
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new efficient domino approach for the synthesis of pyrazolyl-phthalazine-diones. Antiradical activity of novel phenolic products†
Dušica Simijonović *a,
Zorica D. Petrovića,
Vesna M. Milovanovića,
Vladimir P. Petrovića and
Goran A. Bogdanovićb
*a,
Zorica D. Petrovića,
Vesna M. Milovanovića,
Vladimir P. Petrovića and
Goran A. Bogdanovićb
aUniversity of Kragujevac, Faculty of Science, Department of Chemistry, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: dusicachem@kg.ac.rs; Tel: +381-34-336-223 Tel: +381-34-335-039
bVinča Institute of Nuclear Sciences, University of Belgrade, P. O. Box 522, 11001 Belgrade, Serbia
First published on 4th May 2018
Abstract
Pyrazolyl-phthalazine-dione derivatives (PPDs) were synthetized in the ionic liquid catalyzed one-pot multicomponent reaction of acetylacetone, 2,3-dihydrophthalazine-1,4-dione, and different aldehydes in moderate to good yields. Six new PPDs were obtained, and the crystal structure of 2-acetyl-1-(4-fluorophenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-4) was determined. The most interesting structural features of the novel PPD-4 is the formation of a rather short intermolecular distance between the F atom of one molecule and the midpoint of the neighbouring six-membered heterocyclic ring. This interaction arranges all molecules into parallel supramolecular chains. UV-Vis spectra of all PPDs were acquired and compared to the simulated ones obtained with TD-DFT. All synthetized compounds were subjected to evaluation of their in vitro antioxidative activity using a stable DPPH radical. It was shown that PPD-7, with a catechol motive, is the most active antioxidant, while PPD-9, with two neighbouring methoxy groups to the phenolic OH, exerted a somewhat lower, but significant antioxidative potential. The results of DFT thermodynamical study are in agreement with experimental findings that PPD-7 and PPD-9 should be considered as powerful radical scavengers. In addition, the obtained theoretical results (bond dissociation and proton abstraction energies) specify SPLET as a prevailing radical scavenging mechanism in polar solvents, and HAT in solvents with lower polarity. On the other hand, the obtained reaction enthalpies for inactivation of free radicals suggest competition between HAT and SPLET mechanisms, except in the case of the ˙OH radical in polar solvents, where HAT is labeled as prefered.
Introduction
The synthesis of different bioactive nitrogen-containing heterocyclic compounds has always been a topic of synthetic organic chemistry. Among a huge number of nitrogen-containing heterocyclic compounds, heterocycles with phthalazine and pyrazole moieties are especially recognized in the fields of medicine and pharmacy as the integral parts of some drugs.1 Such compounds express a broad spectrum of biological activities, such as anticancer,2 anticonvulsant,3 anti-inflammatory,1b,4 vasorelaxant,5 antitubercular,6 antihypertensive,7 and antimicrobial activities.5,8a,b Budralazine, hydralazine, azelastine, and zaleplon are some of commercially available drugs used in the treatment of vasorelaxation, hypertension, allergic rhinitis, and insomnia (Fig. 1).1 Additionally, these compounds serve as new luminescence materials or fluorescence probes.9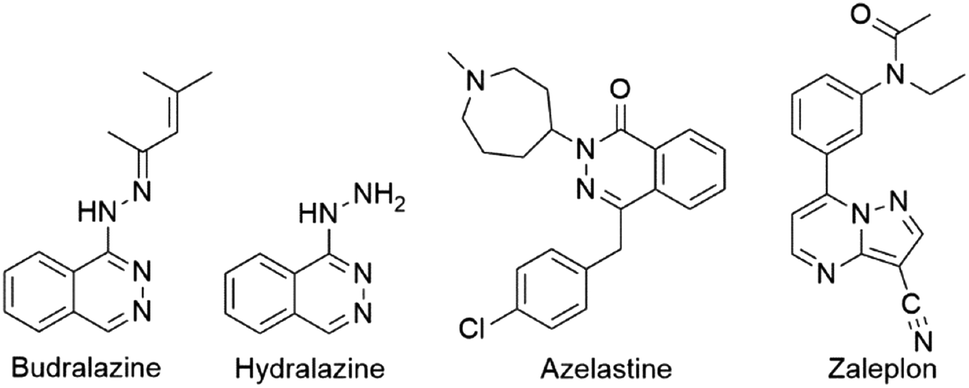 | ||
| Fig. 1 Structure of some commercially available drugs with phthalazine and pyrazolo moieties. | ||
In view of their importance, several procedures have been reported for the synthesis of phthalazine derivatives, including the reaction of (i) phthalhydrazide and dialkyl acetylenedicarboxylates in the presence of N-heterocycles,10 (ii) phthalhydrazide, aromatic aldehydes, and malononitrile,11 and (iii) palladium-catalyzed 1,3-dipolar cycloaddition of phthalhydrazide with methylenecyclopropanes, vinylidenecyclopropane, and methylenecyclobutane.12 Procedures for the synthesis of indazolyl-phthalazine-trione and pyrazolyl-phthalazine-dione derivatives (PPDs), starting from phthalhydrazide, aromatic or aliphatic aldehydes and cyclic or acyclic 1,3-diketones can be found in the literature, but procedures for the synthesis of indazole derivatives prevail.13 Various catalytic systems, such as phosphomolybdic acid-silica,13c Ce(SO4)2·4H2O,13d dodecylphosphonic acid,13e heteropoly acids,13f solid acids,13g N-halosulfonamides,13h silica-sulfuric acid,13i and p-TSA13j have been used for these reactions. However, the methodologies based on the using these catalysts have a number of serious disadvantages such as toxicity, harsh reaction conditions, and difficulty in the separation of products. Therefore, the synthesis of the complex pyrazolyl-phthalazine derivatives in the presence of ionic liquids (ILs) could be a promising alternative.9,14 Due to their ecological acceptability, polarity, and good dissolution ability for a wide range of compounds, ILs are used as catalysts in different organic reactions.15 Also, results regarding the usage of economical one-pot multicomponent reactions (MCRs) in the synthesis of PPDs are scarce.16 In addition, reactions with acyclic 1,3-diketones for the synthesis of phenolic PPDs cannot be found in the literature. Taking all above stated into account, we wanted to fulfil that gap and design an efficient synergistic protocol for the preparation of different PPDs. With that goal, one-pot three-component reaction was done using reusable diethanolammonium chloroacetate ([HDEA][ClAc]) as IL catalyst, and acetylacetone as acyclic diketone. All obtained compounds were characterized using NMR, IR, and UV-Vis spectroscopy, as well as with TD-DFT. Supramolecular structure of PPD-4 was elucidated using single-crystal X-ray diffraction analysis. Estimation of antioxidative potential of obtained phenolic PPDs was explored experimentally. In addition, preferred radical scavenging mechanism was investigated from thermodynamical aspect, using density functional theory (DFT).
Results and discussion
Bearing in mind that [HDEA][ClAc] exerted the best catalytic performance in the Mannich reaction,17 we assumed that this catalyst could be efficient for a new, domino, approach for the synthesis of PPDs, starting from acetylacetone, 2,3-dihydrophthalazine-1,4-dione and different aldehydes. It is worth pointing out that detailed characterization of this IL was presented before.18 To test efficiency of PPDs preparation, one-pot reaction of acetylacetone with 2,3-dihydrophthalazine-1,4-dione and benzaldehyde was selected as model reaction. Initially, the model reaction was performed without catalyst, and with using diethanolammonium acetate [HDEA][Ac] or [HDEA][ClAc] as catalysts. The influence of different amounts of catalysts (10, 15, and 20 mol%) and different reaction temperatures (100, 120, 140, and 160 °C) were investigated, Table 1. It is worth pointing out, that formation of the product was not observed in uncatalyzed reaction. In the presence of 10 mol% of catalyst ([HDEA][Ac] or [HDEA][ClAc]) product was formed in low to moderate yield and increase of the catalyst amount to 15 mol% increased the yield. Further increase of the catalyst amount to 20 mol% did not significantly affect the yield of the reactions. Furtherly, influence of the reaction temperature was investigated. Reactions performed at lower temperatures (100 and 120 °C) resulted of high amount of the starting material. Increase of temperature to 140 °C led to the improved yield of the desired product, while further increase of the temperature (160 °C) did not provide better yields. The optimal time for conversion of the starting material was 6 h, and further prolongation did not affect production of the PPDs.| Entry | Conditions | Yield % |
|---|---|---|
| 1 | No catalyst, 100 °C | — |
| 2 | No catalyst, 120 °C | — |
| 3 | No catalyst, 140 °C | — |
| 4 | No catalyst, 160 °C | — |
| 5 | [HDEA][Ac]/[HDEA][ClAc] 10 mol%, 100 °C | 20/52 |
| 6 | [HDEA][Ac]/[HDEA][ClAc] 10 mol%, 120 °C | 24/58 |
| 7 | [HDEA][Ac]/[HDEA][ClAc] 10 mol%, 140 °C | 35/65 |
| 8 | [HDEA][Ac]/[HDEA][ClAc] 10 mol%, 160 °C | 33/64 |
| 9 | [HDEA][Ac]/[HDEA][ClAc] 15 mol%, 100 °C | 38/70 |
| 10 | [HDEA][Ac]/[HDEA][ClAc] 15 mol%, 120 °C | 43/75 |
| 11 | [HDEA][Ac]/[HDEA][ClAc] 15 mol%, 140 °C | 48/82 |
| 12 | [HDEA][Ac]/[HDEA][ClAc] 15 mol%, 160 °C | 46/80 |
| 13 | [HDEA][Ac]/[HDEA][ClAc] 20 mol%, 100 °C | 45/72 |
| 14 | [HDEA][Ac]/[HDEA][ClAc] 20 mol%, 120 °C | 50/74 |
| 15 | [HDEA][Ac]/[HDEA][ClAc] 20 mol%, 140 °C | 55/82 |
| 16 | [HDEA][Ac]/[HDEA][ClAc] 20 mol%, 160 °C | 57/78 |
Since catalytic performance of [HDEA][ClAc] was much better, it was used as catalyst for further reactions. Optimized reaction conditions, i.e. 15 mol% of [HDEA][ClAc], 140 °C, and 6 h were used for all investigated reactions, Scheme 1, Table 2. All PPDs were obtained in moderate to good yields. For their isolation column chromatography was used. Ethyl acetate and hexane were used as eluents and environmental friendly organic solvents with low toxicity. Six of ten PPDs are newly synthetized compounds (PPD-4 and PPD-6–10). Among them, only crystals of PPD-4 were suitable for single-crystal X-ray diffraction analysis.
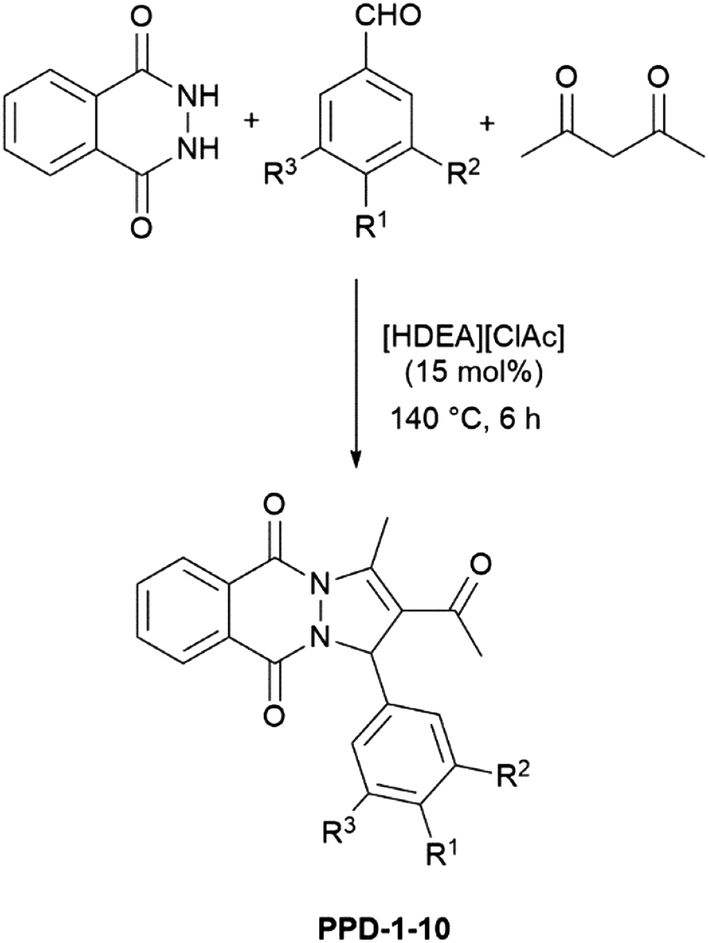 | ||
| Scheme 1 General synthesis of PPD-1–10. | ||
| Entry | Product | R1 | R2 | R3 | Yield (%) |
|---|---|---|---|---|---|
a Reaction conditions: acethylacetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2,3-dihydrophthalazine-1,4-dione:aromatic aldehyde = 2.5:1:1 (molar ratio); catalyst – [HDEA][ClAc] (15 mol%); temperature – 140 °C; time – 6 h. :2,3-dihydrophthalazine-1,4-dione:aromatic aldehyde = 2.5:1:1 (molar ratio); catalyst – [HDEA][ClAc] (15 mol%); temperature – 140 °C; time – 6 h. |
|||||
| 1 | PPD-1 | H | H | H | 82 |
| 2 | PPD-2 | CH3 | H | H | 80 |
| 3 | PPD-3 | Cl | H | H | 89 |
| 4 | PPD-4 | F | H | H | 87 |
| 5 | PPD-5 | NO2 | H | H | 90 |
| 6 | PPD-6 | OH | H | H | 80 |
| 7 | PPD-7 | OH | OH | H | 77 |
| 8 | PPD-8 | OH | OCH3 | H | 80 |
| 9 | PPD-9 | OH | OCH3 | OCH3 | 70 |
| 10 | PPD-10 | OCH3 | OCH3 | OCH3 | 72 |
All compounds were characterized with NMR, IR, and UV-Vis spectra, elemental analysis, and melting points. The signals observed in the 1H NMR spectra of synthesized compounds, related to the PPD skeleton, are presented in Table 3, and corresponding atoms labelling in Fig. 2. Chemical shifts in 1H NMR spectra of all PPDs are very similar. Singlet originating from protons of C6 methyl group appears around 2 ppm. It is important to emphasize that, in place of expected singlets originating from protons of C1–H and of C4 methyl group, quartet around 6.5 ppm with J = 1.4 Hz, and doublet about 3 ppm with J = 1.5 Hz appeared. These peaks represent result of the long-range coupling of proton of asymmetric C1 with protons of C4 methyl group. The protons attached to the aromatic carbons C8–C13 and C15–C20 resonated as multiplets in the range of 7.76–8.39 ppm and 6.66–7.92, while in the case of PPD-9 and PPD-10, the aromatic protons from ring C15–C20 appeared as singlets at 6.70 and 6.68 ppm.
| Product | C1–H | C4–H | C6–H | ArH | |
|---|---|---|---|---|---|
| C8–C13 | C15–C20 | ||||
| PPD-1 | 3.08 | 6.50 | 2.09 | 7.79–8.36 | 7.30–7.48 |
| PPD-2 | 3.07 | 6.46 | 2.08 | 7.77–8.37 | 7.15–7.39 |
| PPD-3 | 3.07 | 6.47 | 2.13 | 7.76–8.36 | 7.31–7.47 |
| PPD-4 | 3.08 | 6.50 | 2.12 | 7.78–8.37 | 6.99–7.53 |
| PPD-5 | 3.11 | 6.57 | 2.24 | 8.16–8.39 | 7.67–7.92 |
| PPD-6 | 3.07 | 6.44 | 2.09 | 7.78–8.40 | 6.66–7.30 |
| PPD-7 | 3.00 | 6.33 | 2.05 | 7.77–8.32 | 6.77–6.91 |
| PPD-8 | 3.07 | 6.45 | 2.10 | 7.76–8.36 | 6.87–7.02 |
| PPD-9 | 3.08 | 6.44 | 2.09 | 7.81–8.38 | 6.70 |
| PPD-10 | 3.07 | 6.45 | 2.12 | 7.81–8.38 | 6.68 |
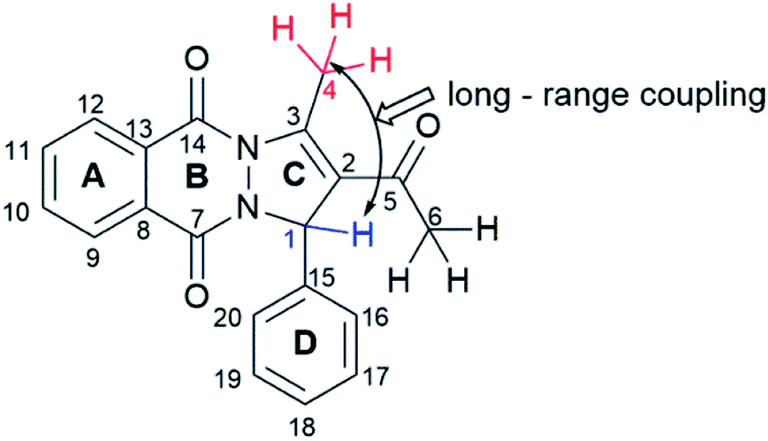 | ||
| Fig. 2 Skeleton of PPD compounds. | ||
The mechanism for the formation of 2H-indazolo[2,1-b]phthalazine-triones can be found in the literature,13b but not for the formation of pyrazolo[1,2-b]phthalazine-dione. Bearing in mind that the only difference between these reactions is in the usage of diketone, it can be expected that they will obey similar reaction mechanism, Scheme 2. The reaction starts with the IL catalysed enolization of diketone, and protonation of aromatic aldehyde.
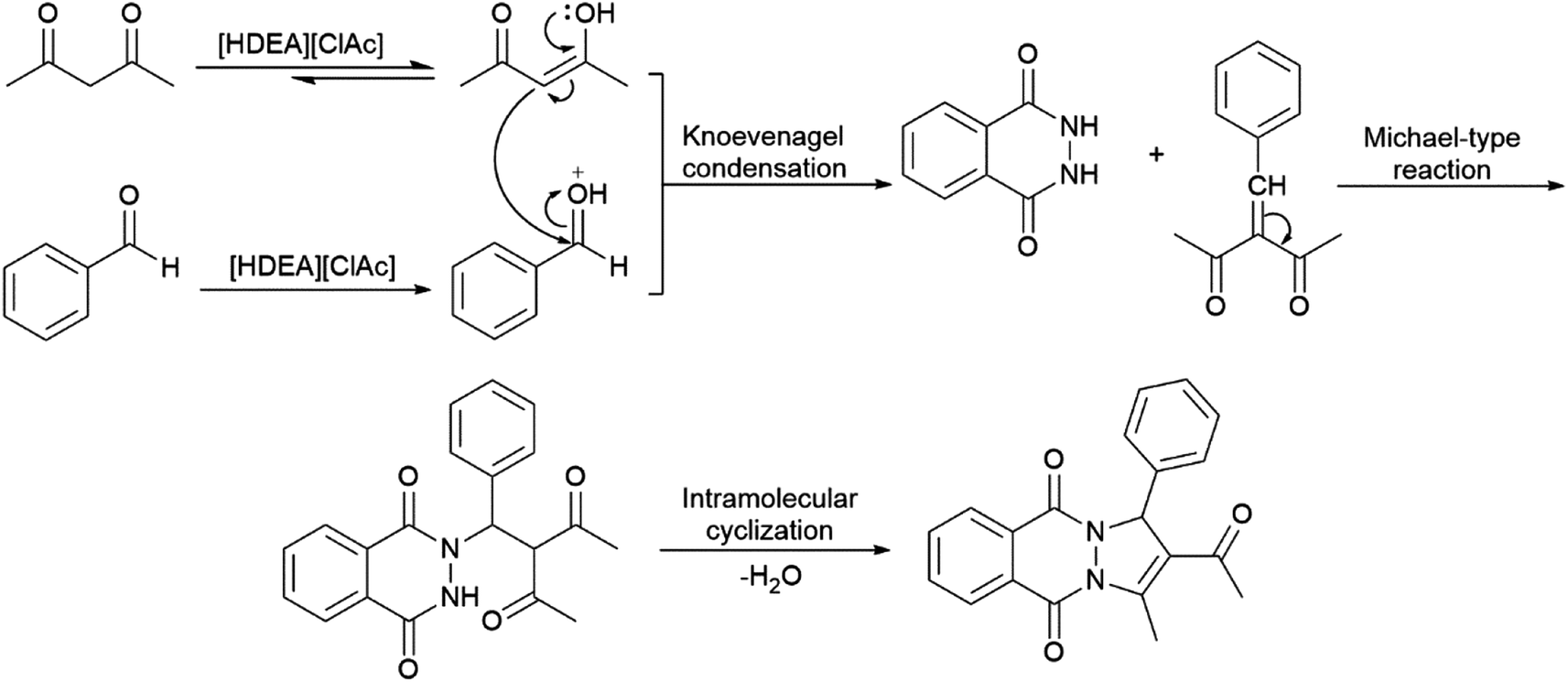 | ||
| Scheme 2 The suggested mechanism for the considered reaction. | ||
Based on the proposed mechanism, electronic effects of the aldehyde substituents influence on the reaction flow, as well as on the products yield. The nucleophilic attack of the enol form of ketone to the carbonyl group of aldehyde would be facilitated with electron-withdrawing groups positioned on the aromatic ring of the aldehyde (Table 2, entries 3–5) enabling quick formation of the Knoevenagel adduct. Next step of the reaction, i.e. Michael type addition of 2,3-dihydrophthalazine-1,4-dione to the Knoevenagel adduct, will be also favoured with the same type of substitution. After intramolecular cyclization of the Michael adduct, the appropriate PPD is formed. The proposed mechanism agrees with the obtained yields which are reduced in the case of benzaldehyde and electron-donating substituted benzaldehyde (Table 2, entries 1, 2, and 7–10).
UV-Vis spectral characterization of PPD-4
In all experimental and simulated spectra two major absorption bands appear around 220 nm and 360 nm, with shoulder appearing around 250 nm Fig. 3 and S1.† The only exception is in the case of PPD-5, where additional band appears around 300 nm. To be precise, this band is present in absorption spectra of all compounds, but at lower wavelength (around 280 nm), as shoulder and with low oscillator strength. It is worth pointing out, that all simulated absorption bands are somewhat redshifted, with the most significant deviation for weak experimental band around 280 nm, which appears around 310 nm in simulated ones.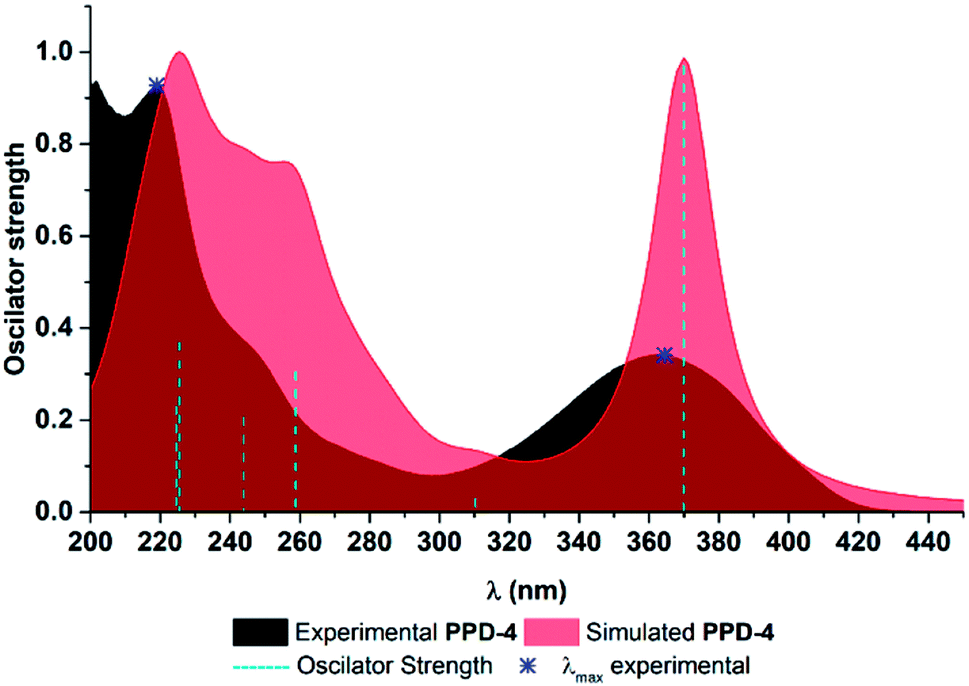 | ||
| Fig. 3 UV-Vis spectra of PPD-4. | ||
To explore which transitions are responsible for the appearance of each absorption band, Kohn–Sham orbitals were constructed, Fig. S2–11,† while electron transitions and corresponding orbital energies are provided in Tables S1 and S2.† For the sake of clarity, parts of all compounds were labelled as shown in Fig. 2. Here, spectrum of PPD-4 will be discussed in detail, while all relevant data for other investigated PPDs are provided in ESI.† Band at the lowest wavelength (219 nm in experimental) is a consequence of several electron transitions with bands positioned at 224.6 and 225.5 nm in simulated spectra, Fig. 3. The first one is caused by electron transition from HOMO−3 to LUMO+2 orbital, as well as HOMO−7 and HOMO−8 transitions to LUMO+1, while the second one by HOMO−5 and HOMO−7 to LUMO+1 electron transitions, Fig. S5.† At molecular level, the band at 224.6 nm is enabled by electron transition from ring C to the rings A and B, from the rings B and D to the rings A, B, C, and acyl group of ring C, and from nitrogen and oxygen atoms of ring B, ring C to the rings A, B, C. The band at 225.5 nm is a consequence of electron transition from rings A and oxygens of ring B (HOMO−5) and from ring C and oxygens of ring B (HOMO−7) to the rings A, B, and C. Shoulder appearing around 250 nm in experimental spectrum is consequence of multiple electron transitions presented with two absorption bands at 244 and 259 nm in simulated spectrum. Lower wavelength band is product of transitions from HOMO−6 to LUMO and LUMO+1 (nitrogen and oxygen atoms of ring B, ring C to the rings A, B, C, and acyl group of ring C, as well as to the rings A, B, and C), and HOMO−8 to LUMO electron transition (rings B and C to the rings A, B, and C), while the higher wavelength is caused by HOMO−5 to LUMO (ring A and oxygen atoms of ring B to the to the rings A, B) and HOMO−1 to LUMO+1 electron transition (ring D and oxygen and nitrogen atom of ring B to the rings A, B, and C). Weak intensity shoulder around 280 nm appears due to HOMO to LUMO+1 electron transition (from rings B, C, and acyl group of ring C to the rings A, B, and C). The highest absorption band around 360 nm in experimental spectra is a consequence of HOMO to LUMO electron transition (from rings B, C, and acyl group of ring C to the rings A, B, and C). Based on the obtained results, one can see that lower wavelength bands (bellow 280 nm in experimental spectra) are appearing owing to relatively large energetical but relatively small spatial separation, while the higher wavelengths are consequence of relatively smaller energy separation of corresponding HOMO and LUMO. It is worth pointing out that bands in UV-Vis spectra of all other PPDs are consequence of similar electron transitions. As mentioned above, the only deviation is in the case of UV-Vis spectra of PPD-5, where the presence of electron withdrawing nitro group on the ring D is responsible for redshifted appearance of this band as one peak around 300 nm (instead of small shoulder like in other cases). Namely, in the spectra of PPD-5, this band is enabled by electron transition from HOMO−2 to LUMO (rings A and D to the ring D including nitro group), which is obviously favoured by electron accepting nature of nitro group.
Crystal structure of PPD-4
Single-crystal X-ray diffraction analysis showed that PPD-4 crystallizes with two crystallographically independent molecules in an asymmetric unit of the centrosymmetric Pbca space group. These two geometrically very similar molecules are designated in Fig. 4 as molecules A and B.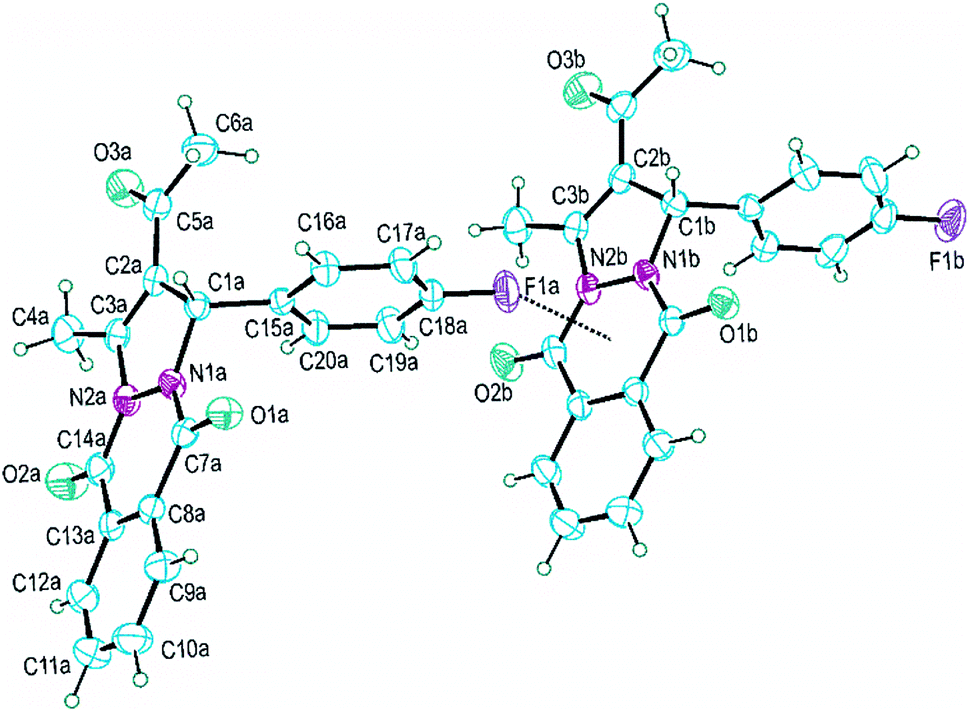 | ||
| Fig. 4 Molecular structure and atom-numbering scheme of two independent molecules of PPD-4 (molecule A left, molecule B right). Displacement ellipsoids are drawn at the 30% probability level. Dotted line represents F1a⋯Cg1 interaction (Cg1 is midpoint of the N1–C7–C8–C13–C14–N2 ring). | ||
Corresponding bond lengths in two molecules are mutually similar as presented in Table 4. The only chiral atom in PPD-4 forms the C1–C15 (1.52 Å) and C1–C2 (1.51 Å) bonds which are the longest C–C bonds in both molecules. The C2–C3 bond with bond length of 1.34 Å is the shortest C–C bond and can be assigned as only localized C–C double bond in PPD-4, Table 4. It is interesting to compare four N–C bonds since they exhibit rather wide range of values (from 1.34 to 1.48 Å) and considering that the N1 and N2 atoms form the N1–N2 bond and have very similar neighbourhood, Fig. 4. The nitrogen atoms are directly linked to two equivalent and geometrically equally placed carbonyl groups (O1–C7 and O2–C14) but in both molecules the N1–C7 bond is somewhat shorter than corresponding N2–C14, Table 4. Bond length difference between the N1–C1 and corresponding N2–C3 bond is larger and can be explained by different character of the C1 and C3 atoms.
| Molecule A | Molecule B | |
|---|---|---|
| F1–C18 | 1.366(2) | 1.365(3) |
| N1–N2 | 1.411(2) | 1.410(2) |
| N1–C1 | 1.475(3) | 1.473(3) |
| N1–C7 | 1.344(3) | 1.344(3) |
| N2–C3 | 1.402(3) | 1.401(3) |
| N2–C14 | 1.376(3) | 1.380(3) |
| O1–C7 | 1.227(2) | 1.229(2) |
| O2–C14 | 1.220(3) | 1.222(3) |
| O3–C5 | 1.219(3) | 1.221(3) |
| C1–C2 | 1.514(3) | 1.509(3) |
| C1–C15 | 1.518(3) | 1.519(3) |
| C2–C3 | 1.345(3) | 1.343(3) |
| C2–C5 | 1.467(3) | 1.474(3) |
| C7–C8 | 1.476(3) | 1.468(3) |
| C13–C14 | 1.461(3) | 1.463(3) |
All non-hydrogen atoms with exception of the substituted C15–C20 phenyl ring are approximately coplanar. Therefore, nineteen C, O and N atoms form very large planar system regardless of the fact that many of them form single bonds. Thus, the heterocyclic N1–C7–C8–C13–C14–N2 ring exhibits root-mean-square deviation of fitted atoms from mean plane less than 0.03 Å in both molecules. Dihedral angle between this heterocyclic ring and the C8–C13 phenyl ring is 1.2(2) and 1.1(2)° for molecules A and B respectively. The five-membered heterocyclic ring is the most puckering ring in PPD-4, but it also could be accepted as approximately planar since that torsion angles within the ring do not exceed the value of 5°.
Although on first view two molecules have very similar conformation there are some structural differences, Fig. 4. The largest difference is in the orientation of the C15–C20 phenyl ring regarding to the rest of molecule and it can be illustrated by the N1–C1–C15–C20 torsion angle; 65.4(3) and 56.8(3)° for molecules A and B respectively. Also, the C7–N1–N2–C14 angles have significantly different values; −9.1(3) and −2.8(3)° for A and B respectively.
The most interesting structural characteristic in crystal structure of PPD-4 is the formation of rather short F1a⋯Cg1b and F1b⋯Cg1a intermolecular distances between F atom and neighbouring six-membered heterocyclic ring with two carbonyl groups (Cg1 is midpoint of the N1–C7–C8–C13–C14–N2 ring), Fig. 4 and 5. These intermolecular interactions should not be treated as coincidental since the F1⋯Cg1 distance is shorter than 3 Å (2.93 and 2.88 Å for F1a and F1b respectively) and all molecules form extended chains along c-crystallographic axis using this interaction, Fig. 5. Regardless of the presence of two aromatic rings, the molecules of PPD-4 do not participate in π–π interactions but they form several weak C–H⋯O intermolecular hydrogen bonds shown in Table 5. The O3 carbonyl atom in both independent molecules forms C4–H⋯O3 intramolecular H-bond, Table 5.
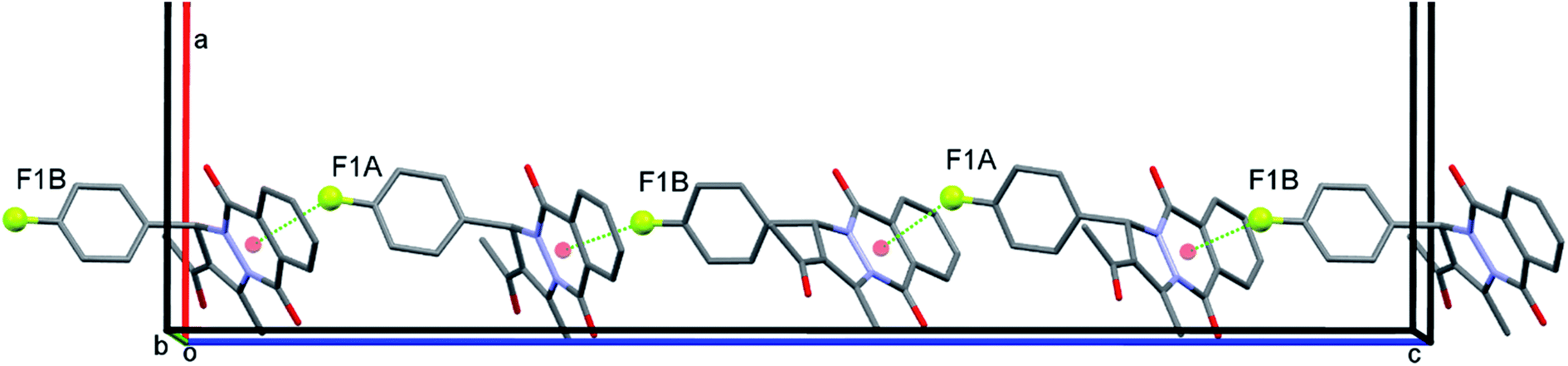 | ||
| Fig. 5 Crystal lattice fragment of PPD-4 representing formation of supramolecular chain along c-crystallographic axis using F1⋯Cg1 intermolecular interactions. The Cg1 (labelled by red circle) is midpoint of six-membered heterocyclic ring. H atoms are omitted for clarity. | ||
| C–H⋯O | C–H (Å) | C⋯O (Å) | H⋯O (Å) | C–H⋯O (°) | Symmetry code for O |
|---|---|---|---|---|---|
| C1a−H1a⋯O1a | 0.98 | 3.452(3) | 2.49 | 166 | −x + 0.5, y + 0.5, z |
| C9a−H9a⋯O1a | 0.93 | 3.234(3) | 2.54 | 132 | −x + 0.5, y − 0.5, z |
| C12b−H12b⋯O2a | 0.93 | 3.203(3) | 2.47 | 136 | −x, y − 0.5, −z + 1.5 |
| C4a−H4a1⋯O3a | 0.96 | 2.936(3) | 2.23 | 130 | x, y, z |
| C4a−H4a3⋯O3a | 0.96 | 3.293(3) | 2.59 | 130 | −x, y − 0.5, −z + 1.5 |
| C1b−H1b⋯O1b | 0.98 | 3.421(3) | 2.45 | 173 | −x + 0.5, y + 0.5, z |
| C9b−H9b⋯O1b | 0.93 | 3.347(3) | 2.58 | 140 | −x + 0.5, y − 0.5, z |
| C4b−H4b1⋯O3b | 0.96 | 2.943(3) | 2.23 | 130 | x, y, z |
DPPH radical scavenging activity
All synthetized compounds PPD-1–10 were subjected to evaluation of their in vitro antioxidative activity. The radical scavenging ability of the compounds was tested against the 1,1-diphenyl-2-picryl-hydrazyl (DPPH) stable free radical, Tables 6 and S3.† Among the tested substances, compounds PPD-7 and PPD-9, obtained from protocatechualdehyde and syringaldehyde, exhibited the best scavenging activities, with IC50 values of 4.1 and 14.6 μM, Table 6. PPD-8, obtained from vanillin, exerted lower activity, while PPD-1–6 and PPD-10 didn't express DPPH scavenging activity.| PPD-7 | PPD-8 | PPD-9 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HAT | SET-PT | SPLET | HAT | SET-PT | SPLET | HAT | SET-PT | SPLET | |||||||
| a Results represent mean values ± standard deviation (SD) of three independent measurements. | |||||||||||||||
| IC50 (μM) | 4.1 ± 0.2a | 135.6 ± 0.9a | 14.6 ± 2.4a | ||||||||||||
|
|||||||||||||||
| Thermodynamical parameters (kJ mol−1) | |||||||||||||||
| BDE | IP | PDE | PA | ETE | BDE | IP | PDE | PA | ETE | BDE | IP | PDE | PA | ETE | |
| 328 | 474 | 16 | 133 | 357 | 334 | 466 | 30 | 149 | 347 | 316 | 455 | 22 | 147 | 330 | |
| 327 | 15 | 132 | 357 | ||||||||||||
|
|||||||||||||||
| Reaction enthalpies (kJ mol−1) | |||||||||||||||
| Radical | ΔHBDE | ΔHIP | ΔHPDE | ΔHPA | ΔHETE | ΔHBDE | ΔHIP | ΔHPDE | ΔHPA | ΔHETE | ΔHBDE | ΔHIP | ΔHPDE | ΔHPA | ΔHETE |
| ˙OCH3 | −99 | 96 | −195 | −78 | −21 | −93 | 89 | −182 | −63 | −30 | −111 | 78 | −189 | −64 | −47 |
| −100 | −197 | −80 | −20 | ||||||||||||
| ˙OC(CH3)3 | −108 | 97 | −204 | −87 | −20 | −101 | 89 | −191 | −72 | −30 | −120 | 78 | −198 | −73 | −47 |
| −109 | −205 | −89 | −20 | ||||||||||||
| ˙OH | −171 | 18 | −188 | −71 | −99 | −164 | 10 | −175 | −56 | −109 | −183 | −1 | −182 | −57 | −126 |
| −172 | −189 | −73 | −99 | ||||||||||||
| ˙OOH | −31 | 119 | −150 | −33 | 2 | −25 | 111 | −136 | −17 | −7 | −43 | 100 | −143 | −19 | −25 |
| −32 | −151 | −34 | 2 | ||||||||||||
| ˙OOCH3 | −24 | 127 | −151 | −34 | 10 | −18 | 120 | −137 | −18 | 1 | −36 | 108 | −145 | −20 | −17 |
| −25 | −152 | −35 | 10 | ||||||||||||
˙OO–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
−23 | 106 | −129 | −12 | −11 | −17 | 98 | −115 | 4 | −20 | −35 | 87 | −122 | 2 | −38 |
| −24 | −130 | −13 | −11 | ||||||||||||
| DPPH | 8 | 96 | −88 | 29 | −21 | 15 | 89 | −74 | 45 | −30 | −4 | 78 | −82 | 43 | −47 |
| 7 | −89 | 28 | −20 | ||||||||||||
| O2˙− | 41 | 285 | −244 | 39 | 2 | 48 | 278 | −231 | 55 | −7 | 29 | 267 | −238 | 54 | −25 |
| 40 | −245 | 38 | 2 | ||||||||||||
Obtained results are in accordance with the literature data that the antioxidative capacity of phenolic compounds depends on the type, number, and position of neighbouring groups (OH, OR, NH2) to the phenolic hydroxy group.19 Resonance and electron donating effects of these groups increase the stability of the formed phenoxy radical. This stabilisation effect is the most pronounced in catechol-like compounds, where the hydroxy group contributes to the homolytic cleavage of the neighbouring O–H bond, and has the ability to form a hydrogen bond with formed phenoxy radical.20 In addition, experimental and theoretical studies have shown that stabilization by H-bonding is more pronounced in formed o-semiquinone radical than in parent catechol.21 Our results of antioxidative capacity of PPDs are in perfect agreement with above mentioned findings. Namely, PPD-7, with catechol motive, is the most active antioxidant, while PPD-9, with two neighbouring methoxy groups to the phenolic OH, exerted somewhat lower, but significant antioxidative potential. Compound PPD-8, with one methoxy group in ortho-position, expressed moderate activity.
Antioxidative mechanisms and free radical scavenging mechanisms of PPDs with different free radicals
It is well known that there are several possible mechanisms of radical scavenging: Hydrogen Atom Transfer (HAT), Single Electron–Proton Transfer (SET-PT), and Sequential Proton-Loss Electron-Transfer (SPLET). To determine the most probable mechanism of radical scavenging of phenolic compounds PPD-7–9, DFT and thermodynamic approach were employed. Insight into the most probable reaction pathway in the absence of free radicals from the thermodynamic point of view can be accessed by calculating Bond Dissociation Enthalpy (BDE), Ionisation Potential (IP), and Proton Abstraction (PA) energies.22 On the other hand, the scavenging mechanisms are highly influenced by the electronic properties of the scavenged free radical species.22g Therefore, the reaction enthalpies (ΔrH) were calculated for the reactions of phenolic PPDs with each of the eight selected free radicals: hydroxy (˙OH), hydroperoxy (˙OOH), methylperoxy (CH3–O–O˙), superoxide radical anion (O2˙−), methoxy (˙OCH3), tert-butoxy (˙OC(CH3)3), vinyl peroxy (CH2CH–O–O˙), and DPPH. Obtained values for ΔHBDE, ΔHIP and ΔHPDE, ΔHPA and ΔHETE indicate which of HAT, SET-PT, and SPLET mechanism prevails, respectively. Details on calculation of thermodynamic parameters in the absence and in the presence of free radicals are well known in the literature.22 The thermodynamic parameters were obtained by optimization of all relevant species in three solvents. Here we present results obtained in methanol (Table 6), since it was used for experimental DPPH assay, while results regarding benzene (to mimic non-polar environment) and water are provided in Tables S4 and S5 of ESI.† Selection of radicals has been made on their presence in living cell, and their chemical behaviour within the cell.23 Namely, O2˙− is formed as a product of respiration in living cells,24 and is susceptible to protonation at pH lower than 4.8, yielding more reactive ˙OOH radical. In addition, O2˙− undergoes dismutation to give hydrogen peroxide (H2O2), which via Fenton and Haber–Weiss reactions produces the ˙OH radical. This powerful oxidant, in the presence of air, reacts with hydrocarbons producing peroxy radicals, which are furtherly transformed into the alkoxy radicals. DPPH was selected because it was used in experimental assay.
Inspection of Tables 6, S4, and S5† undoubtedly provided evidence that one can eliminate SET-PT as working mechanism in the absence and in the presence of free radicals, and in all investigated solvents. Obtained values for IP and ΔHIP are significantly higher than those obtained for thermodynamic parameters and reaction enthalpies of HAT and SPLET mechanisms. On the other hand, BDE is considerably larger than PA in polar solvents, pointing out that all investigated PPDs will obey SPLET mechanism, while in environment with low polarity HAT. This is consistent with plausible heterolytic bond dissociation in polar solvents and homolytic in non-polar.25 Namely, product of HAT mechanism is radical formed from each of PPDs, while in the case of SPLET, the first step is proton abstraction and formation of corresponding anions.
Similarly to the case of mechanism of action in the absence of free radicals, in the presence of free radicals preferred route of radical scavenging is highly influenced by solvent polarity. At first glance, based on ΔrHBDE and ΔrHPA enthalpies obtained in polar solvents (methanol and water), HAT seems to be preferred mechanism, while in the low polar media SPLET. However, deeper insight in reaction enthalpies for hydrogen atom and proton abstraction in Tables 6, S4, and S5† proved this to be correct only in the case of ˙OH radical, and only in the case of polar solvents. Here, difference between ΔrHBDE and ΔrHPA values is significant, designating HAT as preferred mechanism. In the case of alkoxy radicals (˙OCH3 and ˙OC(CH3)3) and peroxy radicals (˙OOH, CH3–O–O˙, and CH2CH–O–O˙) this difference is less pronounced, indicating that there is competition between HAT and SPLET mechanisms. In the case of DPPH radical, low positive values of reaction enthalpies suggest slow interaction of examined compounds with this radical. In addition, ΔrHPA values are higher than ΔrHBDE in all solvents and for all PPDs, indicating HAT as less endothermic and therefore preferred scavenging route of this radical. In the case of CH2CH–O–O˙ radical, positive values of ΔrHPA for the reactions in methanol and water, express endothermic nature of the SPLET mechanism, while ΔrHBDE are negative, impaling HAT as more probable mechanism. It is worth pointing out, that in all examined cases and in all solvents, enthalpies of the reactions with O2˙− are the highest, suggesting that this radical will be scarcely quenched.26
Conclusions
A new efficient, ionic liquid [HDEA][ClAc] catalysed, synthesis of PPDs has been realized. The products were obtained in moderate to good yields, depending on the aldehyde substitution. Namely, electron-withdrawing groups provided higher yields of obtained products PPD-3–5. Six PPDs (4, 6, 7, 8, 9, and 10) are reported here for the first time. All obtained compounds were characterized using NMR, IR, and UV-Vis spectroscopy, and for PPD-4 the crystal structure was determined using single-crystal X-ray diffraction analysis. It was found that this compound crystallizes with two crystallographically independent molecules and their structural properties are analysed in detail. The most interesting structural characteristic in the crystal structure of PPD-4 is the formation of rather short intermolecular distances (2.93 and 2.88 Å for F1a and F1b respectively) between F atom and midpoint of neighbouring six-membered heterocyclic ring, which is the cause of the formation of parallel one-dimensional supramolecular chains.All synthetized PPD derivatives were screened for DPPH radical scavenging potential. PPD-7 and PPD-9, with catechol and syringic moiety, excreted excellent antioxidant activity, while compound PPD-8 with vanillic moiety moderate. Thermodynamical data obtained from DFT study specify SPLET as prevailing mechanism in polar solvents, while in environment with low polarity HAT. On the other hand, pronounced differences in reaction enthalpies for inactivation of ˙OH radical in polar solvents denote HAT as dominant mechanism. However, in all other cases and in solvents with high and low polarity, obtained reaction enthalpies suggest competition between HAT and SPLET mechanisms.
Experimental
The compounds acetic acid, chloroacetic acid, 2,3-dihydrophthalazine-1,4-dione, benzaldehyde, 4-fluorobenzaldehyde, 4-chlorobenzaldehyde, 4-methylbenzaldehyde, 4-nitrobenzaldehyde, 4-hydroxybenzaldehyde, 3,4-dihydroxybenzaldehyde, 3,4,5-trimethoxybenzaldehyde, vanillin, syringaldehyde and acetylacetone were obtained from Aldrich Chemical Co. Diethanolamine (DEA) was purchased from Fluka. All common chemicals were of reagent grade. The UV-Vis spectra were measured at room temperature within the 200–500 nm range on the Agilent Technologies, Cary 300 Series UV-Vis Spectrophotometer. A solution of 2.5 × 10−5 M of each compound was prepared in methanol and then 2 mL of the corresponding solution was injected into the 10 mm quartz cell and recorded spectrum. The IR spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrometer using the thin film technique and KBr plates. The 1H NMR and 13C NMR spectra were run in CDCl3 and CD3OD on a Varian Gemini 200 MHz spectrometer. Melting points were determined on a Mel-Temp capillary melting points apparatus, model 1001. Elemental microanalysis for carbon, hydrogen, and nitrogen were performed at the Faculty of Chemistry, University of Belgrade.Synthesis of [HDEA][Ac] and [HDEA][ClAc]
The procedure for synthesis of the [HDEA][Ac] and [HDEA][ClAc], as well as their corresponding spectral characterization are given in ref. 18.General procedure for synthesis of PPDs
A mixture of acethylacetone (2.5 mmol), 2,3-dihydrophthalazine-1,4-dione (1 mmol), aromatic aldehyde (1 mmol), and 15 mol% [HDEA][ClAc] was heated at 140 °C for 6 h. Reaction progress was monitored using thin layer chromatography (TLC). After completion of the reaction, the resulting mixture was dissolved in ethyl acetate and washed with water. The residues obtained by ethyl acetate evaporation were purified by silica gel chromatography using ethyl acetate/hexane (2:1) as eluent. All products (PPD-1-10) were characterized with elemental analysis, melting point, 1HNMR, 13CNMR and IR spectra. The characterizations of the new compounds are given in main part of the manuscript, while for known compounds in ESI,† as well as 1H and 13C NMR spectra for all compounds.
Recycling experiment
After completion of the reaction, the mixture was dissolved in ethyl acetate. Resulting ethyl acetate mixture was washed several times with water. Upon this, separated water parts were combined and evaporated. After water evaporation, [HDEA][ClAc] residue was reused directly without further purification. So recovered catalyst was reused four times in new experiments, without significant decrease of the product yields.Density functional theory calculations
The Gaussian 09 program package was used to perform all calculations.27 The equilibrium geometries of all compounds, its radical cation, radicals and anions, as well as all other species that participate in the reactions of all studied mechanisms were calculated using the B3LYP functional in conjunction with the 6-311+G(d,p) basis set.28 The influence of methanol (ε = 32.6), water (ε = 78.4), and benzene (ε = 2.3) as solvents was estimated, using SMD model as implemented in Gaussian 09.27,29 To confirm that all structures are local minima, frequency calculations were done. The optimized geometries were confirmed by the absence of any imaginary frequency. Simulations of UV-Vis spectra were performed using TD-DFT and structures optimized in methanol, since experimental spectra were acquired using this solvent. For the calculations of open-shell systems unrestricted spins were used. All relative enthalpies were calculated at 298.15 K. The values of the solvation enthalpies of proton and electron in methanol were used from literature.302-Acetyl-1-(4-fluorophenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-4)
Yellow crystals (ethyl acetate/ethanol (1:2)) – mp 208–210 °C; 1H NMR (200 MHz, CDCl3) δ: 2.12 (s, 3H), 3.08 (d, J = 1.4 Hz, 3H), 6.50 (q, J = 1.3, 1H), 7.12–6.99 (m, 2H), 7.53–7.42 (m, 2H), 7.89–7.78 (m, 2H), 8.26–8.22 (m, 1H), 8.37–8.33 (m, 1H); 13C NMR (50 MHz, CDCl3) δ: 14.5, 30.5, 65.3, 115.6, 116.1, 118.8, 127.3, 128.0, 128.7, 129.5, 129.9, 130.2, 132.3, 133.4, 134.2, 146.1, 154.2, 156.2, 160.4, 165.4, 192.9; IR (cm−1): 3581, 3059, 2992, 2917, 1691, 1645, 1599, 1509, 1468, 1415, 1355, 1619, 1291, 1274, 1219, 1108, 961, 842, 701, 555; C20H15FN2O3 (FW = 350.35): C, 68.57; N, 8.00; H, 4.32%; found: C, 68.35; N, 7.97; H, 4.33%;
2-Acetyl-1-(4-hydroxyphenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-6)
Yellow powder – mp 205–206 °C; 1H NMR (200 MHz, CDCl3) δ: 2.09 (s, 3H), 3.07 (d, J = 1.5 Hz, 3H), 6.44 (d, J = 1.5 Hz, 1H), 6.73–7.816.64 (m, 2H), 7.25 (s, 1H), 7.30 (s, 1H), 7.88–7.78 (m, 2H), 8.26–8.21 (m, 1H), 8.40–8.30 (m, 1H); 13C NMR (50 MHz, CDCl3) δ: 14.5, 30.5, 65.9, 116.0, 119.1, 127.3, 127.8, 128.1, 128.8, 129.7, 133.5, 134.3, 145.9, 154.3, 156.2, 156.8, 193.7; IR (cm−1): 3370, 2922, 2851, 1647, 1602, 1516, 1419, 1354, 1318, 1290, 1275, 1105, 1015, 961, 837, 791, 697, 563; C20H16N2O4 (FW = 348.36): C, 68.96; N, 8.04; H, 4.63%; found: C, 68.69; N, 8.07; H, 4.65%;2-Acetyl-1-(3,4-dihydroxyphenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-7)
Yellow powder – mp 241–243 °C; 1H NMR (200 MHz, CDCl3 and CD3OD) δ: 2.05 (s, 3H), 3.00 (d, J = 1.5 Hz, 3H), 6.33 (q, J = 1.4 Hz, 1H), 6.77 (d, J = 1.7 Hz, 2H), 6.91–6.88 (m, 1H), 7.83–7.77 (m, 2H), 8.19–8.14 (m, 1H), 8.32–8.27 (m, 1H); 13C NMR (50 MHz, CDCl3 and CD3OD) δ: 13.8, 29.8, 65.7, 114.9, 115.1, 119.1, 120.0, 126.7, 127.2, 127.6, 128.5, 129.2, 133.4, 134.1, 144.9, 145.5, 145.7, 154.1, 156.1, 194.5; IR (cm−1): 3483, 3223, 1687, 1637, 1598, 1518, 1470, 1375, 1356, 1289, 1200, 1106, 962, 891, 824, 764, 692, 579; C20H16N2O5 (FW = 364.36): C, 65.93; N, 7.69; H, 4.43%; found: C, 65.75; N, 7.66; H, 4.41%;2-Acetyl-1-(4-hydroxy-3-methoxyphenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-8)
Yellow powder – mp 230–231 °C; 1H NMR (200 MHz, CDCl3) δ: 2.10 (s, 3H), 3.07 (d, J = 1.5 Hz, 3H), 3.90 (s, 3H), 5.80 (br, s, 1H), 6.45 (q, J = 1.4 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.94 (dd, J = 8.2, 1.9 Hz, 1H), 7.02 (d, J = 1.8 Hz, 1H), 7.87–7.76 (m, 2H), 8.30–8.19 (m, 1H), 8.36–8.31 (m, 1H); 13C NMR (50 MHz, CDCl3) δ: 14.4, 30.5, 56.1, 65.9, 111.4, 114.7, 118.8, 121.2, 127.3, 127.9, 128.1, 128.9, 129.6, 133.4, 134.1, 145.9, 146.5, 146.7, 154.3, 156.3, 193.5; IR (cm−1): 3418, 2921, 2850, 1687, 1641, 1518, 1433, 1371, 1349, 1276, 1196, 1109, 1040, 959, 791, 696, 586; C21H18N2O5 (FW = 378.38): C, 66.66; N, 7.40; H, 4.80%; found: C, 66.71; N, 7.42; H, 4.82%.2-Acetyl-1-(4-hydroxy-3,5-dimethoxyphenyl)-3-methyl-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-9)
Brown solid – mp 95–96 °C; 1H NMR (200 MHz, CDCl3) δ: 2.09 (s, 3H), 3.08 (d, J = 1.1 Hz, 3H), 3.89 (s, 6H), 5.57 (br, s, 1H), 6.44 (d, J = 1.3 Hz, 1H), 6.70 (s, 2H), 7.85–7.81 (m, 2H), 8.30–8.21 (m, 1H), 8.38–8.30 (m, 1H); 13C NMR (50 MHz, CDCl3) δ: 14.4, 30.5, 56.5, 66.3, 105.6, 118.6, 127.2, 127.3, 127.9, 128.9, 129.6, 133.4, 134.2, 135.7, 145.9, 147.3, 154.5, 156.3, 193.6; IR (cm−1): 3420, 2939, 2842, 1721, 1687, 1646, 1602, 1516, 1465, 1430, 1355, 1306, 1111, 697; C22H20N2O6 (FW = 408.41): C, 64.70; N, 6.86; H, 4.94%; found: C, 64.45; N, 6.89; H, 4.96%;2-Acetyl-3-methyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrazolo[1,2-b]phthalazine-5,10-dione (PPD-10)
Yellow solid – mp 167–169 °C; 1H NMR (200 MHz, CDCl3) δ: 2.12 (s, 3H), 3.07 (d, J = 1.1 Hz, 3H), 3.81 (s, 3H), 3.86 (s, 6H), 6.45 (d, J = 1.5 Hz, 1H), 6.68 (s, 2H), 7.86–7.81 (m, 2H), 8.29–8.25 (d, J = 1.8 Hz, 1H), 8.38–8.33 (m, 1H); 13C NMR (50 MHz, CDCl3) δ: 14.4, 30.5, 56.3, 60.7, 66.2, 105.9, 118.7, 127.3, 128.0, 128.9, 129.6, 131.7, 133.5, 134.2, 138.9, 145.9, 153.9, 154.5, 156.3, 193.4; IR (cm−1): 3452, 2927, 1655, 1682, 1599, 1504, 1465, 1421, 1353, 1315, 1255, 1199, 1128, 1042, 835, 786, 698; C23H22N2O6 (FW = 422.44): C, 65.40; N, 6.63; H, 5.25%; found: C, 65.26; N, 6.65; H, 5.23%;X-ray crystal structure determination of PPD-4
Single-crystal X-ray diffraction data for PPD-4 were collected at an Oxford Gemini S diffractometer equipped with a CCD detector, using monochromatized MoKα radiation (λ = 0.71073 Å). Data reduction and empirical absorption correction were performed with CrysAlisPRO.31 The structure was solved by direct methods using SHELXS and refined on F2 by full-matrix least-squares using SHELXL.32 All non-hydrogen atoms were refined anisotropically. H atoms were placed in geometrically calculated positions and refined using the riding model with Uiso values constrained to 1.2 Ueq or 1.5 Ueq of the parent C atoms. The PARST33 and PLATON34 software were used to perform geometrical calculation, while ORTEP3 (ref. 35) and was employed for molecular graphics. Crystallographic details for structure analysis of the PPD-4 are summarized in Table S6.†CCDC 1584133 contains the supplementary crystallographic data for compound PPD-4.
DPPH free radical scavenging assay
The free radical scavenging activity of the examined compounds was performed using the DPPH method, according to ref. 36. DPPH solution (1 mL, 0.05 mM) in methanol was mixed with the tested compound (20 μL of compound solution in DMSO and 980 μL of methanol). The reaction mixture was allowed to stand at room temperature for 20 and 60 min. After incubation the absorbance was determined spectrophotometrically at 517 nm. As control solution, methanol was used. IC50 values represent the concentration necessary to obtain 50% of a maximum scavenging capacity. NDGA was used as positive control. All measurements were performed on three replicates. The results presented as mean values ± standard deviation (SD) of three independent measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This investigation was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia project No. 172016 and 172014.Notes and references
- (a) M. Asif, Curr. Med. Chem., 2012, 19, 2984 CrossRef CAS PubMed; (b) A. Marella, M. Shaquiquzzaman, M. Akhter, G. Verma and M. M. Alam, J. Enzyme Inhib. Med. Chem., 2015, 30, 597 CrossRef CAS PubMed; (c) K. R. A. Abdellatif, M. T. Elsaady, S. A. Abdel-Aziz and A. H. A. Abusabaa, J. Enzyme Inhib. Med. Chem., 2016, 31, 1545 CrossRef CAS PubMed; (d) M. J. Naim, O. Alam, F. Nawaz, M. J. Alam and P. Alam, J. Pharm. BioAllied Sci., 2016, 8, 2 CrossRef CAS PubMed.
- (a) J. Li, Y. F. Zhao, X. Y. Yuan, J. X. Xu and P. Gong, Molecules, 2006, 11, 574 CrossRef CAS PubMed; (b) I. A. Al-Suwaidan, N. I. Abdel-Aziz, A. S. El-Azab, M. A.-A. El-Sayed, A. M. Alanazi, M. B. El-Ashmawy and A. A.-M. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2015, 30, 679 CrossRef CAS PubMed.
- L. Zhang, L. P. Guan, X. Y. Sun, C. X. Wei, K. Y. Chai and Z. S. Quan, Chem. Biol. Drug Des., 2009, 73, 313 CAS.
- J. Sinkkonen, V. Ovcharenko, K. N. Zelenin, I. P. Bezhan, B. A. Chakchir, F. Al-Assar and K. Pihlaja, Eur. J. Org. Chem., 2002, 6, 2046 CrossRef.
- C. K. Ryu, R. E. Park, M. Y. Ma and J. H. Nho, Bioorg. Med. Chem. Lett., 2007, 17, 2577 CrossRef CAS PubMed.
- S. B. Jadhava, S. Fatemaa, M. A. I. Patela and M. Farooqui, Chem. Biol. Interface, 2017, 7, 134 Search PubMed.
- K. Abouzid, M. A. Hakeem, O. Khalil and Y. Maklad, Bioorg. Med. Chem., 2008, 16, 382 CrossRef CAS PubMed.
- (a) S. S. El-Saka, A. H. Soliman and A. M. Imam, Afinidad, 2009, 66, 167 Search PubMed; (b) M. Abbasi, S. M. R. Nazifi, Z. S. Nazifi and A. R. Massah, J. Chem. Sci., 2017, 129, 1257 CrossRef CAS.
- D. S. Raghuvanshi and K. N. Singh, Tetrahedron Lett., 2011, 52, 5702 CrossRef CAS.
- R. Ghahremanzadeh, S. Ahadi, M. Sayyafi and A. Bazgir, Tetrahedron Lett., 2008, 49, 4479 CrossRef CAS.
- (a) R. Ghahremanzadeh, G. I. Shakibaei and A. Bazgir, Synlett, 2008, 1129 CAS; (b) M. R. Nabid, S. J. T. Rezaei, R. Ghahremanzadeh and A. Bazgir, Ultrason. Sonochem., 2010, 17, 159 CrossRef CAS PubMed.
- L.-P. Liu, J.-M. Lu and M. Shi, Org. Lett., 2007, 9, 1303 CrossRef CAS PubMed.
- (a) B. Maleki and S. S. Ashrafi, J. Mex. Chem. Soc., 2014, 58, 159 CAS; (b) A. R. Kiasat, S. Noorizadeh, M. Ghahremani and S. J. Saghanejad, J. Mol. Struct., 2013, 1036, 216 CrossRef CAS; (c) G. Sabitha, C. Srinivas, A. Raghavendar and J. S. Yadav, Helv. Chim. Acta, 2010, 93, 1375 CrossRef CAS; (d) E. Mosaddegh and A. Hassankhani, Tetrahedron Lett., 2011, 52, 488 CrossRef CAS; (e) M. Kidwai, R. Chauhan and A. Jahan, Chin. Sci. Bull., 2012, 57, 2273 CrossRef; (f) H. J. Wang, X. N. Zhang and Z. H. Zhang, Monatsh. Chem., 2010, 141, 425 CrossRef CAS; (g) A. Corma and H. Garcia, Catal. Today, 1997, 38, 257 CrossRef CAS; (h) R. G. Vaghei, R. K. Nami, Z. K. Semiromi, M. Amiri and M. Ghavidel, Tetrahedron, 2011, 67, 1930 CrossRef; (i) H. R. Shaterian, M. Ghashang and M. Feyzi, Appl. Catal., A, 2008, 345, 128 CrossRef CAS; (j) M. Sayyafi, M. Sayyedhamzeh, H. R. Khavasi and A. Bazgir, Tetrahedron, 2008, 64, 2375 CrossRef CAS.
- J. M. Khurana and D. Magoo, Tetrahedron Lett., 2009, 50, 7300 CrossRef CAS.
- (a) G. Zhao, T. Jiang, H. Gao, B. Han, J. Huang and D. Sun, Green Chem., 2004, 6, 75 RSC; (b) S. Sahoo, T. Joseph and S. B. Halligudi, J. Mol. Catal. A: Chem., 2006, 244, 179 CrossRef CAS; (c) A. Kumar and S. S. Pawar, J. Org. Chem., 2004, 69, 1419 CrossRef CAS PubMed; (d) A. Aggarwal, N. L. Lancaster, A. R. Sethi and T. Welton, Green Chem., 2002, 4, 517 RSC; (e) D. Fang, K. Gong, D. Zhang and Z. Liu, Monatsh. Chem., 2009, 140, 1325 CrossRef CAS; (f) D. Yin, C. Li, L. Tao, N. Yu, S. Hu and D. Yin, J. Mol. Catal. A: Chem., 2006, 245, 260 CrossRef CAS.
- (a) M. Sayyafi, M. Seyyedhamzeh, H. R. Khavasi and A. Bazgir, Tetrahedron, 2008, 64, 2375 CrossRef CAS; (b) J. Davarpanahab and A. R. Kiasat, RSC Adv., 2015, 5, 7986 RSC; (c) A. R. Kiasat and J. Davarpanah, J. Mol. Catal. A: Chem., 2013, 373, 46 CrossRef CAS.
- V. P. Petrović, D. Simijonović, M. N. Živanović, J. V. Košarić, Z. D. Petrović, S. Marković and S. D. Marković, RSC Adv., 2014, 4, 24635 RSC.
- D. Simijonović, Z. D. Petrović and V. P. Petrović, J. Mol. Liq., 2013, 179, 98 CrossRef.
- L. Valgimigli, R. Amorati, M. G. Fumo, G. A. DiLabio, G. F. Pedulli, K. U. Ingold and D. A. Pratt, J. Org. Chem., 2008, 73, 1830 CrossRef CAS PubMed.
- (a) A. T. De Pinedo, P. Peňalver and J. C. Morales, Food Chem., 2007, 103, 55 CrossRef; (b) M. C. Foti, E. R. Johnson, M. R. Vinqvist, J. S. Wright, L. R. C. Barclay and K. U. Ingold, J. Org. Chem., 2002, 67, 5190 CrossRef CAS PubMed; (c) M. C. Foti, L. R. C. Barclay and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 12881 CrossRef CAS PubMed.
- (a) J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173 CrossRef CAS PubMed; (b) M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 928 CrossRef CAS PubMed.
- (a) A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335 CrossRef CAS PubMed; (b) A. Galano, J. Mex. Chem. Soc., 2015, 59, 231 Search PubMed; (c) A. Pé rez-Gonzá lez, A. Galano and J. Raú Alvarez-Idaboy, New J. Chem., 2014, 38, 2639 RSC; (d) J. Rimarčỉk, V. Lukeš, E. Klein and M. Ilčin, J. Mol. Struct.: THEOCHEM, 2010, 952, 25 CrossRef; (e) E. Klein, V. Lukeš and M. Ilčin, Chem. Phys., 2007, 336, 51 CrossRef CAS; (f) Z. Marković, J. Đorović, Z. D. Petrović, V. Petrović and D. Simijonović, J. Mol. Model., 2015, 21, 293 CrossRef PubMed; (g) J. Xie and K. M. Schaich, J. Agric. Food Chem., 2014, 62, 4251 CrossRef CAS PubMed; (h) Z. D. Petrović, D. Simijonović, J. Đorovi, V. Milovanović, Z. Marković and V. P. Petrović, ChemistrySelect, 2017, 2, 11187 CrossRef.
- A. Amić, Z. Marković, J. M. D. Marković, S. Jeremić, B. Lučić and D. Amić, Comput. Biol. Chem., 2016, 65, 45 CrossRef PubMed.
- H.-D. Belitz, W. Grosch and P. Schieberle, Food Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009 Search PubMed.
- Z. Marković, D. Milenković, J. Đorović, J. M. Dimitrić Marković, V. Stepanić, B. Lučić and D. Amić, Food Chem., 2012, 134, 1754 CrossRef PubMed.
- (a) X. Li, P. Fang, J. Mai, E. T. Choi, H. Wang and X. Yang, J. Hematol. Oncol., 2013, 6, 19 CrossRef CAS PubMed; (b) F. Muller, J. Am. Aging Assoc., 2000, 23, 227 CAS.
- M. J. Frisch, W. G. Trucks, B. H. Schlegel, E. G. Scuseria, A. M. Robb, R. J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, A. G. Petersson, H. Nakatsuji, M. Caricato, X. Li, P. H. Hratchian, F. A. Izmaylov, J. Bloino, G. Zheng, L. J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, A. J. Montgomery Jr., E. J. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, N. K. Kudin, N. V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, C. J. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, E. R. Stratmann, O. Yazyev, J. A. Austin, R. Cammi, C. Pomelli, W. J. Ochterski, L. R. Martin, K. Morokuma, G. V. Zakrzewski, A. G. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, D. A. Daniels, O. Farkas, B. J. Foresman, V. J. Ortiz, J. Cioslowski and J. D. Fox, Gaussian 09 Rev C, Gaussian Inc., Wallingford, 2009 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 2009, 113, 4538 CrossRef CAS PubMed.
- Z. Marković, J. Tošović, D. Milenković and S. Marković, Comput. Theor. Chem., 2016, 1077, 11 CrossRef.
- CrysAlisPRO Software system, Agilent Technologies UK Ltd., Oxford, England, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- C. Kontogiorgis and D. Hadjipavlou-Litina, J. Enzyme Inhib. Med. Chem., 2003, 18, 63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of compounds, 1H NMR and 13C NMR spectra, melting points, Kohn–Sham orbitals of PDDs, Experimental and simulated UV-Vis spectra. CCDC 1584133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02702a |
| This journal is © The Royal Society of Chemistry 2018 |