
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo decades of advances in diterpenoid alkaloids with cytotoxicity activities
Xiaoxia Liang†
 *,
Yingying Gao† and
Shangxian Luan
*,
Yingying Gao† and
Shangxian Luan
Natural Medicine Research Center, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu 611130, P. R. China. E-mail: liangxiaoxial@sicau.edu.cn; Tel: +86-028-8629-1162
First published on 2nd July 2018
Abstract
The important pharmacological activities and structural complexity of diterpenoid alkaloids have long stimulated strong scientific interest; some of these naturally abundant compounds have been reported to be highly promising for treating cancer. From 2008 to 2018, the cytotoxicity activities of more than 250 diterpenoid alkaloids were tested against several cancer cell lines. This review focuses on the progress of diterpenoid alkaloids with different structures derived from Ranunculaceae plants and some of their derivatives with potential anticancer activities. Then, we discuss the application prospects and development of active diterpenoid alkaloids.
1. Introduction
Throughout medical history, the majority of drugs used in cancer chemotherapy have been categorized as plant alkaloids, alkylating agents, antimetabolites, antibiotics, topoisomerase inhibitors, monoclonal antibodies, and other antitumor agents.1–9 Due to their important pharmacological activities and structural complexity, the phytochemistry, synthesis, and medicinal chemistry of diterpenoid alkaloids have stimulated strong scientific interest for a long time; however, little information on their cytotoxic properties has been reported.Recently, numerous diterpenoid alkaloids have been isolated from various species of Aconitum, Consolida, and Delphinium (Ranunculaceae); they are classified according to their chemical structures as C18-, C19-, or C20-diterpenoid alkaloids (Fig. 1).10,11 Their therapeutic potentials are being extensively studied: multiple data on their analgesic and anti-inflammatory activities, their effects on the central nervous system, and their arrhythmogenic, antiarrhythmic, antiparasite and anticancer properties are being continuously reported. These compounds still represent an active area of research. Accordingly, a number of reviews on various aspects of diterpenoid alkaloids have been published in the past decades, such as Pelletier's review in 1999,12 Wang and Liang's contributions in 2002 and 2009,13,14 and Wang's latest work in 2010.10,11 Those reviews mostly cover the classification, distribution and occurrence, biosynthesis, spectroscopy, chemical reactions and stereochemistry, and pharmacological activity of diterpenoid alkaloids.
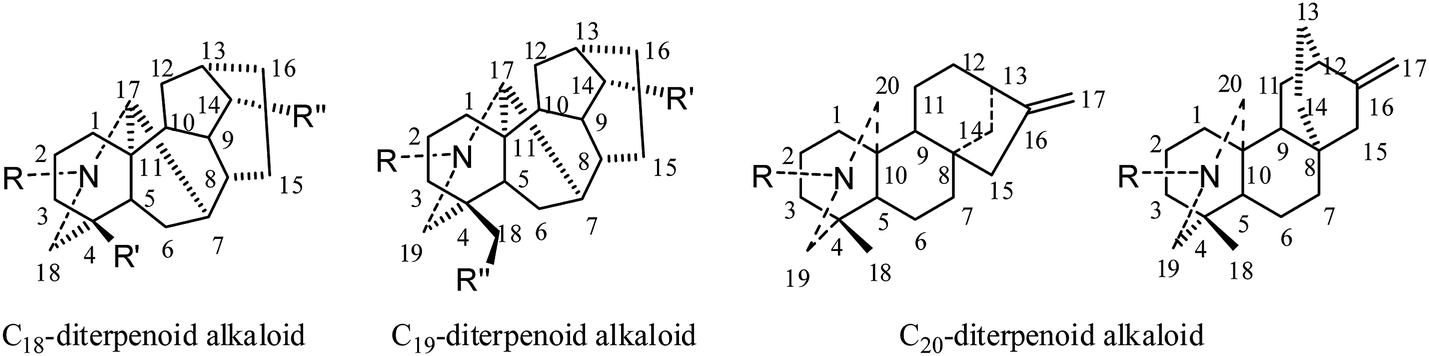 | ||
| Fig. 1 General structures and numbering systems for C18-, C19-, and C20-diterpenoid alkaloids. | ||
However, fewer reviews have focused on the cytotoxicity of these compounds. In this paper, we will review advances in the cytotoxicity activities of diterpenoid alkaloids in the past two decades (from 2008 to 2018) in order to determine promising areas for therapeutic applications.
The first reported antiproliferative activity of 8-O-azeloyl-14-benzoylaconine (1) (Fig. 2), an aconitine-type C19-diterpenoid alkaloid (IC50 values against HCT-15, A549 and MCF-7 ranging from 10 to 20 μM), was published in 2005.15 In the same year, a second report on a C18-derivative, lappaconitine hydrobromide (2), showed efficient antitumor effects in mice, with inhibition rates ranging from 11.20% to 53.08% for liver tumor growth and from 29.81% to 53.96% for S180 tumor growth.16 A third report in 2006 described the cytotoxic effects of various C19-diterpenoid alkaloids against tumor cell lines,17 among which neoline (3), 8-O-methylcolumbianine (4), 1,14-diacetylcardiopetaline (5), 18-O-demethylpubescenine (6), 14-deacetylpubescenine (7), pubescenine (8), 14-deacetylajadine (9), lycoctonine (10), browniine (11), delphatine (12), dehydrotakaosamine (13), and ajadelphinine (14) (Fig. 2) exhibited selective cytotoxicity to cancerous versus non-cancerous cells. In an additional report by Kashiwakura et al. in 2007, the cytotoxicities of ten C19-diterpenoid alkaloids and thirty-three C20-diterpenoid alkaloids against A172 (human malignant glioma cells) were examined; three C20-diterpenoid alkaloids (15a–c) were found to be the most potent cytotoxic agents. The other compounds had very weak activities.18 Since 2007, the cytotoxicities of many C19- and C20-diterpenoid alkaloids as well as their semisynthetic derivatives against different human tumor cell lines have been evaluated by various assays, including cell growth, cell cycle distribution, and cell cycle-related assays.19–45
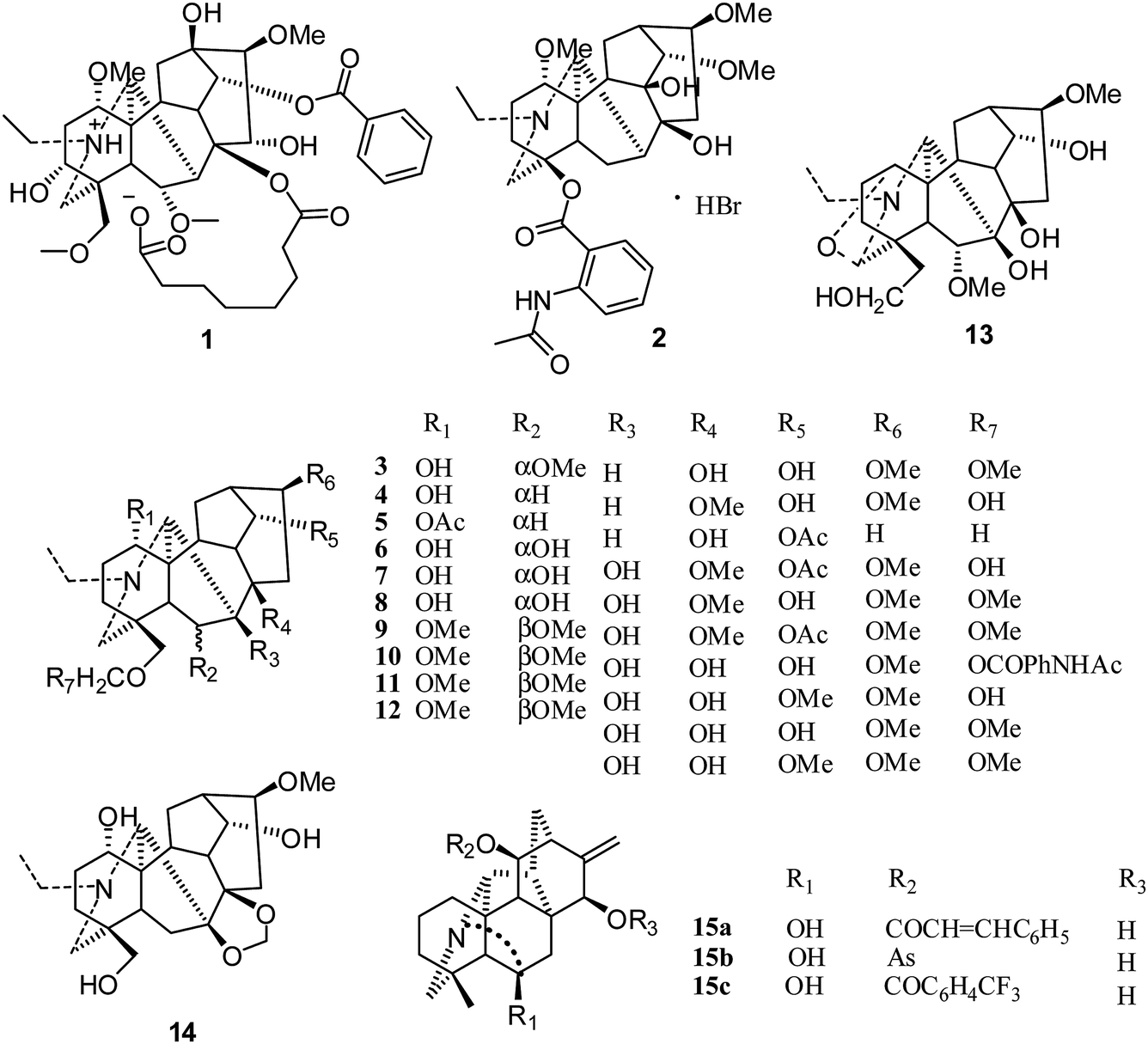 | ||
| Fig. 2 Early cytotoxicity studies of diterpenoid alkaloids (before 2008). | ||
2. Progress in cytotoxicity research of diterpenoid alkaloids
2.1 C18-diterpenoid alkaloids with cytotoxicity activities
Lappaconitine (16) (Fig. 3), a typical C18-diterpenoid alkaloid, is commonly used as postoperative analgesia and relief for clinical cancer pain as a non-addictive analgesic.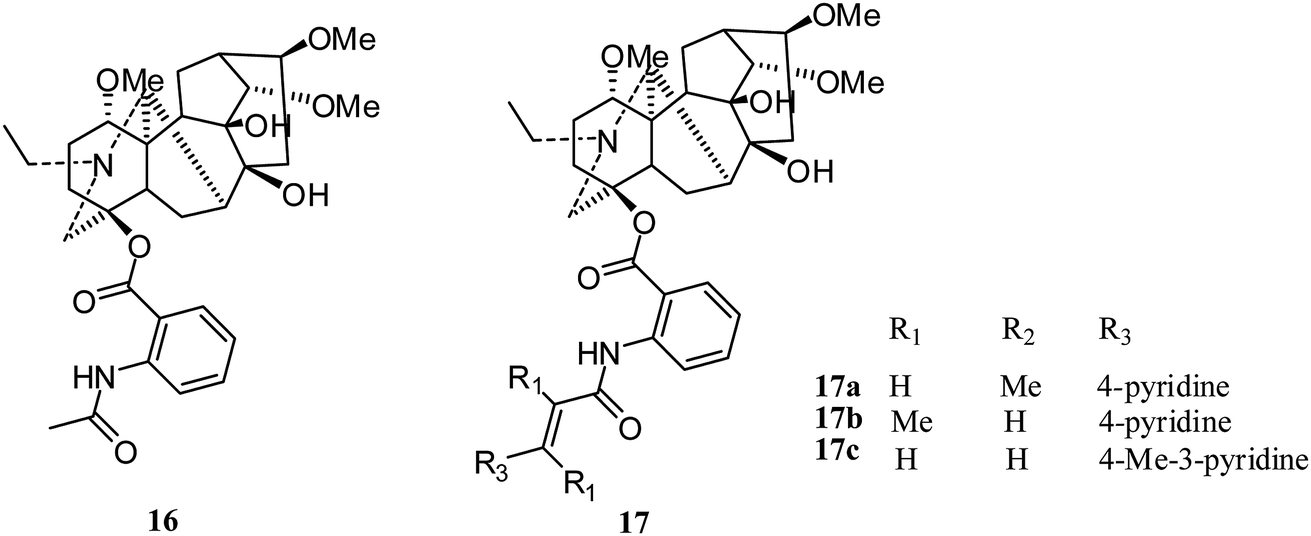 | ||
| Fig. 3 Lappaconitine and its derivatives (16–17) with cytotoxicity activities. | ||
After the first report of lappaconitine hydrobromide in 2005, Xiong and coworkers observed its dose-dependent inhibition of the proliferation of A549 human non-small cell lung cancer cells in 2010 and 2014.19,20 With increasing lappaconitine concentration, the proportion of A549 cells in G1 + G0 phase increased gradually and the proportion in S and G2 + M phases decreased; the apoptosis rate increased with down-regulated expression of cyclin E1. Also, the expression of VEGF-A was inhibited, and a combination of lappaconitine and oxaliplatin arrested cells in G1/G0 phase and inhibited the expression of cyclin E1.20
In 2018, Liang's group reported nineteen lappaconitine aza-cinnamic acid derivatives (17) (Fig. 3) with cytotoxicity activities;21 only 17a–c exhibited inhibited effects on tumor cells, with equal or better activity than 5-fluorouracil against the human prostate cancer cell line (PC-3), human gastric adenocarcinoma cell line (SGC-7901) and human lung cancer cell line (A-549).
2.2 C19-Diterpenoid alkaloids with cytotoxicity activities
The C19-diterpenoid alkaloids are usually divided into six types: lycoctonine, aconitine, pyro, lactone, 7,17-seco, and rearranged.10,11 Most of the isolated C19-diterpenoid alkaloids are lycoctonine- and aconitine-type.Lycoctonine-type. In 2010, Gao and coworkers reported the cytotoxic activities of delpheline (18), delbrunine (19), and delectinine (20) against the MCF-7 and A549 cell lines (Fig. 4).22 Compound 19 showed moderate activities against both cell lines, with IC50 values of 16.5 and 10.6 μM, respectively; meanwhile, compound 18 was only active against MCF-7 cells (IC50 17.3 μM). In contrast, compound 20 was inactive for both tumor cell lines (IC50 > 50 μM).
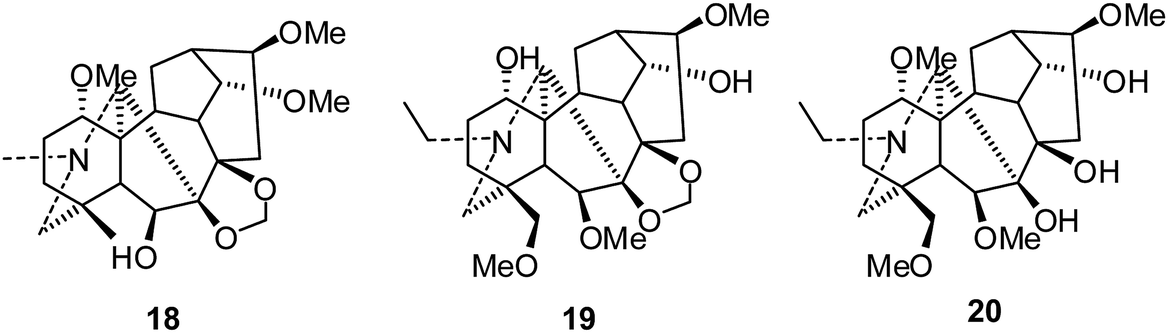 | ||
| Fig. 4 The structures of C19-diterpenoid alkaloids 18–20. | ||
Meanwhile, Lee's group synthesized twenty-four new derivatives (21–47) (Fig. 5 and 6) of delpheline (18) and evaluated their cytotoxic activities against lung (A549), prostate (DU145), nasopharyngeal (KB), and vincristine-resistant nasopharyngeal (KB-VIN) cancer cell lines. Acylation of C-6 was found to be critical for cytotoxic activity.27 Compounds 31, 36, 37, and 39 displayed the greatest potency to the four tested cell lines. Compounds 31, 32, 35, and 38 also exhibited greater inhibitory activities against drug-resistant KB-VIN cells (2.15 to 2.57 fold) than the parental KB cells. Moreover, compounds 21, 22, 24, 25, 28, 29, and 32 showed tumor-selective activities, while others showed inactive potency against the four cell lines. Lee and coworkers also isolated three new delpheline analogues (45–47) (Fig. 6) from Delphinium elatum in 2015; these showed inactivity against the A549, DU145, KB, and KB-VIN cancer cell lines.28 Furthermore, Lee et al. tested the antiproliferative activities of 108 diterpenoid alkaloids against four tumor cell lines (A549, DU145, KB and KB-VIN); thirty-six of these were C19-diterpenoid alkaloids, including neoline (3), delpheline (18), 6-(3-trifluoro-methylbenzoyl)-delpheline (28), aconitine (48), deoxyaconitine (52), hypaconitine (53), and mesaconitine (54).25 However, only 28 exhibited moderately average IC50 values (10 to 20 μM).29
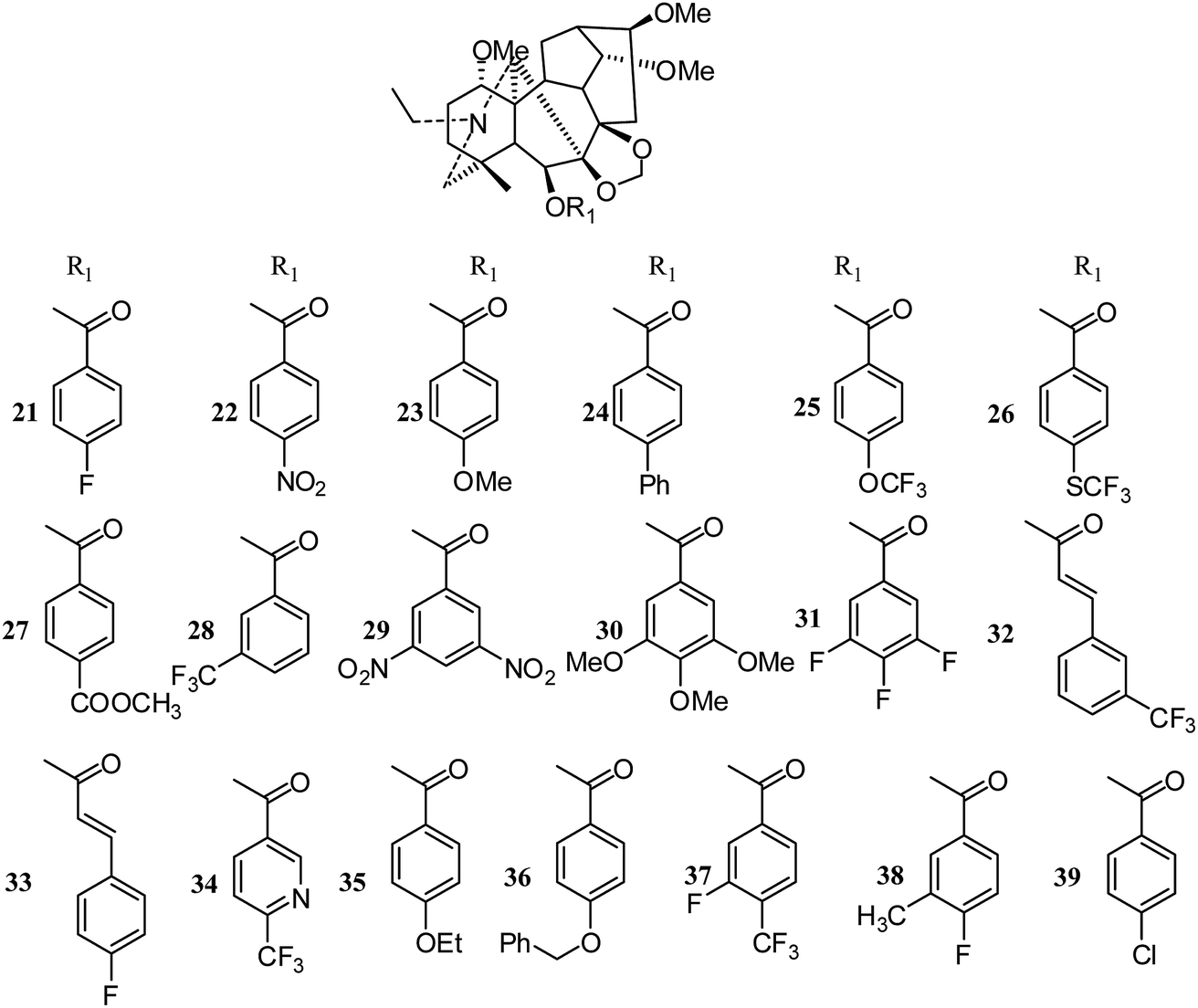 | ||
| Fig. 5 New analogues (21–39) of delpheline (18) by Lee's group (I). | ||
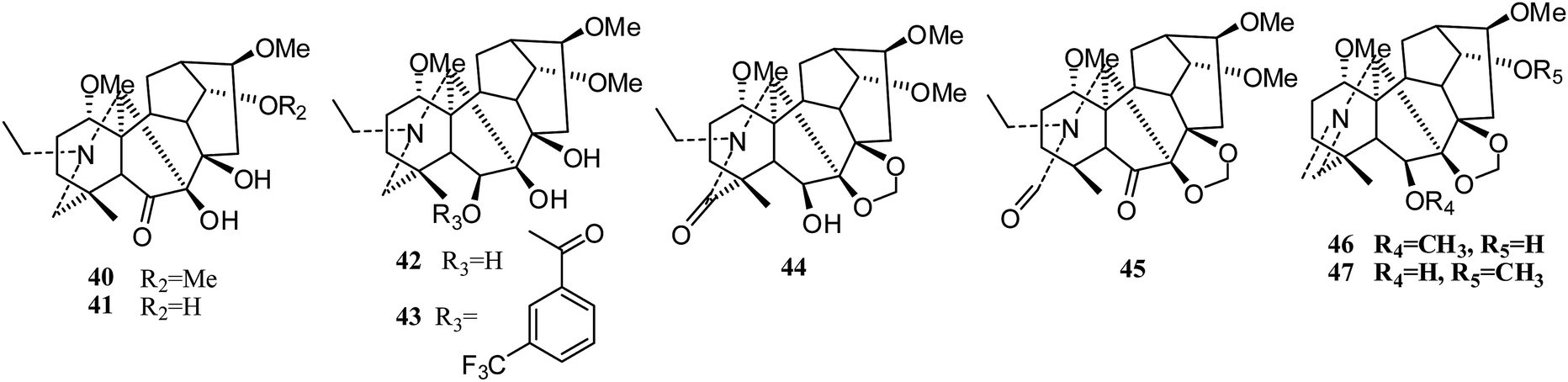 | ||
| Fig. 6 New analogues (40–47) of delpheline (18) by Lee's group (II). | ||
Aconitine-type. In 2012, eight C19-diterpenoid alkaloids, aconitine (48), chasmanine (49), crassicauline A (50), oxonitine (51), deoxyaconitine (52), hypaconitine (53), mesaconitine (54), and senbusine A (55) (Fig. 7), were tested against several cell lines by the MTT method;23 compounds 18 and 20–25 exhibited moderate to strong cytotoxic activities against the HCT8, MCF7 and HePG2 cell lines. Among these, aconitine (48), oxonitine (51) and deoxyaconitine (52) showed the strongest cytotoxic activities against the HCT8, MCF7 and HePG2 cancer cell lines, respectively. Moreover, in an earlier report, the reversal of multidrug resistance of aconitine (48) was evaluated in drug-resistant human oral squamous cell carcinoma (KBv200); it showed a slight inhibitory effect on the growth of KBv200 (IC50 = 224.91 μg ml−1). However, it was considered to be a multidrug resistant reversing agent for increasing the sensitivity of vincristine to kill tumor cells.24
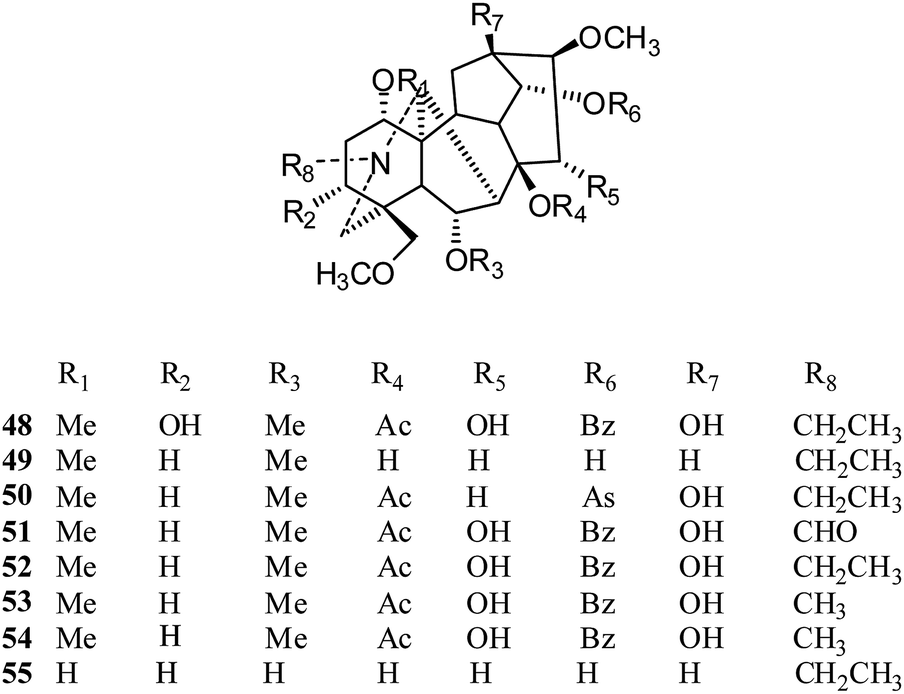 | ||
| Fig. 7 The structures of C19-diterpenoid alkaloids 48–55. | ||
In the same year, a series of mono-[O-(14-benzoylaconine-8-yl)]esters (56a–h) (Fig. 8), built from the 8-O-azeloyl-14-benzoylaconine scaffold and differing in the lengths of their alkyl linker chains, were synthesized and evaluated against a panel of human tumor cell lines: A-549, MCF-7 and HCT-15.25 The results showed that none of the mono-[O-(14-benzoylaconine-8-yl)]esters displayed in vitro cytotoxicity activity (IC50 > 60 μM).
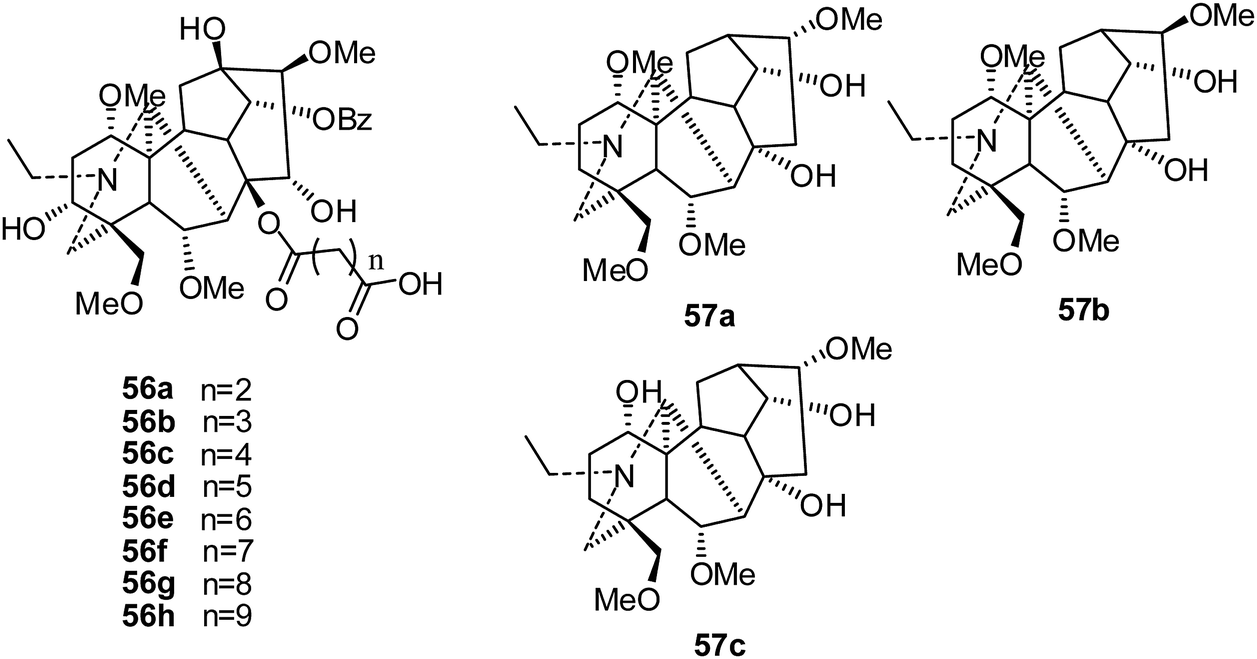 | ||
| Fig. 8 The structures of C19-diterpenoid alkaloids 56–57. | ||
In 2014, three C19-diterpenoid alkaloids, 57a–c (Fig. 8), were isolated from Aconitum taipeicum by Guo's group, and their cytotoxicities against HL-60 and K562 cells were assayed; 57a exhibited stronger cell growth inhibition than adriamycin.30 Further study showed that 57a inhibited the proliferation of HepG2 cells in a dose- and time-dependent manner and also inhibited the invasiveness of HepG2 cells. It also induced significant apoptosis of tumor cells at high dosages.31
After that, in 2017, Shen's group isolated three new C19-diterpenoid alkaloids; only 15-hydroxyldelphisine (58) (Fig. 9) showed inhibition of the cancer cell line SK-OV-3 (IC50 43.78 μM).32 Later in 2017, Liang and coworkers reported the cytotoxic activities of three lipo-alkaloids (59–61) (Fig. 9) from Aconitum sinchiangense W. T. Wang; 59 and 60 showed significant cytotoxicity activities against tumor cells (HL-60, A-549, SMCC-7721, MCF-7 and SW480), with IC50 values comparable to that of cisplatin.33,34
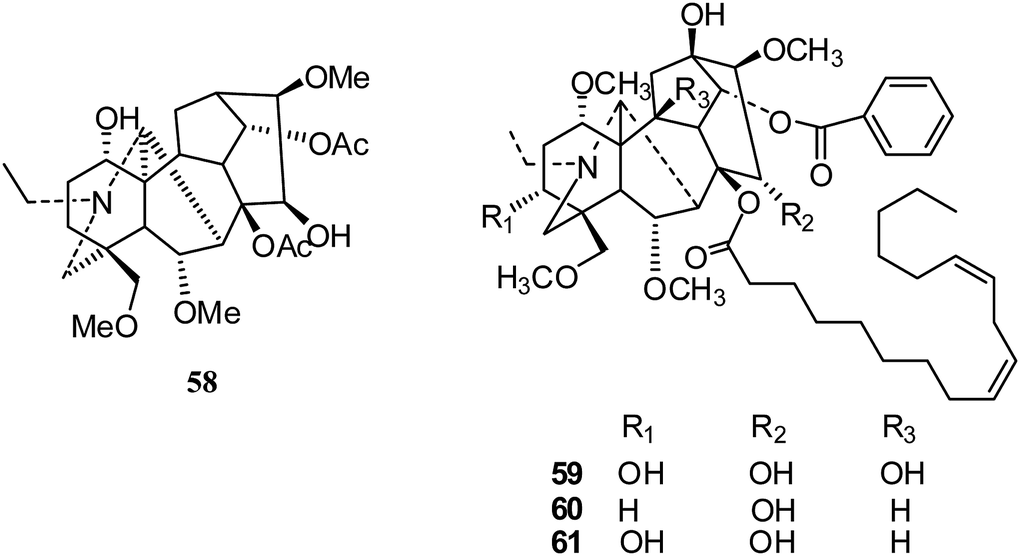 | ||
| Fig. 9 The structures of C19-diterpenoid alkaloids 58–61. | ||
According to their early report in 2005, Robert et al. determined that the C-8 group may contribute to enhanced cytotoxic activity.15 In 2007, although Kashiwakura et al. did not obtain C19-diterpenoid alkaloids with high cytotoxic activities, they found that slight growth inhibitory activities were effected in the presence of a hydroxyl group (ether group or methoxy) or an N–H group (N-ethyl) substituent at C-1.18 Then, from the present reports, Gao et al. concluded that the number of ester groups of alkaloids has an extraordinarily important influence on their cytotoxicity.23 Obviously, the cytotoxicities of the C19-diterpenoid alkaloids containing two ester groups, such as aconitine (48), crassicauline A (50), oxonitine (51), deoxyaconitine (52), hypaconitine (53) and mesaconitine (54), were markedly stronger than those of chasmanine (49) and senbusine A (55), which have no ester group substituents. From Lee's work, 6-acylation appears to be critical for producing cytotoxic activity.27 According to Liang's work, the hydroxyl group at C-3 and the long-chain ester group at C-8 may increase cytotoxic activity.33 Thus, in conclusion, the number and position of the hydroxyl and ester groups as well as the variety of ester groups require further study to identify more potent antitumor C19-diterpenoid alkaloids. Specially, substitutions at C-1, C-3, C-6, and C-8 should be paid more attention.
In 2014, Guo and co-workers found that 57a blocked the cell cycle at the G1/S phase; further mechanism studies revealed that it may upregulate the protein expression of Bax and caspase-3 and downregulate the protein expression of Bcl-2 and CCND1.31
2.3 C20-Diterpenoid alkaloids with cytotoxicity activities
The C20-diterpenoid alkaloids are usually divided into four classes (atisane, kaurene, rearranged and bisditerpenoid classes) and nineteen types: atisine, denudatine, spireine, hetidine, cardionidine, albovionitine, hetisine, vakognavine, veatchine, napelline, anopterine, delnudine, kusnesoline, racemulosine, atisine–hetidine, rearranged atisine–hetidine, denudatine–denudatine, and heteratisine–denudatine.11,13 Most of the isolated C20-diterpenoid alkaloids are atisine-, hetisine-, and napelline-types.Atisine-type. In 2010, Gao's group reported a new diterpene alkaloid, delphatisine C (62) (Fig. 10), with significant cytotoxic activity (IC50 2.36 μM) against the A549 cell line.22 In the following year, a new diterpene alkaloid, honatisine (63) (Fig. 10), was isolated from Delphinium honanense and showed significant cytotoxic activity (IC50 3.16 μM) against the MCF-7 cell line by SRB assay.38
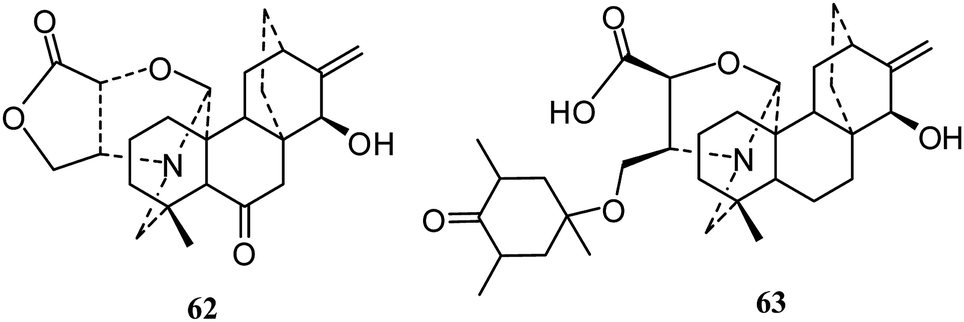 | ||
| Fig. 10 The structures of atisine-type alkaloids 62 and 63. | ||
Hetisine-type. Twenty-two C20-diterpenoid alkaloids, including hetisine type and veatchine type, were tested for their effects on the growth of the A172, A549, HeLa and Raji tumor cell lines in 2009.37 Only six hetisine-type derivatives, 64a–f (Fig. 11), showed significant suppressive effects; 64c showed strong effects for the four cell lines, and 64d showed the best effects against the Raji cell line. The IC50 values of 64a–f against A549 cells were 4.4, 3.2, 1.7, 3.5, 3.5 and 5.1 μM, respectively.
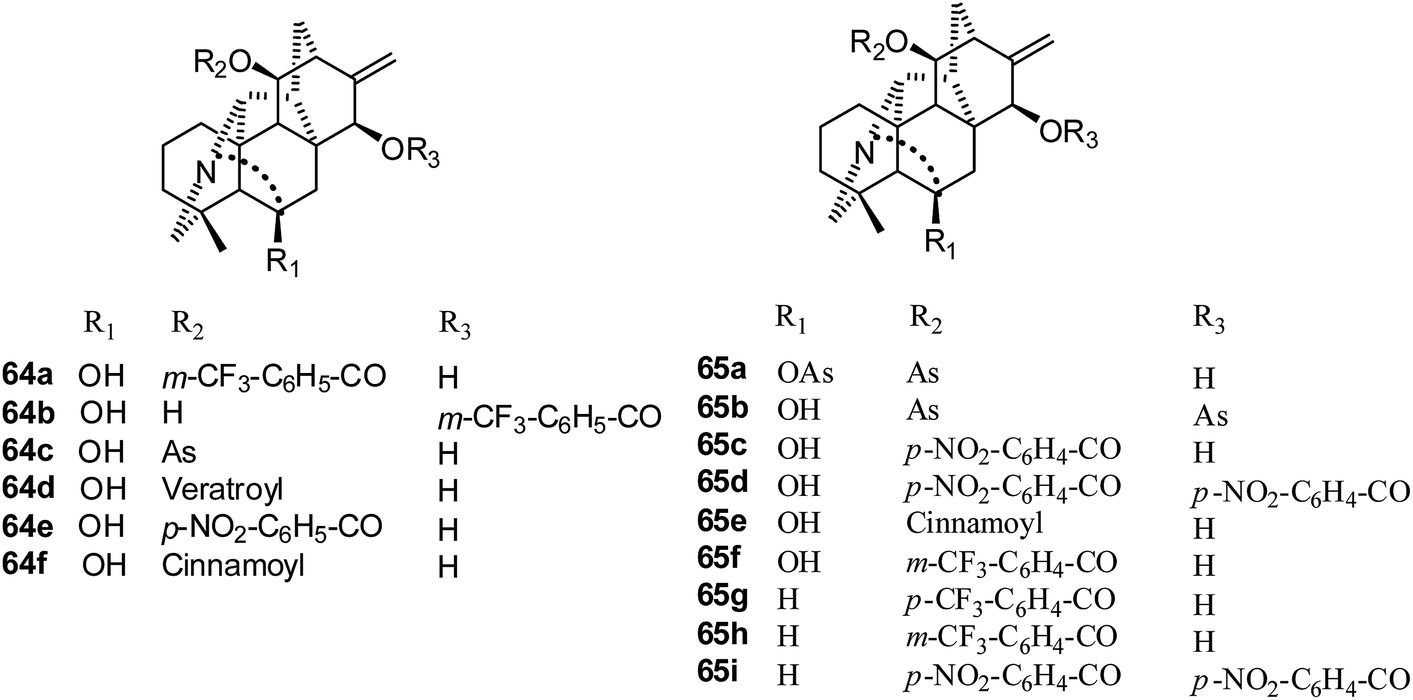 | ||
| Fig. 11 The structures of hetisine-type alkaloids 64–65. | ||
In 2011, thirty-nine hetisine type alkaloids were reported by Kashiwakura's group;39,40 among these, eleven acyl derivatives (64c–d, 65a–i) (Fig. 11) exhibited good cytotoxic activities, and 11,15-dianisoylpseudokobusine (65b) was found to be the most potent cytotoxic agent. Their IC50 values against A549 cells ranged from 1.72 to 5.44 μM.39 11-Anisoyl (As)-pseudokobusine (64c) and 11-m-trifluorometylbenzoyl (Mb)-pseudokobuisne (65f) also showed significant suppressive effects against Raji cells; their IC50 values were 2.2 μg ml−1 and 2.4 μg ml−1, respectively.40 Interestingly, no significant suppressive effects on the growth of human CD34 + hematopoietic stem/progenitor cells (HSPC) were observed for 11-Mb-pseudokobusine (65f), whereas 11-As-pseudokobusine (64c) showed significant suppressive effects on the growth of HSPC.
In 2014, Zhu and coworkers isolated five hetisine-type C20-diterpenoid alkaloids, trichodelphinines A–E (66a–e), from Delphinium trichophorum Franch (Fig. 12). Their cytotoxic activities against A549 cancer cells were evaluated using the MTT method, and the IC50 values ranged from 12.03 to 52.79 μM.41 The most active compounds, 66b and 66e, had lower IC50 values of 18.64 and 12.03 μM, respectively.
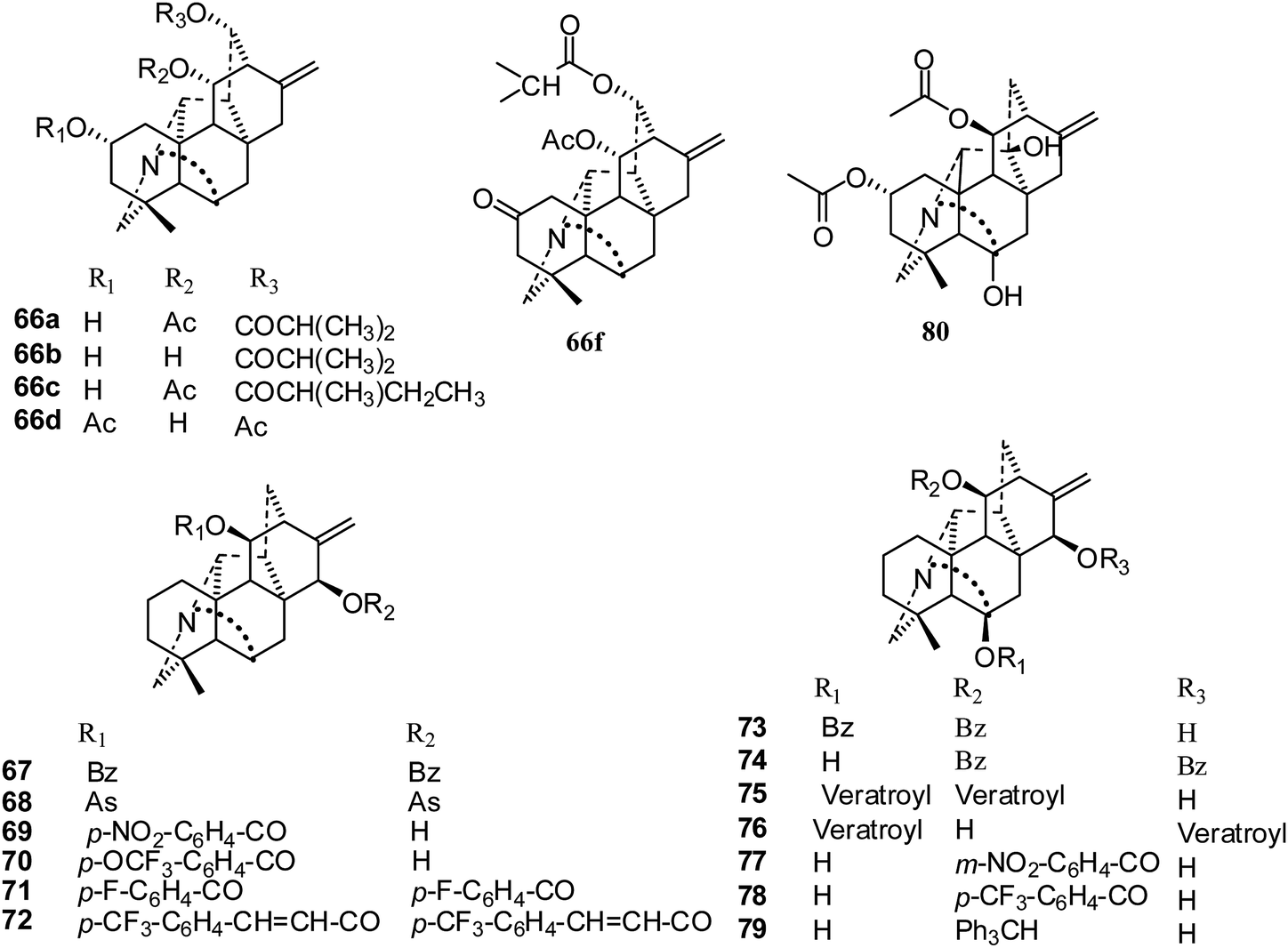 | ||
| Fig. 12 The structures of hetisine-type alkaloids 66–80. | ||
In 2015, the cytotoxic activities of seventy-two C20-diterpenoid alkaloids were determined against the A549, DU145, KB, and KB-VIN cell lines (Fig. 12).29 Fifty-two compounds were non-toxic (IC50 > 20 μM), and nine derivatives (61c–d, 65g, 69, 70, 73, 75, 76, 78) showed mild antiproliferative effects (IC50 = 10 to 20 μM). It was found that eleven derivatives (64a, 65c, 65e, 65i, 67, 68, 71, 72, 74, 77, 79), which are acylated or tritylated at the C-11 hydroxyl, exhibited the greatest potency over all four tested cell lines, including multidrug-resistant KB-VIN (IC50 < 10 μM). Also, all of these were hetisine-type C20-diterpenoid alkaloids with two different substitution patterns: C-11 and C-11, 15. The most potent compound, pseudokobusine 11,30-trifluoromethylbenzoate (64a), is a promising new lead that merits additional evaluation against multidrug-resistant tumors.29
The most recent report was published in 2017, where Shen et al. reported a new C20-diterpenoid alkaloid, 80 (Fig. 12), that showed only low inhibition of the SK-OV-3 cancer cell line of 24% (IC50 32.14 μM).32
Anopterine-type. In 2015, eight trifluoroacetate salts of anopterine-type diterpenoid alkaloids, 6α-acetoxyanopterine (81), 4′-hydroxy-6α-acetoxy-anopterine (82), 4′-hydroxyanopterine (83), 11α-benzoyl-anopterine (84), anopterine (85), 7β-hydroxyanopterine (86), 7β,4′-dihydroxyanopterine (87), and 7β-hydroxy-11α-benzoyl-anopterine (88) (Fig. 13), were reported by Davis's group; they appeared to be highly active toward prostate cancer cell lines, with IC50 values < 400 nM. Compounds 81, 82, and 88 were the most active, with IC50 values of 3.1, 10.9, and 6.7 nM against LNCaP cells, respectively.42 Furthermore, live-cell imaging assays on 81–88 showed concentration- and time-dependent effects on the cell morphology and proliferation of LNCaP cells.
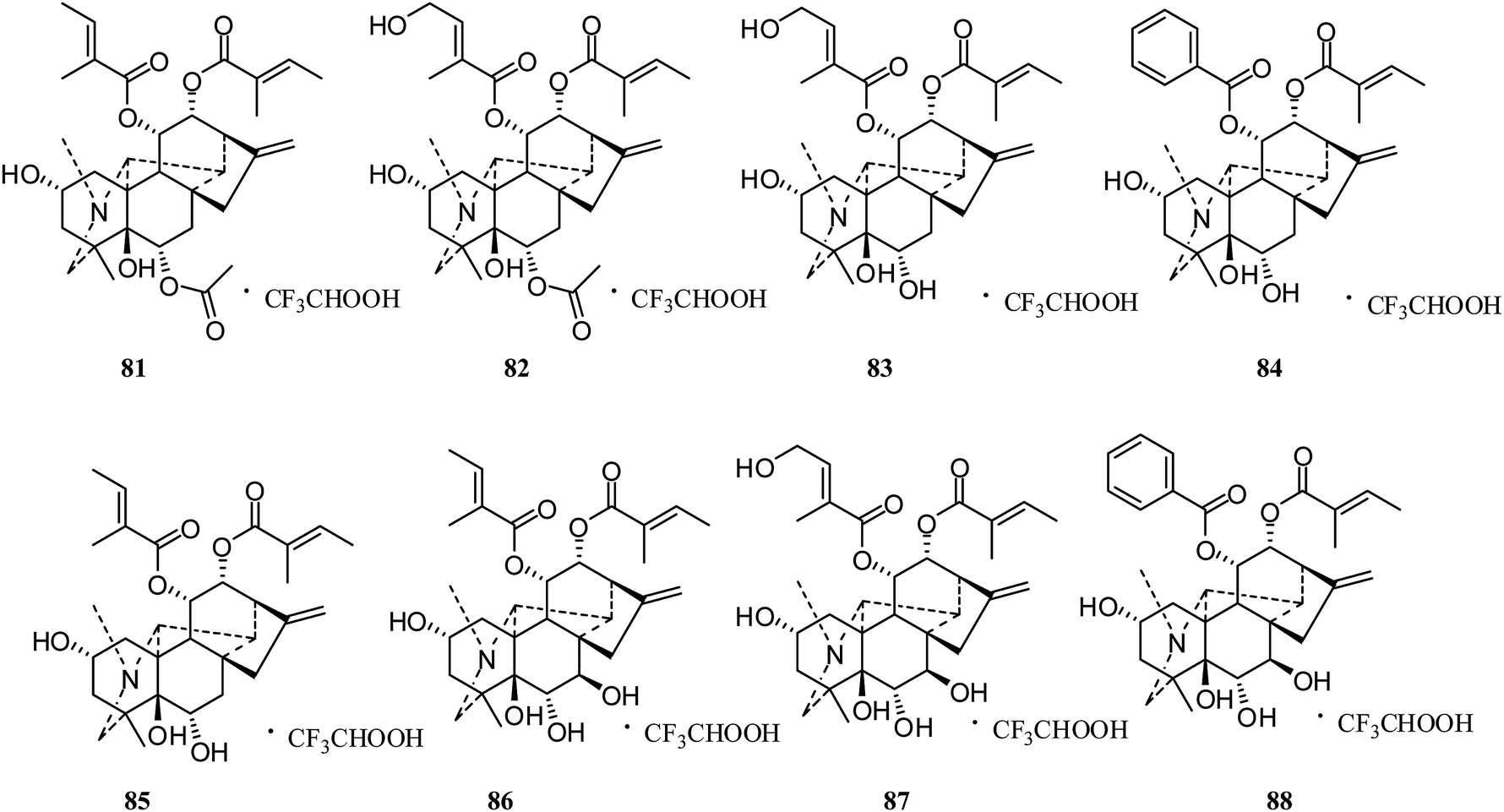 | ||
| Fig. 13 The structures of anopterine-type alkaloid trifluoroacetate salts 81–88. | ||
Delnudine-type. In 2014, Zhu and coworkers isolated one delnudine-type C20-diterpenoid alkaloid, trichodelphinine F (89), from Delphinium trichophorum Franch (Fig. 14).41 Its cytotoxic activity against A549 cancer cells was evaluated using the MTT method, with an IC50 value of 16.55 μM.
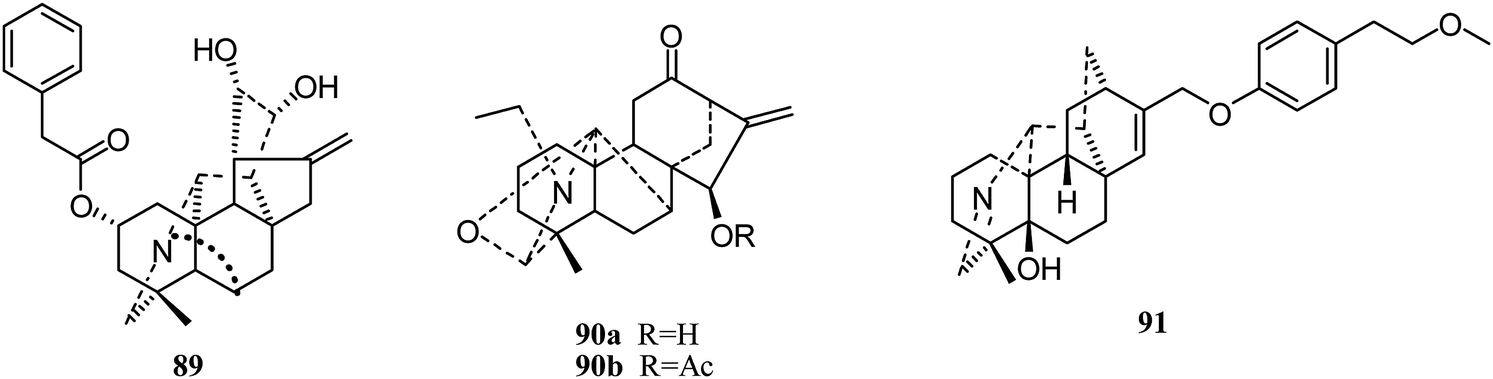 | ||
| Fig. 14 The structures of other type C20-alkaloids 89–91. | ||
Napelline-type. In 2012, Gao et al. also reported two napelline-type C20-diterpenoid alkaloids, songoramine (90a) and 15-cetylsongoramine (90b) (Fig. 14);23 the latter showed lower cytotoxicity against HePG2 cells (IC50 58.55 ± 9.17 μM).
Hetidine-type. In 2017, a new hetidine-type alkaloid, navicularine C (91), was isolated from the ground parts of Aconitum naviculare. However, it showed no cytotoxic activities against five tumor cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW480).43
In the cytotoxic effects of hetisine-type alkaloids, the hetisine-type structure, which is characteristic of C20-diterpenoid alkaloids, plays a very important role in their pharmacological properties.40 The results of the first cytotoxicity report of C20-diterpenoid alkaloids suggested that the hydroxyl groups at C-6 and C-15 of hetisine-type compounds are necessary to their inhibitory effects. In particular, the C-11 residues are an important component for their anti-tumor properties and for their lower toxicity to hematopoiesis.40 Replacement by an acyl group at both C-11 and C-15 resulted in enhanced activity of the parent alkaloids, and the presence of a hydroxy group at the C-6 position was required for the cytotoxic effects.39 The identity of the acyl group had varying effects. Simple benzoyl ester derivatives were generally less active. Cinnamoyl, p-nitrobenzoyl, m-trifluoromethylbenzoyl, and veratroyl substitutions were effective. Anisoate esters were generally found to be more potent than the other esters tested.18,44 After further analysis of Lee's work,29 it was further confirmed that all of the more active hetisine-type compounds had an ester or ether group on the C-11 hydroxyl and were either 11,15-diester analogs or 11-monoester/11,15-diester analogs.
For napelline-type, Gao and coworkers found that a compound with an additional acetyl group at C-15, 15-acetylsongoramine (90b), was more potent than songoramine (90a) against several human cancer cell lines; this shows that the ester groups play a considerably important role in the anticancer potency of these diterpenoid alkaloids.23
According to the work of Kashiwakura's group, a mechanism study indicated that hetisine derivatives inhibit cell growth through G1 arrest. Furthermore, 11-Mb-pseudokobusine (65f) and 78 clearly inhibited the phosphorylation of extracellular signal-regulated kinase, induced enhanced phosphoinositide 3 kinase phosphorylation and led to subsequent accumulation of G1 and/or sub G1 phase in Raji cells.29,39,40
2.4 Bis-diterpenoid alkaloids
Only a few reports have focused on the cytotoxicity activities of bis-diterpenoid alkaloids.In 2012, a series of bifunctional acyl compounds (92a–f) (Fig. 15) were also synthesized from the 8-O-azeloyl-14-benzoylaconine scaffold; their cytotoxicity activities were evaluated against a panel of human tumor cell lines, A-549, MCF-7 and HCT-15.25 Surprisingly, three bis-[O-(14-benzoyl-aconine-8-yl)] esters (92d–f) bearing 7, 8 and 9 carbon atoms between the two aconitine moieties presented noticeable in vitro cytotoxic activities, with IC50 values ranging between 4 and 28 μM; this highlights the influence of the length of the alkyl linker. The most active compound, bis[O-(14-benzoylaconine-8-yl)]suberate (92e), was then evaluated in vivo in immunodeficient mice bearing human tumor xenografts originating from MCF-7 and HCT-15 cells. Antitumor activity was clearly detected at a dose of 10 mg kg−1, which is far below the maximum tolerated dose (15 mg kg−1).25 Further studies on 92e showed higher than average activities against leukemia and melanoma cell lines, especially SK-MEL-5 and SK-MEL-28, and against the COLO-205 and HT-29 (colorectal) and MDA-MB-468 (breast) cancer cell lines.26 Also, it was implied that 92e blocks the cell cycle at G2/M phase, displaying a mechanism of action related to that of nitrosoureas.26
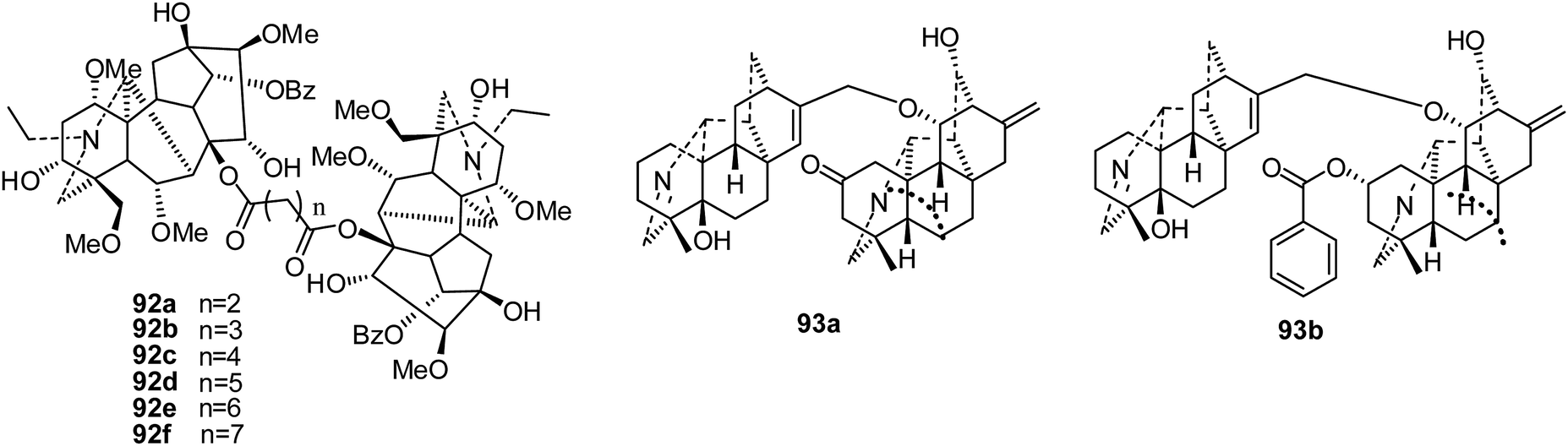 | ||
| Fig. 15 The structures of bis-diterpenoid alkaloids 92–93. | ||
In 2017, two new bisditerpenoid alkaloids, navicularines A and B (93a–b), were isolated from the ground parts of Aconitum naviculare (Fig. 15). Only navicularine B (93b) exhibited certain cytotoxic activities in vitro, with IC50 values of 13.50, 18.52, 17.22, 11.18, and 16.36 μM, respectively, against five cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW480).43
3. Conclusions
Research on the cytotoxicity activities of more than 250 diterpenoid alkaloids has been reported in the past two decades. Some have shown effective anticancer properties in various cancer cell lines. These properties include inhibiting cell growth, inducing apoptosis, interfering with the cell cycle, and altering MDR.In general, C20-diterpenoid alkaloids have been studied more deeply than C19-diterpenoid alkaloids. More derivatives of C20-diterpenoid alkaloids have shown notable anticancer potential, such as 81–88, with IC50 values < 400 nM. Meanwhile, some natural and synthesized C19-diterpenoid alkaloids have shown anticancer effects, such as compounds 28, 57a, and 60 (with strong cytotoxicity activities comparable to those of adriamycin or cisplatin). Some also exert noteworthy anticancer effects in animal models.25 Furthermore, many diterpenoid alkaloids tend to exhibit improved activity after simple structural modification;27 many structures may affect the activity of a compound, such as the type and position of substituents and the linker-chain length.45
In brief, diterpenoid alkaloids have great potential as new drugs for treating cancer. However, the present research in this field is limited. Much additional research is required before these compounds can be applied as antitumor drugs (Table 1).
| Diterpenoid alkaloid type/class | Natural compounds | Plant source | Ref. |
|---|---|---|---|
| C18 | Lappaconitine (16) | A. sinomontanum Nakai | 20 |
| Lycoctonine-type C19 | Delpheline (18), delbrunine (19), delectinine (20), | Delphinium chrysotrichum | 22 |
| Lycoctonine-type C19 | N-Formyl-4,19-secopacinine (45), iminoisodelpheline (46), iminodelpheline (47) | Delphinium elatum | 28 |
| Aconitine-type C19 | Aconitine (48), chasmanine (49), crassicauline A (50), oxonitine (51), deoxyaconitine (52), hypaconitine (53), mesaconitine (54), senbusine A (55) | Aconitum carmichaelii Debx. | 23 |
| Aconitine-type C19 | Taipeinine A (57a), taipeinine B (57b), taipeinine C (57c) | Aconitum taipeicum | 30 |
| Aconitine-type C19 | 15-Hydroxyldelphisine (58) | Aconitum nagarum var. heterotrichum | 32 |
| Aconitine-type C19 | Sinchiangensine A (59), lipodeoxyaconitine (60) | Aconitum sinchiangense W. T. Wang | 33 |
| Atisine-type C20 | Delphatisine C (62) | Delphinium chrysotrichum | 22 |
| Atisine-type C20 | Honatisine (63) | Delphinium honanense | 38 |
| Hetisine-type C20 | Trichodelphinine A (66a), trichodelphinine B (66b), trichodelphinine C (66c), trichodelphinine D (66d), trichodelphinine E (66e) | Delphinium trichophorum Franch | 41 |
| Hetisine-type C20 | 14-Hydroxyl-2-acetoxy-spiradine C (80) | Aconitum nagarum var. heterotrichum | 32 |
| Anopterine-type C20 | 6α-Acetoxyanopterine (81), 4′-hydroxy-6α-acetoxyanopterine (82), 4′-hydroxyanopterine (83), 11α-benzoylanopterine (84), anopterine (85), 7β-hydroxyanopterine (86), 7β,4′-dihydroxyanopterine (87), 7β-hydroxy-11α-benzoylanopterine (88) | Anopterus macleayanus | 42 |
| Delnudine-type C20 | Trichodelphinine F (89) | Delphinium trichophorum Franch | 41 |
| Napelline-type C20 | Songoramine (90a), 15-cetylsongoramine (90b) | Aconitum carmichaelii Debx. | 23 |
| Hetidine-type C20 | Navicularine C (91), navicularines A and B (93a–b) | Aconitum naviculare | 43 |
3.1 Selectivity between tumor cells and normal cells
As far as we know, most diterpenoid alkaloids with both active and toxic components are obtained from Ranunculaceae plants. Thus, the most important and urgent studies should focus on increasing the selectivity of tumor cells and normal cells to decrease their toxicity to the human body. However, work in this field is still lacking.3.2 SAR study
The structure–cytotoxicity activity relationships (SAR) of diterpenoid alkaloids have not been deeply researched, particularly for C19-diterpenoid alkaloids. In order to synthesize more potential active diterpenoid alkaloid derivatives, detailed SAR studies, such as quantitative structure–activity relationship (QSAR) studies, should be performed.3.3 Further mechanism studies
The existing cytotoxicity mechanism studies for C19- and C20-diterpenoids are superficial; both mechanisms require full elucidation, which can also provide precise targets for further computer-aided design.3.4 In vitro study to in vivo study
Finally and most importantly, the present reports are mostly restricted to in vitro tests. In vivo activity has been reported for only in vivo activities have been reported for only two compounds, 2 and 92e. One compound, 92e. Thus, a great step from in vitro to in vivo tests should be taken in the very near future to develop new pre-clinical antitumor diterpenoid alkaloid agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (No. 81703387).References
- H. M. Ling, J. S. Zhang and J. Ding, Biochem. Pharmacol., 2001, 62, 733–741 CrossRef.
- C. Qing, C. Jiang, J. S. Zhang and J. Ding, Anti-Cancer Drugs, 2001, 12, 51–56 CrossRef PubMed.
- C. Didelot, J. F. Mirjolet, M. Barberi-Heyob, C. Ramacci and J. L. Merlin, Can. J. Physiol. Pharmacol., 2002, 80, 638–643 CrossRef PubMed.
- M. Baumann and M. Krause, Radiother. Oncol., 2004, 72, 257–266 CrossRef PubMed.
- J. Haveman, N. C. Kreder, H. M. Rodermond, C. V. Bree, N. A. P. Franken, L. J. A. Stalpers, M. Z. Zdzienicka and G. J. Peters, Oncol. Rep., 2004, 12, 187–192 Search PubMed.
- J. Sonnemann, V. Gekeler, K. Ahlbrecht, K. Brischwein, L. Chao, P. Bader, C. Muller, D. Niethammerc and J. F. Beck, Cancer Lett., 2004, 209, 177–185 CrossRef PubMed.
- M. Zhang, M. Boyer, L. Rivory, A. Hong, S. Clarke, G. Stevens and K. Fife, Int. J. Radiat. Oncol., Biol., Phys., 2004, 58, 353–360 CrossRef.
- L. K. Kvols, J. Nucl. Med., 2005, 46, 187S–190S Search PubMed.
- B. Pauwels, A. E. C. Korst, V. Andriessen, M. F. D. Baay, G. G. O. Pattyn, H. A. J. Lambrechts, C. M. J. D. Pooter, F. Lardon and J. B. Vermorken, Radiat. Res., 2005, 164, 642–650 CrossRef PubMed.
- F. P. Wang and Q. H. Chen, The Alkaloids, 2010, vol. 69 Search PubMed.
- F. P. Wang, Q. H. Chen and X. Y. Liu, Nat. Prod. Rep., 2010, 27, 529–570 RSC.
- B. S. Joshi and S. W. Pelletier, Alkaloids: Chemical and Biological Perspectives, Elsevier Science Ltd, Langford Lane Kidlington, UK, 1999, p. 289 Search PubMed.
- F. P. Wang and X. T. Liang, Alkaloids Chemistry & Biology, 2002, vol. 59, pp. 1–280 Search PubMed.
- F. P. Wang, Q. H. Chen and X. T. Liang, The Alkaloids, 2009, pp. 1–78 Search PubMed.
- A. Chodoeva, J. J. Bosc, J. Guillon, A. Decendit, M. Petraud, C. Absalon, C. Vitry, C. Jarryc and J. Robert, Bioorg. Med. Chem., 2005, 13, 6493–6501 CrossRef PubMed.
- L. Lin, L. Y. Xiao, P. Y. Lin, D. Zhang and Q. W. Chen, Traditional Chinese Medicinal Research, 2005, 18, 16–18 Search PubMed.
- C. D. Inés, M. Reina, J. A. Gavín and A. González-Coloma, Z. Naturforsch., 2006, 61c, 11–18 Search PubMed.
- K. Wada, M. Hazawa, K. Takahashi, T. Mori, N. Kawahara and I. Kashiwakura, J. Nat. Prod., 2007, 7, 1854–1858 CrossRef PubMed.
- L. R. Sheng, M. Xu, L. Q. Xu and F. Xiong, J. Chin. Med. Mater., 2014, 37, 840–843 Search PubMed.
- L. R. Sheng, Master thesis, Jinan University, Guangzhou, China, 2010.
- C. Y. Liang, X. Ju, L. Tian, M. Y. Jia, D. N. Tian and S. J. Ding, US Pat., US20180093966, 2018.
- Y. Q. He, Z. Y. Ma, X. M. Wei, B. Z. Du, Z. X. Jing, B. H. Yao and L. M. Gao, Fitoterapia, 2010, 81, 929–931 CrossRef PubMed.
- F. Gao, Y. Y. Li, D. Wang, X. Huang and Q. Liu, Molecules, 2012, 17, 5187–5194 CrossRef PubMed.
- X. Q. Liu, X. Y. Chen, Y. Z. Wang, S. J. Yuan and Y. Tang, Chin. J. Basic Med. Tradit. Chin. Med., 2004, 10, 55–57 Search PubMed.
- A. Chodoeva, J. J. Bosc, J. Guillon, P. Costet, A. Decendit, J. M. Merillon, J. M. Leger, C. Jarry and J. Robert, Eur. J. Med. Chem., 2012, 54, 343–351 CrossRef PubMed.
- A. Chodoeva, J. J. Bosc, L. Lartigue, J. Guillon, C. Auzanneau, P. Costet, A. Zurdinov, C. Jarry and J. Robert, Invest. New Drugs, 2014, 32, 60–67 CrossRef PubMed.
- K. Wada, E. Ohkoshi, S. L. Morris-Natschke, K. F. Bastow and K. H. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 249–252 CrossRef PubMed.
- K. Wada, R. Chiba, R. Kanazawa, K. Matsuoka, M. Suzuki, M. Ikuta, M. Goto, H. Yamashita and K. H. Lee, Phytochem. Lett., 2015, 12, 79–83 CrossRef.
- K. Wada, E. Ohkoshi, Y. Zhao, M. Goto, S. L. Morris-Natschke and K. H. Lee, Bioorg. Med. Chem. Lett., 2015, 25, 1525–1531 CrossRef PubMed.
- Z. J. Guo, Y. Xu, H. Zhang, M. Y. Li and K. Xi, Nat. Prod. Res., 2014, 28, 164–168 CrossRef PubMed.
- H. Zhang, Z. Guo, L. Han, X. You and Y. Xu, J. BUON, 2014, 19, 705–712 Search PubMed.
- D. K. Zhao, X. Q. Shi, L. M. Zhang, D. Q. Yang, H. C. Guo, Y. P. Chen and Y. Shen, Chin. Chem. Lett., 2017, 28, 358–361 CrossRef.
- X. X. Liang, L. Chen, L. Song, W. B. Fei, M. He, C. L. He and Z. Q. Yin, Nat. Prod. Res., 2017, 31, 2016–2023 CrossRef PubMed.
- X. X. Liang, N. Chen, Y. Y. Gao, C. L. He, Z. Q. Yin, Y. F. Zou, L. Z. Yin and X. Song, Chinese Patent, 201610776382.6, 2016.
- L. Hou, X. Y. Liu, X. Y. Chen, K. T. Zhang, Y. Z. Wang and X. M. Wang, Chin. J. Inf. Tradit. Chin. Med., 2005, 12, 34–36 Search PubMed.
- S. D. Tian, X. Q. Liu, X. M. Wang, Y. Tang and X. Y. Chen, Chin. Arch. Tradit. Chin. Med., 2006, 24, 55–56 Search PubMed.
- M. Hazawa, K. Wada, K. Takahashi, T. Mori, N. Kawahara and I. Kashiwakura, Invest. New Drugs, 2009, 27, 111–119 CrossRef PubMed.
- Y. Q. He, Z. Y. Ma, X. M. Wei, D. J. Liu, B. Z. Dua, B. H. Yao and L. M. Gao, Chem. Biodiversity, 2011, 8, 2104–2109 CrossRef PubMed.
- K. Wada, M. Hazawa, K. Takahashi, T. Mori, N. Kawahara and I. Kashiwakura, J. Nat. Med., 2011, 65, 43–49 CrossRef PubMed.
- M. Hazawa, K. Takahashi, K. Wada, T. Mori, N. Kawahara and I. Kashiwakura, Invest. New Drugs, 2011, 29, 1–8 CrossRef PubMed.
- C. Z. Lin, Z. X. Zhao, S. M. Xie, J. H. Mao, C. C. Zhu, X. H. Li, B. Zeren-dawa, K. Suolang-qimei, D. Zhu, T. Q. Xiong and A. Z. Wu, Phytochemistry, 2014, 97, 88–95 CrossRef PubMed.
- C. Levrier, M. C. Sadowski, C. C. Nelson and R. A. Davis, J. Nat. Prod., 2015, 78, 2908–2916 CrossRef PubMed.
- J. B. He, J. Luan, X. M. Lv, D. Y. Rui, J. Tao, B. Wang, Y. F. Niu and H. P. Ju, Fitoterapia, 2017, 120, 142–145 CrossRef PubMed.
- K. Wada, Chemistry and Biological Activity of Diterpenoid Alkaloids, in Studies in Natural Products Chemistry, Elsevier Science Ltd, Langford Lane Kidlington, UK, 2012, p. 191–223 Search PubMed.
- M. Y. Ren, Q. T. Yu, C. Y. Shi and J. B. Luo, Molecules, 2017, 22, 267 CrossRef PubMed.
Footnote |
| † X. X. L and Y. Y. G. contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |