemia, the slipknot was released, and the myocardium reperfused for 6 h (for analyses of myocardial apoptosis and measuring the size of the myocardial infarct) and 72 h (for determination of cardiac function). Rats in the sham group underwent the same surgical procedures except that the suture passing under the left coronary artery was left untied. Before and during the surgical procedure, animals received different treatments.Echocardiography
Two-dimensional (2D) and M-mode echocardiographic measurements were conducted in a standard setting using a 10S scan head (Vivid I; GE Healthcare, Piscataway, NJ, USA) 72 h after MI/R. After a short-axis 2D image of the left ventricle had been obtained at the level of the papillary muscles, 2D-guided M-mode images were acquired at a sweep speed of 100 mm s−1 and stored digitally. The left ventricular internal dimension at diastole (LVIDd) and left ventricular internal dimension at systole (LVIDs) were measured, allowing calculation of left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF; calculated using Simpson's rule). All parameters were measured over three consecutive cardiac cycles. Measurements were made by an experienced echocardiographer blinded to the treatment protocol.Determination of the size of the myocardial infarct
At the end of 6 h of reperfusion, hearts were excised rapidly. Determination of infarct size was undertaken according to a method described previously with minor modification.34 Ventricle tissues were cut into five transverse slices, and the latter incubated in 0.5% nitrotetrazolium blue chloride for 20 min. Myocardial infarction was distinguished by the different color (pale for ischemic myocardium and dark-red for non-ischemic myocardium). The ischemic myocardium was cut and weighed. The infarct size as a percentage of the ventricular mass was calculated as the weight of the ischemic zone/total weight of ventricle × 100%. The size of the myocardial infarct was expressed as a percentage of the ventricle area.Determination of myocardial apoptosis
To measure the DNA nicks that reflect the extent of myocardial apoptosis, the TUNEL assay was carried out.35 Heart slices were fixed in 10% formalin solution, embedded in paraffin, and sectioned on slides at 5 μm thickness. TUNEL staining with paraffin sections was done according to manufacturer instructions. The apoptosis index was expressed as the number of apoptotic cardiomyocytes/total number of cardiomyocytes × 100%.Determination of serum activities of CK-MB, AST and LDH
Blood samples were drawn 6 h after reperfusion. Collected blood samples were clotted for 2 h at room temperature, and centrifuged at 900 × g for 15 min. The supernatant serums were separated and stored at −80 °C. The activities of CK-MB, AST and LDH were measured using diagnostic kits according to manufacturer instructions.Quantification of superoxide production
Superoxide production in tissue was measured by lucigenin-enhanced chemiluminescence, as described previously.36 One piece of cardiac tissue was weighed precisely. Phosphate-buffered saline containing phosphatase and protease inhibitors (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 w/v) was added. After homogenization, the mixture was centrifuged at 145 × g for 10 min at room temperature. Then, 1 mmol L−1 of NADPH and 50 μmol L−1 of luster concentrate were added into the collected supernatant. A chemiluminescence analyzer was employed to measure the activity of ligninase. Superoxide production was expressed as relative light units per second per milligram heart weight (RLU mg−1 s−1).
:9 w/v) was added. After homogenization, the mixture was centrifuged at 145 × g for 10 min at room temperature. Then, 1 mmol L−1 of NADPH and 50 μmol L−1 of luster concentrate were added into the collected supernatant. A chemiluminescence analyzer was employed to measure the activity of ligninase. Superoxide production was expressed as relative light units per second per milligram heart weight (RLU mg−1 s−1).
Determination of tissue MDA, SOD, CAT and GSH-PX
The tissues of ventricles were excised and homogenized with ice-cold buffer in a homogenizer. The homogenate was centrifuged at 1780 × g for 15 min at room temperature. The supernatant was taken to measure the activities of SOD, CAT and GSH-PX and MDA the content using diagnostic kits according to manufacturer instructions.Detection of tissue levels of IL-1β, IL-6 and TNF-α
Rats were sacrificed after 6 h of reperfusion. Heart tissue was dissected rapidly for measurement of the mRNA levels and activities of IL-1β, IL-6 and TNF-α. Total RNA was isolated from heart tissue using TRIzol™ reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer instructions. RNA was quantified by optical-density measurements at 260 nm and 280 nm. Synthesis of complementary DNA and qPCR was carried out using TransScript® Green Two-Step qRT-PCR SuperMix (TransGen Biotech, Beijing, China). qPCR was done with a reaction mixture (total volume = 20 μL) that consisted of 2× Trans Start Top Green qPCR SuperMix, passive reference dye, double-distilled water, cDNA templates, as well as forward and reverse primers. The amount of mRNA from IL-1β, IL-6 and TNF-α was normalized to β-actin expression. All primers were ordered or custom-made from Beijing Dingguo Changsheng Biotechnology (Table 1). Relative fold-changes in expression of the target gene in control and other groups were determined using the 2−ΔΔCT method. Activities of IL-1β, IL-6 and TNF-α in myocardial tissue homogenates and cardiomyocyte supernatants were detected in strict accordance with manufacturer instructions.37,38 A BCA kit was used to quantify protein.| Gene | Primer sequence (5′–3′) | Accession number |
|---|---|---|
| IL-1β | Forward: GCAATGGTCGGGACATAGTT | NM_031512.2 |
| Reverse: AGACCTGACTTGGCAGAGG | ||
| IL-6 | Forward: AGTGCATCATCGCTGTTCATACA | NM_012589.2 |
| Reverse: ATATGTTCTCAGGGAGATCTTGGAA | ||
| TNF-α | Forward: GTCGTAGCAAACCACCAAGC | NM_012675.3 |
| Reverse: TGTGGGTGAGGAGCACGTAG | ||
| β-Actin | Forward: GATCAAGATCATTGCTCCTCCTG | NM_031144.3 |
| Reverse: AGGGTGTAAAACGCAGCTCA |
Western blotting
Western blotting was used for detecting proteins from SIRT1, Ac-p53, gp91phox, procaspase-3, procaspase-9, Bcl-2 and Bax. Rats were sacrificed after 6 h of reperfusion, and heart tissue was removed for protein extraction. The protein concentration was determined using the BCA protein assay kit. Then, the extracted protein was loaded onto 12% polyacrylamide–sodium dodecyl sulfate gel. After electrophoresis, the gel was blotted onto a polyvinylidene difluoride (PVDF) membrane and blocked with 5% (w/v) non-fat milk for 1 h. The transferred PVDF membrane was incubated with appropriate primary antibodies at 4 °C overnight. Binding of primary antibody was detected with a secondary antibody conjugated to horseradish peroxidase, and visualized using ECL chemiluminescence. β-Actin was used as the internal loading control. Results are expressed by grayscale value analyzed by Image J v1.5.0.26 (NIH).Statistical analyses
Experiments were carried out at least thrice in duplicate/triplicate. Results are the mean value ± S.D. One-way ANOVA/Bonfferroni and individual t-tests were conducted using Prism v5.04 (GraphPad, San Diego, CA, USA). P < 0.05 was considered significant.Results
Effects of 20(S)-ginsenoside Rg2 on cardiac function in MI/R-induced injury
After 72 h of reperfusion, echocardiography showed that cardiac function in the MI/R group had deteriorated remarkably, as evidenced by increased LVIDd, LVIDs and decreased LVFS and LVEF compared with the sham group (P < 0.01) (Fig. 2). Pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) ameliorated myocardial dysfunction significantly, which was verified by increased LVEF, LVFS and decreased LVIDd and LVIDs in MI/R animals (P < 0.05 or P < 0.01) (Fig. 2). Our results suggested that pretreatment with 20(S)-ginsenoside Rg2 improved cardiac function in MI/R-induced injury.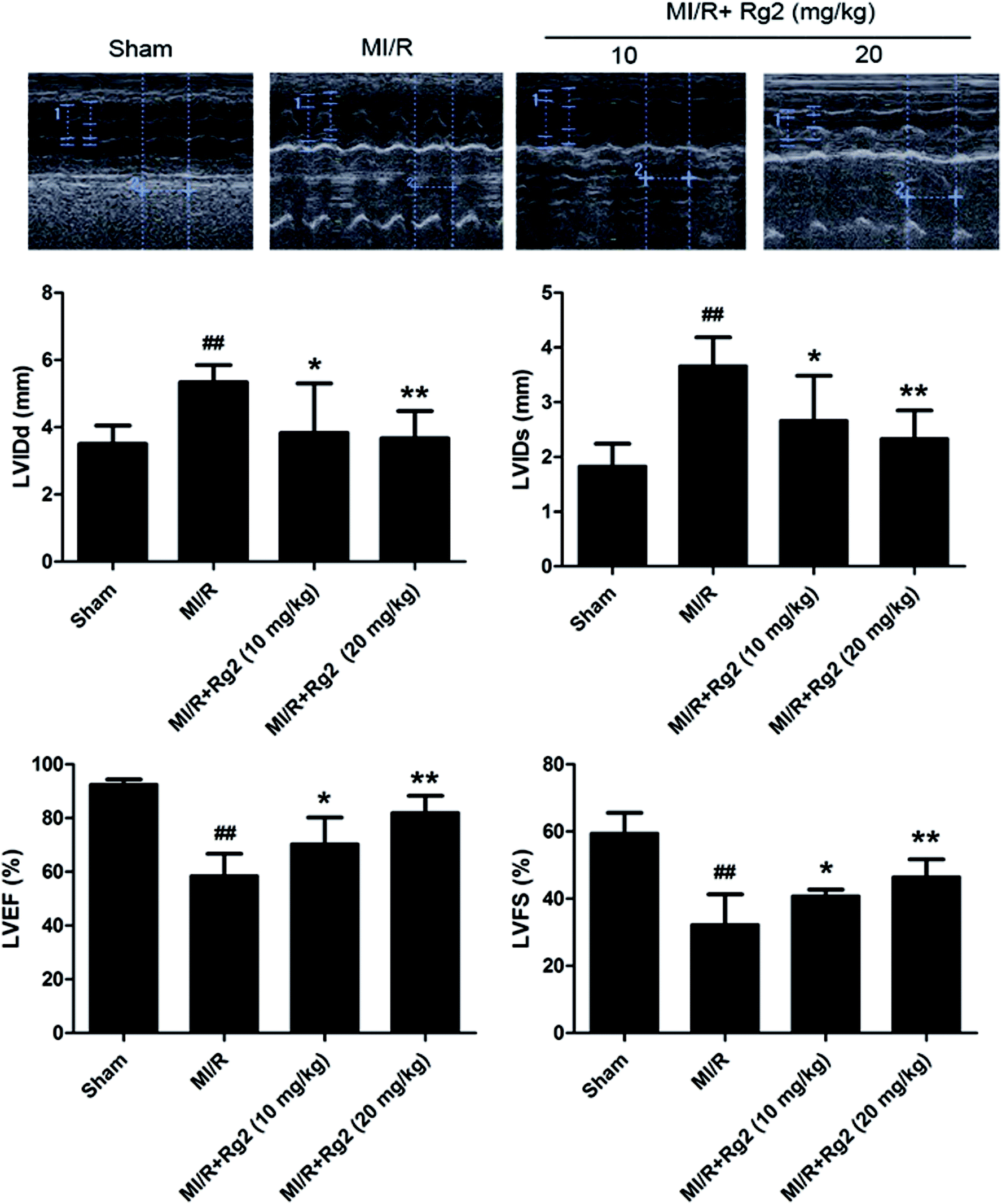 | ||
| Fig. 2 Effects of 20(S)-ginsenoside Rg2 on cardiac function in MI/R-induced injury. Echocardiographic measurement was carried out after 72 h of reperfusion. Representative M-mode images are shown. Evaluation of left ventricular internal dimension at diastole (LVIDd), left ventricular internal dimension at systole (LVIDs), left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) are shown. Data are the mean ± S.D. ##P < 0.01 vs. sham group; *P < 0.05, **P < 0.01 vs. MI/R group. | ||
Effects of 20(S)-ginsenoside Rg2 on the apoptotic index, infarct size, and serum activities of CK-MB, AST and LDH in MI/R-induced injury
After 6 h of reperfusion, pretreatment with 20(S)-ginsenoside Rg2 decreased cardiac apoptosis and necrosis by reducing the apoptotic index and infarct size compared with the MI/R group (P < 0.05 or P < 0.01) (Fig. 3A and B). We evaluated further the activities of CK-MB, AST and LDH in serum, which reflected cellular injury or tissue necrosis and membrane permeability. Compared with the sham group, the activities of CK-MB, AST and LDH were increased in the MI/R group. However, pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) decreased the activities of CK-MB, AST and LDH (P < 0.05 or P < 0.01) (Fig. 3C). These results provided direct evidence that 20(S)-ginsenoside Rg2 could attenuate post-MI/R apoptosis and necrosis.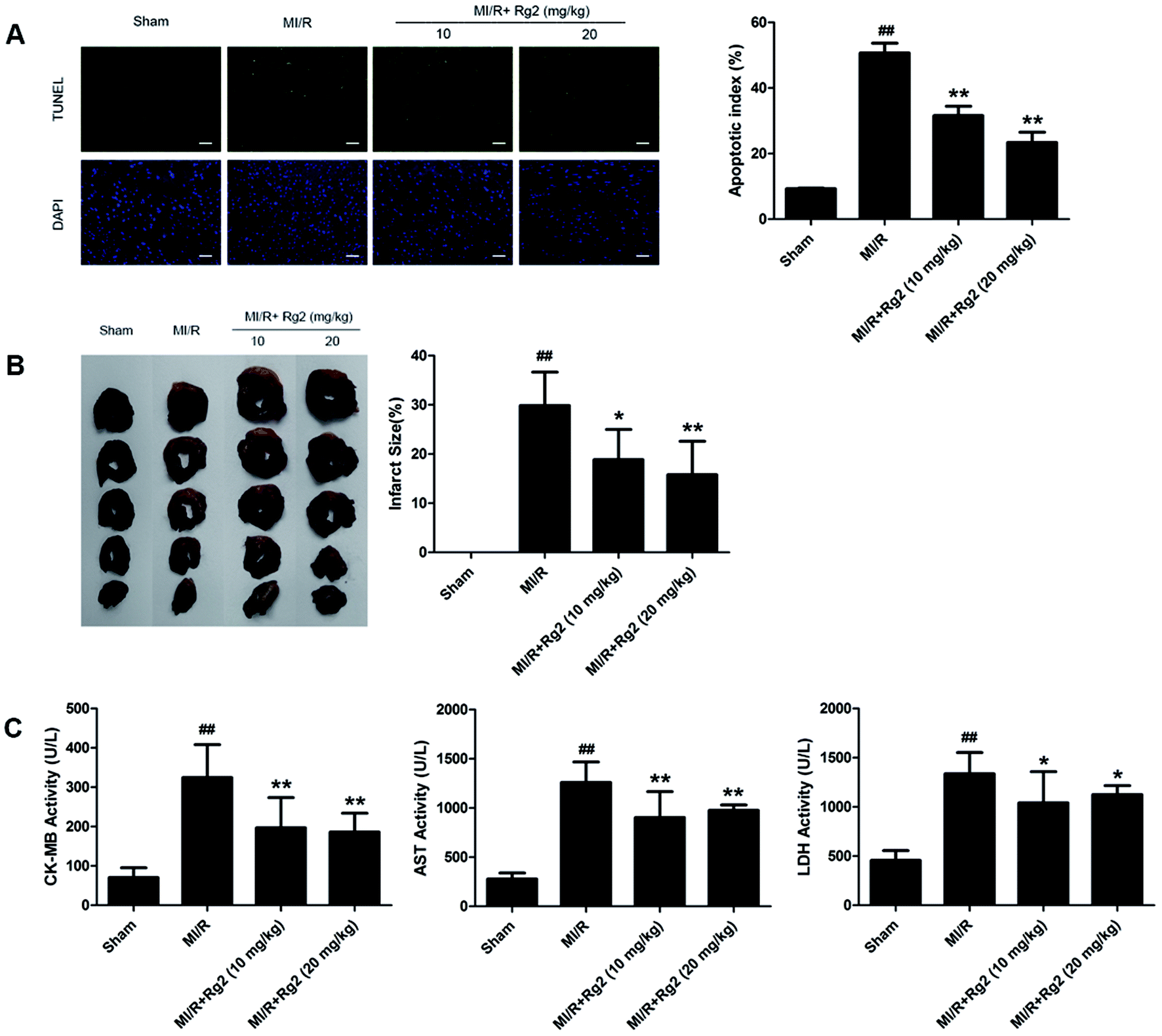 | ||
| Fig. 3 Effects of 20(S)-ginsenoside Rg2 on the apoptotic index, infarct size, and serum activities of CK-MB, AST and LDH in MI/R-induced injury. Apoptotic index, infarct size, and serum levels of creatine-MB (CK-MB), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were measured after 6 h of reperfusion. (A) Representative images of in situ detection of apoptotic cardiomyocytes by TUNEL staining. Green fluorescence shows TUNEL-positive nuclei; blue fluorescence shows nuclei of total cardiomyocytes (scale bar: 100 μm). (B) Representative photographs of heart sections. Pale color indicates an ischemic region and dark-red represents a non-ischemic region. The infarct size was calculated as a percentage of the ventricular mass: weight of ischemic zone/total weight of ventricle × 100%. The size of the myocardial infarct is expressed as a percentage of the ventricle area. (C) Serum activities of CK-MB, AST and LDH after 6 h of reperfusion are shown. Data are the mean ± S.D. ##P < 0.01 vs. sham group; *P < 0.05, **P < 0.01 vs. MI/R group. | ||
Effects of 20(S)-ginsenoside Rg2 on oxidative stress in MI/R-induced injury
Acute reperfusion enhances ROS generation. Therefore, we examined cardiac superoxide generation, expression of gp91phox protein, and oxidative parameters in heart homogenates. The MI/R group showed a significant increase in superoxide content compared with the sham group (Fig. 4A), and pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) decreased superoxide accumulation significantly (P < 0.05 or P < 0.01). Then, we measured expression of gp91phox, a major component of NADPH oxidase that is the most important superoxide-producing enzyme in the ischemic reperfused heart.24 Compared with the MI/R group, pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) decreased gp91phox expression markedly (P < 0.01) (Fig. 4B). The content of MDA (an index of lipid peroxidation) was increased significantly (Fig. 4C), whereas the activities of the antioxidant enzymes SOD, CAT and GSH-PX were decreased significantly in the MI/R group compared with the sham group (P < 0.01). Pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) could decrease MDA content, and increase the activities of SOD, CAT, and GSH-PX (P < 0.01). These results showed that pretreatment with 20(S)-ginsenoside Rg2 decreased superoxide production and reduced oxidative stress in MI/R-induced injury.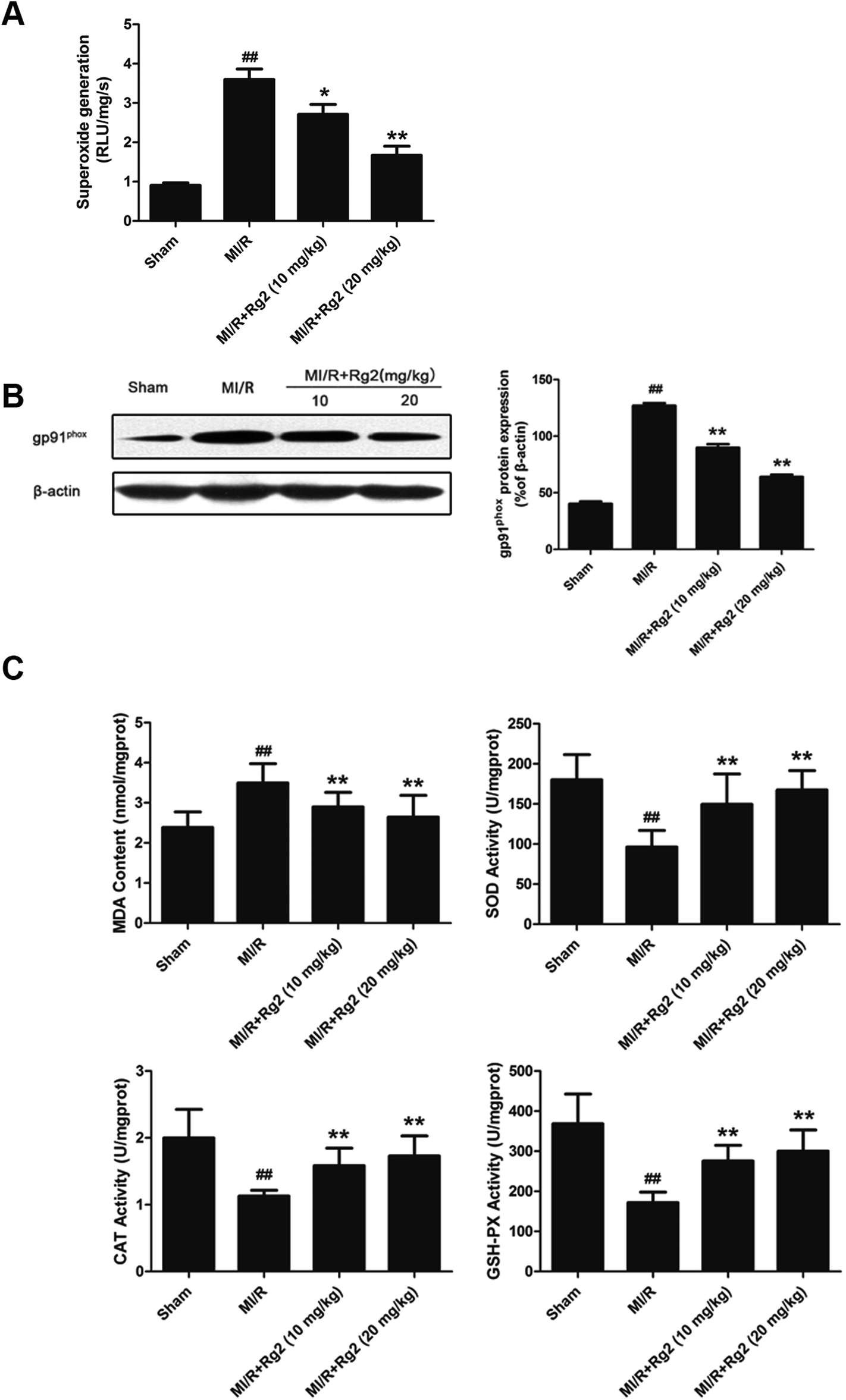 | ||
| Fig. 4 Effects of 20(S)-ginsenoside Rg2 on oxidative stress in MI/R-induced injury. The level of cardiac oxidative stress was measured after 6 h of reperfusion. (A) Cardiac superoxide generation and (B) gp91phox expression were evaluated. (C) MDA content and the activities of SOD, CAT and GSH-PX in the myocardium were measured. Data are the mean ± S.D. ##P < 0.01 vs. sham group; *P < 0.05, **P < 0.01 vs. MI/R group. | ||
Effects of 20(S)-ginsenoside Rg2 on the inflammatory response in MI/R-induced injury
Pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) play an important part in the inflammatory response to MI/R injury. After 6 h of reperfusion, all the levels of pro-inflammatory factors were increased significantly in the MI/R group compared with the sham group (P < 0.01) (Fig. 5). Compared with the MI/R group, pretreatment with 20(S)-ginsenoside Rg2 inhibited the increase in the mRNA levels and activities of IL-1β, IL-6 and TNF-α markedly (P < 0.05 or P < 0.01). These results showed that pretreatment with 20(S)-ginsenoside Rg2 reduced the inflammatory response in MI/R-induced injury.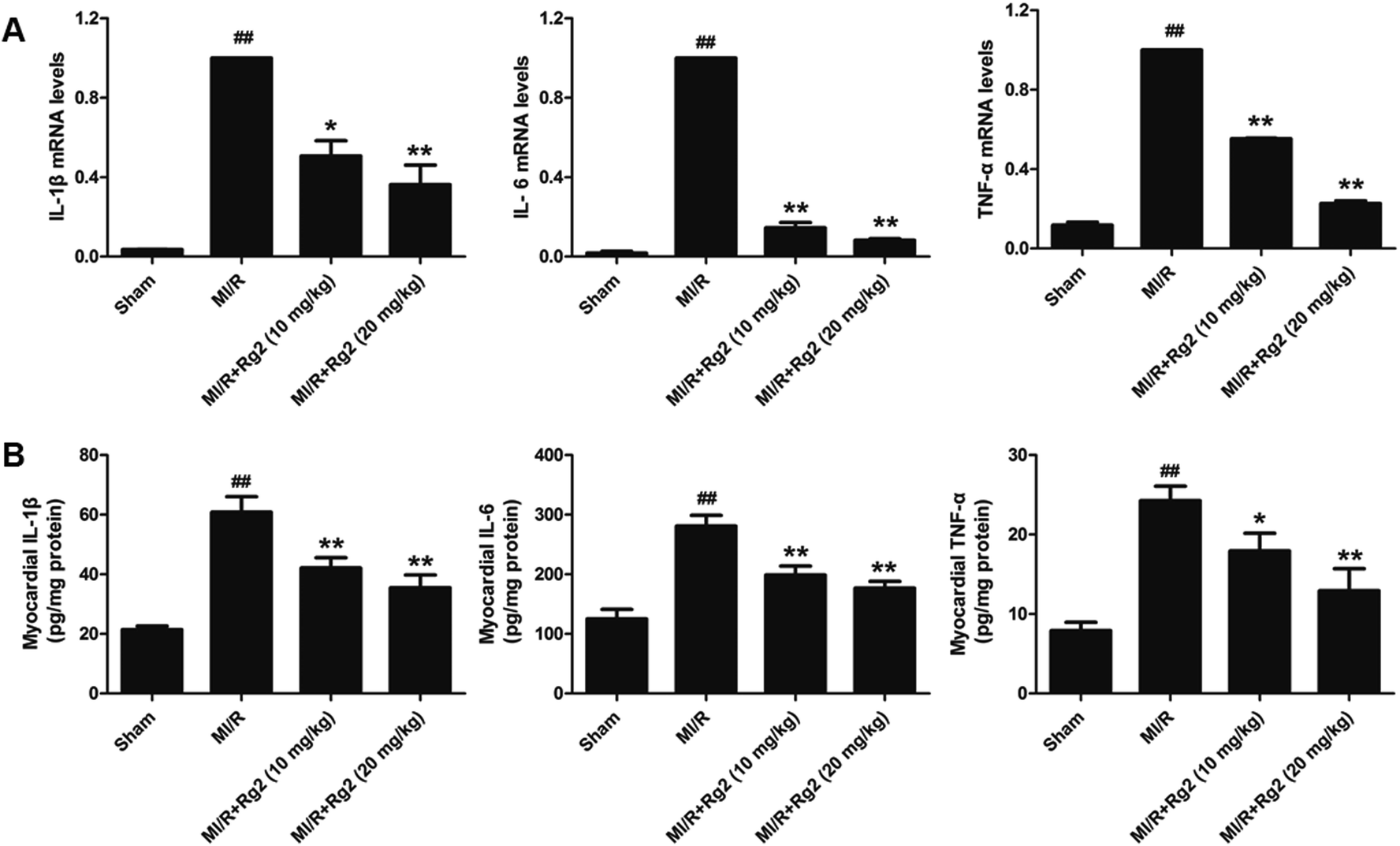 | ||
| Fig. 5 Effects of 20(S)-ginsenoside Rg2 on the inflammatory response in MI/R-induced injury. (A) mRNA levels and (B) activities of IL-1β, IL-6 and TNF-α in the myocardium were measured after 6 h of reperfusion. Data are the mean ± S.D. ##P < 0.01 vs. sham group; *P < 0.05, **P < 0.01 vs. MI/R group. | ||
Effects of 20(S)-ginsenoside Rg2 on expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bcl-2 and Bax in MI/R-induced injury
To examine the potential signaling pathways involved in the protective effects of 20(S)-ginsenoside Rg2, we measured expression of SIRT1 and Ac-p53. SIRT1 expression was decreased significantly and Ac-p53 expression was increased significantly in the MI/R group compared with the sham group (P < 0.01) (Fig. 6). However, pretreatment with 20(S)-ginsenoside Rg2 increased SIRT1 expression and decreased Ac-p53 expression compared with the MI/R group (P < 0.01). Next, we evaluated expression of apoptosis-related proteins. MI/R decreased the expression of procaspase-3, procaspase-9 and Bcl-2 but increased the expression of Bax compared with the sham group (P < 0.01). In contrast, pretreatment with 20(S)-ginsenoside Rg2 (10 and 20 mg kg−1) decreased the expression of Bax and increased the expression of procaspase-3, procaspase-9 and Bcl-2 compared with the MI/R group (P < 0.05 or P < 0.01). These results suggested that pretreatment with 20(S)-ginsenoside Rg2 can activate SIRT1 expression, thus decreasing Ac-p53 expression, and inhibit an apoptotic signaling pathway.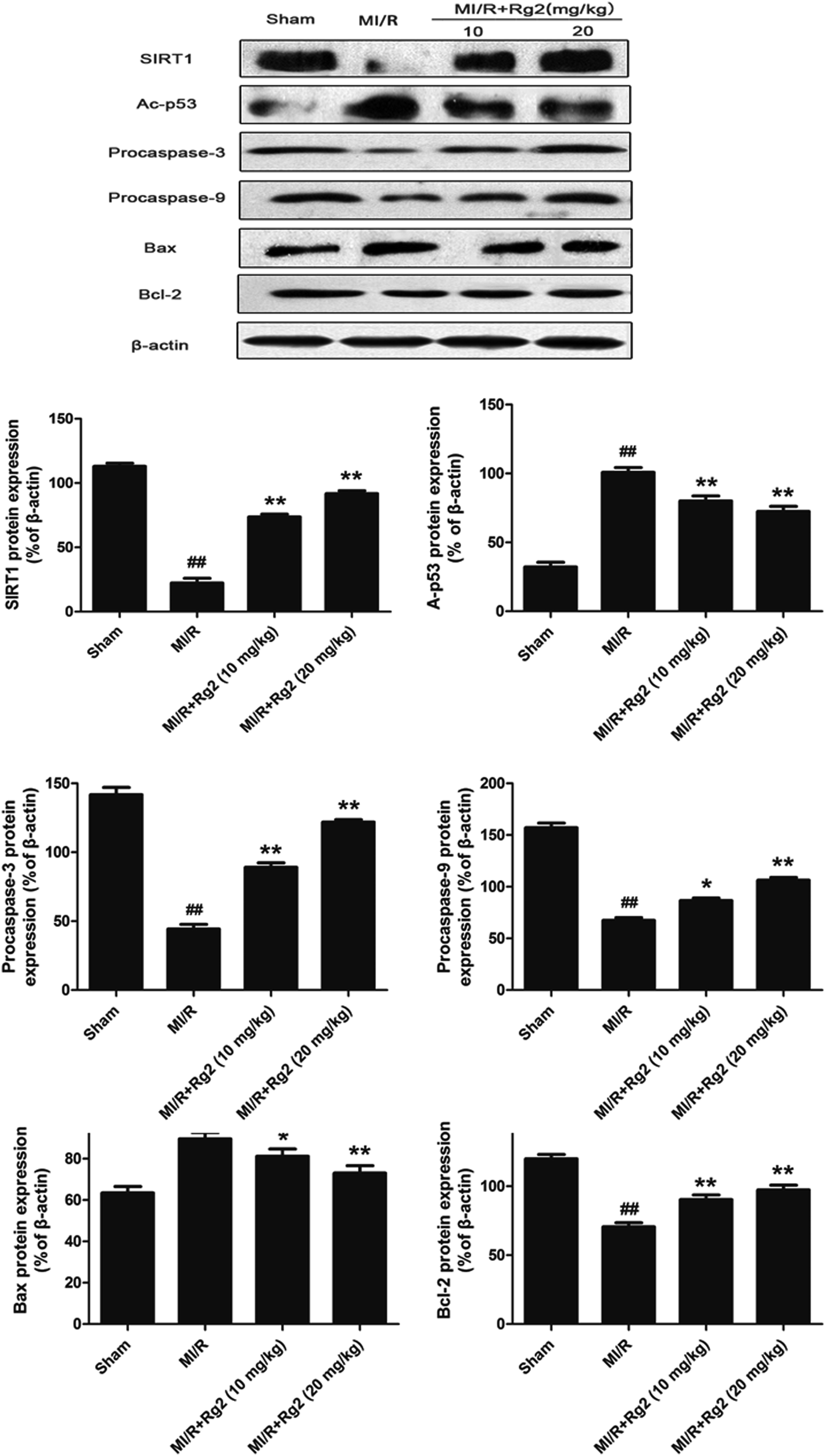 | ||
| Fig. 6 Effects of 20(S)-ginsenoside Rg2 on expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bax and Bcl-2 in MI/R-induced injury. SIRT1-related signaling and apoptosis-related protein levels were measured after 6 h of reperfusion. Representative blots of expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bax and Bcl-2 are shown. Expression of the indicated proteins are shown as the mean ± S.D. ##P < 0.01 vs. sham group; *P < 0.05, **P < 0.01 vs. MI/R group. | ||
Effects of 20(S)-ginsenoside Rg2 and EX527 on cardiac function in MI/R-induced injury
The results shown above suggested that the cardioprotective effect of 20(S)-ginsenoside Rg2 was associated with SIRT1 signaling. To investigate further the mechanisms underlying 20(S)-ginsenoside Rg2-induced cardioprotective effects, we introduced EX527, a selective SIRT1 inhibitor,32 to our study. Pretreatment with 20(S)-ginsenoside Rg2 improved cardiac function significantly, as demonstrated by increased LVEF, LVFS and decreased LVIDd and LVIDs compared with the MI/R group (P < 0.01) (Fig. 7). However, EX527 treatment abolished the protective effect of 20(S)-ginsenoside Rg2 by decreasing LVIDd, LVIDs, LVEF and LVFS compared with the MI/R + Rg2 group (P < 0.05 or P < 0.01), which suggested that the cardioprotective effect of 20(S)-ginsenoside Rg2 may involve a SIRT1-related signaling pathway.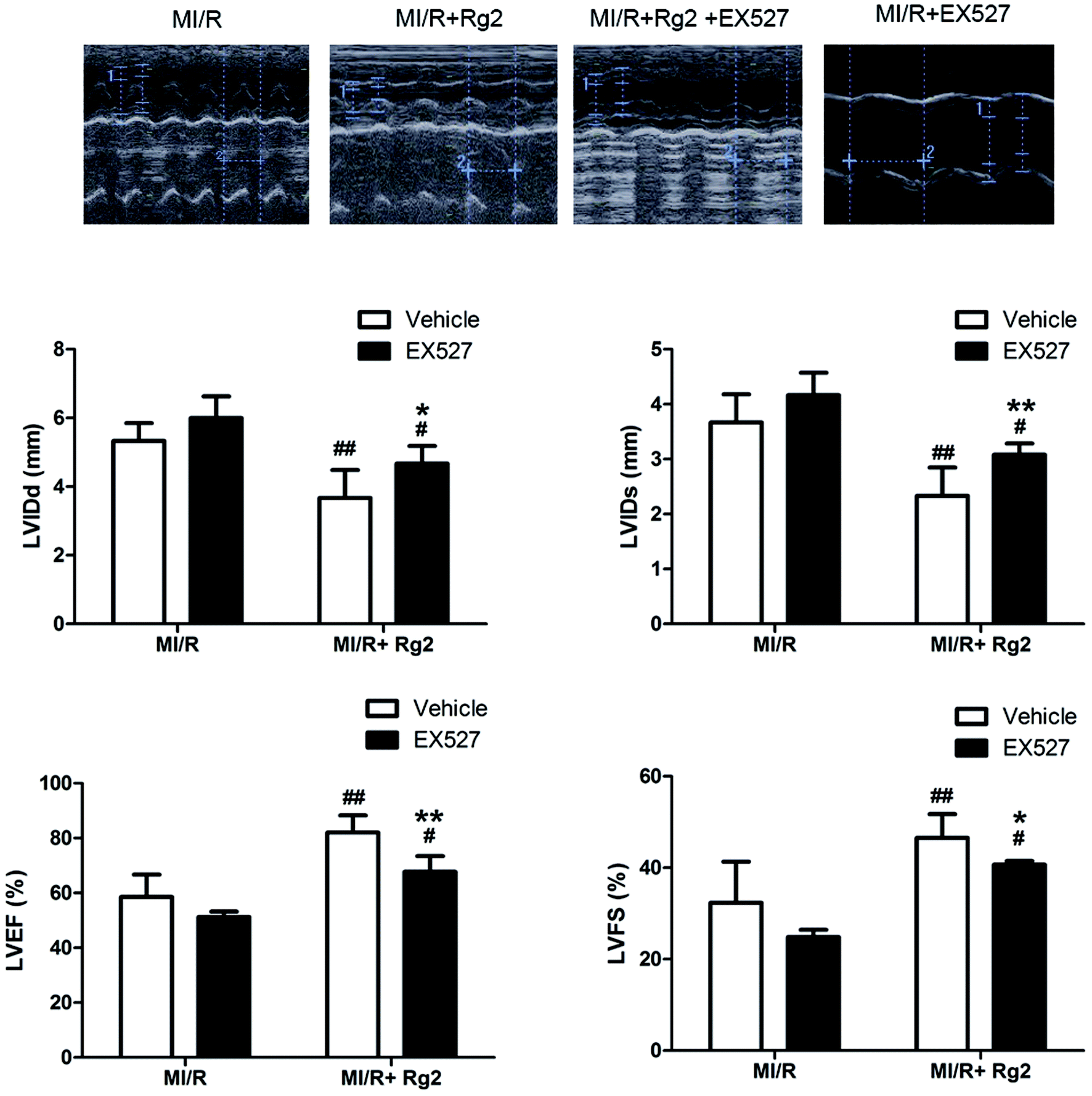 | ||
| Fig. 7 Effects of 20(S)-ginsenoside Rg2 and EX527 on cardiac function in MI/R-induced injury. Echocardiographic measurement was carried out after 72 h of reperfusion. Representative M-mode images are shown. Evaluation of LVIDd, LVIDs, LVEF and LVFS is shown. Images of the MI/R group and MI/R + Rg2 group are shared with Fig. 2. Data are the mean ± S.D. #P < 0.05, ##P < 0.01 vs. MI/R group and *P < 0.05, **P < 0.01 vs. MI/R + Rg2 group. | ||
Effects of 20(S)-ginsenoside Rg2 and EX527 on the apoptotic index, infarct size, and serum activities of CK-MB, AST and LDH in MI/R-induced injury
We determined the apoptotic index, infarct size, and activities of CK-MB, AST, and LDH in serum. Pretreatment with 20(S)-ginsenoside Rg2 could attenuate post-MI/R apoptosis and necrosis, which was demonstrated by reduction in the apoptotic index, infarct size, and activities of CK-MB, AST and LDH compared with the MI/R group (P < 0.01) (Fig. 8). However, the presence of EX527 abolished the protective effect of 20(S)-ginsenoside Rg2 by increasing the apoptotic index, infarct size, the activities of CK-MB, AST and LDH in serum compared with the MI/R + Rg2 group (P < 0.05 or P < 0.01). Our results suggested that the anti-post-MI/R effect of 20(S)-ginsenoside Rg2 on apoptosis and necrosis was associated with activation of SIRT1 signaling.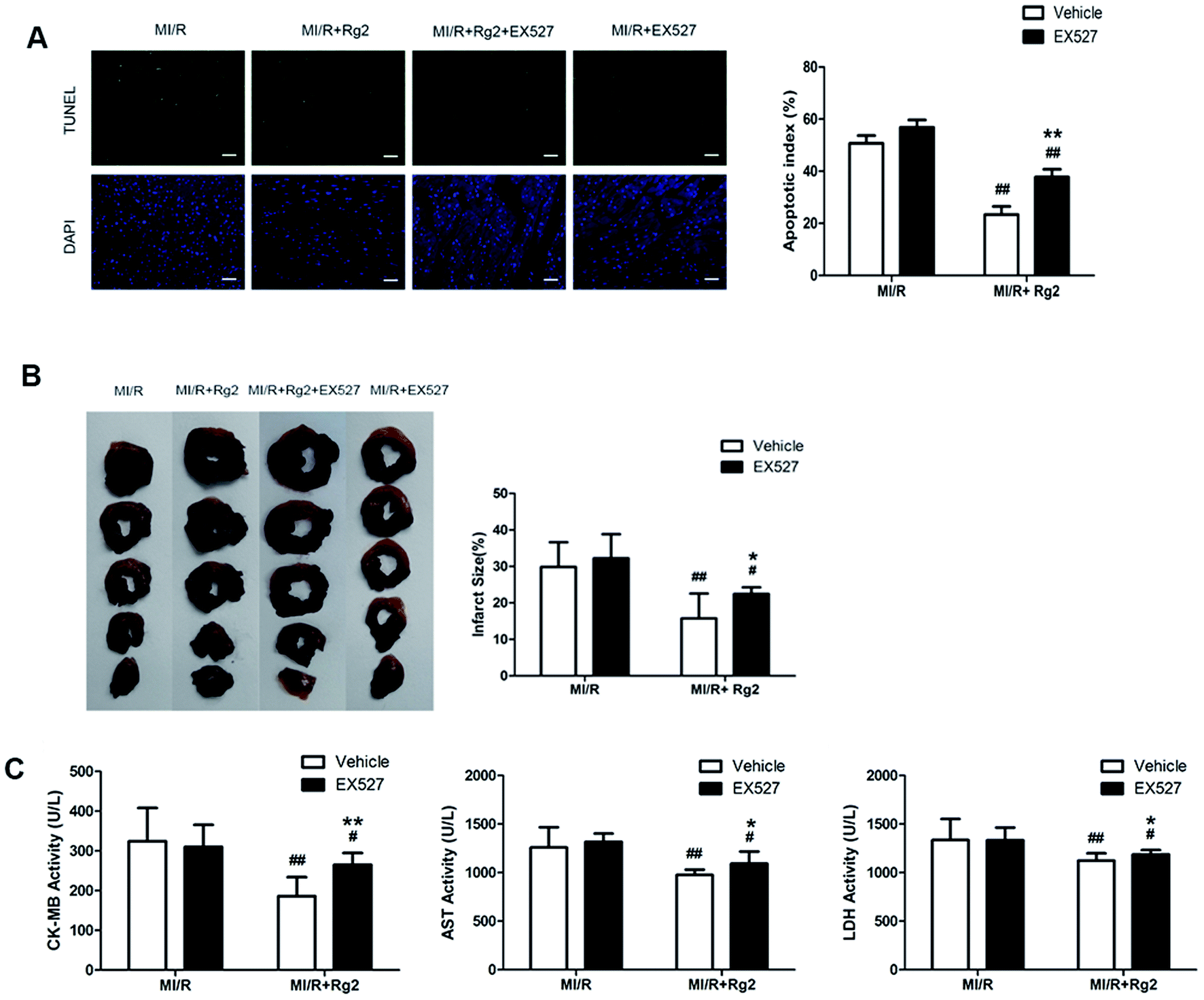 | ||
| Fig. 8 Effects of 20(S)-ginsenoside Rg2 and EX527 on the apoptotic index, infarct size, and serum activities of CK-MB, AST and LDH in MI/R-induced injury. Apoptotic index, infarct size, and serum activities of CK-MB, AST and LDH were measured after 6 h of reperfusion. (A) Representative images of in situ detection of apoptotic cardiomyocytes by TUNEL staining. Green fluorescence shows TUNEL-positive nuclei; blue fluorescence shows nuclei of total cardiomyocytes (scale bar: 100 μm). (B) The infarct size was calculated as a percentage of the ventricular mass: weight of ischemic zone/total weight of ventricle × 100%. The size of the myocardial infarct is expressed as a percentage of the ventricle area. (C) The serum activities of CK-MB, AST and LDH after 6 h reperfusion are shown. Images from the MI/R group and MI/R + Rg2 group are shared with Fig. 3. Data are the mean ± S.D. #P < 0.05 vs. MI/R group and *P < 0.05, **P < 0.01 vs. MI/R + Rg2 group. | ||
Effects of 20(S)-ginsenoside Rg2 and EX527 on oxidative stress in MI/R-induced injury
As shown above, we demonstrated that pretreatment with 20(S)-ginsenoside Rg2 decreased superoxide production and reduced oxidative stress in MI/R hearts. We studied further the effect of EX527 on 20(S)-ginsenoside Rg2 treatment. 20(S)-Ginsenoside Rg2-induced reductions in superoxide generation, gp91phox expression and MDA content were abolished upon EX527 treatment (P < 0.05 or P < 0.01) (Fig. 9). The presence of EX527 also abolished the protective effect of 20(S)-ginsenoside Rg2 by reducing the activities of SOD, CAT and GSH-PX in the myocardium compared with the MI/R + Rg2 group (P < 0.05 or P < 0.01). Together with previous data obtained by our research team,18 these results suggested that 20(S)-ginsenoside Rg2 reduced post-MI/R oxidative stress, thereby improving cardiac function via a SIRT1-related signaling pathway.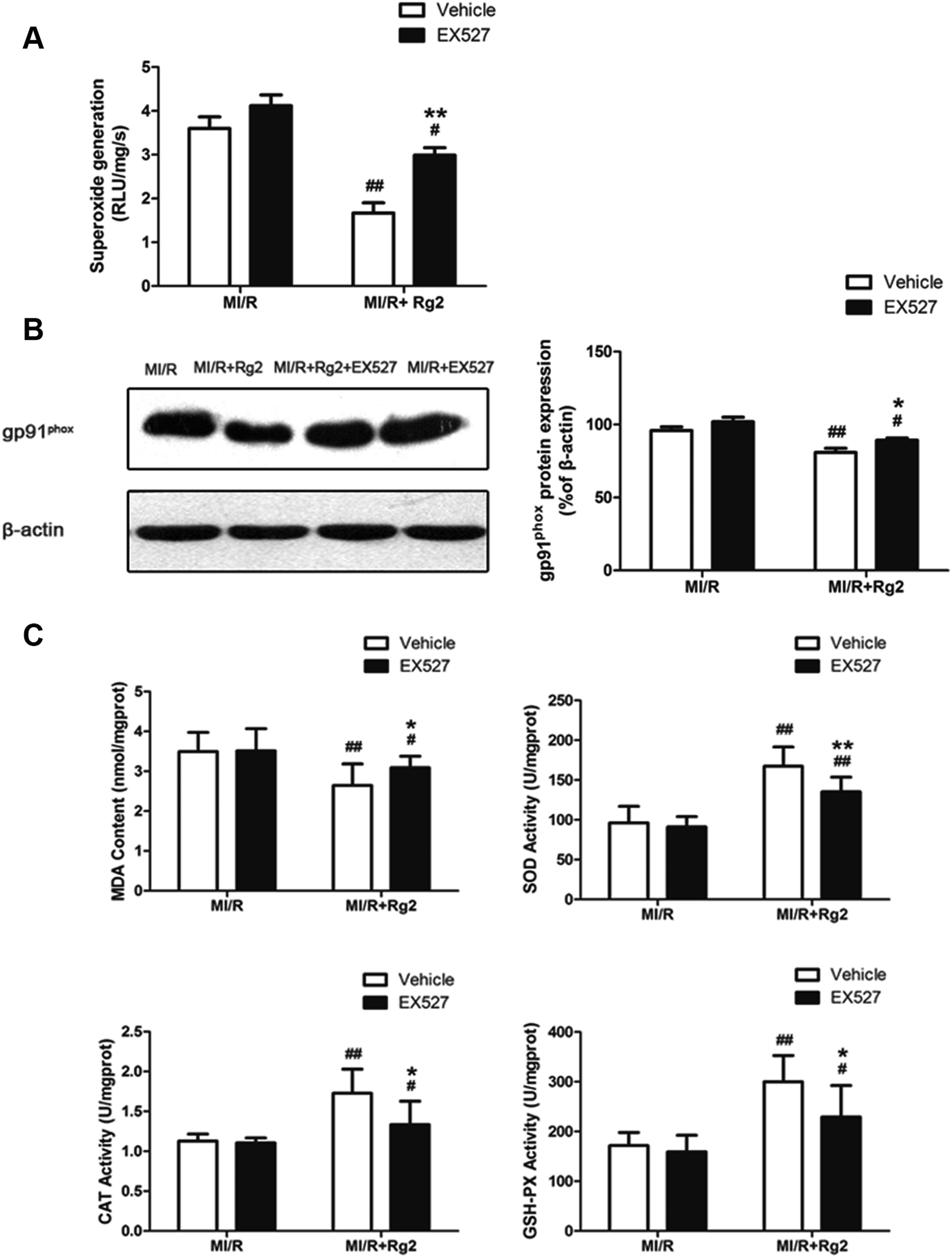 | ||
| Fig. 9 Effects of 20(S)-ginsenoside Rg2 and EX527 on oxidative stress in MI/R-induced injury. Cardiac oxidative stress level was measured after 6 h of reperfusion. (A) Cardiac superoxide generation and (B) gp91phox expression were evaluated. (C) MDA content and activities of SOD, CAT and GSH-PX in the myocardium were measured. Data are the mean ± S.D. #P < 0.05 vs. MI/R group and *P < 0.05, **P < 0.01 vs. MI/R + Rg2 group. | ||
Effects of 20(S)-ginsenoside Rg2 and EX527 on the inflammatory response in MI/R-induced injury
It has been demonstrated that MI/R induces high expression of IL-1β, IL-6 and TNF-α in the myocardium, which aggravates myocardial damage. Pretreatment with 20(S)-ginsenoside Rg2 inhibited the increase in the mRNA levels and activities of pro-inflammatory cytokines markedly compared with the MI/R group (P < 0.05 or P < 0.01) (Fig. 10). However, EX527 treatment abolished the protective effect of 20(S)-ginsenoside Rg2 by increasing the mRNA levels and activities of IL-1β, IL-6 and TNF-α compared with the MI/R + Rg2 group (P < 0.05). These results suggested that 20(S)-ginsenoside Rg2 attenuated inflammation via a SIRT1 signaling pathway.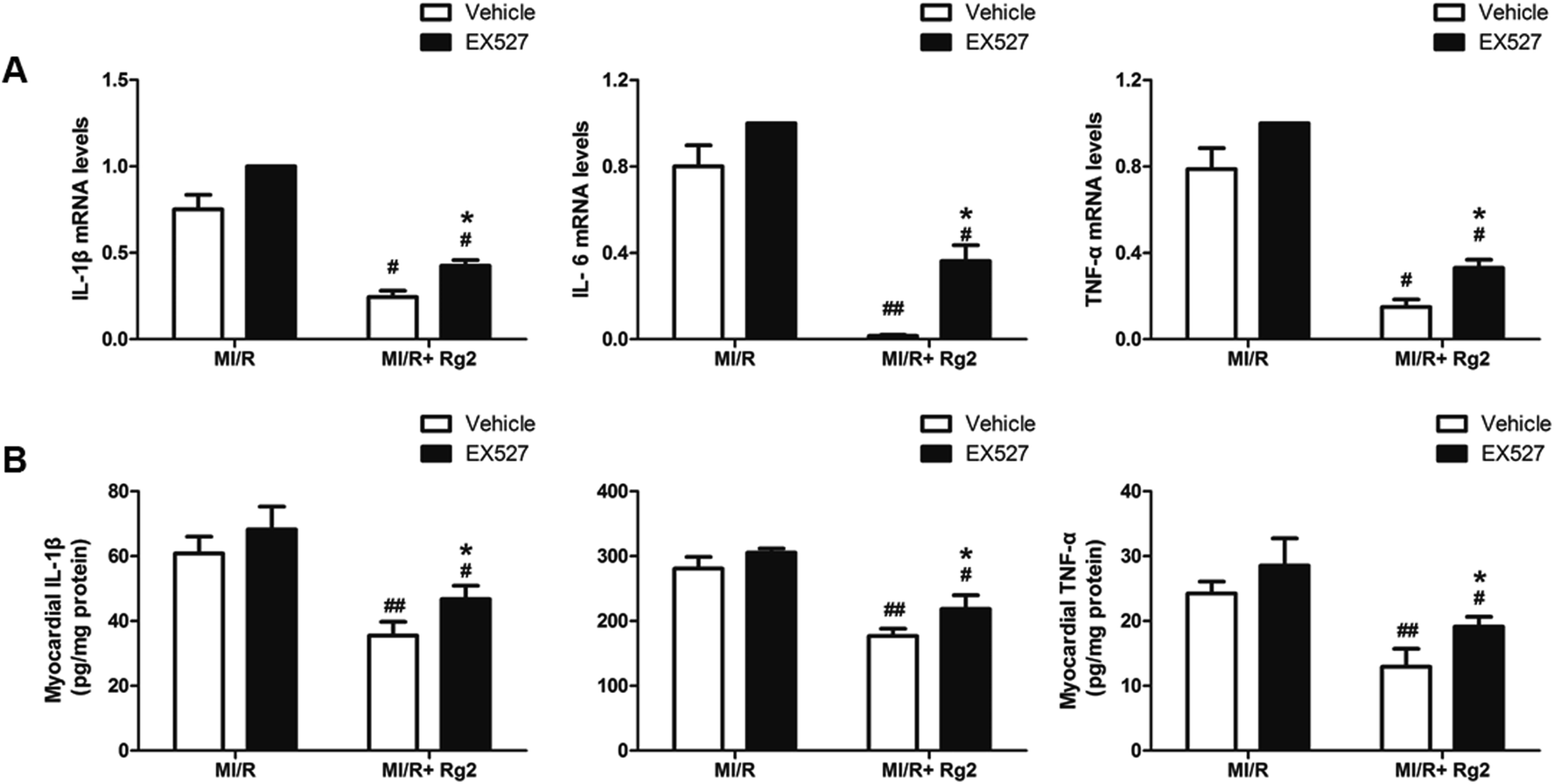 | ||
| Fig. 10 Effects of 20(S)-ginsenoside Rg2 and EX527 on the inflammatory response in MI/R-induced injury. (A) mRNA levels and (B) activities of IL-1β, IL-6 and TNF-α in the myocardium were measured after 6 h of reperfusion. Data are the mean ± S.D. #P < 0.05, ##P < 0.01 vs. MI/R group and *P < 0.05 vs. MI/R + Rg2 group. | ||
Effects of 20(S)-ginsenoside Rg2 and EX527 on expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bcl-2 and Bax in MI/R-induced injury
To establish a more distinct relationship between 20(S)-ginsenoside Rg2 and its anti-apoptotic property in MI/R injury, we investigated if EX527 administration could abolish the anti-apoptotic effect of 20(S)-ginsenoside Rg2. Pretreatment with 20(S)-ginsenoside Rg2 decreased the expression of Bax and increased the expression of procaspase-3, procaspase-9 and Bcl-2 compared with the MI/R group (P < 0.01) (Fig. 11). However, EX527 treatment abolished the protective effect of 20(S)-ginsenoside Rg2 by increasing Ac-p53 expression while decreasing SIRT1 expression compared with the MI/R + Rg2 group (P < 0.05 or P < 0.01). Consistent with previous observations, EX527 treatment enhanced apoptotic signaling by decreasing expression of procaspase-3, procaspase-9 and Bcl-2 while increasing Bax expression compared with the MI/R + Rg2 group (P < 0.05 or P < 0.01). These results suggested that 20(S)-ginsenoside Rg2 reduced apoptosis (at least in part) by upregulating SIRT1 expression.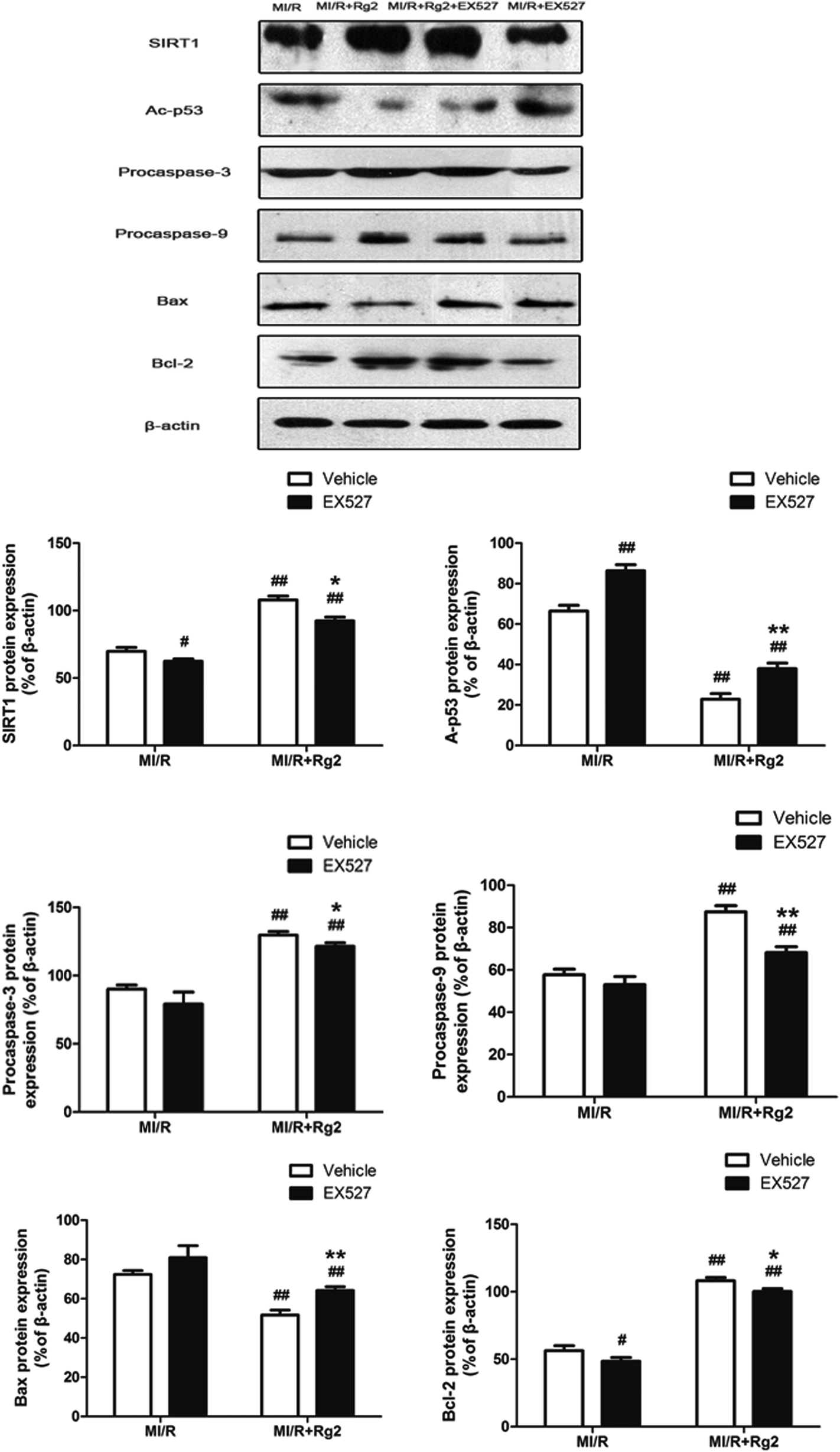 | ||
| Fig. 11 Effects of 20(S)-ginsenoside Rg2 and EX527 on expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bax and Bcl-2 in MI/R-induced injury. SIRT1-related signaling and apoptosis-related proteins were measured after 6 h of reperfusion. Representative blots of expression of SIRT1, Ac-p53, procaspase-3, procaspase-9, Bax, and Bcl-2 and their evaluation are shown. Data are the mean ± S.D. #P < 0.05, ##P < 0.01 vs. MI/R group and *P < 0.05, **P < 0.01 vs. MI/R + Rg2 group. | ||
Discussion
Here, we demonstrated that: (i) 20(S)-ginsenoside Rg2 protected against MI/R-induced injury by reducing oxidative stress and inflammation; (ii) the cardioprotective effect of 20(S)-ginsenoside Rg2 may (at least in part) be mediated by SIRT1 signaling. This is the first report demonstrating that 20(S)-ginsenoside Rg2 protects against MI/R-induced injury by anti-oxidative and anti-inflammatory actions, and that these effects are associated with SIRT1 signaling.20(S)-Ginsenoside Rg2 is a major active component in the roots and stem leaves of ginseng. 20(S)-Ginsenoside Rg2 has a wide range of pharmacologic effects.17,39–42 Our previous study demonstrated that 20(S)-ginsenoside Rg2 can protect cardiomyocytes from H2O2-induced injury by inhibiting ROS production, increasing intracellular levels of antioxidants, and attenuating apoptosis.18 In the present study, we also found that pretreatment with 20(S)-ginsenoside Rg2 conferred cardioprotective effects, as evidenced by improved post-MI/R cardiac function, attenuated oxidative stress and inflammation, as well as reduced myocardial apoptosis and necrosis.
We demonstrated that pretreatment with 20(S)-ginsenoside Rg2 exerted antioxidative effects by decreasing cardiac superoxide generation, gp91phox expression, MDA content, as well as increasing the activities of SOD, CAT and GSH-PX. The mechanisms involved may be attributed to the scavenging of free radicals, increasing the concentration of antioxidants, and inhibiting cardiomyocyte apoptosis. In addition, pretreatment with 20(S)-ginsenoside Rg2 exerted anti-inflammatory effects by decreasing the levels of IL-1β, IL-6 and TNF-α. The mechanisms involved may be cell damage caused by the release of oxygen free radicals and cytotoxic substances. These released inflammatory mediators can damage vascular endothelial cells, increase vascular permeability, and further activate inflammatory cells, which increases the inflammatory response.43 Inhibition of myocardial oxidative stress and inflammation is very important for developing cardioprotective strategies against MI/R injury.44
SIRT1 is a member of the class-III group of histone deacetylase, the activity of which is dependent on the intracellular ratio of NAD+/NADH. In MI/R injury, the deacetylase activity of SIRT1 has a key role in cardioprotection.22,45–47 It has been demonstrated that SIRT1 expression is reduced after MI/R-induced injury, and that activation of SIRT1 signaling with curcumin, melatonin or berberine confers protection against MI/R-induced injury by suppressing apoptosis and oxidative stress.22–24,48 The present study is in accordance with our previous study in vitro (data not shown) demonstrating that 20(S)-ginsenoside Rg2 can increase SIRT1 activity. It is also in accordance with a study showing that 20(S)-ginsenoside Rg2 and some of its metabolites produced by liver microsomes in rats can activate SIRT1.31 However, whether the protective effect of 20(S)-ginsenoside Rg2 is mediated by SIRT1 requires further investigation, so we employed a SIRT1 inhibitor. EX527 is a highly specific inhibitor of SIRT1, and it was used to suppress the change wrought by a potential activator of SIRT1: 20(S)-ginsenoside Rg2. If EX527 could reverse the protective effects of 20(S)-ginsenoside Rg2, SIRT1 would be the curtailing protein that mediates the process. By employing EX527, the improvement in cardiac function provided by 20(S)-ginsenoside Rg2 was abolished. EX527 also abrogated the protective effect of 20(S)-ginsenoside Rg2 by increasing the apoptotic index, infarct size, as well as serum activities of CK-MB, AST and LDH, which aggravated apoptosis and necrosis.
Oxidative stress is a major contributor to primary and secondary MI/R injury.49 SIRT1 activity is dependent upon the intracellular ratio of NAD+/NADH, which triggers it to respond to the high level of oxidants.50 The transcription regulation and deacetylation observed upon SIRT1 administration can alleviate NADPH/NADH oxidase-derived ROS generation, ischemia and reperfusion.50,51 We showed that EX527 increased superoxide generation, gp91phox expression and MDA content, but also decreased the activities of the antioxidant enzymes SOD, CAT and GSH-PX compared with the MI/R + Rg2 group. These results suggest that 20(S)-ginsenoside Rg2 reduces post-MI/R oxidative stress, thereby improving cardiac function via a SIRT1-mediated network.
MI/R injury is also probably related to myocardial inflammatory response due to the released pro-inflammatory mediators. The latter cause damage to vascular endothelial cells, increased vascular permeability, and activate immune cells which aggravate the inflammatory response.52 Studies have shown that SIRT1 activation reduces inflammatory damage.53,54 Here, we illustrated that pretreatment with 20(S)-ginsenoside Rg2 reduced post-MI/R inflammation by decreasing the mRNA levels and activities of pro-inflammatory cytokines. Furthermore, EX527 abolished the effects of 20(S)-ginsenoside Rg2 on inflammation. Taken together with our previous study, SIRT1 is a curtailing mediator of 20(S)-ginsenoside Rg2 that inhibits expression of pro-inflammatory cytokines.
SIRT1 regulates p53 acetylation, thereby inhibiting apoptosis of cardiomyocytes.19 Here, we found that 20(S)-ginsenoside Rg2 induced a significant increase in SIRT1 expression and p53 deacetylation in the MI/R myocardium. Furthermore, 20(S)-ginsenoside Rg2 increased expression of the anti-apoptotic factors Bcl-2, procaspase-3 and procaspase-9, and decreased expression of the pro-apoptotic factor Bax in MI/R-induced injury. However, the protective effects of 20(S)-ginsenoside Rg2 were abolished by EX527. Therefore, 20(S)-ginsenoside Rg2 confers a protective effect against MI/R injury via SIRT1 by alleviating oxidative stress and reducing myocardial inflammation (Fig. 12).
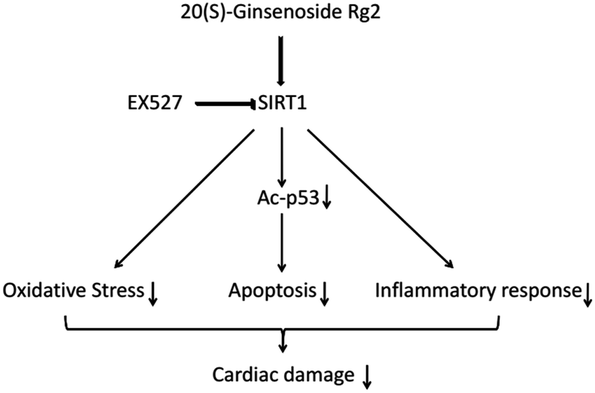 | ||
| Fig. 12 Proposed cardioprotective signaling pathway of 20(S)-ginsenoside Rg2. | ||
Conclusions
20(S)-Ginsenoside Rg2 exerted a profound cardioprotective effect against MI/R injury. This protection appeared to be due largely to activation of SIRT1 signaling and attenuation of oxidative stress and inflammation. Hence, 20(S)-ginsenoside Rg2 may be a promising agent for the treatment of MI/R injury in cardiac surgery and ischemic heart diseases.Conflicts of interest
The authors declare no conflict of interests.Funding
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (81473378) and Natural Science Foundation of Jilin Province (20170101002JC).Acknowledgements
The authors thank Dr Yifa Zhou for providing 20(S)-ginsenoside Rg2 and Dr Tian Wang for his critical reading and language editing of this manuscript.References
- A. Sekikawa, B. Y. Horiuchi, D. Edmundowicz, H. Ueshima, J. D. Curb, K. Sutton-Tyrrell, T. Okamura, T. Kadowaki, A. Kashiwagi, K. Mitsunami, K. Murata, Y. Nakamura, B. L. Rodriguez and L. H. Kuller, Heart, 2003, 89, 255–257 CrossRef PubMed.
- S. Sanada, I. Komuro and M. Kitakaze, Am. J. Physiol.: Heart Circ. Physiol., 2011, 301, H1723–H1741 CrossRef PubMed.
- K. Matsumura, R. W. Jeremy, J. Schaper and L. C. Becker, Circulation, 1998, 97, 795–804 CrossRef PubMed.
- J. L. Zweier and M. A. Talukder, Cardiovasc. Res., 2006, 70, 181–190 CrossRef PubMed.
- Z. Q. Zhao, Curr. Opin. Pharmacol., 2004, 4, 159–165 CrossRef PubMed.
- R. A. Gottlieb, J. Cardiovasc. Pharmacol. Ther., 2011, 16, 233–238 CrossRef PubMed.
- G. Petrosillo, F. M. Ruggiero, N. Di Venosa and G. Paradies, FASEB J., 2003, 17, 714–716 CrossRef PubMed.
- D. B. Zorov, C. R. Filburn, L. O. Klotz, J. L. Zweier and S. J. Sollott, J. Exp. Med., 2000, 192, 1001–1014 CrossRef PubMed.
- C. Maack and B. O'Rourke, Basic Res. Cardiol., 2007, 102, 369–392 CrossRef PubMed.
- H. Tsutsui, T. Ide and S. Kinugawa, Antioxid. Redox Signaling, 2006, 8, 1737–1744 CrossRef PubMed.
- K. W. Leung and A. S. Wong, Chin. Med., 2010, 5, 20 CrossRef PubMed.
- S. Chae, K. A. Kang, W. Y. Chang, M. J. Kim, S. J. Lee, Y. S. Lee, H. S. Kim, D. H. Kim and J. W. Hyun, J. Agric. Food Chem., 2009, 57, 5777–5782 CrossRef PubMed.
- W. C. Cho, W. S. Chung, S. K. Lee, A. W. Leung, C. H. Cheng and K. K. Yue, Eur. J. Pharmacol., 2006, 550, 173–179 CrossRef PubMed.
- W. Wang, E. R. Rayburn, M. Hao, Y. Zhao, D. L. Hill, R. Zhang and H. Wang, Prostate, 2008, 68, 809–819 CrossRef PubMed.
- C. Mancuso and R. Santangelo, Food Chem. Toxicol., 2017, 107, 362–372 CrossRef PubMed.
- X. W. Yang, L. Y. Ma, Q. L. Zhou, W. Xu and Y. B. Zhang, Bioorg. Med. Chem. Lett., 2017, 28, 240–243 CrossRef PubMed.
- Y. Jiang, W. Liu, X. M. Wang, G. G. Zhong, W. J. Zhang, L. Chen, S. Zhan, H. Qi, C. Y. Zhao, X. Y. Ma, S. J. Yang and H. Li, Zhongguo Yaoli Xuebao, 1996, 17, 138–141 Search PubMed.
- W. Fu, D. Sui, X. Yu, D. Gou, Y. Zhou and H. Xu, Int. J. Clin. Exp. Med., 2015, 8, 19938–19947 Search PubMed.
- R. R. Alcendor, L. A. Kirshenbaum, S. Imai, S. F. Vatner and J. Sadoshima, Circ. Res., 2004, 95, 971–980 CrossRef PubMed.
- C. P. Hsu, P. Y. Zhai, T. Yamamoto, Y. Maejima, S. Matsushima, N. Hariharan, D. Shao, H. Takagi, S. Oka and J. Sadoshima, Circulation, 2010, 122, 2170–U2193 CrossRef PubMed.
- C. Tong, A. Morrison, S. Mattison, S. Qian, M. Bryniarski, B. Rankin, J. Wang, D. P. Thomas and J. Li, FASEB J., 2013, 27, 4332–4342 CrossRef PubMed.
- Y. Yang, W. Duan, Y. Lin, W. Yi, Z. Liang, J. Yan, N. Wang, C. Deng, S. Zhang, Y. Li, W. Chen, S. Yu, D. Yi and Z. Jin, Free Radical Biol. Med., 2013, 65, 667–679 CrossRef PubMed.
- L. Yu, Q. Li, B. Yu, Y. Yang, Z. Jin, W. Duan, G. Zhao, M. Zhai, L. Liu, D. Yi, M. Chen and S. Yu, Oxid. Med. Cell. Longevity, 2016, 2016, 1689602 Search PubMed.
- L. Yu, Y. Sun, L. Cheng, Z. Jin, Y. Yang, M. Zhai, H. Pei, X. Wang, H. Zhang, Q. Meng, Y. Zhang, S. Yu and W. Duan, J. Pineal Res., 2014, 57, 228–238 CrossRef PubMed.
- D. H. Kim, C. H. Park, D. Park, Y. J. Choi, M. H. Park, K. W. Chung, S. R. Kim, J. S. Lee and H. Y. Chung, Arch. Pharmacal Res., 2014, 37, 813–820 CrossRef PubMed.
- Z. Song, Y. Liu, B. Hao, S. Yu, H. Zhang, D. Liu, B. Zhou, L. Wu, M. Wang, Z. Xiong, C. Wu, J. Zhu and X. Qian, PLoS One, 2014, 9, e112699 CrossRef PubMed.
- Y. Y. Lee, J. S. Park, E. J. Lee, S. Y. Lee, D. H. Kim, J. L. Kang and H. S. Kim, J. Agric. Food Chem., 2015, 63, 3472–3480 CrossRef PubMed.
- Y. Wang, Y. Chen, H. Wang, Y. Cheng and X. Zhao, Anal. Chem., 2015, 87, 5046–5049 CrossRef PubMed.
- J. L. Yang, T. K. Q. Ha, B. Dhodary, K. H. Kim, J. Park, C. H. Lee, Y. C. Kim and W. K. Oh, J. Nat. Prod., 2014, 77, 1615–1623 CrossRef PubMed.
- J. Y. Han, S. Lee, J. H. Yang, S. Kim, J. Sim, M. G. Kim, T. C. Jeong, S. K. Ku, I. J. Cho and S. H. Ki, J. Ginseng Res., 2015, 39, 105–115 CrossRef PubMed.
- L. Y. Ma, Q. L. Zhou, X. B. Yang, H. P. Wang and X. W. Yang, Molecules, 2016, 21, 757 CrossRef PubMed.
- J. M. Solomon, R. Pasupuleti, L. Xu, T. McDonagh, R. Curtis, P. S. DiStefano and L. J. Huber, Mol. Cell. Biol., 2006, 26, 28–38 CrossRef PubMed.
- Y. H. Jia, Z. Zheng, Y. C. Wang, Q. Zhou, W. X. Cai, W. B. Jia, L. L. Yang, M. L. Dong, X. X. Zhu, L. L. Su and D. H. Hu, PLoS One, 2015, 10, e0120849 CrossRef PubMed.
- X. Ji, B. K. Tan, Y. C. Zhu, W. Linz and Y. Z. Zhu, Life Sci., 2003, 73, 1413–1426 CrossRef PubMed.
- S. H. Lim and J. Lee, Prev. Nutr. Food Sci., 2012, 17, 177–183 CrossRef PubMed.
- H. Su, L. Ji, W. Xing, W. Zhang, H. Zhou, X. Qian, X. Wang, F. Gao, X. Sun and H. Zhang, J. Cell. Mol. Med., 2013, 17, 181–191 CrossRef PubMed.
- J. Lin, H. Wang, J. Li, Q. Wang, S. Zhang, N. Feng, R. Fan and J. Pei, Cytokine, 2013, 61, 842–848 CrossRef PubMed.
- X. Wu, B. Zhang, R. Fan, L. Zhao, Y. Wang, S. Zhang, A. D. Kaye, L. Huang and J. Pei, Cytokine, 2011, 56, 503–507 CrossRef PubMed.
- S. Choi, S. Y. Jung, J. H. Lee, F. Sala, M. Criado, J. Mulet, L. M. Valor, S. Sala, A. G. Engel and S. Y. Nah, Eur. J. Pharmacol., 2002, 442, 37–45 CrossRef PubMed.
- K. Kudo, E. Tachikawa, T. Kashimoto and E. Takahashi, Eur. J. Pharmacol., 1998, 341, 139–144 CrossRef PubMed.
- F. Sala, J. Mulet, S. Choi, S. Y. Jung, S. Y. Nah, H. Rhim, L. M. Valor, M. Criado and S. Sala, J. Pharmacol. Exp. Ther., 2002, 301, 1052–1059 CrossRef PubMed.
- E. Tachikawa, K. Kudo, T. Kashimoto and E. Takahashi, J. Pharmacol. Exp. Ther., 1995, 273, 629–636 Search PubMed.
- X.-L. M. A. M. Lefer, A. Weyrich and D. J. Lefer, Agents Actions Suppl., 1993, 41, 127–135 Search PubMed.
- Y. Tian, H. Li, P. Liu, J. M. Xu, M. G. Irwin, Z. Xia and G. Tian, Mediators Inflammation, 2015, 2015, 819232 Search PubMed.
- C. J. Chen, W. Yu, Y. C. Fu, X. Wang, J. L. Li and W. Wang, Biochem. Biophys. Res. Commun., 2009, 378, 389–393 CrossRef PubMed.
- S. Chung, H. Yao, S. Caito, J. W. Hwang, G. Arunachalam and I. Rahman, Arch. Biochem. Biophys., 2010, 501, 79–90 CrossRef PubMed.
- C. P. Hsu, P. Zhai, T. Yamamoto, Y. Maejima, S. Matsushima, N. Hariharan, D. Shao, H. Takagi, S. Oka and J. Sadoshima, Circulation, 2010, 122, 2170–2182 CrossRef PubMed.
- L. Yu, F. Li, G. Zhao, Y. Yang, Z. Jin, M. Zhai, W. Yu, L. Zhao, W. Chen, W. Duan and S. Yu, Apoptosis, 2015, 20, 796–810 CrossRef PubMed.
- H. Li, Y. Bian, N. Zhang, J. Guo, C. Wang, W. B. Lau and C. Xiao, Cardiovasc. Diabetol., 2013, 12, 91 CrossRef PubMed.
- S. Y. Shin, T. H. Kim, H. Wu, Y. H. Choi and S. G. Kim, Eur. J. Pharmacol., 2014, 727, 115–124 CrossRef PubMed.
- T. Zhang, J. G. Berrocal, K. M. Frizzell, M. J. Gamble, M. E. DuMond, R. Krishnakumar, T. L. Yang, A. A. Sauve and W. L. Kraus, J. Biol. Chem., 2009, 284, 20408–20417 CrossRef PubMed.
- A. M. Lefer, X. L. Ma, A. Weyrich and D. J. Lefer, Agents Actions Suppl., 1993, 41, 127–135 Search PubMed.
- S. Winnik, J. Auwerx, D. A. Sinclair and C. M. Matter, Eur. Heart J., 2015, 36, 3404–3412 CrossRef PubMed.
- Y. Zhang, X. X. Liu, Z. G. She, D. S. Jiang, N. A. Wan, H. Xia, X. H. Zhu, X. Wei, X. D. Zhang and H. L. Li, Basic Res. Cardiol., 2014, 109, 434 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |