
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWaste wool derived nitrogen-doped hierarchical porous carbon for selective CO2 capture†
Yao Li ac,
Ran Xua,
Xin Wangb,
Binbin Wang*b,
Jianliang Caod,
Juan Yang*ac and
Jianping Wei*ac
ac,
Ran Xua,
Xin Wangb,
Binbin Wang*b,
Jianliang Caod,
Juan Yang*ac and
Jianping Wei*ac
aSchool of Safety Science and Engineering, Henan Polytechnic University, Jiaozuo, Henan 454000, China. E-mail: yangjuanhpu@163.com; hpuwjp@163.com
bSchool of Materials Science and Engineering, Henan Polytechnic University, Jiaozuo, Henan 454000, China. E-mail: wangbb580@aliyun.com
cState Key Laboratory Cultivation Base for Gas Geology and Gas Control, Henan Polytechnic University, Jiaozuo 454000, China
dSchool of Chemistry and Chemical Engineering, Henan Polytechnic University, Jiaozuo 454000, China
First published on 30th May 2018
Abstract
The goal of this research is to develop a low-cost porous carbon adsorbent for selective CO2 capture. To obtain advanced adsorbents, it is critical to understand the synergetic effect of textural characteristics and surface functionality of the adsorbents for CO2 capture performance. Herein, we report a sustainable and scalable bio-inspired fabrication of nitrogen-doped hierarchical porous carbon by employing KOH chemical activation of waste wool. The optimal sample possesses a large surface area and a hierarchical porous structure, and exhibits good CO2 adsorption capacities of 2.78 mmol g−1 and 3.72 mmol g−1 at 25 °C and 0 °C, respectively, under 1 bar. Additionally, this sample also displays a moderate CO2/N2 selectivity, an appropriate CO2 isosteric heat of adsorption and a stable cyclic ability. These multiple advantages combined with the low-cost of the raw material demonstrate that this sample is an excellent candidate as an adsorbent for CO2 capture.
1. Introduction
As industry and society are developing rapidly, the use of fossil fuels will continue to dominate worldwide, and carbon dioxide (CO2) emissions are expected to continue increasing.1,2 CO2 emissions have become a serious environmental issue of modern civilization because of their effects on global warming and the associated consequences.3 Accordingly, carbon capture and storage (CCS) is considered to be a useful approach to mitigate CO2 emissions.4 Chemical absorption methods using aqueous amine solutions for CO2 capture have been widely used in industry,5 but they are a double-edged sword, as having strong chemical interactions between CO2 and amines results in a high capacity, but they also make the equipment suffer from serious corrosion and the system experiences a high energy penalty.6 However, solid amines, systems attaching alkylamine groups on highly hierarchical porous supports, exhibit very high CO2 adsorption capacities with simulated flue gas and practical conditions with a lower energy penalty.7,8 The beauty of these systems is that they can work under real flue gas conditions to capture very low concentrations of CO2 in the mixture gas, associated with moisture, and at elevated temperatures. However the long-term cyclic stabilities are still a concerning problem. In comparison to chemical absorption, physical adsorption based on solid adsorbents is regarded as an alternative method. Varieties of solid absorbents including silica,9 metal–organic frameworks (MOFs),10–12 organic polymers,13 zeolites14 and porous carbon materials15,16 have become the focus for CO2 capture research. Among them, porous carbon materials have attracted much attention due to their features of large surface area, high porosity, chemical and thermal stability, hydrophobicity and no toxicity, and they are regarded as the most promising CO2 capture materials.15–19As is well known, carbon adsorbents with remarkable CO2 capacity are commonly those possessing large specific surface areas and hierarchical porous architectures with abundant fine micropores (<1 nm) and complementary mesopores, in which the fine micropores (<1 nm) are important for CO2 adsorption, while the mesopores are useful for CO2 diffusion.20–22 Meanwhile, due to the fact that CO2 is weakly acidic, nitrogen elements can be introduced into the carbon frameworks. Nitrogen-doped (N-doped) sites can act as basic active sites to enhance the interactions between CO2 molecules and the carbon surface via base–acid interactions, and can subsequently improve the adsorption capacity and selectivity for CO2.22–24 At present, numerous N-doped porous carbon materials have been reported for the capture of CO2, and they are prepared by the carbonation of nitrogen-containing organic polymers.25–28 Unfortunately, the high cost of some polymer precursors and the complicated earlier synthetic processes make it hard to scale up production of these porous carbon materials and their practical applications. For reducing the cost and making practical applications viable whilst simultaneously considering environmental factors, biomass materials, especially biomass waste, have been widely used as carbon sources because of their low cost, sustainability, and abundance.29–32 In addition, biomass precursors naturally contain some useful heteroatoms such as nitrogen that will be anchored on the surface of the carbon framework, and this will enhance the CO2 adsorption capability of the carbon materials.32–34 Meanwhile, chemical activation with KOH is found to be a very common and useful method for the preparation of porous carbon materials,35–37 especially biomass-derived porous carbon materials.30–34,38 For instance, by means of KOH activation, Guo et al.38 successfully transformed waste coffee grounds into an effective porous carbon sorbent material for CO2 capture. Chen et al.30 and Yang et al.31 used KOH to activate carbonized coconut shells and develop porous carbon adsorbents for CO2 capture. Liang et al.32 reported the synthesis of N-doped microporous carbon by employing the steam-explosion of popcorn and subsequent KOH activation, in which the resulting carbon material was employed to give a high performance of CO2 capture. Zhu et al.33 synthesized N-doped nanoporous carbon from pine cones by means of chemical activation with KOH, and it exhibited a high CO2 capture capacity. Alabadi et al.34 obtained highly porous carbon from biomass carbonization and subsequent KOH activation, and it displayed a high CO2 capture capacity.
Herein, with comprehensive considerations, we report a sustainable and scalable synthesis method by employing waste wool as the raw material. Pre-carbonization and subsequent KOH activation were used to prepare the N-doped hierarchical porous carbon. We chose waste wool as the carbon precursor mainly due to the following: (I) short wool fibres are generally discarded as waste due to their unsuitability for clothing applications, which wastes a resource;39,40 (II) wool is mostly composed of amino acids that are unique renewable natural materials with high nitrogen content;40,41 (III) functionalized porous carbon can be fabricated from biomass precursors to meet various demands. The resulting waste wool derived N-doped hierarchical porous carbon prepared by chemical activation with the moderate ratio of KOH/carbon = 3 (designated as WNPC-3) possessed a large specific surface area of 1351 m2 g−1, a hierarchical porous structure with a total pore volume of 0.78 m3 g−1, a micropore volume as high as 0.54 m3 g−1 and a fine micropore volume up to 0.30 m3 g−1, and a nitrogen content of 4.41 wt%. The CO2 adsorption capacity of WNPC-3 reached 2.78 mmol g−1 at 25 °C and 3.73 mmol g−1 at 0 °C, under atmospheric pressure (1 bar). Furthermore, WNPC-3 exhibited a moderate CO2/N2 selectivity for CO2 capture from flue gas, an appropriate CO2 isosteric heat of adsorption in the range of physical adsorption, and a stable CO2 adsorption capacity after several cycles. Our study may provide a new method for the large-scale production of promising porous carbon adsorbents for selective CO2 capture.
2. Experimental
2.1 Materials
Waste wool was obtained from Henan, China. Potassium hydroxide (KOH) and hydrochloric acid (HCl) were purchased from Sinopharm Chemical Reagent Co. Ltd. China and used as received without any further purification.2.2 Sample preparation
The waste wool was thoroughly washed with distilled water and dried at 60 °C. The cleaned wool was pre-carbonized at 300 °C for 2 h with a heating rate of 3 °C min−1 under a N2 atmosphere, and was then cooled down to room temperature naturally. To simplify, the pre-carbonized wool was denoted WNC. Hereafter, in a typical KOH-activated process, the WNC (0.2 g) was mixed with the KOH activation agent (0.6 g) in 2.5 ml distilled water with a KOH/WNC mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and then the resulting mixture was treated with ultrasound for 10 min to obtain a uniform carbon suspension in the KOH solution. Subsequently, the mixture was activated under N2 protection at 600 °C for 1 h in a tubular furnace with a heating rate of 3 °C min−1. After complete activation, the as-prepared sample was washed with suitable amounts of 0.1 M HCl solution and distilled water until neutral, and then dried at 60 °C for 12 h to obtain the final product. To investigate the effect of KOH dosage on the final product, a sequence of contrast experiments were carried out under the same conditions. For the convenience of the following discussion, the wool derived N-doped porous carbon materials prepared under different KOH dosages were designated as WNPC-x where x represents the weight ratio of KOH/WNC. For comparison, the WNC directly carbonized at 600 °C for 1 h without the addition of KOH was designated as WNPC.
:1, and then the resulting mixture was treated with ultrasound for 10 min to obtain a uniform carbon suspension in the KOH solution. Subsequently, the mixture was activated under N2 protection at 600 °C for 1 h in a tubular furnace with a heating rate of 3 °C min−1. After complete activation, the as-prepared sample was washed with suitable amounts of 0.1 M HCl solution and distilled water until neutral, and then dried at 60 °C for 12 h to obtain the final product. To investigate the effect of KOH dosage on the final product, a sequence of contrast experiments were carried out under the same conditions. For the convenience of the following discussion, the wool derived N-doped porous carbon materials prepared under different KOH dosages were designated as WNPC-x where x represents the weight ratio of KOH/WNC. For comparison, the WNC directly carbonized at 600 °C for 1 h without the addition of KOH was designated as WNPC.
2.3 Characterization
Field emission scanning electron microscopy (FE-SEM, Hitachi S-4800) with an energy dispersive X-ray spectrometer (EDS) and transmission electron microscopy (TEM, JEOL-21) were employed to examine the morphology, microstructure and elemental mapping of the carbon samples. Powder X-ray diffraction (XRD) was performed on a Bruker D8 advanced X-ray diffractometer using Cu Kα radiation (λ = 0.15406 nm). Raman spectra were collected at an excitation wavelength of 514 nm using a Renishaw inVia spectrometer. Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet iS10 FTIR spectrometer. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALAB250 equipped with an Al Kα excitation source. Elemental analysis (C, H and N) was performed on a dry basis using a VarioEL III Elemental Analyzer. Nitrogen adsorption–desorption isotherms were measured on a Quantachrome NOVA1000e sorption analyzer by nitrogen physisorption at −196 °C using the conventional volumetric technique. The specific surface area (SBET) was calculated according to the multipoint Brunauer–Emmett–Teller (BET) method based on adsorption data in the relative pressure range between 0.005 to 0.05, and the total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure of about 0.99. The pore size distribution (PSD) was determined by non-local density functional theory (NLDFT) using nitrogen adsorption data and assuming a slit-shaped pore model. The volume of micropores (pores below 2 nm) and the volume of fine micropores (pores below 1 nm) were calculated based on PSD curves obtained using the NLDFT method.2.4 CO2 adsorption measurements
The CO2 adsorption isotherms of the carbon samples were determined from 0 to 1 bar using an intelligent gravimetric analyzer (IGA-003) at 0 °C and 25 °C. The sample tube was immersed in an ice-water bath for the 0 °C test and a thermostatic water bath for the 25 °C test. Prior to the CO2 adsorption test, the sample was dried and degassed in a vacuum at 200 °C for 3 h to remove any guest-molecules. After the sample was cooled down to the adsorption temperature, the dry mass was set and CO2 gas was introduced into the system.3. Results and discussion
3.1 Physicochemical characterization
In our synthetic process, two procedures including pre-carbonization and the subsequent KOH activation were carried out for the efficient fabrication of wool derived N-doped porous carbon with a hierarchical structure and high surface area; hence, the resulting carbon can be effectively used for selective CO2 capture, as schematically presented in Fig. 1. The morphology and porous structure of representative sample WNPC-3 were studied using scanning electron microscopy (SEM). As shown in Fig. 2a, the SEM image reveals that the carbon sample possesses a sponge-like morphology characterized by many nested net cavities. The surface of WNPC-3 exhibits an extremely 3D porous structure formed by KOH activation (Fig. 2b). The micropores and mesopores with different pore-sizes of WNPC-3 are interconnected, and can be widely used for CO2 capture (Fig. 2c). The overall morphology and porous structure of sample WNPC-3 were further studied using transmission electron microscopy (TEM). As can be seen in Fig. 2d and e, the low magnification view confirms that WNPC-3 maintains a continuous sponge-like morphology with a cross-linked porous texture, which is in agreement with previous SEM results. The high-resolution transmission electron microscopy (HR-TEM) image (Fig. 2f) shows that the randomly distributed worm-like micropores are formed by the stacking of disordered graphene layers.16 The selected area electron diffraction (SAED) pattern of WNPC-3 (inset in Fig. 2f) exhibits typical diffuse rings, reflecting the amorphous nature of carbon.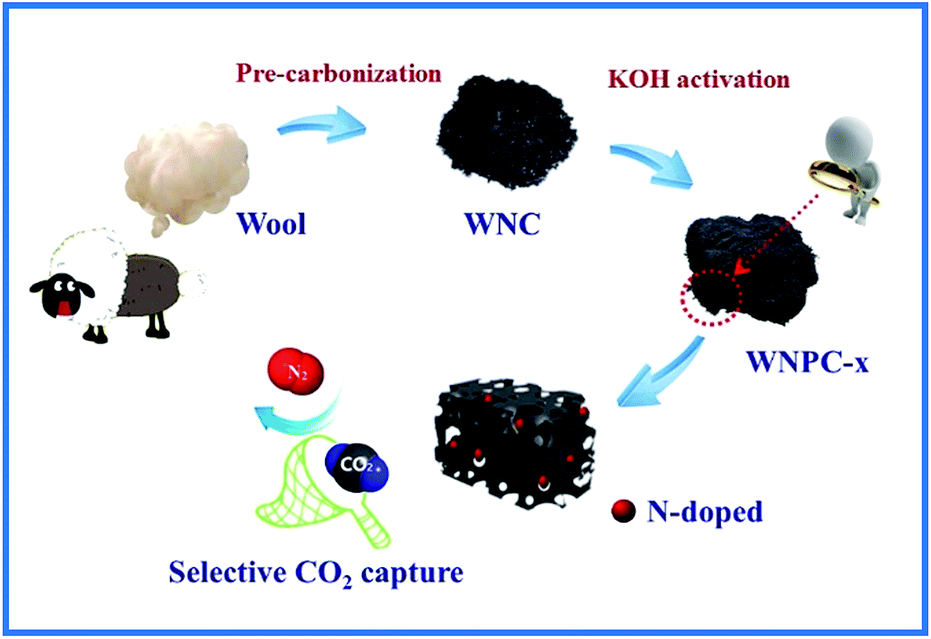 | ||
| Fig. 1 Schematic illustrating the fabrication of WNPC-x and uses in selective CO2 capture. | ||
 | ||
| Fig. 2 (a–c) FE-SEM images, (d and e) TEM images and (f) HR-TEM image of the sample WNPC-3. The inset shows the SAED pattern. | ||
To determine the crystallite structure of a sample of WNPC-3, XRD and Raman spectra were recorded. The two broad peaks at around 22.5° and 43° in the XRD spectrum (Fig. 3a) correspond to typical reflections of the (002) and (100) planes of graphite, indicating the amorphous structure of WNPC-3.42,43 The Raman spectrum (Fig. 3b) exhibits two characteristic peaks for WNPC-3 at around 1330 cm−1 (D-band) and 1590 cm−1 (G-band), which correspond to disordered carbon and the ordered graphite lattice, respectively.42–44 The intensity ratio of the two bands (ID/IG) is known to be proportional to the number of defect sites in the graphitic carbon, that is, the higher the ID/IG ratio is, the lower the graphitization is.43,44 The calculated ID/IG ratio of WNPC-3 is 1.12, confirming that many defects exist in the carbon structure, which is consistent with the XRD results. The chemical bonding information of WNPC-3 was investigated using Fourier transform infrared (FT-IR) spectroscopy (Fig. 3c). The appearance of a broad absorption band at around 3430 cm−1 can be assigned to N–H and/or O–H stretching vibrations.34 The existence of a band at 1620 cm−1 indicates the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in aromatic rings for the activated carbon.27 The peak at around 1200–1000 cm−1 relates to the stretching vibrations of C–N and/or C–O.25 The weak peak between 900 and 650 cm−1 can be assigned to N–H and/or C–H out-of-plane deformation vibrations.25,26 Therefore, the FT-IR spectra preliminarily confirm that N–H and C–N species are in WNPC-3.
C stretching vibrations in aromatic rings for the activated carbon.27 The peak at around 1200–1000 cm−1 relates to the stretching vibrations of C–N and/or C–O.25 The weak peak between 900 and 650 cm−1 can be assigned to N–H and/or C–H out-of-plane deformation vibrations.25,26 Therefore, the FT-IR spectra preliminarily confirm that N–H and C–N species are in WNPC-3.
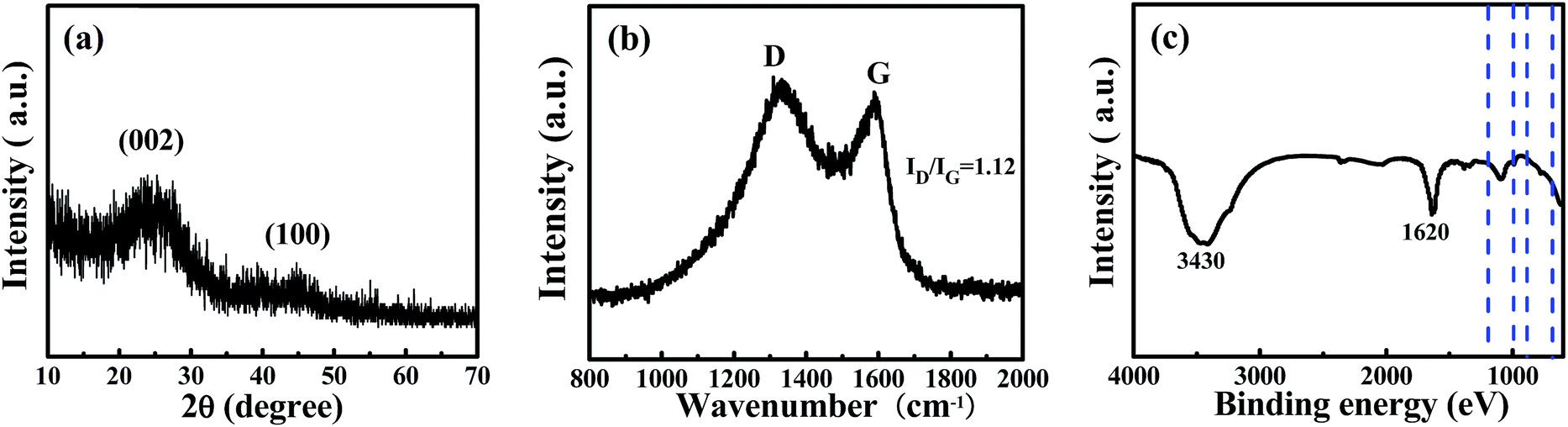 | ||
| Fig. 3 (a) XRD, (b) Raman and (c) FT-IR spectra of the sample of WNPC-3. | ||
To further analyze the surface chemical states and the composition of WNPC-3, X-ray photoelectron spectroscopy (XPS) was employed to systematically investigate the intrinsic nature of the C, N and O species in WNPC-3. The full survey spectrum clearly shows the existence of three typical peaks for C 1s, N 1s and O 1s in the sample of WNPC-3 (Fig. 4a). The representative spectrum of C 1s can be deconvoluted into five peaks (Fig. 4b). The peak at about 285.9 eV confirms the presence of C–N in WNPC-3, while the dominant peak at 284.7 eV could be attributed to C–C.26,45 The peaks at about 285.4 eV, 286.7 eV and 287.8 eV correspond to C–OH,26 C–O34 and CO,41 respectively. The high resolution N 1s spectrum reveals the chemical states of nitrogen atoms in WNPC-3 (Fig. 4c). The deconvoluted spectrum displays three nitrogen species with binding energies of 400.1 eV, 398.5 eV and 402.8 eV, which can be attributed as pyrrolic-N/pyridonic-N, pyridinic-N and pyridine N-oxides, respectively.25,26,41,45 Pyrrolic-N and pyridonic-N are hard to distinguish from each other within the accuracy of XPS measurements, however, considering the greater stability of pyridonic-N compared to pyrrolic-N, pyridonic-N is more likely to be present in WNPC-3.25–27 The corresponding peak areas analysis shows that the amount of pyridonic-N is higher that of the two others, and it has been reported that pyridonic-N acts as a main anchor for CO2 capture.16,25,26 Three characteristic peaks are observed at 531.3 eV, 533.1 eV and 534.9 eV in the deconvoluted O 1s spectrum (Fig. 4d), which can be ascribed to OC, O–C, and chemisorbed O2 and/or H2O bound to WNPC-3 respectively.26,41,45
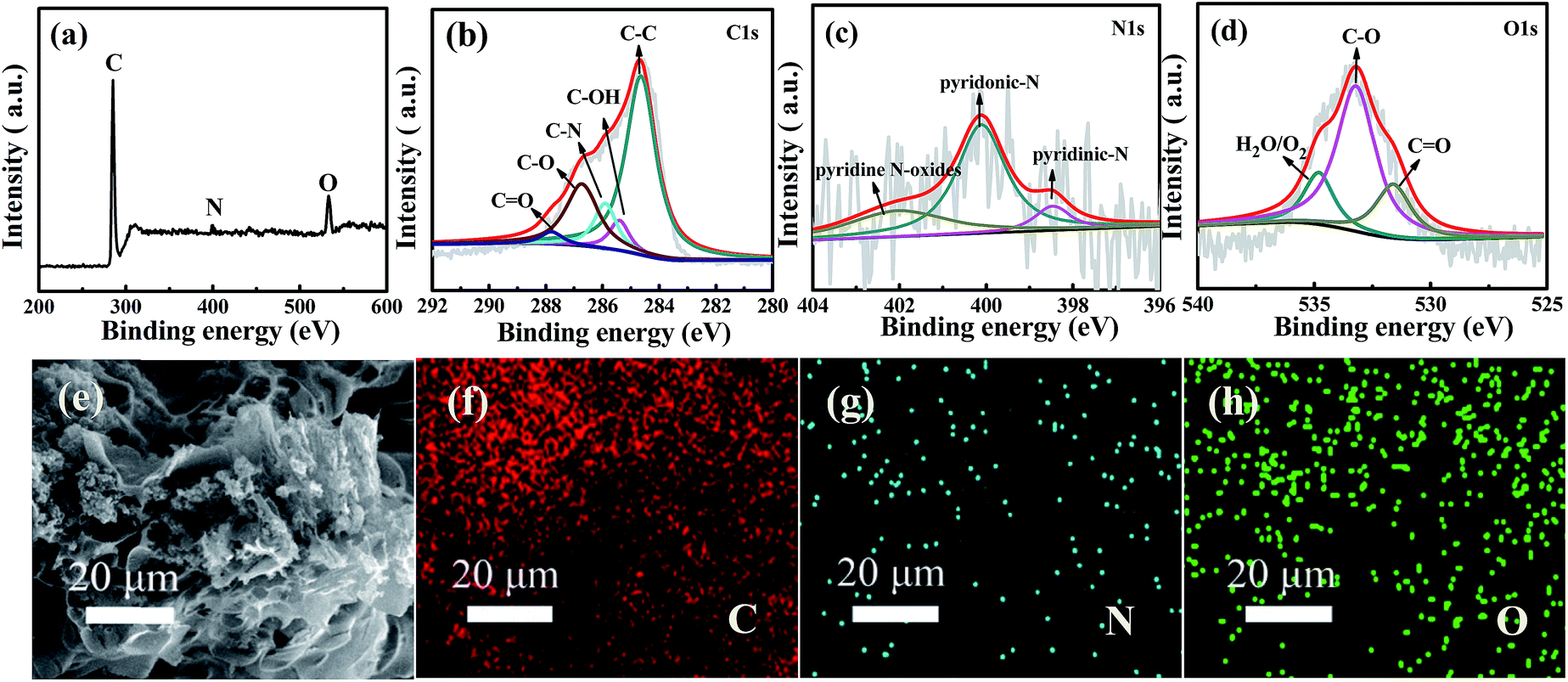 | ||
| Fig. 4 XPS spectra of WNPC-3: (a) high-resolution full survey spectrum of (b) C 1s, (c) N 1s and (d) O 1s; (e–h) SEM image and the corresponding EDS element mappings of (f) C, (g) N and (h) O for WNPC-3. | ||
As previously reported,46,47 the C–O and CO functional groups may play a important role in CO2 adsorption and CO2/N2 selectivity, due to enhanced interaction forces between the O-decorated carbon framework and CO2 molecules. Additionally, the SEM energy dispersive X-ray spectroscopy (EDS) mapping analysis for WNPC-3 shows that the elements of C, N and O are uniformly distributed in the sample (Fig. 4e–h).
The chemical compositions of a sample of WNPC-3 as well as the other samples of WNPC, WNPC-1 and WNPC-5 were further investigated by C, N and O elemental analysis, as presented in Table 1. As can be seen, the nitrogen content decreases from 11.25 wt% (un-activated WNPC) to 3.70 wt% (strongly activated WNPC-5). Approximately 4.57 wt% of nitrogen is detected for the low-activated WNPC-1, while about 4.14 wt% is detected for moderately activated WNPC-3. The nitrogen content decreases with the increasing KOH/WNC weight ratio, which is consistent with the previously reported literature.23–27 There is a rapid drop in nitrogen content from un-activated WNPC to the activated WNPC-1. The nitrogen content decreases slowly when the KOH/WNC ratio increases from 1 to 5, especially with a KOH/WNC ratio from 1 to 3. However, with the nitrogen content decrease, the carbon content also decreases concomitantly with the KOH activation, while the hydrogen content appears to be relatively unchanged. Furthermore, the oxygen content is increased with the increase in the KOH/WNC ratio in the samples, from WNCP to WNCP-1 to WNCP-3 to WNCP-5, because the KOH activation promotes the oxidation process of the carbon.48 The KOH activated carbon materials contain more O atoms, which could be attributed to the reaction of carbon with CO2 evolved from K2CO3 and K2O, as well as oxygen chemisorption from air.49,50 The increased oxygen content from WNCP to WNCP-5 should be due to the increased KOH/WNC ratio, which is consistent with the behavior of KOH activated carbon materials we have reported previously.26,45 Therefore the activated carbon materials can preserve more oxygen functional groups such as CO and C–O.46,51
The porosities of WNPC and the WNPC-x series were investigated by nitrogen adsorption–desorption isothermal analysis, as presented in Fig. 5, and the textural characteristics are shown in Table 2. The salt concentration (such as the KOH amount used in activation) can alter the shape of the adsorption–desorption isotherm, and thus alter the specific surface area and especially the pore-size distribution of the activated carbon materials.36–38 As can be seen from Fig. 5a, the un-activated sample WNPC has a very low nitrogen adsorption in the low pressure range and does not display a hysteresis loop in the middle pressure range, which reveals that only a small number of micropores exist in the WNPC. In contrast, all of the activated WNPC-x series samples display significantly enhanced nitrogen adsorption, exhibiting typical type-I/IV isothermal curves with sharp increases at low pressure and obvious hysteresis loops at middle pressures, which implies that micropores and mesopores all exist in WNPC-x. As the ratio of KOH/WNC increased from 1 to 3 and 5, the adsorption curve increased steeply at low pressure, indicating the development of the specific surface area and microporosity.36,38 Moreover, with the increasing KOH/WNC ratio, the isotherm knees of the activated samples are gradually widened to some extent, indicating the generation of some large micropores and/or small mesopores in the carbon framework.52,53 Fig. 5b shows the corresponding pore size distribution (PSD) curves of all samples, and these were calculated using the NLDFT model. The pore size of WNPC mainly centers at 0.93 nm indicating a microporous structure. The PSD curves of the activated WNPC-x series are distributed in the range of 0.50–4.00 nm indicating hierarchical porous structures. Interestingly, all activated samples (WNPC-1, WNPC-3 and WNPC-5) possess an important fraction of fine micropores (<1 nm). In particular, the moderately activated WNPC-3 exhibits an extraordinarily strong peak at 0.52 nm, indicating a large number of fine micropores (<1 nm) in the carbon framework. Furthermore, WNPC-3 also has two moderately strong peaks at 0.84 nm and 1.19 nm, and one weak peak at around 3.5 nm. In contrast, the low-activated WNPC-1 only exhibits two moderately strong peaks at 0.50 nm and 1.39 nm, and one weak peak at about 2.5 nm. Similarly, the strongly activated WNPC-5 only has two moderately strong peaks at 0.57 nm and 1.41 nm, and two weak peaks at around 2.5 nm and 4 nm. However, these PSD curves are slightly enlarged with the increasing KOH/WNC ratio, and this is probably caused by merging of neighboring small micropores into mesopores or larger ones due to the continuous KOH activation.38,52–54 The pore structures can be developed by the evolution of gaseous by-products during KOH activation.36,38 To the best of our knowledge, the chemical reaction between KOH and carbon proceeds as follows: 6KOH + C → 2K + 3H2 + 2K2CO3, followed by K2CO3 decomposition and the reaction of K/K2CO3/CO2 with carbon.36,55 Thus with increasing KOH activation, pore widening is observed. The progressive KOH etching on the carbon results in a gradual increase in both the specific surface area and the total pore volume. From Table 2, it can be observed that the strongly activated WNPC-5 has the largest specific area (SBET) and total pore volume (Vtotal) of 1420 m2 g−1 and 0.86 m3 g−1, respectively, which are larger than those of un-activated WNPC (SBET = 447 m2 g−1 and Vtotal = 0.20 m3 g−1), low-activated WNPC-1 (SBET = 1010 m2 g−1 and Vtotal = 0.57 m3 g−1) and moderately activated WNPC-3 (SBET = 1352 m2 g−1 and Vtotal = 0.78 m3 g−1). However, to the best of our knowledge, besides the specific surface area and total pore volume, the micropore volume (Vmicro) and fine micropore volume (PV1 nm) are all very important for CO2 capture.53,54,56–58 By analyzing the cumulative pore volume against the pore size (Fig. 5c), it is worth noting that WNPC-3 possesses the largest Vmicro and PV1 nm of 0.54 m3 g−1 and 0.30 m3 g−1, respectively, which are much larger than those of WNPC (Vmicro = 0.18 m3 g−1 and PV1 nm = 0.11 m3 g−1), WNPC-1 (Vmicro = 0.37 m3 g−1 and PV1 nm = 0.15 m3 g−1) and WNPC-5 (Vmicro = 0.52 m3 g−1 and PV1 nm = 0.18 m3 g−1), as can be seen in Table 2. Furthermore, the percentage of microporosity (Vmicro/Vtotal) for WNPC-3 can reach 69%, which is higher than other activated carbon samples (WNPC-1 is 64% and WNPC-5 is 60%). Although the surface area and total pore volume of WNPC-3 are smaller than those of WNPC-5, the CO2 adsorption capability requires the co-operation of the micropore volume, the fine micropore volume and the percentage of microporosity, which can effectively guarantee the CO2 capture performance for WNPC-3.
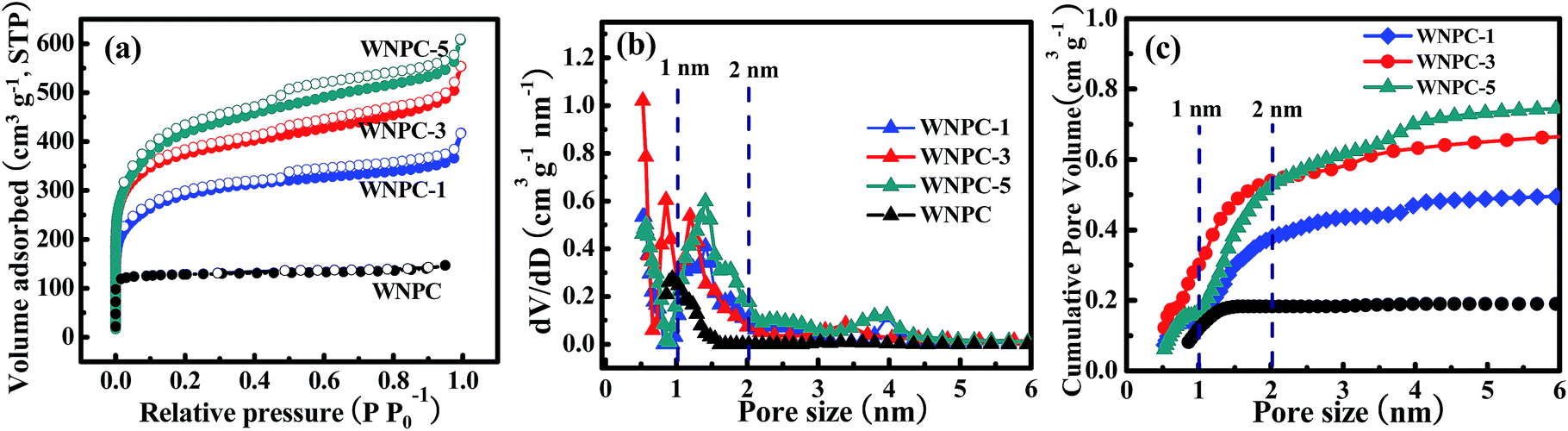 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherms, (b) PSDs and (c) cumulative pore volumes against pore sizes for wool-derived carbon materials. | ||
| Sample | Textural characteristics | CO2 uptake | |||
|---|---|---|---|---|---|
| SBETa | Vtotalb | Vmicroc | PV1 nmd | 25 °C (mmol g−1) | |
| a SBET is the specific surface area calculated by BET equations based on the adsorption data in the P/P0 range from 0.005 to 0.05.b Vtotal is the total pore volume obtained at P/P0 ∼ 0.99.c Vmicro is the cumulative micropore volume (pore size < 2 nm) analyzed using the NLDFT method.d PV1 nm is the cumulative fine micropore volume (pore size < 1 nm) analyzed using the NLDFT method. | |||||
| WNCP | 447 | 0.22 | 0.18 | 0.11 | 1.48 |
| WNCP-1 | 1010 | 0.57 | 0.37 | 0.15 | 2.33 |
| WNCP-3 | 1352 | 0.78 | 0.54 | 0.30 | 2.78 |
| WNCP-5 | 1420 | 0.86 | 0.52 | 0.18 | 2.35 |
3.2 Adsorption performance
The CO2 adsorption isotherms for the carbon samples were investigated at 25 °C under atmospheric pressure (1 bar), as shown in Fig. 6a. As Gadipelli et al.35,36,59 previously reported, the low pressure (∼1 bar) adsorption behavior is mainly dependent on the narrow pore size distribution in the micropore region, due to the caging effect, particularly for those pores smaller than 1 nm.20,59–61 WNPC-3 exhibits a significant fine micropore volume (Table 2), which can be attributed to its large number of fine micropores (centred on 0.52 nm and 0.84 nm). Therefore, WNPC-3 demonstrates a good CO2 adsorption capacity of 2.78 mmol g−1 (at 1 bar and 25 °C), which can be compared with those porous carbon materials with similar specific surface areas and/or pore-size distribution structures (Table S1†). Among the WNPC and WNPC-x samples, the corresponding relationship between CO2 uptake and fine micropore volume can be clearly seen (Fig. 6b). WNPC-3 exhibits an obviously better CO2 uptake (2.78 mmol g−1) than those of WNCP (1.48 mmol g−1), WNPC-1 (2.33 mmol g−1) and WNPC-5 (2.35 mmol g−1) (Table 2). The presence of a large number of fine micropores is interesting for gas storage applications.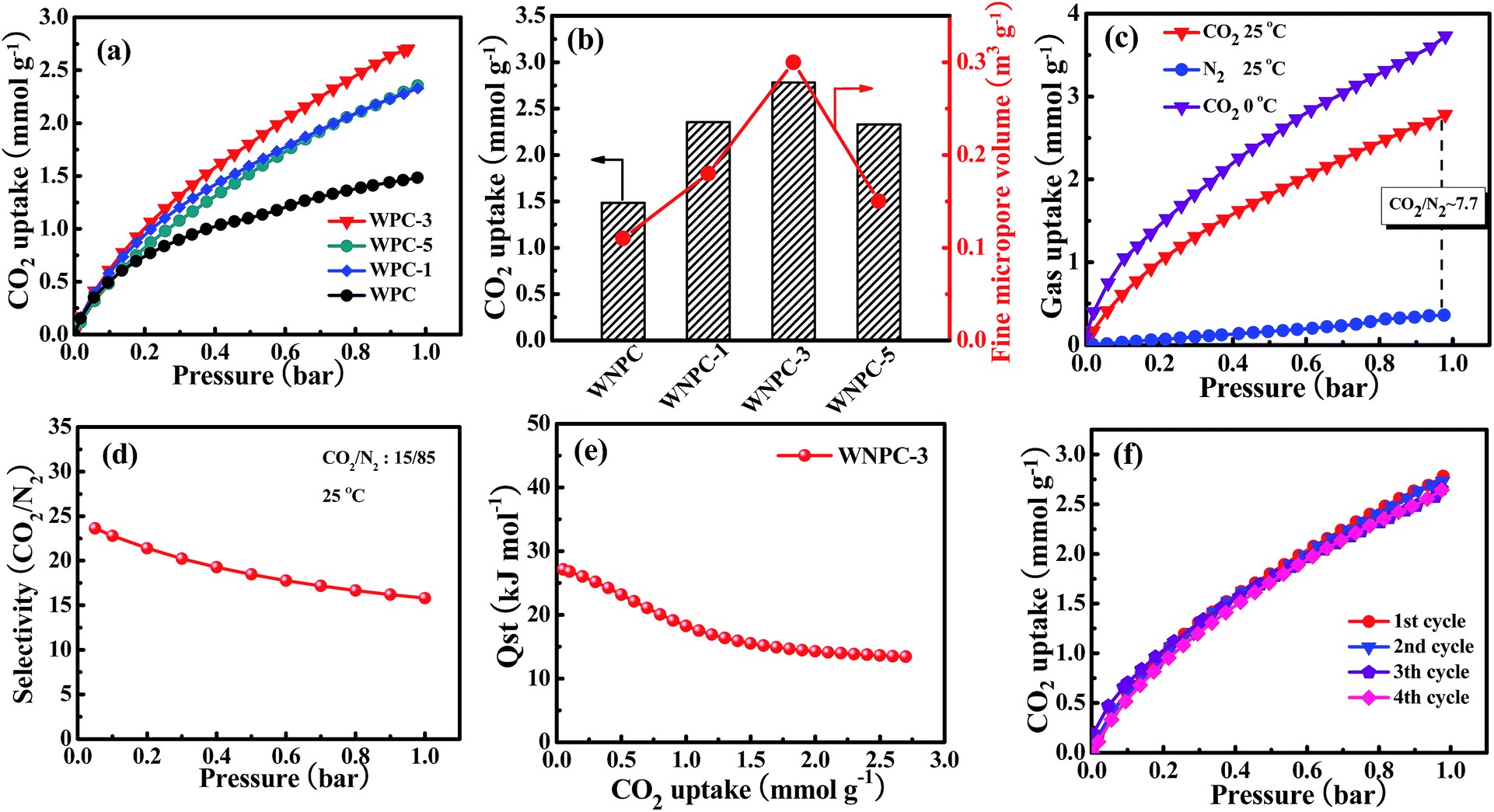 | ||
| Fig. 6 (a) CO2 adsorption isotherms at 25 °C and (b) corresponding relationship of CO2 uptake and fine micropore volume for the carbon samples; (c) CO2 and N2 adsorption isotherms at 25 °C and 0 °C, (d) IAST selectivity for CO2 over N2 collected at 25 °C (assuming CO2/N2 is 15/85), (e) CO2 isosteric heats of adsorption and (f) CO2 multicycle adsorption isotherms at 25 °C for WNPC-3. | ||
Therefore, taking WNPC-3 as the representative sample, the CO2 adsorption performance was measured at 0 °C and 1.0 bar, and it exhibited a larger CO2 adsorption capacity of 3.72 mmol g−1 than that at 25 °C (Fig. 6c), indicating that the CO2 adsorption is an exothermic process. As can be seen, the CO2 uptake increases steadily without a distinct plateau in the isotherm for the entire pressure range, indicating that more CO2 can be adsorbed at higher pressures. Thus, the CO2 capture performance for WNPC-3 was further measured at a high pressure of 10 bar (under 0 °C), and it could reach up to 10.39 mmol g−1 (Fig. S1†).
Besides the high uptake of CO2, an excellent CO2 absorbent should also demonstrate high CO2 selectivity against other gases such as N2. As shown in Fig. 6c, the adsorption capacity of N2 by WNPC-3 was also measured at 25 °C under 1 bar pressure. Clearly, the uptake of N2 is just 0.36 mmol g−1, which is much lower than the uptake of CO2 under the same conditions. In fact, the CO2 uptake is about 7.7 times the N2 adsorption (Fig. 6c), and such a property is essential for CO2 capture. The CO2/N2 selectivity was measured using Henry’s law constants, which can be calculated from the ratio of the initial slopes for the pure gas isotherms.62–64 According to the initial slope calculation, the selectivity for CO2 over N2 for WNPC-3 is about 23 at 25 °C (Fig. S2†), indicating a high selectivity towards CO2. In addition, we further used ideal adsorbed solution theory (IAST)65–68 to predict CO2/N2 selectivity in binary gas mixtures using only pure gas isotherms measured at 25 °C. In the calculations, the ratio of CO2/N2 is assumed to be 15/85, which is a typical component of flue gas.6,37,66,67,69 Herein, the dual-site Langmuir model (DL) and the single-site Langmuir model (L) were chosen to fit the CO2 and N2 adsorption isotherms, respectively, and then DL/L-IAST was utilized to estimate the CO2/N2 selectivity of WNPC-3 (Fig. S3†). As depicted in Fig. 6d, the IAST selectivity for CO2/N2 of WNPC-3 can reach 16 at 25 °C and 1 bar, indicating that it is an excellent prospect for industrial applications. To the best of our knowledge, CO2 exhibits a higher quadrupole moment and polarizability than N2, and in comparison, N2 is much more chemically inert and exhibits lower polarizability than CO2.68 Therefore, the selectivity is influenced by the presence of N in the carbon framework, because N will create some electrostatic microdomains within the pore volume, which can play an important role in segregating the quadrupolar fluids N2 and CO2 through dipole and quadrupole moments.37 Furthermore, the preserved oxygen functional groups in the carbon framework also play a crucial role in CO2/N2 selectivity, due to the enhanced interaction forces between the O-decorated carbon framework and CO2 molecules.46,47 However, comparing with some other carbon materials reported in the literature (Table S1†), the CO2/N2 selectivity of WNPC-3 is just moderate. The moderate CO2/N2 selectivity can be explained by the large number of micropores (ca. 0.52–1.19 nm), in which N2 may experience higher binding energies from the carbon surface through van der Waals’ or electrostatic forces.37 In addition, the selectivity of WNPC-3 was obtained from the pure gas adsorption isotherms, which assumes the interactions between the fluid molecules are negligible; therefore, the selectivity value can be regarded as a practical maximum.37
As we know, the overall effects of micropore size and N-/O-dopants in the porous carbon materials are to increase the adsorbent–adsorbate interaction energy, which may incur a greater energy penalty for regeneration.38,68 Therefore, to understand the interaction strength of CO2 molecules with WNPC-3, the isosteric heat of adsorption (Qst) for WNPC-3 was calculated using a variant of the Clausius–Clapeyron equation,38,63,66,70 which was accomplished by collecting CO2 adsorption isotherms at 0 °C and 25 °C and fitting the data to the DL model (Fig. S3†). As displayed in Fig. 6e, the calculated Qst for WNPC-3 is in the range of 27–13 kJ mol−1, with the CO2 uptake varying from 0.05 to 2.7 mmol g−1. At low CO2 loading, the high initial Qst value leads to a preferential adsorption of CO2 over N2, which can be attributed to the CO2 molecules being selectively adsorbed on the surface active nitrogen sites15,16 and the multiple pore wall interactions with CO2 molecules.6,13 At high CO2 loading, as the active nitrogen sites and microporous surface became gradually saturated, the availability of sorption sites progressively decreased, leading to a lowering in the adsorption heat.16,24 We note that the Qst of WNPC-3 decreases mildly with the increasing CO2 loading until a near-plateau is achieved, which suggests that the binding energies of CO2 in the pores are heterogeneous.30,31 However, the Qst value for WNPC-3 is comparable to that for un-doped carbon,38 which is much below the energy of covalent bonds and consistent with the fully reversible CO2 isotherm, suggesting that the CO2 release can be achieved by a pressure drop without heating.13,30,31
From the viewpoint of practical applications, the recyclability of an adsorbent is important, and thus, the multicycle adsorptions of CO2 on WNPC-3 were conducted at 25 °C. As can be seen in Fig. 6e, the CO2 uptake remains almost unchanged in four cycles, indicating that WNPC-3 has excellent recyclability and stability. It is worth noting that, after each adsorption cycle, the saturated WNPC-3 released CO2 by a pressure drop without heating, which should be attributed to its appropriate Qst values.
4. Conclusions
In summary, N-doped hierarchical porous carbon has been successfully synthesized in this work using renewable waste wool as the raw material. The resulting optimal sample WNPC-3 was endowed with a high specific surface area of 1351 m2 g−1, a hierarchical porous structure with a total pore volume of 0.78 m3 g−1, a micropore volume up to 0.54 m3 g−1 and a fine micropore volume as high as 0.30 m3 g−1, and a certain number of nitrogen and oxygen functional groups. The conspicuous synergetic effect of the textural characteristics and functional groups provides an excellent guarantee for CO2 capture performance. We demonstrate that micropores are principally responsible for CO2 capture at low pressure (1 bar). Remarkably, WNPC-3 exhibits a good CO2 adsorption capability of 3.72 mmol g−1 under 0 °C at atmospheric pressure (1 bar). Furthermore, an appropriate Qst leads to WNPC-3 possessing an excellent CO2/N2 selectivity and a significant regenerability. More significantly, here the strategy we used provides an excellent method to make best use of the low-cost yet abundant resources endowed by nature to fabricate sustainable carbon adsorbents for selective CO2 capture, thus opening new ways to set up an economical platform for practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Henan Province Colleges and Universities Key Research Project (18A620002, 18A150005), China Postdoctoral Science Foundation (2018M632775), the Henan Polytechnic University Doctor Foundation (660107/017), the Academician Workstation Innovation Foundation (13160093/010), the Program for Innovative Research Team in University of Ministry of Education of China (IRT_16R22) and the NSFC (No. U1704146).Notes and references
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef PubMed.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef PubMed.
- J. Kou and L. B. Sun, Ind. Eng. Chem. Res., 2016, 55, 10916–10925 CrossRef.
- S. Gadipelli, H. A. Patel and Z. Guo, Adv. Mater., 2015, 27, 4903–4909 CrossRef PubMed.
- S. Gadipelli, Y. Lu, N. T. Skipper, T. Yildirim and Z. Guo, J. Mater. Chem. A, 2017, 5, 17833–17840 Search PubMed.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 Search PubMed.
- J. Yu, L. H. Xie, J. R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef PubMed.
- S. Gadipelli, W. Travis, W. Zhou and Z. Guo, Energy Environ. Sci., 2014, 7, 2232–2238 Search PubMed.
- S. Gadipelli and Z. Guo, Chem. Mater., 2014, 26, 6333–6338 CrossRef.
- T. Islamoglu, S. Behera, Z. Kahveci, T. D. Tessema, P. Jena and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2016, 8, 14648–14655 Search PubMed.
- T. H. Nguyen, S. Kim, M. Yoon and T. H. Bae, ChemSusChem, 2016, 9, 455–461 CrossRef PubMed.
- N. Fu, H. M. Wei, H. L. Lin, L. Li, C. H. Ji, N. B. Yu, H. J. Chen, S. Han and G. Y. Xiao, ACS Appl. Mater. Interfaces, 2017, 9, 9955–9963 Search PubMed.
- M. Yang, L. Guo, G. Hu, X. Hu, J. Chen, S. Shen, W. Dai and M. Fan, Ind. Eng. Chem. Res., 2016, 55, 757–765 CrossRef.
- A. S. Jalilov, G. Ruan, C. C. Hwang, D. E. Schipper, J. J. Tour, Y. Li, H. Fei, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 1376–1382 Search PubMed.
- J. Zhang, X. Wang, G. Qi, B. Li, Z. Song, H. Jiang, X. Zhang and J. Qiao, Carbon, 2016, 96, 864–870 CrossRef.
- H. Wei, W. Qian, N. Fu, H. Chen, J. Liu, X. Jiang, G. Lan, H. Lin and S. Han, J. Mater. Sci., 2017, 52, 10308–10320 CrossRef.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 Search PubMed.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 Search PubMed.
- J. W. F. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W. G. Bae, L. Pan, J. B. H. Tok, J. Wilcox and Z. Bao, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef PubMed.
- G. Sethia and A. Sayari, Carbon, 2015, 93, 68–80 CrossRef.
- L. Liu, Z. H. Xie, Q. F. Deng, X. X. Hou and Z. Y. Yuan, J. Mater. Chem. A, 2017, 5, 418–425 Search PubMed.
- X. Fan, L. Zhang, G. Zhang, Z. Shu and J. Shi, Carbon, 2013, 61, 423–430 CrossRef.
- Y. Li, B. Zou, C. Hu and M. Cao, Carbon, 2016, 99, 79–89 CrossRef.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef.
- D. Lee, C. Z. Zhang, C. Wei, B. L. Ashfeld and H. Gao, J. Mater. Chem., 2013, 1, 14862–14867 RSC.
- P. Wang, Y. Guo, C. Zhao, J. Yan and P. Lu, Appl. Energy, 2017, 201, 34–44 CrossRef.
- J. Chen, J. Yang, G. Hu, X. Hu, Z. Li, S. Shen, M. Radosz and M. Fan, ACS Sustainable Chem. Eng., 2016, 4, 1439–1445 CrossRef.
- M. Yang, L. Guo, G. Hu, X. Hu, L. Xu, J. Chen, W. Dai and M. Fan, Environ. Sci. Technol., 2015, 49, 7063–7070 CrossRef PubMed.
- T. Liang, C. Chen, X. Li and J. Zhang, Langmuir, 2016, 32, 8042–8049 CrossRef PubMed.
- B. Zhu, C. Shang and Z. Guo, ACS Sustainable Chem. Eng., 2016, 4, 1050–1057 CrossRef.
- A. Alabadi, S. Razzaque, Y. Yang, S. Chen and B. Tan, Chem. Eng. J., 2015, 281, 606–612 CrossRef.
- S. Gadipelli and Z. X. Guo, Prog. Mater. Sci., 2015, 69, 1–60 CrossRef.
- S. Gadipelli, J. Burressa and T. Yildirim, Energy Environ. Sci., 2012, 5, 6453–6459 Search PubMed.
- K. V. Kumar, S. Gadipelli, K. Preuss, H. Porwal, T. Zhao, Z. X. Guo and M. M. Titirici, ChemSusChem, 2017, 10, 199–209 CrossRef PubMed.
- W. Travis, S. Gadipelli and Z. Guo, RSC Adv., 2015, 5, 29558–29562 RSC.
- G. Cui, Y. Dong, B. Li, Y. Li and P. Wang, Fibers Polym., 2017, 18, 713–719 CrossRef.
- G. Cui, Y. Dong, Y. Li, W. Shen and Z. Chen, Color. Technol., 2017, 133, 200–208 Search PubMed.
- L. Zhou, H. Cao, S. Zhu, L. Hou and C. Yuan, Green Chem., 2015, 17, 2373–2382 RSC.
- H. Wei, H. Chen, N. Fu, J. Chen, G. Lan, W. Qian, Y. Liu, H. Lin and S. Han, Electrochim. Acta, 2017, 231, 403–411 CrossRef.
- D. Liu, W. Zhang, H. Lin, Y. Li, H. Lu and Y. Wang, RSC Adv., 2015, 5, 19294–19300 RSC.
- T. Wei, X. Wei, Y. Gao and H. Li, Electrochim. Acta, 2015, 169, 186–194 CrossRef.
- Y. Li and M. Cao, Chem.–Asian J., 2015, 10, 1496–1504 CrossRef PubMed.
- J. Wang, R. Krishna, X. Wu, Y. Sun and S. Deng, Langmuir, 2015, 31, 9845–9852 CrossRef PubMed.
- J. Wang, R. Krishna, J. Yang and S. Deng, Environ. Sci. Technol., 2015, 49, 9364–9373 CrossRef PubMed.
- Y. Sudaryanto, S. B. Hartono, W. Irawaty, H. Hindarso and S. Ismadji, Bioresour. Technol., 2006, 97, 734–739 CrossRef PubMed.
- R. Q. Sun, L. B. Sun, Y. Chun and Q. H. Xu, Carbon, 2008, 46, 1757–1764 CrossRef.
- S. J. Park and W. Y. Jung, J. Colloid Interface Sci., 2002, 250, 93–98 CrossRef PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef.
- J. Cai, J. Qi, C. Yang and X. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 3703–3711 Search PubMed.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 82, 297–303 CrossRef.
- C. Zhang, W. Song, Q. Ma, L. Xie, X. Zhang and H. Guo, Energy Fuels, 2016, 30, 4181–4190 CrossRef.
- M. A. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41, 267–275 CrossRef.
- P. Cheng, T. Li, H. Yu, L. Zhi, Z. Liu and Z. Lei, J. Phys. Chem. C, 2016, 120, 2079–2086 Search PubMed.
- N. P. Wickramaratne and M. Jaroniec, Adsorption, 2014, 20, 287–293 CrossRef.
- N. P. Wickramaratne, J. Xu, M. Wang, L. Zhu, L. Dai and M. Jaroniec, Chem. Mater., 2014, 26, 2820–2828 CrossRef.
- S. Gadipelli, V. Krungleviciute, Z. X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335–342 Search PubMed.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523–2529 RSC.
- V. Presser, J. McDonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 Search PubMed.
- J. Wang, I. Senkovska, M. Oschatz, M. R. Lohe, L. Borchardt, A. Heerwig, Q. Liu and S. Kaskel, ACS Appl. Mater. Interfaces, 2013, 5, 3160–3167 Search PubMed.
- S. M. Mahurin, J. Gorka, K. M. Nelson, R. T. Mayes and S. Dai, Carbon, 2014, 67, 457–464 CrossRef.
- R. Narasimman, S. Vijayan and K. Prabhakaran, RSC Adv., 2014, 4, 578–582 RSC.
- T. Islamoglu, M. G. Rabbani and H.M. El-Kaderi, J. Mater. Chem. A, 2013, 1, 10259–10266 Search PubMed.
- S. Bandyopadhyay, A. G. Anil, A. James and A. Patra, ACS Appl. Mater. Interfaces, 2016, 8, 27669–27678 Search PubMed.
- Y. Shi, J. Zhu, X. Liu, G. Geng and L. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20340–20349 Search PubMed.
- S. Gadipelli and Z. X. Guo, ChemSusChem, 2015, 8, 2123–2132 CrossRef PubMed.
- R. Li, X. Ren, X. Feng, X. Li, C. Hu and B. Wang, Chem. Commun., 2014, 50, 6894–6897 RSC.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CO2 adsorption data, CO2/N2 selectivity ratios, ideal adsorption solution theory calculations, isosteric heat of adsorption calculations and additional figures. See DOI: 10.1039/c8ra02701c |
| This journal is © The Royal Society of Chemistry 2018 |