
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of core–shell N-TiO2@CuOx with enhanced visible light photocatalytic performance†
Shu Wang,
Rufei Huo,
Rui Zhang,
Yuchuan Zheng,
Changjiang Li and
Le Pan *
*
College of Chemistry and Chemical Engineering, Huangshan University, Huangshan 245041, China. E-mail: panle_hs@163.com
First published on 11th July 2018
Abstract
In this paper, a core–shell N-TiO2@CuOx nanomaterial with increased visible light photocatalytic activity was successfully synthesized using a simple method. By synthesizing ammonium titanyl oxalate as a precursor, N-doped TiO2 can be prepared, then the core–shell structure of N-TiO2@CuOx with a catalyst loading of Cu on its surface was prepared using a precipitation method. It was characterized in detail using XRD, TEM, BET, XPS and H2-TPR, while its photocatalytic activity was evaluated using the probe reaction of the degradation of methyl orange. We found that the core–shell N-TiO2@CuOx nanomaterial can lessen the TiO2 energy band-gap width due to the N-doping, as well as remarkably improving the photo-degradation activity due to a certain loading of Cu on the surfaces of N-TiO2 supports. Therefore, a preparation method for a novel N, Cu co-doped TiO2 photocatalyst with a core–shell structure and efficient photocatalytic performance has been provided.
Introduction
Metallic oxides are important and widely used in catalysis, environmental science, ceramics, electronic devices and materials chemistry.1–4 However, there are some disadvantages to single metallic oxides.5–7 For example, anatase TiO2 as a photocatalyst has many advantages, such as: cheapness, nontoxicity, stable chemical properties, degradation of organic matter and so on.8–13 In the past twenty years, it has received wide attention. However, the band gap of 3.2 eV can only respond to UV light with a wavelength less than 387 nm.14–17 The energy of ultraviolet light in sunlight only accounts for about 5%, so this seriously restricts the application of this photocatalyst to solar energy. If the scope of TiO2 light absorption can be effectively expanded to visible light, its application will be greatly expanded, so this photocatalyst can make full use of solar energy, and the two major problems facing humanity, the energy crisis and environmental pollution, will be effectively solved.Since Asahi18 reported the semiconductor TiO2 photocatalytic degradation of organic pollutants, crucial progress has been made by the doping of the element N, which causes the decrease of the band-gap of TiO2 because of the element N. The amount of research on TiO2 doped with N has been increasing.19–23 In the process of the preparation of TiO2, a variety of physical and chemical methods are needed to prepare N-doped TiO2. These methods, without exception, require nitrogen sources. On this basis, Fang24 found that a N-doped TiO2 photocatalyst could be obtained using ammonium titanyl oxalate as the precursor, and the efficiency of the process was greatly improved.
At the same time, the precious metals that have been reported with TiO2 to improve the photocatalytic performance include precious metals such as Pt, Ag, Ir, Au, Ru, Pd, Rh and so on, among which there are the most reports about Pt, followed by Pd and Au.25–28 The photocatalytic performance after Pt modification is the best, but comes with a higher cost. Ag modification leads to relatively less toxicity and has a lower cost. Therefore, the preparation of highly active noble metal doped photocatalysts by Ag deposition and modification is considered to be one of the important directions to improve photocatalytic activity in the future. Cu, Ag and Au elements belong to the same group of the periodic table IB, which means they may have a similar catalytic activity in the oxidation colour-fading reaction. However, the catalytic activity of CuOx was obviously lower than those of AgOx and AuOx due to the catalytic reaction on the surface of CuOx which has been shown to have a higher activation energy in previous research.29–31
In this paper, from the perspective of energy utilization and catalytic activity, we composed a new TiO2 compound with N and Cu co-doped by a two-step method. When TiO2 was synthesized, N doping was completed simultaneously using a suitable synthesis in the first step, which is different to the traditional method. There are many benefits to employing the new synthetic method of N-doped TiO2, such as: excellent reproducibility of sample synthesis, a better N-doped effect, and outstanding stability of catalytic activity based on “zero” loss of the N element. In the second step, the core–shell structure N-TiO2@CuOx catalyst was prepared by a precipitation method. The light energy which was absorbed by N doped TiO2 could be translated into the internal energy which was employed across the high energy barrier mentioned above. Then, their structures were examined with XRD, TEM, BET, and XPS. Lastly, we compared their catalytic activities using methyl orange degradation experiments. Therefore, an N, Cu co-doped TiO2 photocatalyst has been synthesized using a simple and low-cost process, and its core–shell structure and efficient photocatalytic performance have been studied.
Results and discussion
Fig. S1† displays the concentration of methyl orange as a function of the reaction time over the different amounts of copper oxide in the catalytic system of TiO2@CuOx and N-TiO2@CuOx catalysts in UV, all wavelengths, and visible light respectively. This shows that the adsorption performance decreased and the catalytic activity increased as the CuOx loading increased. Considering the above conditions, the total degradation performances of TiO2@0.1CuOx and N-TiO2@0.1CuOx showed maximum activities in the respective catalytic systems. Therefore, 0.1CuOx was chosen in the later text and we do not specifically point out “0.1”.Fig. 1 displays the XRD patterns of the TiO2 (A), N-TiO2 (B), TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts. The XRD patterns of the four catalysts all clearly show anatase TiO2 diffraction peaks, while only in the XRD pattern of TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts can we find Cu2O diffraction peaks. These indicate the amount of Cu2O nanoparticles formed when the copper nitrate was deposited on the surface of TiO2 and N-TiO2.
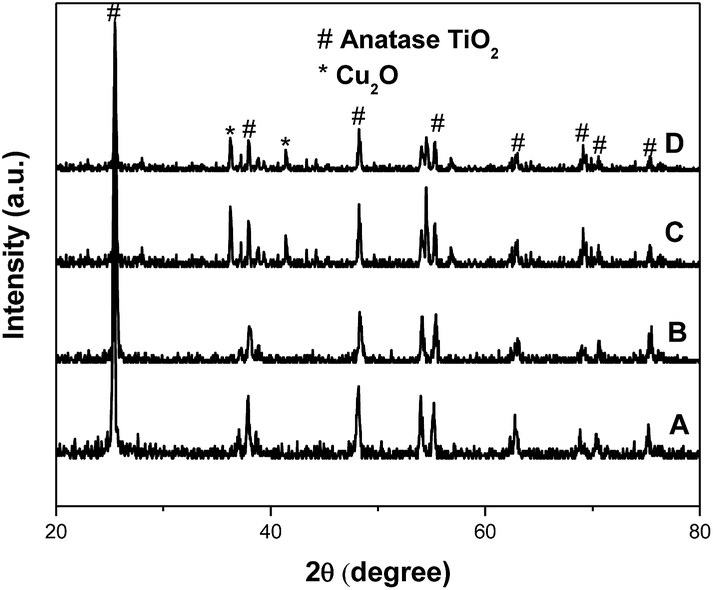 | ||
| Fig. 1 XRD patterns of the TiO2 (A), N-TiO2 (B), TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts. | ||
Fig. 2 shows representative transmission electron microscopy (TEM) images of TiO2 (A), N-TiO2 (B), TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts. The TEM images of the four catalysts all clearly show the core and shell structures in the TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts, but not in the TiO2 (A) and N-TiO2 (B) catalysts.
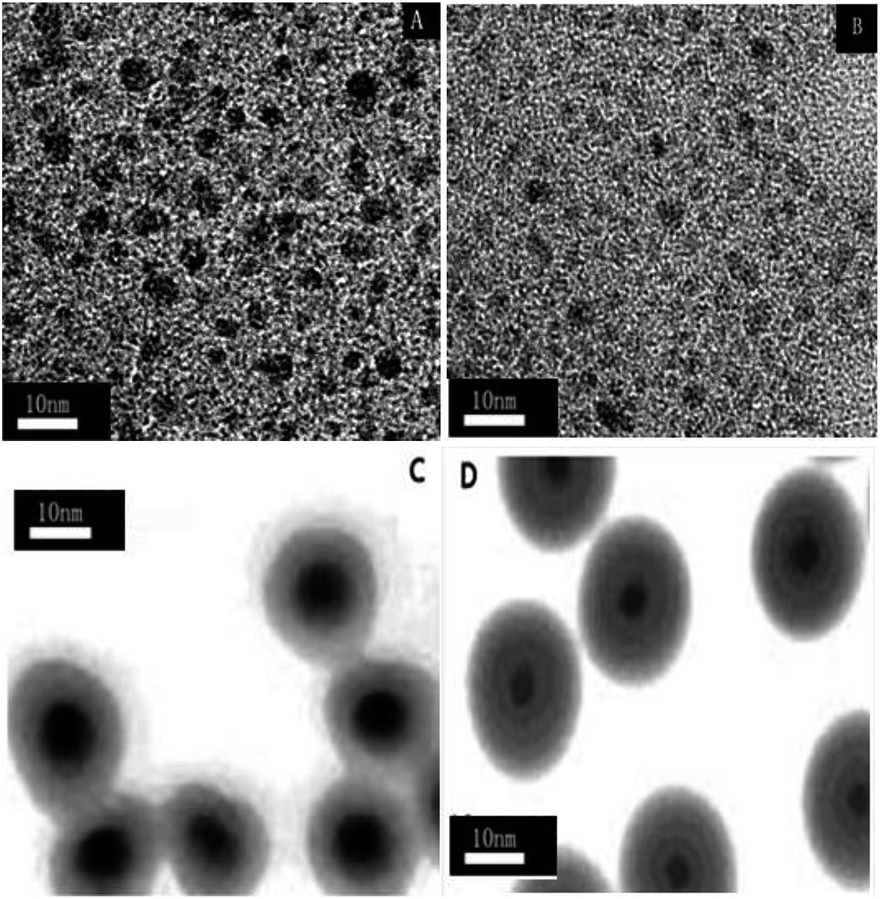 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of the TiO2 (A), N-TiO2 (B), TiO2@CuOx (C) and N-TiO2@CuOx (D) catalysts. | ||
Fig. 3 displays Ti 2p XPS patterns of the TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts. The Ti 2p XPS spectra of TiO2 and N-TiO2 can be nicely fitted with one component centring at 458.5 eV, which can be attributed to Ti4+ on the surface.32 With the Cu loading on TiO2 and N-TiO2, the Ti 2p peak shifts from 458.5 eV to 459.0 eV. Therefore, we hypothesize that the interaction between Ti and Cu on the interface contributes to this shift. While the Ti 2p XPS spectra of TiO2@CuOX and N-TiO2@CuOX fit with another component centring at 459.0 eV, the formation of the chemical bonds increases the binding energy of Ti,17 which can be attributed to the complex species on the surfaces of core–shell structures.
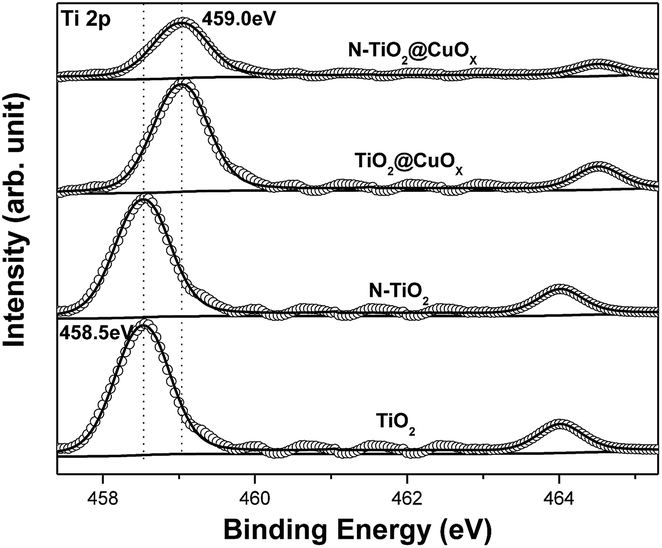 | ||
| Fig. 3 Ti 2p XPS patterns of the TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts. Scattered circles and solid lines indicate the experimental data and fitting spectra, respectively. | ||
Fig. 4 shows the Cu 2p3/2 XPS patterns of the TiO2@CuOx and N-TiO2@CuOx catalysts. Table 1 shows the peak-fitting results of the Cu XPS spectra. We can observe that the TiO2@CuOx and N-TiO2@CuOx catalysts both show double peaks which are split in the Cu 2p3/2 XPS patterns. As the binding energy is almost the same for Cu0 and Cu+ in XPS,33 we cannot differentiate between the two peaks at low binding energies in the XPS spectra. As the 941–944 eV shake-up34 is unique for Cu2+ in XPS spectra, and the binding energy of Cu2+ increases, we can confirm that it is the Cu2+ that caused the peak at 935.3 eV. The difference between the Cu 2p3/2 XPS spectra of the two samples is great, which indicates the different speciation which exists on their surfaces.
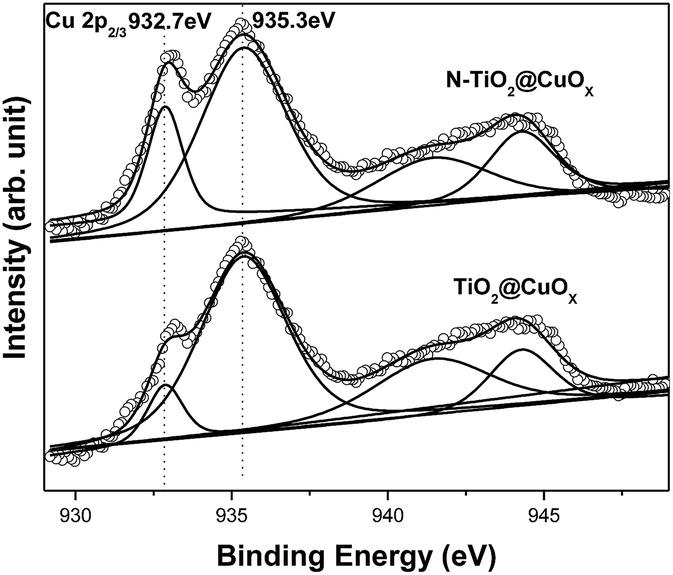 | ||
| Fig. 4 Cu 2p3/2 XPS patterns of the TiO2@CuOx and N-TiO2@CuOx catalysts. Scattered circles and solid lines indicate the experimental data and fitting spectra, respectively. | ||
| Catalyst | Component 1 | Component 2 | ||
|---|---|---|---|---|
| BE (eV) | Fraction | BE (eV) | Fraction | |
| TiO2@CuOx | 932.7 | 9% | 935.3 | 91% |
| N-TiO2@CuOx | 932.7 | 25% | 935.3 | 75% |
The following conclusions can be reached by analyzing the above results. When the copper nitrate was separately deposited on the surfaces of TiO2 and N-TiO2, the content of Cu0 and Cu+ obtained on the surfaces of TiO2 and N-TiO2 was consistent with previous XRD results. Furthermore, we cannot find a peak due to CuO in the XRD patterns, but it is obvious in the XPS patterns. Therefore it can be hypothesized that the CuO on the surface of the catalysts was amorphous.
The N 1s XPS peak could be adequately fitted by two components with the N 1s binding energies at 399.5 and 401.5 eV in Fig. 5. We attribute the binding energy at 401.5 eV to the substitution with N in O–Ti–N,35 while we attribute the binding energy at 399.5 eV to doped N in interstitial lattice sites, which can decrease the band gap from 3.2 eV to 2.4 eV.22
 | ||
| Fig. 5 N 1s XPS patterns of the N-TiO2 and N-TiO2@CuOx catalysts. Scattered circles and solid lines indicate the experimental data and fitting spectra, respectively. | ||
Fig. 6 and Table 2 show the N2 adsorption–desorption isotherms and textural parameters of TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx respectively. The N2 adsorption–desorption isotherms of TiO2 and N-TiO2@CuOx are similar with H2 hysteresis loops according to IUPAC, which illustrate that the mesoporous structure exists on the surface of the catalysts.36 We attribute the difference in absorption and desorption mechanisms to the “ink-bottle” pore.37 The N2 adsorption–desorption isotherms of N-TiO2 show that the hysteresis loop is very wide with a high specific surface area and small size. The N2 adsorption–desorption isotherm of TiO2@CuOx shows that the hysteresis loop shifts to the higher relative pressure (P/P0 = 0.9) between the H2 and H3 hysteresis loops according to the BDDT classification, which illustrates that the macro-pore structure exists on the surface of the support with a low specific surface area and big size.
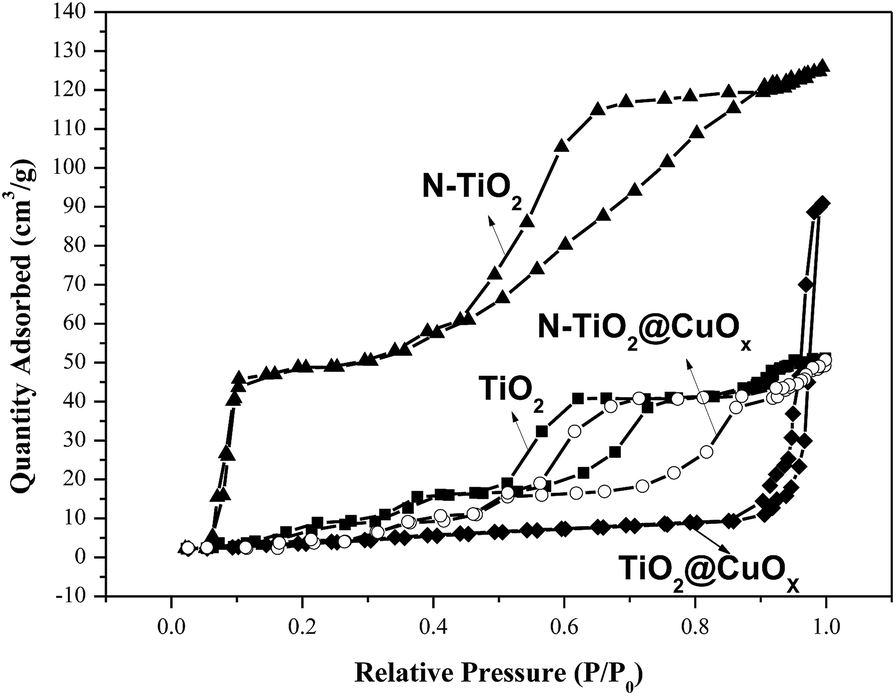 | ||
| Fig. 6 The N2 adsorption–desorption isotherms for TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx. | ||
| Support | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| TiO2 | 38.2 | 0.195 | 22.4 |
| N-TiO2 | 174 | 0.294 | 15.4 |
| TiO2@CuOx | 16.5 | 0.115 | 62.7 |
| N-TiO2@CuOx | 29.4 | 0.181 | 48.7 |
Fig. S2† shows the concentration of methyl orange as a function of the reaction time over the TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts in UV light, all wavelengths and visible light. The degradation properties are composed of two parts, adsorption activity and photocatalytic activity, and the degradation of methyl orange as the probe reaction was employed to respond to these two parts of the activity under different conditions. The results before the light illumination were characterized as adsorption activity, and after the light illumination as photocatalytic activity. Considering the results of BET and the adsorption and activity performance, we find that the adsorption performance is closely correlated to the specific surface area and size. Among the catalysts, N-TiO2 has the best adsorption and TiO2@CuOx has the worst, while N-TiO2@CuOx is close to TiO2 which is in the middle. We can conclude from the results of XPS, TEM and the absorption data that N-doping can effectively improve the absorption by the catalyst, but the core–shell structure may have an adverse effect.
Fig. 7 and 8 display the photo-degradation rate and total degradation rate of the TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts in UV light, all wavelengths and visible light. The catalytic activity of N-TiO2 is much better than that of TiO2 in visible light, and also of N-TiO2@CuOx and TiO2@CuOx. We can make a conclusion that N-doping can effectively reduce the band gap of energy of TiO2 and improve the use of visible light.
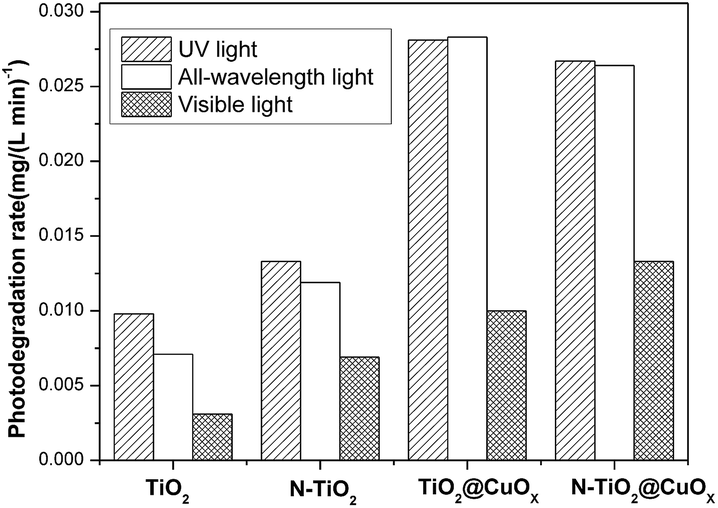 | ||
| Fig. 7 Photo-degradation rate of the TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts in UV light, all wavelengths and visible light. | ||
 | ||
| Fig. 8 Total degradation rate of TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx catalysts in UV light, all wavelengths and visible light respectively. | ||
Fig. 9 shows the UV-vis diffuse reflection spectra of N-TiO2 and N-TiO2@CuOx. The N-TiO2 and N-TiO2@CuOx catalysts exhibit a pale yellow colour. The UV-vis diffuse reflection spectra of these two catalysts show a continuous and tailing absorption in the visible region. The band-gap energies of the N-TiO2 and N-TiO2@CuOx catalysts are 2.83 and 2.80 eV, respectively, which were determined by the plot of the modified Kubelka–Munk function (αhν)1/2 versus photon energy assuming indirect transitions.38 Based on the above data, we can make a conclusion that N-doping can effectively reduce the band gap of energy of TiO2 in N-TiO2 and N-TiO2@CuOx catalysts.
 | ||
| Fig. 9 UV-vis diffuse reflection spectra for the catalysts N-TiO2 and N-TiO2@CuOx. | ||
Fig. 10 shows the H2-TPR spectra of the catalysts of TiO2@CuOx and N-TiO2@CuOx. The H2-TPR of CuO supported on CeO2 has been extensively investigated:39–42 CuO is reduced below 300 °C and the reduction temperature of different CuO species on CeO2 follows the order: finely dispersed CuO < large CuO crystallites < bulk CuO. Therefore, the reduction peak at 221 °C is assigned to the reduction of finely dispersed CuO, and that at 258 °C to the reduction of large CuO crystallites and bulk CuO.43
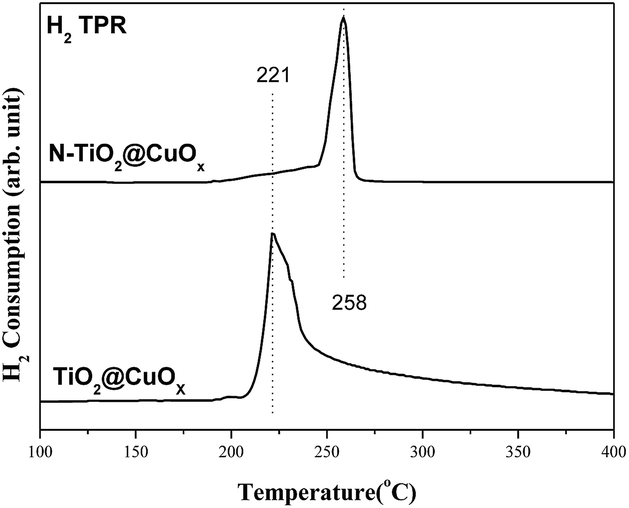 | ||
| Fig. 10 H2-TPR spectra of the catalysts TiO2@CuOx and N-TiO2@CuOx. | ||
Considering the results from H2-TPR and the photo-degradation performance of TiO2@CuOx and N-TiO2@CuOx catalysts in UV and all-wavelength light, the activity increases as the size of CuO decreases on the surface. However, the reason for the activity of TiO2@CuOx being worse than N-TiO2@CuOx in visible light can be attributed to the decrease in efficiency for visible light.
On the basis of TEM, BET, H2-TPR, XPS, the catalytic activity performance and the stability results, the interfacial structures of TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx and their responses to different wavelengths of light are schematically illustrated in Fig. 11. These results demonstrate that the interfacial reaction mechanisms on the surfaces of TiO2@CuOx and N-TiO2@CuOx are the same. The light energy which was absorbed by TiO2 and N-doped TiO2 is translated into the internal energy which was employed to overcome the energy barrier of the reaction on the CuOx surface. The difference is simply the wavelengths of light which are responded to by TiO2 and N-doped TiO2.
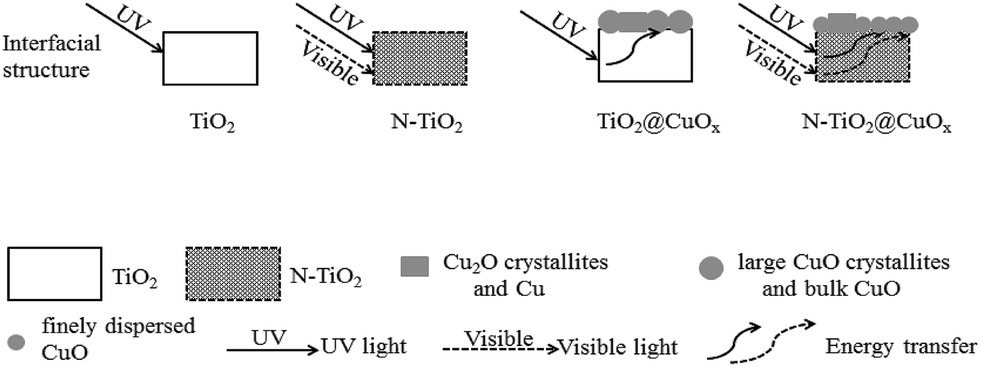 | ||
| Fig. 11 Schematic illustration of the interfacial structures of TiO2, N-TiO2, TiO2@CuOx and N-TiO2@CuOx and their responses to different wavelengths of light. | ||
Fig. 12 shows the stability of N-TiO2@CuOx in UV light, all wavelengths, and visible light, and obviously shows that there is hardly any absorption performance, although the activity performances are almost unchanged during the 40 hours. We consider that the N-TiO2@CuOx catalyst is a kind of reusable material to degrade the methyl orange.
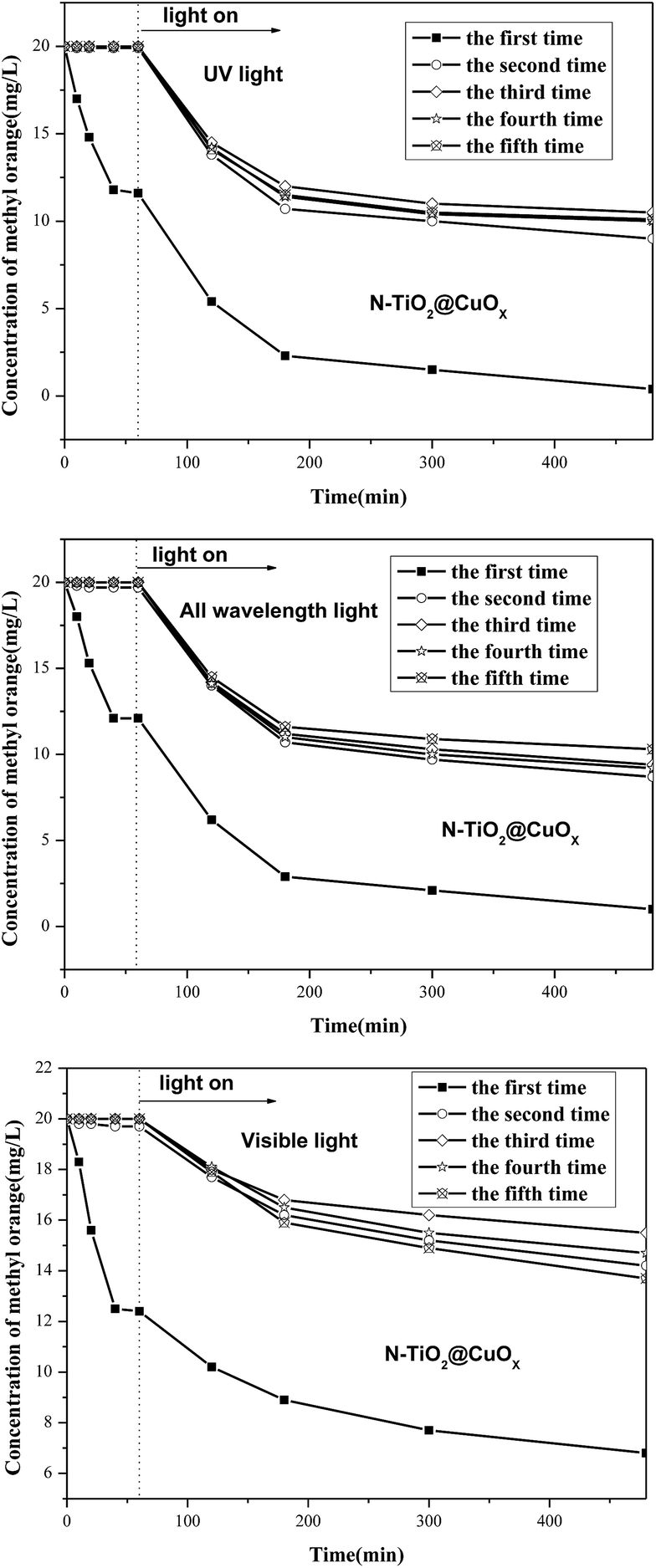 | ||
| Fig. 12 Stability of the N-TiO2@0.1CuOx in UV light, all wavelengths, and visible light. | ||
Experimental
Catalyst preparation
We prepared two solutions by adding 20.0 g oxalic acid to 200 mL distilled water for both, then dripped 10 mL titanium tetrachloride into both of them dropwise, and stirred them evenly. After the drops were added, we added sodium hydroxide and ammonia solution respectively to adjust the pH values of the solutions to 8–9. We continuously stirred the above solutions for 8 h, after which white precipitates were produced. After the agitation, we aged them for 8 h, and then filtered out these white precipitates. These white precipitates were washed with distilled water and ethanol several times until there was no chloride in the filtrate, then dried at 70 °C and calcined for 2 h at 400 °C in sequence. These two obtained products were recorded as TiO2 and N-TiO2, respectively.The above TiO2 (5 g) and N-TiO2 (5 g) were ground to powders and each was put into a three-neck bottle. Then we added 5 mL copper nitrate solution (1.06 mol L−1) and distilled water to both of them and stirred them at 70 °C for 30 min. Then we added sodium hydroxide and ammonia solution respectively to adjust the pH values of the solutions to 8–9 separately, and stirred them at 70 °C for 24 h. Then we filtered these solids, washed them with distilled water and ethanol several times, dried them at 70 °C for 12 h, and calcined them in air at 400 °C for 4 h. These two obtained products were recorded as TiO2@CuOx and N-TiO2@CuOx respectively.
Catalyst characterization
Powder X-ray diffraction (XRD) patterns were acquired on a Philips X’Pert PRO SUPER X-ray diffractometer with a Ni-filtered Cu Kα X-ray source operating at 40 kV and 50 mA. X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALAB 250 electron spectrometer using a monochromatized Al Kα excitation source (hν = 1486.6 eV). Transmission electron microscopy (TEM) measurements were carried out on a JEOL-2100F transmission electron microscope at an accelerating voltage of 200 kV. N2 adsorption–desorption isotherms (BET) were acquired on a Beckman Coulter SA3100 surface area analyzer, in which the sample was degassed at 300 °C for 30 min under a nitrogen atmosphere prior to the measurements. The reduction behaviors of the catalysts were measured using H2 temperature-programmed reduction (H2-TPR) techniques. UV-vis absorption spectra were recorded using a SolidSpec-DUV-3700 DUV-vis-NIR spectrometer.Degradation property measurements
The degradation property is composed of two parts, the adsorption activity and the photocatalytic activity. We employed the degradation of methyl orange as the probe reaction. We added 0.12 g catalyst to 20 mg L−1 methyl orange aqueous solution (40 mL), and stirred it for 1 h whilst avoiding light. We subjected the catalyst to the different wavelengths of irradiation, and took the supernatant to measure the concentration of methyl orange at different time points. The results before the light illumination were characterized as adsorption activity, and after the light illumination as photocatalytic activity.Conclusions
A novel N, Cu co-doped TiO2 photocatalyst with a core–shell structure has been prepared using a simple method and the structure–adsorption–photo-degradation activity relationships of TiO2 base catalysts were examined using the probe reaction of the degradation of methyl orange. The results suggested that the N-TiO2@CuOx nanomaterial can reduce the band gap width of TiO2 by N-doping. Meanwhile, we found the photo-degradation rate of catalysts clearly improved with a certain loading of Cu on the surfaces of N-TiO2. Consequently, we synthesized an efficient N-doped TiO2@CuOx photocatalyst with a core–shell structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21401065), the Undergraduate Training Programs for Innovation, Entrepreneurship of China (201710375026, 201710375012) and the Natural Science Foundation of Huangshan University (2011xkj014, 2015xkj003).Notes and references
- Q. Hua, T. Cao, X. K. Gu, J. Q. Lu, Z. Q. Jiang, X. R. Pan, L. F. Luo, W. X. Li and W. X. Huang, Angew. Chem., Int. Ed., 2014, 53, 4856–4861 CrossRef PubMed.
- Q. Hua, T. Cao, H. Z. Bao, Z. Q. Jiang and W. X. Huang, ChemSusChem, 2013, 6, 1966–1972 CrossRef PubMed.
- H. B. Tao, L. W. Fan, J. Z. Chen, H. B. Yang, J. J. Gao, J. W. Miao, S. L. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef PubMed.
- C. Y. Hou, M. W. Zhang, T. S. Kasama, C. Engelbrekt, L. L. Zhang, H. Z. Wang and Q. J. Chi, Adv. Mater., 2016, 28, 4097–4104 CrossRef PubMed.
- N. Serpone, I. Texier, A. V. Emerline, P. Pichat, H. Hidaka and J. Zhao, J. Photochem. Photobiol., A, 2000, 136, 145–246 CrossRef.
- M. M. Aboelnga and J. W. Gauld, J. Phys. Chem. B, 2017, 121, 6163–6174 CrossRef PubMed.
- A. Dawson and V. K. Prashant, J. Phys. Chem. B, 2001, 105, 960–966 CrossRef.
- W. Z. Fang, M. Y. Xing and J. L. Zhang, J. Photochem. Photobiol., C, 2017, 32, 21–39 CrossRef.
- F. Jiang, Z. Zheng, Z. Y. Xu, S. R. Zheng, Z. B. Guo and L. Q. Chen, J. Hazard. Mater., 2006, 134, 94–103 CrossRef PubMed.
- G. Colon, M. C. Hidalgo and J. A. Navio, Appl. Catal., B, 2003, 45, 39–50 CrossRef.
- P. Mohapatra, S. K. Samantaray and K. Parida, J. Photochem. Photobiol., A, 2005, 170, 189–194 CrossRef.
- A. Nakajima, H. Obata, Y. Kameshima and K. Okada, Catal. Commun., 2005, 6, 716–720 CrossRef.
- X. C. Wang, J. C. Yu, P. Liu, X. X. Wang, W. Y. Su and X. Z. Fu, J. Photochem. Photobiol., A, 2006, 179, 339–347 CrossRef.
- F. Nemati and A. Elhampour, Res. Chem. Intermed., 2016, 42, 7611–7624 CrossRef.
- J. Fang, F. C. Shi, J. J. Ding, S. T. Xu, J. Bao, Y. S. Ma, J. Q. Jiang, W. P. Zhang and W. X. Huang, J. Phys. Chem. C, 2010, 114, 7940–7948 CrossRef.
- J. Fang, H. Z. Bao, B. He, F. Wang, D. J. Si, Z. Q. Jiang, Z. Y. Pan, S. Q. Wei and W. X. Huang, J. Phys. Chem. C, 2007, 111, 19078–19085 CrossRef.
- J. Fang, X. Z. Bi, D. J. Si, Z. Q. Jiang and W. X. Huang, Appl. Surf. Sci., 2007, 253, 8952–8961 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef PubMed.
- X. W. Cheng, H. L. Liu, Q. H. Chen, J. J. Li and P. Wang, Electrochim. Acta, 2013, 103, 134–142 CrossRef.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef.
- O. Diwald, T. L. Thompson and T. Zubkov, J. Phys. Chem. B, 2004, 108, 6004–6008 CrossRef.
- O. Diwald, T. L. Thompson and E. G. Goralski, J. Phys. Chem. B, 2004, 108, 52–57 CrossRef.
- S. Yin, H. Yamaki and M. Komatsu, J. Mater. Chem., 2003, 13, 2996–3001 RSC.
- J. Fang, F. Wang, K. Qian, H. Z. Bao, H. X. Sun, Z. Q. Jiang and W. X. Huang, J. Phys. Chem. C, 2008, 112, 18150–18156 CrossRef.
- D. M. Blake, NREL/TP-430-6084, National Renewal Energy Laboratory, Golden, Co, 1994 Search PubMed.
- D. M. Blake, NREL/TP-340-22197, National Renewal Energy Laboratory, Golden, Co, 1997 Search PubMed.
- D. M. Blake, NREL/TP-570-26797, National Renewal Energy Laboratory, Golden, Co, 1999 Search PubMed.
- D. M. Blake, NREL/TP-640-28297, National Renewal Energy Laboratory, Golden, Co, 2002 Search PubMed.
- B. White, M. Yin, A. Hall, D. Le, S. Stolbov, T. Rahman, N. Turro and S. O’Brien, Nano Lett., 2006, 6, 2095–2098 CrossRef PubMed.
- M. F. Luo, J. M. Ma, J. Q. Lu, Y. P. Song and Y. J. Wang, J. Catal., 2007, 246, 52–59 CrossRef.
- G. Avgouropoulos and T. Ioannides, Appl. Catal., B, 2006, 67, 1–11 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Eden Prairie, MN, 1992 Search PubMed.
- G. Schon, Surf. Sci., 1973, 35, 96–108 CrossRef.
- C. C. Chusuei, M. A. Brookshier and D. W. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef.
- X. Chen and C. Burda, J. Phys. Chem. B, 2004, 108, 15446–15449 CrossRef.
- X. Yang, Y. H. Wang, L. L. Xu, X. D. Yu and Y. H. Guo, J. Phys. Chem. C, 2008, 112, 11481–11489 CrossRef.
- H. G. Yu, S. C. Lee, J. G. Yu and C. H. Ao, J. Mol. Catal. A: Chem., 2006, 246, 206–211 CrossRef.
- I. Motoki, Y. Keiji, K. Tsutomu, O. P. M. Orlando, O. Bunsho and W. Hisanobu, Appl. Catal., B, 2013, 132–133, 39–44 Search PubMed.
- M. F. Luo, J. M. Ma, J. Q. Lu, Y. P. Song and Y. J. Wang, J. Catal., 2007, 246, 52–59 CrossRef.
- H. B. Zou, X. F. Dong and W. M. Lin, Appl. Surf. Sci., 2006, 253, 2893–2898 CrossRef.
- T. Caputo, L. Lisi, R. Pirone and G. Russo, Appl. Catal., A, 2008, 348, 42–53 CrossRef.
- X. L. Tang, B. C. Zhang, Y. Li, Y. D. Xu, Q. Xin and W. J. Shen, Appl. Catal., A, 2005, 288, 116–125 CrossRef.
- K. Qian, Z. X. Qian, Q. Hua, Z. Q. Jiang and W. X. Huang, Appl. Surf. Sci., 2013, 273, 357–363 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02708k |
| This journal is © The Royal Society of Chemistry 2018 |