
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtrazine exposure improves the proliferation of H22 cells in vitro and in vivo
Yong Tian†
ab,
Jingchun He†bd,
Nan Liu†eg,
Di Huangbf,
Zhuo Liue,
Yanrong Yangb,
Junyu Chenc,
Benzheng Zhaoc,
Shuhua Zhao*c and
Bing Liang *a
*a
aSchool of Nursing, Jilin University, Changchun 130021, Jilin, China
bBasic Medical College, Jilin University, Changchun 130021, Jilin, China
cThe Second Affiliate Hospital, Jilin University, Changchun 130021, Jilin, China
dThe 4th Center Clinical College, Tianjin Medical University, Tianjin 300140, China
eChina-Japan Union Hospital, Jilin University, Changchun 130021, Jilin, China
fTongji Medical College, Huazhong University of Science & Technology, Wuhan 430000, Hubei, China
gQian Wei Hospital of Jilin Province, Changchun 130021, Jilin, China
First published on 13th June 2018
Abstract
Atrazine (ATZ), a widely used triazine herbicide, has been detected in the surface and ground water even far from where it is applied. Recently, the biotoxicity of atrazine to the immune, reproductive and endocrine systems has been preliminarily observed in laboratory experiments and epidemiological research studies. In order to further comprehend the carcinogenic nature of ATZ, in vitro and in vivo models were established in this study to explore the effects of ATZ exposure on hepatocellular carcinoma. The results showed that after being treated with ATZ, the proliferation of H22 cells increased, and the tumor volume and amount of ascites were significantly increased in an in situ transplantation tumor model established in C57BL/6 mice compared to the control group. The expression of p53 was down-regulated, while the expression of cyclin-D1, VEGF, MMP2, Stat3 and C-myc was up-regulated in the ATZ-treated groups compared to the control group. These results indicate that ATZ might activate the Stat3 signaling pathway and promote the proliferation and invasion of hepatocellular carcinoma cells.
1. Introduction
The triazine herbicide atrazine (ATZ) has been used throughout the world to control broadleaf weeds and raise agricultural production, but the widespread application of ATZ has resulted in increased contamination of surface and ground water. The toxicity of ATZ to the endocrine, reproductive and immune systems has been discussed.1–4 However, the specific association between atrazine and cancer risk needs to be carefully and thoroughly explored. In early studies, Van Leeuwen confirmed an association between ATZ contamination levels and stomach cancer incidence,5 but Sathiakumar N comprehensively evaluated the epidemiologic evidence regarding the human carcinogenic potential of triazine and proposed that the data were inadequate for determining the association between ATZ and cancer risk.6 Similarly, Boffetta concluded that ATZ is unlikely to pose a cancer risk to humans through epidemiological research.7 Moreover, there are other epidemiological research studies that failed to identify links between ATZ and various cancer types, including prostate cancer, ovarian cancer and breast cancer.8,9However, laboratory research has provided evidence linking atrazine to a higher risk of various cancers.10 Meanwhile, in vitro and in vivo studies have revealed the connections between ATZ and various signaling pathways in different systems. There have been results linking ATZ to the alteration of immune function and consequent disease resistance in a B6C3F10 tumor model.11 An in vivo study showed that long-term dietary administration of atrazine at the 400 ppm level to Fisher 334 and Sprague–Dawley female rats caused a prolonged estrous cycle and earlier onset of mammary and pituitary tumors, but not an increased incidence of either.12 A recent study verified a novel mechanism in which estrogen receptor α and G protein-coupled receptor 30 might be involved. This study indicated that ATZ can stimulate these pathways and exert effects in cancer cells and cancer-associated fibroblasts, thus inducing the proliferation of tumor cells.13,14 Besides, it has also been verified that atrazine can reduce metabolic activity in fish and induce various dysfunctions in exposed male frogs, including nuclear DNA damage, tumorigenesis and hermaphroditism.15
Generally speaking, the specific association of the effects of atrazine with cancer risks needs to be further explored and discussed. In this study, to discuss the possible connection between ATZ and liver cancer, H22 cells and hepatoma tumor model mice were exposed to atrazine, and the expression of genes associated with proliferation, apoptosis and metastasis was analyzed. Both in vitro and in vivo experiments showed the up-regulation of proliferation and metastasis-associated genes including p53, cyclin-D1, VEGF and MMP2, while no statistical changes were observed in the apoptosis-associated genes Bax and caspase-3. Furthermore, two upstream cancer genes, Stat3 and C-myc, were up-regulated. The above molecular changes can well verify the association between atrazine and tumor progression, and the mechanism via which ATZ exerts its pro-cancer effects.
2. Materials and methods
2.1. Cell lines and cell culture
Mouse H22 hepatoma cells, obtained from the Shanghai Institute of Cell Biology (Shanghai, China), were cultured in IMDM containing 10% fetal bovine serum. The cells were randomly divided into three groups (0 μM, 0.01 μM and 0.1 μM) according to the concentration of atrazine administered. All culture media were purchased from Hyclone (Logan, UT, USA).2.2. C57BL/6 mice
C57BL/6 mice, weighing 18–22 g, were purchased from the Beijing Institute for Experimental Animals. All animal procedures were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals. Also, the mouse experiments received approval from the Medical Ethics Committee of Basic Medical College of Jilin University.2.3. Reagents
Atrazine (99% purity), SDS, TEMED, acrylamide, N,N-dimethyl-bis-acrylamide, DTT and PMSF were obtained from Sigma Chemical Company (St. Louis, MO, USA). ATZ solutions were prepared by dissolving ATZ in DMSO (for the in vitro experiments) or corn oil (for the in vivo experiments). All of the solutions were kept at 4 °C for a maximum of 1 week. Rabbit anti-primary monoclonal antibodies, and HRP-labeled anti-rabbit IgG secondary antibodies were acquired from Proteintech Group (Wuhan, China). Pierce ECL Plus Kits were acquired from Thermo Fisher Scientific Inc. (Rockford, IL, USA).2.4. MTT assay
H22 cells in the exponential growth phase were digested with 0.25% trypsin, and seeded into 96-well plates at a density of 4 × 103 cells per well in 100 μl of IMDM containing 10% FBS. ATZ was diluted to 0.01 μM and 0.1 μM respectively, and the control wells were treated with blank storage solution. All three groups were incubated for 0 h, 24 h, 48 h and 72 h. 10 μl of tetrazolium (MTT, 5 mg ml−1) was pipetted into each well and incubated for 4 h at 37 °C, the medium was removed and 150 μl of DMSO was added to dissolve the violet-crystals. The OD was measured at 570 nm by a microplate reader. Five wells were used for each group.2.5. Cell cycle distribution
The cells were harvested and permeabilized after being treated with ATZ for 48 h. Nuclear DNA was labeled with propidium iodide (PI) using a Cycle Test Plus DNA reagent kit (Becton, Franklin Lakes, NJ, USA). Flow cytometry was conducted, and the Cell Quest software was used to determine the relative DNA content.2.6. Establishment of an orthotopically implanted hepatocarcinoma tumor model
In all groups, 0.2 ml (1 × 108 cells per ml) of H22 cell solution was subcutaneously inoculated to form a solid tumor. After 10 days, the tumors, which were 3–5 mm in diameter, were removed and sheared into small pieces of 1 mm3 under sterile conditions. C57BL/6 mice were anesthetized by coelio-injection of pentobarbitone (70 mg kg−1) and a laparotomy was performed. Under sterile conditions the right lobe of the liver was punctured to form a 3 mm long sinus tract and a small piece of tumor tissue was inserted into each sinus tract, and thus the orthotopic transplantation tumor model of HCC was established.2.7. Animal treatment
Seven days after orthotopic tumor implantation, the mice were randomly divided into 3 groups (n = 10 per group). Before treatment, all mice were starved overnight and then they were orally given 20 mg kg−1 or 100 mg kg−1 ATZ each time, while 100 μl of coin oil was given to the mice in the control group. The animals were sacrificed 14 days after the first administration. The orthotopically transplanted tumors were excised and weighed. The HCC tissues, and adjacent non-cancerous liver tissues were sampled and fixed in 10% formalin to determine metastases.2.8. RNA isolation and quantitative RT-PCR
Total RNA was isolated from the cells using Trizol reagent (Gibco, Grand Island, NY, USA) as described by the manufacturer. The cDNA was synthesized using a Takara RNA PCR Kit (TaKaRa, Japan). The sequences of the qRT-PCR primers were as follows: Stat3 (forward, 5′-TTG CCA GTT GTG ATC-3′ and reverse, 5′-AGA ACC CAG AAG GAG AAGC-3′), Bcl-2 (forward, 5′-ACT TGA CAG AAG ATC ATG CC-3′ and reverse, 5′-GGT TAT CAT ACC CTG TTC TC-3′), MMP9 (forward, 5′-CCC ACT TAC TTT GGA AAC G-3′ and reverse, 5′-GAA GAT GAA TGG AAA TAC GC-3′), MMP2 (forward, 5′-GGA AGC ATC AAA TCG GAC TG-3′ and reverse, 5′-CAC CCT CTT AAA TCT GAA ATC ACC-3′), cyclin-D1 (forward, 5′-CGC CTT CCG TTT CTT ACT TCA-3′ and reverse, 5′-AAC TTC TCG GCA GTC AGG GGA-3′), VEGF (forward, 5′-AGT CCC ATG AAG TGA TCA AGT TC-3′ and reverse, 5′-ATC CGC ATG ATC TGC ATG G-3′), c-Myc (forward, 5′-AGT TGG ACA GTG GCA GGG-3′′ and reverse, 5′-ACA GGA TGT AGG CGG TGG-3′), Bax (forward, 5-′AGG GTT TCA TCC AGG ATC GAG C-3′ and reverse, 5-′AGG CGG TGA GGA CTC CAG CC-3′), p53 (forward, 5′-GGC CAT CTA CAA GCA GTC ACA G-3′ and reverse, 5′-AGC AAA TCT ACA AGC AGT CAC AG-3′) and β-actin (forward, 5′-ATA TCG CTG CGC TGG TCG TC-3′ and reverse, 5′-AGG ATG GCG TGA GGG AGA GC-3′). Quantitative gene expression was determined by using a SYBR Master Mixture PCR Kit (Takara, Japan). A total of three parallel RT-PCR experiments were performed for each group.2.9. Western blotting
Total proteins were extracted using a protein extraction Kit (Roche, Indianapolis, IN, USA), and quantified using an Enhanced BCA Protein Assay Kit. The proteins were transferred onto PVDF membranes by electrophoretic transfer following electrophoretic separation by SDS-PAGE. The membranes were probed with primary antibodies overnight at 4 °C. After three washes with TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The bands were visualized using enhanced chemiluminescence reagents and analyzed with a Bio-Rad Versa Doc 5000 MP system (Life Science Research, Hercules, CA, USA), and β-actin was used as a loading control.2.10. Cell apoptosis analysis
Apoptosis was assessed using an Annexin-V Apoptosis Detection Kit following the manufacturer's instructions (Sanjian, Tianjin, China). After respective exposure, 5 × 105 cells were incubated with Annexin-V/FITC and propidium iodide (PI) for 5 min at 37 °C in the dark and analyzed by flow cytometry.2.11. HE staining
After being fixed with 10% formalin, the tissues were dehydrated, processed, and embedded in paraffin. Serial sections were prepared and stained with hematoxylin and eosin (H&E) for histopathological evaluation by a light microscope.2.12. Immunohistochemical staining
The tissue sections were deparaffinized, dehydrated and then boiled in 0.01 M citrate buffer (pH 6.0) for 2 min. Hydrogen peroxide (0.3%) was applied to block endogenous peroxidase activity and the sections were incubated with normal goat serum to reduce non-specific binding. The tissue sections were incubated with rabbit primary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :150 dilution). Biotinylated goat anti-rabbit serum IgG was used as a secondary antibody. After washing, the sections were incubated with streptavidin–biotin conjugated with horseradish peroxidase, and then the peroxidase reaction was developed with 3,3′-diaminobenzidine tetrahydrochloride. Counterstaining with hematoxylin was performed, and the sections were dehydrated in ethanol before mounting.
:150 dilution). Biotinylated goat anti-rabbit serum IgG was used as a secondary antibody. After washing, the sections were incubated with streptavidin–biotin conjugated with horseradish peroxidase, and then the peroxidase reaction was developed with 3,3′-diaminobenzidine tetrahydrochloride. Counterstaining with hematoxylin was performed, and the sections were dehydrated in ethanol before mounting.
2.13. Statistical analysis
Data were presented as the mean ± s.d. Statistical analysis was performed for multiple comparisons using one-factor analysis of variance (ANOVA). All experiments were repeated three times. Differences were regarded as statistically significant at p < 0.05 (SPSS17.0 statistical software).3. Results
3.1. ATZ exposure promoted H22 cell proliferation but not apoptosis
To quantify the effects of atrazine on the growth process of H22 cells, an MTT proliferation assay was employed. As the results showed, cells exposed to 0.01 μM ATZ exhibited a significant increase in cell proliferation compared to the control group, while the proliferation of the 0.1 μM exposure group was slightly lower than that of the 0.01 μM group (p < 0.05) (Fig. 1A). To better explain the increase in cell proliferation, flow cytometry was employed to assess both the changes in the cell cycle distribution and the apoptosis of H22 cells after exposure to ATZ for 72 h. The results of PI staining showed that in both the 0.01 μM and 0.1 μM groups, a decrease in the number of cells in the G2 phase and an increase in the number of cells in the S phase was observed compared to the control group. A prolonged S phase and a narrowed G2 phase suggested that ATZ exposure might induce the accumulation of cells in the S phase and mean that H22 cells move quickly through the G2 phase (Fig. 1B and C). The results of Annexin V-FITC and PI staining did not show a statistically significant difference in the percentage of apoptosis cells among the three groups (Fig. 1D and F).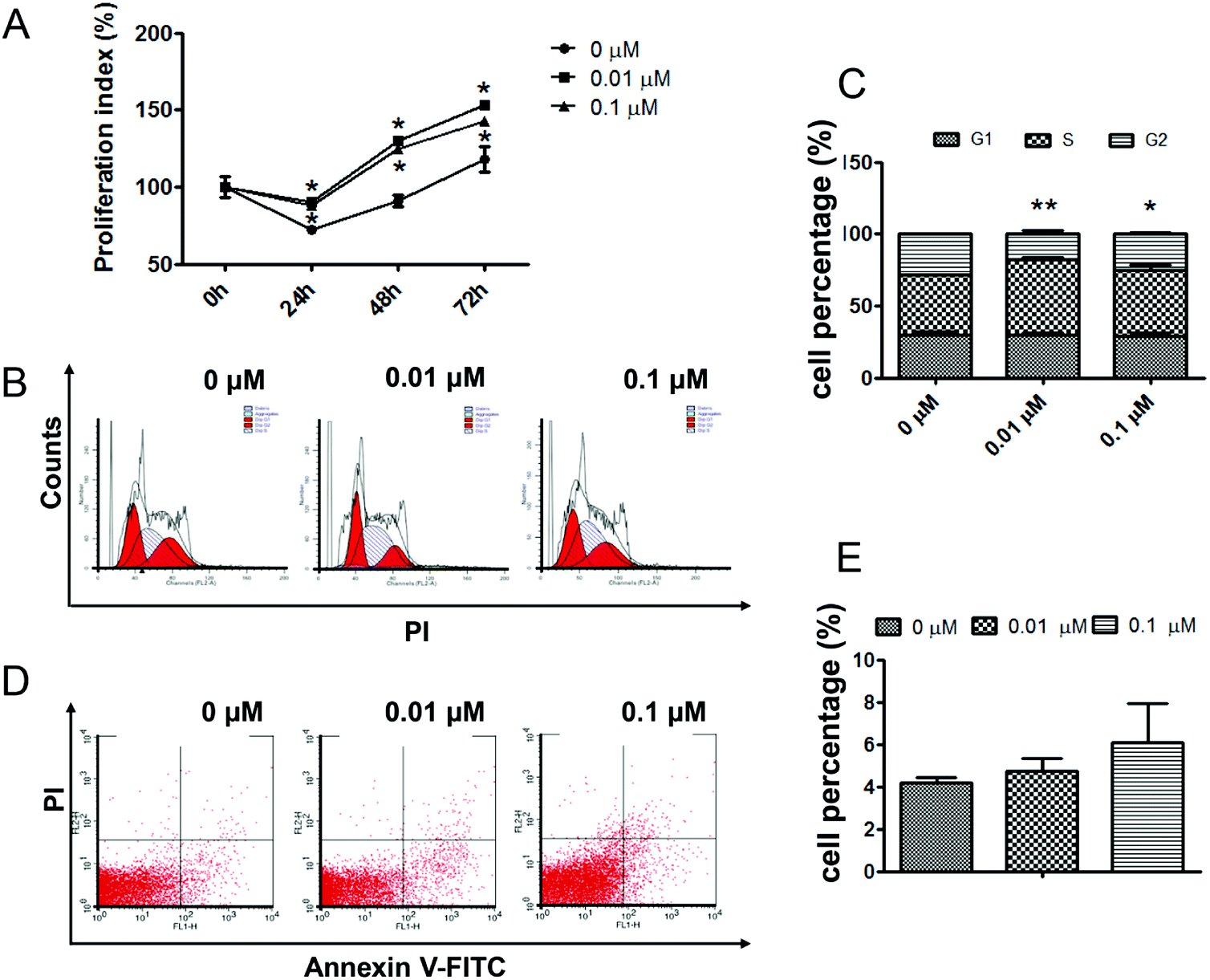 | ||
| Fig. 1 ATZ exposure promoted the proliferation of H22 cells. (A) The number of viable cells was assessed using MTT. (B and C) PI staining and flow cytometry analysis showed the cell cycle in the H22 cells. (D and E) Annexin V-FITC/PI staining and flow cytometry analysis showed apoptosis of the H22 cells. The presence of an asterisk indicates a difference due to ATZ exposure (*p < 0.05, **p < 0.01). | ||
3.2. ATZ up-regulated the expression of genes associated with proliferation and metastasis but not with apoptosis
To verify the effects of ATZ exposure on the genes related to proliferation, metastasis and apoptosis, real time PCR and western blot were employed. To assess the gene and protein level changes pertaining to proliferation and the cell cycle, p53 and cyclin-D1 were assessed. The results showed a significant downregulation of the p53 gene in both the 0.01 μM and 0.1 μM groups, while the upregulation of cyclin-D1 was observed in the 0.1 μM group (Fig. 2A–C). To confirm the similarity in apoptosis between the experimental groups and control group, the transcription and expression of Bax and caspase-3 were detected. No statistically significant differences were detected both in the gene and the protein (Fig. 2A, F and G). Furthermore, to evaluate the influence of ATZ exposure on tumor malignancy and invasion, VEGF, MMP2 and MMP9 were assessed. As Fig. 2A shows, notable upregulation of MMP2 and VEGF was observed, while the change in MMP9 was not statistically significant compared to the control group. The expressions of MMP2 protein and VEGF protein were upregulated after exposure to ATZ in a dose-dependent manner (p < 0.01) (Fig. 2D and E). For further comprehension of these cellular changes, the C-Myc and Stat3 genes, both of which have been well established as regulators of cell cycle progress and tumorigenesis, were detected. The results showed that C-Myc exhibited a statistically significant increase in expression in the 0.1 μM group, but the difference was not significant in the 0.01 μM group (p < 0.05). Meanwhile, the Stat3 gene was notably upregulated in terms of the transcription and expression level dose-dependently (Fig. 2A, H and I).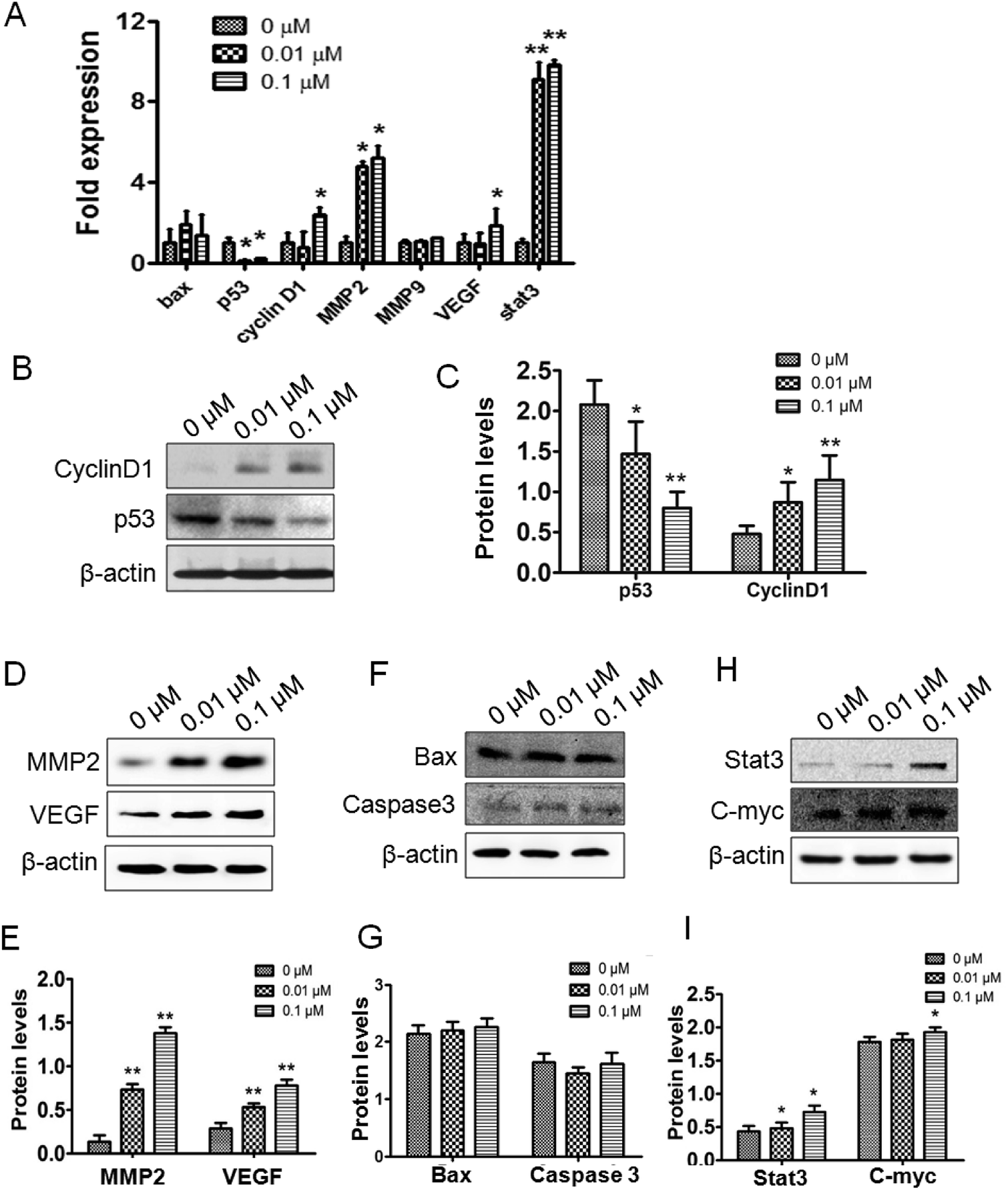 | ||
| Fig. 2 ATZ upregulated the expression of genes associated with proliferation and metastasis in vitro. (A) Transcription of Bax, p53, cyclin-D1, MMP2, MMP9, VEGF and stat3. (B and C) Western blot results of the proliferation and cell cycle-associated proteins cyclin-D1 and p53. (D and E) Western blot results of the metastasis-associated proteins MMP2 and VEGF. (F and G) Western blot results of the apoptosis-associated proteins Bax and caspase-3. (H and I) Western blot results of the upstream proteins Stat3 and C-myc. The presence of an asterisk indicates a difference due to ATZ exposure (*p < 0.05,**p < 0.01). | ||
3.3. ATZ exposure promoted tumor proliferation and metastasis in vivo
In this study, an in situ model of hepatic carcinoma was established in C57BL/6 mice to verify the effects of ATZ exposure on animals. The mean tumor weight (g) in the 20 mg kg−1 and 100 mg kg−1 ATZ groups was 0.21 ± 0.07 and 0.27 ± 0.11 compared to 0.13 ± 0.06 for the control group (Fig. 3C and D) (p < 0.05), implying that ATZ exposure promotes tumor growth and development. In immunohistochemistry staining, the ratio of PCNA positive cells was confirmed to exhibit an evident increase compared to the controls, verifying that ATZ can greatly increase the proliferation of tumor cells in vivo (p < 0.05) (Fig. 3F and G). Increases in body weight were observed with significant abdominal distention (Fig. 3A and B), which implies that severe ascites might be induced by ATZ exposure in vivo. Accordingly, notable invasion to the basement layer of H22 cells can be detected in the 20 mg kg−1 group, and evident malignant invasion was observed in the 100 mg kg−1 group in HE staining (Fig. 3E), which indicated that ATZ exposure can contribute to the metastasis of hepatic carcinoma in situ.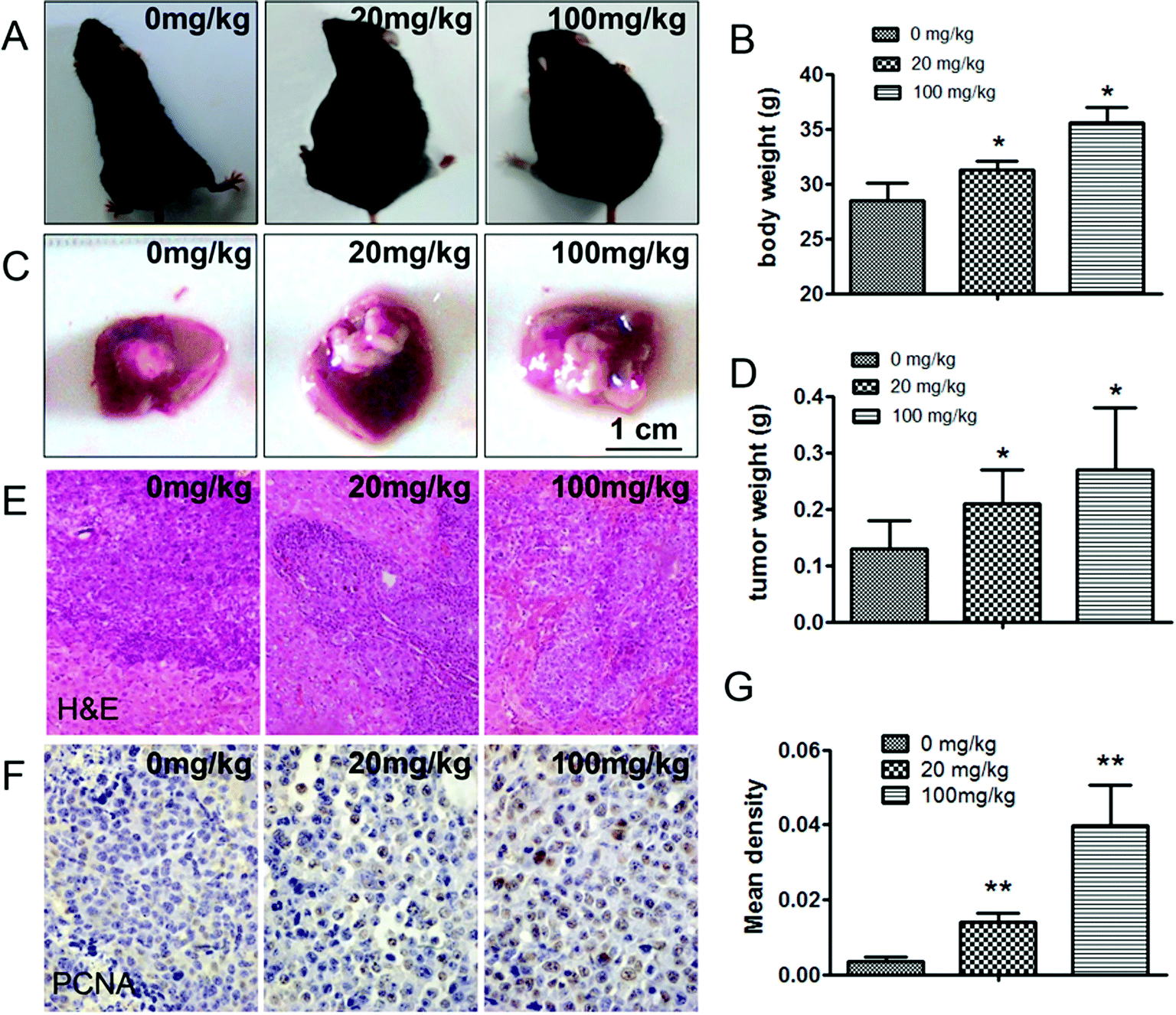 | ||
| Fig. 3 ATZ exposure promoted tumor proliferation and metastasis in vivo. (A and B) The body weight of the mice in the experimental groups was increased with abdominal distention. (C and D) The tumor weight was increased after exposure to ATZ. (E) HE staining showed obvious invasion into the liver membrane in the treatment groups. (F and G) More PCNA positive cells can be observed by immunohistochemical staining. The presence of an asterisk indicates a difference due to ATZ exposure (*p < 0.05, **p < 0.01). | ||
3.4. ATZ exposure affected the gene expression related to proliferation and metastasis but not to apoptosis in vivo
Relevant genes were reviewed in the in vivo studies. The p53 protein was downregulated compared to the control. Meanwhile, cyclin-D1 transcription and expression were observed to be upregulated (Fig. 4A, D and E). In addition, the changes in the p53 and cyclin-D1 proteins in the tumor tissue could be further verified under immunohistochemical staining (Fig. 5A and B). The transcription and expression of Bax, Bcl-2 and caspase-3 showed no statistically significant difference, indicating that apoptosis might not be involved in ATZ toxicity (Fig. 4A–C). To confirm the metastasis in the in vivo studies, the changes in VEGF and MMP2 were assessed. As shown, both the expression and transcription of MMP2 showed a notably strong dose-effect. For VEGF, significant upregulation of transcription and expression can be observed in the 100 mg kg−1 group (Fig. 4A, F and G). The upregulation of VEGF and MMP2 can be detected in immunohistochemical staining (Fig. 5C and D). This variation implies that ATZ exposure can greatly facilitate the metastasis and invasion of tumor cells.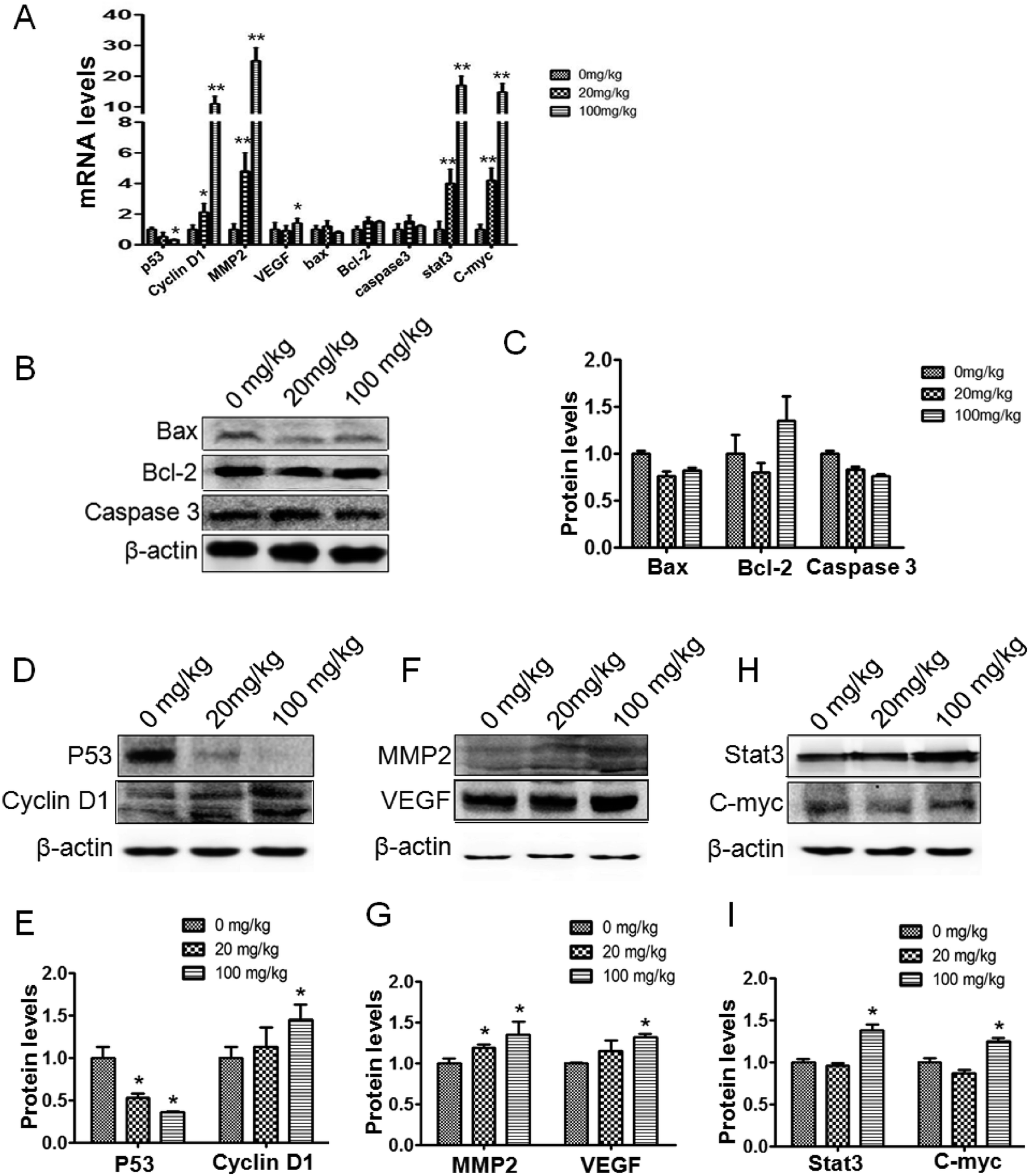 | ||
| Fig. 4 ATZ upregulated the expression of genes associated with proliferation and metastasis in vivo. (A) Real-time PCR results of p53, cyclin-D1, MMP2, VEGF, Bax, Bcl-2, caspase-3, Stat3 and C-myc. (B and C) Western blot results of the apoptosis-associated proteins Bax and caspase-3. (D and E) Western blot results of the proliferation and cell cycle-associated proteins cyclin-D1 and p53. (F and G) Western blot results of the metastasis-associated proteins MMP2 and VEGF. (H and I) Western blot results of the upstream proteins Stat3 and C-myc. The presence of an asterisk indicates a difference due to ATZ exposure (*p < 0.05, **p < 0.01). | ||
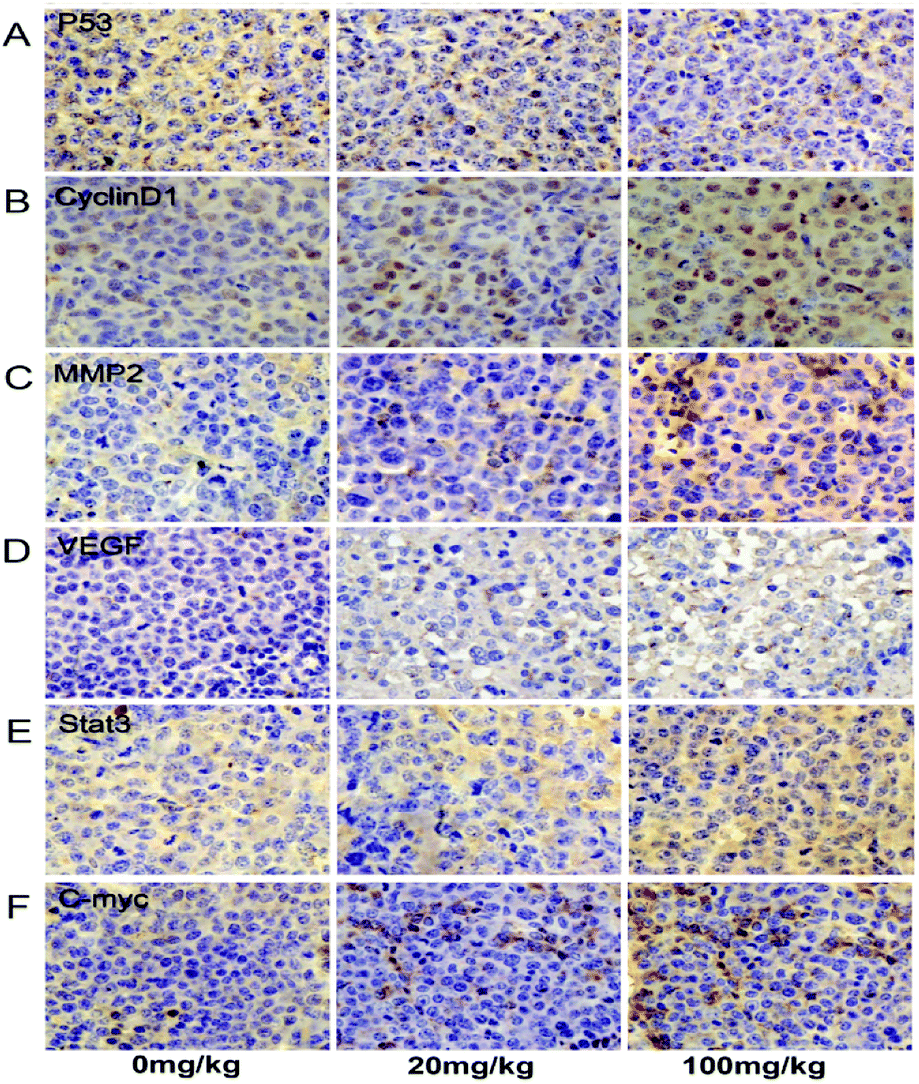 | ||
| Fig. 5 ATZ exposure up-regulated the Stat3/C-myc signaling pathway in vivo. (A–F) The expression of p53, cyclin-D1, MMP2, VEGF, Stat3 and C-myc in tumor specimens was assessed by immunohistochemical staining. | ||
3.5. ATZ exposure up-regulated the Stat3/C-myc signaling pathway in vivo
For further exploration of the underlying mechanism, the Stat3 and C-myc genes were detected. As shown, the transcription of the Stat3 gene was increased in a dose-dependent manner compared to the control group, and the expression of Stat3 protein showed a notable increase in the 100 mg kg−1 group compared to the control group (Fig. 4A, H and I, and 5E). The C-myc gene and protein showed an interestingly similar pattern to Stat3. The transcription of C-myc was upregulated in the 20 mg kg−1 and 100 mg kg−1 group, and the expression of C-myc exhibited a notable increase in the 100 mg kg−1 group (Fig. 4A, H and I, and 5F). These results reveal a possible association between ATZ exposure and the Stat3 pathways.4. Discussion
The effects of ATZ on different organs and systems have been reported, including immune toxicity, endocrine toxicity and reproductive toxicity.2 However, previous studies focus more on in vitro study and epidemiological research. In this paper, both in vitro and in vivo studies are performed to better explore the effects of atrazine exposure on tumor development.Notably increased proliferation was detected in the exposed group, which implies that ATZ exposure might promote the proliferation of tumor cells. Being a vital part in cell progress, the cell cycle determines cell proliferation. As is well-known, the cell cycle can be divided into three periods: interphase, mitotic (M) phase and Gap 2 (G2). The G2 checkpoint ensures that everything is ready to enter the M phase and conduct cell division.16 In this study, the results show that the G2 phase is narrowed compared to the control, implying that ATZ exposure accelerates the cell cycle by promoting H22 cells to rush through the G2 phase, thus increasing cell proliferation.
Furthermore, the transcription and expression of cyclin-D1 and p53, two proliferation-associated genes that regulate the cell cycle, were detected. p53 serves as a tumor suppressor by conserving stability, preventing genome mutation, and inducing cell cycle arrest and apoptosis.16 p53 induces the transcription of the cell cycle-related gene p21. p21 binds to the CDK2/cyclin-E complex and then arrests the progress of the cell cycle.17 The other proliferation-related gene cyclin-D1 is a confirmed cell cycle regulator.18,19 By forming active cyclin-dependent kinases (CDKs), cyclin-D1 promotes the phosphorylation of retinoblastoma protein (Rb), such that the phosphorylated Rb fails to repress the transcription of the genes that are required for DNA synthesis, thus cancelling its growth-repressive functions.16 As is mentioned above, down-regulation of p53 and up-regulation of cyclin-D1 might explain the increased proliferation in H22 cells, and the increased tumor weight and PCNA ratio in mice.
To clarify whether ATZ exposure influences the apoptosis of cancer cells, the transcription and expression of apoptosis-associated genes were detected. The caspase family is a widely recognized series of proteins that execute apoptosis. Caspase-3 is one of the interacting caspase proteins activated in an apoptotic cell,20 and up-regulation of caspase-3 causes indiscriminate cellular mortality.21 Apoptosis-promotor Bax induces intrinsic apoptosis by binding to and antagonizing the Bcl-2 protein.22 Bcl-2 and Bax are two major contrary factors that mediate apoptosis, which can be regulated by a complex network of bimolecular interactions. p53 is also involved in this network by forming an inhibitory complex with Bcl-2 and activating Bax, thus promoting programmed cell death.16 In this study, caspase-3 and Bax showed no signs of being activated in terms of the transcription and expression level compared to the control, implying that ATZ fails to break the homeostasis between Bax and Bcl-2, even with the help of the down-regulation of p53.
Metastasis is the most notable characteristic that differentiates malignant tumors from benign ones. Matrix metalloproteinase-2 (MMP-2)23 and matrix metallopeptidase 9 (MMP-9)24 are involved in the development of various human malignancies, as the degradation of collagen IV in the basement membrane and extracellular matrix can facilitate tumor invasion, metastasis, growth and angiogenesis.25 In addition, MMP9 has been found to contribute to the release of vascular endothelial growth factor (VEGF) and the formation of functional VEGF–VEGFR-2 complexes.26 In this study, notable invasion into the liver membrane, increased body weight and abdominal distention can be observed in the experiment groups compared to the control group, and the related transcription and expression levels of MMP2 and VEGF were increased dose-dependently. These changes indicated that by up-regulating MMP2 and VEGF, ATZ exposure promotes malignancy, especially metastasis and invasion of the tumor.
As a central mediator, C-myc directly regulates various cellular behaviors including apoptosis, metastasis and proliferation. By tethering to transcription promoters, C-myc is involved in the transcription of CDK4, CDC25a, Bax, Bcl-2, etc. Meanwhile, C-myc also participates in transcriptional changes of the Ras/Raf/Erk and Ras/PI3K/Akt pathways,27–30 thus casting light on the full network that is involved in the mechanisms through which ATZ achieves its pro-oncogenic functions. As the major upstream gene of C-myc, Stat3 mediates the expression of a variety of genes, such as C-myc, Bcl-2 and cyclin-D, in response to cell stimuli and transcription activation, and therefore plays a key role in many cellular processes like growth, differentiation and apoptosis.31,32 In this study, C-myc and Stat3 exhibited a higher expression than that in the control group, which confirms that Stat3 is greatly involved in the mechanism network through which ATZ achieves its pro-oncogene effects.
From the results, it is inferred that the up-regulated Stat3 signaling pathway is responsible for the pro-oncogenic behaviors. The high expression of Stat3 up-regulates C-myc, followed by a wide activation of various signaling pathways in the tumor cells. These activated signaling pathways then induce the malignant advance of the tumor, represented as increased proliferation and invasion.
5. Conclusions
In summary, through ATZ-treated H22 cells and C57BL/6 mouse hepatic carcinoma homografts, we find that atrazine at 0.01 μM and 0.1 μM can promote H22 cell proliferation and metastasis, as well as cause C57BL/6 mouse ascites at an early stage. These processes may be regulated by Stat3. The results of this study imply that atrazine biotoxicity can produce a pro-cancer effect through a series of signaling pathways, which requires further exploration.Conflicts of interest
The authors declare that no conflict of interest exists.Acknowledgements
This work was supported by the National Natural Science Foundation of China (31700738) the China Postdoctoral Science Foundation (2016T90262), the Science & Technology Development Planning Project of Jilin Province (20140203013YY and 2014220101001742) and the Project of Jilin Province Development and Reform Commission (2041G073).References
- N. M. Filipov, L. M. Pinchuk, B. L. Boyd and P. L. Crittenden, Toxicol. Sci., 2005, 86, 324–332 CrossRef PubMed.
- L. M. Pinchuk, S. R. Lee and N. M. Filipov, Toxicol. Appl. Pharmacol., 2007, 223, 206–217 CrossRef PubMed.
- A. M. Rowe, K. M. Brundage and J. B. Barnett, Basic Clin. Pharmacol. Toxicol., 2008, 102, 139–145 CrossRef PubMed.
- X. F. Zhang, M. Q. Wang, S. Y. Gao, R. Ren, J. Zheng and Y. Zhang, BMC Med., 2011, 9, 117 CrossRef PubMed.
- J. A. Van Leeuwen, D. Waltner-Toews, T. Abernathy, B. Smit and M. Shoukri, Int. J. Epidemiol., 1999, 28, 836–840 CrossRef PubMed.
- N. Sathiakumar, P. A. MacLennan, J. Mandel and E. Delzell, Crit. Rev. Toxicol., 2011, 41, 1–34 CrossRef PubMed.
- P. Boffetta, H. O. Adami, C. Berry and J. S. Mandel, Eur. J. Cancer Prev., 2013, 22, 169–180 CrossRef PubMed.
- H. A. Young, P. K. Mills, D. G. Riordan and R. D. Cress, J. Occup. Environ. Med., 2005, 47, 1148–1156 CrossRef.
- J. A. Mcelroy, R. E. Gangnon, P. A. Newcomb, M. S. Kanarek, H. A. Anderson, J. V. Brook, A. Trentham-Dietz and P. L. Remington, J. Exposure Sci. Environ. Epidemiol., 2007, 17, 207–214 CrossRef PubMed.
- P. A. MacLennan, E. Delzell, N. Sathiakumar, S. L. Myers, H. Cheng, W. Grizzle, V. W. Chen and X. C. Wu, J. Occup. Environ. Med., 2002, 44, 1048–1058 CrossRef.
- N. A. Karrow, J. A. McCay, R. D. Brown, D. L. Musgrove, T. L. Guo, D. R. Germolec and K. L. White, Toxicology, 2005, 209, 15–28 CrossRef PubMed.
- L. T. Wetzel, L. G. Luempert, C. B. Breckenridge, M. O. Tisdel, J. T. Stevens, A. K. Thakur, P. J. Extrom and J. C. Eldridge, J. Toxicol. Environ. Health, 1994, 43, 169–182 CrossRef PubMed.
- L. Albanito, R. Lappano, A. Madeo, A. Chimento, E. R. Prossnitz, A. R. Cappello, V. Dolce, S. Abonante, V. Pezzi and M. Maggiolini, Environ. Health Perspect., 2015, 123, 493–499 Search PubMed.
- L. Albanito, R. Lappano, A. Madeo, A. Chimento, E. R. Prossnitz, A. R. Cappello, V. Dolce, S. Abonante, V. Pezzi and M. Maggiolini, Environ. Health Perspect., 2014, 122, A42 CrossRef PubMed.
- S. Lim, S. Y. Ahn, I. C. Song, M. H. Chung, H. C. Jang, K. S. Park, K. U. Lee, Y. K. Pak and H. K. Lee, PLoS One, 2009, 4, e5186 Search PubMed.
- H. J. Yun, S. K. Hyun, J. H. Park, B. W. Kim and H. J. Kwon, Mol. Cell. Biochem., 2012, 363, 281–289 CrossRef PubMed.
- G. P. Dotto, BBA, Biochim. Biophys. Acta, Rev. Cancer, 2000, 1471, M43–M56 CrossRef.
- T. Motokura, T. Bloom, H. G. Kim, H. Juppner, J. V. Ruderman, H. M. Kronenberg and A. Arnold, Nature, 1991, 350, 512–515 CrossRef PubMed.
- D. J. Lew, V. Dulic and S. I. Reed, Cell, 1991, 66, 1197–1206 CrossRef PubMed.
- S. Ghavami, M. Hashemi, S. R. Ande, B. Yeganeh, W. Xiao, M. Eshraghi, C. J. Bus, K. Kadkhoda, E. Wiechec, A. J. Halayko and M. Los, J. Med. Genet., 2009, 46, 497–510 CrossRef PubMed.
- P. Moongkarndi, C. Srisawat, P. Saetun, J. Jantaravinid, C. Peerapittayamongkol, R. Soi-Ampornkul, S. Junnu, S. Sinchaikul, S. T. Chen, P. Charoensilp, V. Thongboonkerd and N. Neungton, J. Proteome Res., 2010, 9, 2076–2086 CrossRef PubMed.
- A. Pryczynicz, M. Gryko, K. Niewiarowska, D. Cepowicz, M. Ustymowicz, A. Kemona and K. Guzinska-Ustymowicz, World J. Gastroenterol., 2014, 20, 1305–1310 CrossRef PubMed.
- H. S. T. Kai, G. S. Butler, C. J. Morrison, A. E. King, G. R. Pelman and C. M. Overall, J. Biol. Chem., 2002, 277, 48696–48707 CrossRef PubMed.
- J. Vandooren, P. E. Van den Steen and G. Opdenakker, Crit. Rev. Biochem. Mol. Biol., 2013, 48, 222–272 CrossRef PubMed.
- M. Groblewska, M. Siewko, B. Mroczko and M. Szmitkowski, Folia Histochem. Cytobiol., 2012, 50, 12–19 CrossRef PubMed.
- X. J. Li, L. Claesson-Welsh and M. Shibuya, Methods Enzymol., 2008, 443, 261–284 Search PubMed.
- H. L. Huang, H. Y. Weng, H. Zhou and L. H. Qu, Curr. Pharm. Des., 2014, 20, 6543–6554 CrossRef PubMed.
- P. C. Fernandez, S. R. Frank, L. Q. Wang, M. Schroeder, S. X. Liu, J. Greene, A. Cocito and B. Amati, Genes Dev., 2003, 17, 1115–1129 CrossRef PubMed.
- K. O. Mitchell, M. S. Ricci, T. Miyashita, D. T. Dicker, Z. Y. Jin, J. C. Reed and W. S. El-Deiry, Cancer Res., 2000, 60, 6318–6325 Search PubMed.
- R. Sears, F. Nuckolls, E. Haura, Y. Taya, K. Tamai and J. R. Nevins, Genes Dev., 2000, 14, 2501–2514 CrossRef PubMed.
- J. Lee, J. C. K. Kim, S. E. Lee, C. Quinley, H. Kim, S. Herdman, M. Corr and E. Raz, J. Biol. Chem., 2012, 287, 18182–18189 CrossRef PubMed.
- C. M. Silva, Oncogene, 2004, 23, 8017–8023 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |