
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOsteoinductive 3D scaffolds prepared by blend centrifugal spinning for long-term delivery of osteogenic supplements
Lukasova Vera *abc,
Buzgo Matejb,
Vocetkova Karolinaab,
Kubíková Terezad,
Tonar Zbyněkd,
Doupnik Miroslavb,
Blahnova Veronikaab,
Litvinec Andreja,
Sovkova Veraab,
Voltrová Barboraa,
Staffa Andreab,
Svora Petrb,
Kralickova Milenad,
Amler Evzenbe,
Filova Evaae,
Rustichelli Francoa and
Rampichova Michalaab
*abc,
Buzgo Matejb,
Vocetkova Karolinaab,
Kubíková Terezad,
Tonar Zbyněkd,
Doupnik Miroslavb,
Blahnova Veronikaab,
Litvinec Andreja,
Sovkova Veraab,
Voltrová Barboraa,
Staffa Andreab,
Svora Petrb,
Kralickova Milenad,
Amler Evzenbe,
Filova Evaae,
Rustichelli Francoa and
Rampichova Michalaab
aLaboratory of Tissue Engineering, The Czech Academy of Sciences, Institute of Experimental Medicine, Videnska 1083, 142 20 Prague 4, Czech Republic. E-mail: vera_lukasova@labdemo.cz; Fax: +420 2 9644 2387; Tel: +420 2 9644 2387
bUniversity Center for Energy Efficient Buildings, Czech Technical University in Prague, Trinecka 1024, 273 43, Bustehrad, Czech Republic
cDepartment of Cell Biology, Faculty of Science, Charles University, Prague, Czech Republic
dDepartment of Histology and Embryology and Biomedical Center, Faculty of Medicine in Pilsen, Charles University, Karlovarská 48, 301 00, Pilsen, Czech Republic
eInstitute of Biophysics, 2nd Faculty of Medicine, Charles University, V Uvalu 84, 150 06 Prague 5, Czech Republic
First published on 13th June 2018
Abstract
Bone regeneration is a long-term process requiring proper scaffolding and drug delivery systems. The current study delivers a three-dimensional (3D) scaffold prepared by blend centrifugal spinning loaded with the osteogenic supplements (OS) β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone. The OS were successfully encapsulated into a fibrous scaffold and showed sustained release for 30 days. Furthermore, biological testing showed the osteoinductive properties of the scaffolds on a model of human mesenchymal stem cells and stimulatory effect on a model of osteoblasts. The osteoinductive properties were further proved in vivo in critical size defects of rabbits. The amount of bone trabecules was bigger compared to control fibers without OS. The results indicate that due to its long-term drug releasing properties, single step fabrication process and 3D structure, the system shows ideal properties for use as a cell-free bone implant in tissue-engineering.
1 Introduction
Over the last few years, bone tissue engineering has gained increased attention. Despite the high regeneration potential of bone itself, prevalent cases of traumatic injury, tumor, infection or osteoporosis result in the formation of critical bone defects. The treatment of these complications is connected with serious socioeconomic problems and the risk of long-term disability for patients.1 In cases where the spontaneous healing of bones is often insufficient, reconstruction using natural or artificial grafts is necessary.2,3 Today, plenty of bone grafting methods exist, with autologous bone widely used. However, there remain some drawbacks such as the risk of disease transmission, limited integration of the graft and complex storage conditions.4,5 Thus, alternative routes for the treatment of defects are being investigated. Traditionally, non-degradable metals are used as materials for bone implants. Non-degradable scaffolds have limits in terms of bone ingrowth into the scaffold and the scaffold does not respond to changes in bone organization and shape.6 As a consequence, attention has been centered on biodegradable scaffolds. A number of manufacturing methods for biodegradable implants have recently been described – i.e. scaffolds can take the form of foams, three-dimensional (3D) printed scaffolds, fibrous scaffolds or demineralized extracellular matrix (ECM).7 Bone scaffolds should meet several criteria such as possessing proper mechanics to withstand stress and strain in bone tissue, osteoconductivity for the promotion of proper cellular differentiation and osteoinductivity to enable connection with the surrounding bone.8,9In elderly patients the incidence of fractures has increased significantly.1 Moreover, fracture healing in older patients is slower due to the decreased regeneration capacity of bone. The risks, connected with autologous graft transplantations or the aspiration of the proper amount of autologous cells for cellular based scaffolds are therefore higher with increased patient age. As a solution, non-cellular based scaffolds enriched with bioactive compounds stimulating bone regeneration are attractive alternatives for the treatment of bone fracture healing.10 Since the regeneration process of bone is long, the supplementation of active molecules should be combined with systems prolonging drug delivery. The combination of drugs with drug delivery systems and scaffolds not only prolongs the bioavailability of the drugs, but also results in increased local concentration and stimulates bone healing in a spaciotemporal manner. Suitable bioactive compounds for osteogenesis promoting factors include widely used osteogenic supplements (OS)11,12 e.g. β-glycerol phosphate, ascorbate-2-phosphate, dexamethasone and also growth factors such as bone morphogenetic protein-2 (BMP-2) and -7, insulin growth factor or fibroblast growth factor.13–15
An advantage of drug releasing scaffolds is their active stimulation of the bone healing process. Electrospinning is one of the most utilized technologies for producing biomimetic and drug releasing scaffolds in bone tissue engineering.16 However, despite numerous advantages, electrospun scaffolds have a two-dimensional (2D) morphology, eliminating their practical application in healing large bone defects. For instance, the size of the pores is often insufficient to allow cell penetration.17 The goal of this study is to overcome the inherent properties of fibrous scaffolds to improve the osteogenesis of seeded cells and subsequently to test the properties in vivo. This can be achieved by a centrifugal spinning method, which allows for the production of 3D scaffolds with higher pore diameters.18 In the current work we aim to develop drug releasing 3D fibrous scaffolds by using a blend centrifugal spinning process. The technology of blend centrifugal spinning enables bioactive compounds to be encapsulated into the scaffold and their subsequent gradual release. The present work describes the fabrication, properties and osteoconductive potential of scaffolds prepared from poly-ε-caprolactone (PCL) with encapsulated OS. For in vitro testing of these scaffolds Saos2 osteosarcoma cell line as model of mature osteoblasts and hMSCs as multipotent stem cells were used.
2 Material and methods
2.1 Fibrous scaffold preparation using centrifugal spinning technology
Fibrous meshes were prepared using a centrifugal spinning device (Cyclone 1000 L/M Forcespinning® device, Fiberio, McAllen, TX, USA). 13% PCL (molecular weight 80 kDa, Sigma Aldrich, St. Louis, USA) and 16% PCL (molecular weight 45 kDa, Sigma Aldrich, St. Louis, USA) were dissolved in a mixture of acetic and formic acid in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and blended with OS (β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone). The spinneret was based on a flat disc with three orifices of G30 in size and a rotation speed of 10000 rpm was used to prepare the fibrous meshes. The fibers were deposited on spunbond textile using vacuum-assisted deposition. Fibers with 4 different concentrations of OS were prepared: once concentrated OS (OS1), two times concentrated (OS2), five times concentrated (OS5) and ten times concentrated (OS10) (Table 1). Plain PCL fibers were used for control samples (CGM, COM). The spinning conditions for all groups were constant to evaluate the effect of polymeric blend properties on scaffold performance.
:1 and blended with OS (β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone). The spinneret was based on a flat disc with three orifices of G30 in size and a rotation speed of 10000 rpm was used to prepare the fibrous meshes. The fibers were deposited on spunbond textile using vacuum-assisted deposition. Fibers with 4 different concentrations of OS were prepared: once concentrated OS (OS1), two times concentrated (OS2), five times concentrated (OS5) and ten times concentrated (OS10) (Table 1). Plain PCL fibers were used for control samples (CGM, COM). The spinning conditions for all groups were constant to evaluate the effect of polymeric blend properties on scaffold performance.
| Sample | asc-2-P (μg mL−1) | β-GP (mM) | dex (nM) | Sample | asc-2-P (μg mL−1) | β-GP (mM) | dex (nM) |
|---|---|---|---|---|---|---|---|
| a asc-2-P, ascorbate-2-phosphate; β-GP, β-glycerol phosphate; dex, dexamethasone; OS, osteogenic supplement. | |||||||
| OS1 | 40 | 10 | 100 | OS5 | 200 | 50 | 500 |
| OS2 | 80 | 20 | 200 | OS10 | 400 | 100 | 1000 |
2.2 Characterization of fibrous scaffolds prepared using centrifugal spinning
The prepared fibers were visualized using scanning electron microscopy (SEM) and fiber diameter was determined. Samples were coated with a thin layer of platinum using a Quorum Q 150R S device (3 cycles, Quorum Technologies, Lewes, GB) and visualized using SEM (Tescan Vega 3, Brno, Czech Republic). The acceleration voltage for all samples was 10 kV. Fiber diameter and pore size was analyzed in the ImageJ program. The chemical composition of the scaffolds was analyzed using Fourier Transform Infrared-Attenuated Total Reflectance (FTIR-ATR) spectroscopy. Samples of plain PCL and PCL with OS were pelleted using a manual press. The pellets were analyzed using FTIR-ATR (IRAffinity-1, Shimazu with ZnS ATR). The measurement was performed with Happ-Ganzel apodization, at a resolution of 8 cm−1 and 20 scans per spectrum.2.3 Determination of phosphate using HR-SEM
The samples were observed with a Zeiss scanning electron microscope Merlin equipped with an energy dispersive spectrometer (EDS) by Oxford Instruments with a silicon drifted detector X-Max 150. EDS measurements were carried out with charge compensator (injected medium was nitrogen), accelerating voltage 5 kV, beam current 1 nA, working distance – 8.5 mm. The EDS maps were analyzed with AZTEC software by Oxford Instruments. Samples were mounted on conductive carbon tape. The samples were not coated.2.4 Characterization of phosphate release
To quantify the release of β-glycerol phosphate and ascorbate-2-phosphate from the fibrous scaffolds, we quantified the phosphate using ammonium molybdate. The release experiment was based on testing of samples with a size of 1 cm2. The samples were placed in an Eppendorf tube and 1 mL of TBS was added to each sample. At specific time intervals, the supernatant was collected and frozen until analysis. 1 mL of Tris-buffered saline (TBS) was added to the scaffolds until the next time-interval. After completing the release experiment, the samples were dissolved in chloroform and resuspended in TBS for further analysis.The analysis was performed in Eppendorf tubes. The samples were placed into Eppendorf tubes containing 0.25 mL of 2.5% ammonium molybdate solution followed by 0.25 mL of 10% ascorbic acid solution (freshly prepared), mixed thoroughly, and heated for 5 min in a boiling water bath. After cooling the samples in a cold-water bath for 5 min, the optical density at 820 nm was measured using a Synergy H1 microplate reader. In each series of measurements, parallel determinations were made of the blank values of the reagent solution and a standard preparation. The release experiment was performed in 5 replicates and samples were analyzed as absolute release and cumulative release.
2.5 Saos2 and hMSC seeding of the scaffold
Before cell seeding, PCL nanofibers were cut into round patches 6 mm in diameter and sterilized using ethylene oxide. Samples were placed in the 96-well plate and pre-wet with 20 μL of growth media. Samples were seeded with 25 × 103 human bone marrow-derived mesenchymal stem cells (hMSCs; ScienCell, San Diego, California, USA) or with 10 × 103 Saos2 osteosarcoma cell line (Cell line service, Germany) per well in the 96-well plate. Samples (OS1, OS2, OS5, and OS10) with hMSCs were cultivated in growth medium (MEM supplemented with 10% fetal bovine serum (FBS) and Penicillin/Streptomycin; Sigma Aldrich, USA) and with Saos2, the same samples, were cultivated in growth medium (McCoy's 5A Medium supplemented with 15% FBS and Penicillin/Streptomycin; Sigma Aldrich, USA), without any other supplement. Two types of controls were used; plain PCL fibers cultivated in growth medium (CGM) and plain PCL fibers cultivated in osteogenic medium (COM). Osteogenic medium was growth medium supplemented with 10 mM β-glycerol phosphate, 100 nM dexamethasone and 40 μg mL−1 ascorbate-2-phosphate. One half of the culture medium was changed on days 7 and 14. Minimal changing of medium was used so as to not to wash out the OS released from the scaffolds to the medium.2.6 Cell viability analysis
To determine the metabolic activity of the cells seeded on the prepared scaffolds, the MTS assay (CellTiter96® AQueous One Solution Cell Proliferation Assay, Promega, WI, USA) was used on days 1, 3, 7, 14 and 21 of the experiment. Briefly, the scaffolds were transferred into new wells to prevent the cells adhered to the tissue culture plastic to misrepresent the measured data. Subsequently, 100 μL of fresh media and 20 μL of the MTS substrate were added to each well. After 2 hour incubation at 37 °C, subsequently, 100 μL of the cultured solution was transferred to a new clean well. The absorbance of the media was detected at 490 nm using a microplate reader (Infinite® M200 PRO; Tecan, Switzerland). The background absorbance (690 nm) and the absorbance of the medium without cells were subtracted from the measured absorbance.2.7 Cell proliferation analysis
The proliferation of cells on the scaffolds was determined using a Quant-iT™ dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) from the amount of DNA on days 1, 3, 7, 14 and 21. The scaffolds were put into a vial with 200 μL of cell lysis solution (0.2% v/v Triton X-100, 10 mM Tris (pH 7.0), and 1 mM EDTA) and processed through 3 freeze/thaw cycles; the scaffolds were first frozen at −80 °C and thawed at room temperature. Between each freeze/thaw cycle, the scaffolds were roughly vortexed. A sample (10 μL) was mixed with 200 μL of reagent solution and fluorescence intensity was detected using a multiplate fluorescence reader (Infinite® M200 PRO; Tecan, Switzerland; λex = 485 nm, λem = 525 nm). The DNA content was determined according to the calibration curve using the standards in the kit.2.8 Visualization of cell adhesion and proliferation on scaffolds
DiOC6(3) (3,3′-diethyloxacarbocyanine iodide; Invitrogen, Molecular Probes) staining was used to detect cell adhesion on the scaffolds on days 1, 7, 14 and 21. The samples were fixed with frozen methyl alcohol (−20 °C) for 10 min and rinsed with phosphate buffered saline (PBS; pH 7.4). A fluorescent probe DiOC6(3) at a concentration of 1 μg mL−1 in PBS was added and incubated with the samples for 45 min at room temperature. Subsequently, the samples were rinsed with PBS, and propidium iodide (Sigma Aldrich, USA) at concentration of 5 μg mL−1 in PBS was added for 5 min, followed by rinsing with PBS. The samples were visualized using a ZEISS LSM 5 DUO confocal microscope. λex = 488 and 560 nm and λem = 520 and 580 nm was used for DiOC6(3) and propidium iodide detection, respectively.2.9 Cell visualization on scaffolds using scanning electron microscopy
The hMSC's morphology was evaluated by SEM on days 1 and 14. Scaffolds with hMSCs were washed in PBS and fixed in 2.5% glutaraldehyde for 2 hours at 4 °C. Then the samples were dehydrated in ethanol ranging from 35–100%. To dry the scaffolds hexamethyldisilazane (Sigma-Aldrich) was added. Scaffolds were analyzed using Vega 3 Tescan as described previously.2.10 RNA isolation and qPCR analysis
The total RNA was isolated using a Qiagen RNA mini-kit (Qiagen, Hilden, Germany) according to the protocol provided by the manufacturer. Reverse transcription was performed using a RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). Transcript levels were evaluated using quantitative PCR (qPCR). qPCR was performed using the Light Cycler 480 II real-time PCR system (Roche, Basel, Switzerland) with TaqMan Master Mix and TaqMan probes (Thermo Scientific, Waltham, MA USA) following the protocols from the manufacturer. The genes whose expression was analyzed by qPCR were RunX2 (86 bp, RUNX2, Hs01047973_m1, Thermo Scientific), osteocalcin (138 bp, BGLAP Hs01587814_g1, Thermo Scientific), type I collagen (66 bp, COL1A1 Hs00164004_m1, Thermo Scientific) and eukaryotic elongation factor (EEF1 Hs00265885_g1, Thermo Scientific). All samples were run in triplicates. The thermo cycling parameters were 95 °C for 10 min; 95 °C for 10 s, 60 °C for 10 s (45 cycles); and 40 °C for 1 min. All samples were scaled relative to the median of the EEF1 expression level, which was used as an endogenous control gene. Gene expression data were analyzed using the 2(−ΔCt) method (relative quantification).2.11 Detection of alkaline phosphatase activity
Alkaline phosphatase (ALP) activity was measured using a 1-Step™ PNPP kit (Thermo Scientific, Waltham, MA USA). 100 μL of PNPP was added to each well and incubated for 10 min. Subsequently, the reaction was stopped with 2 N NaOH. Absorbance was measured using a spectrophotometer at 405 nm (Infinite® M200 PRO; Tecan, Switzerland).2.12 Detection of osteocalcin using indirect immunofluorescence staining
The presence of osteocalcin, a marker typical for osteogenic differentiation, was confirmed using indirect immunofluorescence staining on days 7, 14 and 21. Samples were fixed with frozen methanol (−20 °C) for 10 min, washed with PBS and incubated in 3% FBS in PBS/0.1% Triton X-100 for 30 min at room temperature. To detect osteocalcin, primary monoclonal antibody against osteocalcin (Rabbit anti-osteocalcin IgG, Peninsula Laboratories, dilution 1:20, San Carlos, CA USA) was added and the samples were incubated overnight at 2–8 °C. After three washes with PBS/0.05% Tween 20, the samples were incubated with secondary antibody, Alexa Fluor 488 conjugated anti-rabbit antibody (Thermo Scientific), at a dilution of 1:300 for 45 min. Subsequently, a solution of propidium iodide was added for 5 min (5 μg mL−1 in PBS) to visualize the cellular nuclei. Finally, the samples were washed three times with PBS/0.05% Tween 20. A confocal microscope (ZEISS LSM 5 DUO) was used to detect the present osteocalcin (Alexa Fluor 488: λexc = 488 nm, λem = 505–550 nm propidium iodide: λexc = 561 nm, λem = 630–700 nm).
2.13 Surgery and animal care
Scaffolds were implanted into the critical sized defects in the femur of New Zealand white rabbits. 22 male New Zealand white rabbits, 3 months old, weight approximately 3.0 kg, were obtained from a conventional breed (Velaz, Czech Republic) and bred in standard cages without bedding. The rabbits were fed ad libitum using standard granular mixture for rabbits (TM–MaK 1, Bergman, CZ). The Ethical Principles and Guidelines for Scientific Experiments on Animals were respected throughout the study. The maintenance and handling of the experimental animals followed EU Council Directive (86/609 EEC), and the animals were treated in accordance with the principles of Care and Use of Animals. The investigation was approved by the Expert Committee of the Institute of Physiology, The Czech Academy of Sciences, Prague, CZ, and conformed to Czech Animal Protection Law no. 246/92. The surgical procedure was conducted under general anesthesia using Narketan (0.5 mg kg−1) and Xylapan (6 mg kg−1) and subsequently inhalation of O2 + 1.5–2.0% Halothane during surgery. In lateral femoral condyles, a critical size defect 6 mm in diameter and 10 ± 0.5 mm deep was made using a drill. Animals were divided into three groups. At group I (control group I, 7 animals) the defects were filled with PCL scaffolds. Defects of group II (control group II, 8 animals) were filled with PCL scaffolds with encapsulated OS. Defects of group III (tested group, 7 animals) were left empty, without any treatment. Scaffolds were cut into a cylinder shape with 6 mm in diameter and 10 mm in height and sterilized using ethylene oxide. Antibiotics (5 mg per kg per day i.m. of Marbocyl 2%) and analgesics (0.1 mg per kg per day s.c. of Meloxydil 5 ad us.vet.) were administered during the first 4 days. Rabbits were not limited in their movement after surgery. Euthanasia of the rabbits was performed 12 weeks after surgery, and the femoral condyles were examined. All of the harvested samples were fixed in 4% phosphate buffered formaldehyde.2.14 Histological processing
Samples were demineralized in 5% solution of nitric acid (Merci, Brno, Czech Republic). After 7 days, nitric acid was neutralized by 5% solution of sodium sulfate for 1 hour and subsequently immersed in physiological solution for 24 hours. Every sample was cut in the center of the defect perpendicular to its longitudinal axis into two equal parts (13 × 13 × 5 mm). Tissue blocks were processed individually. Eight serial 5 μm thick histological sections were cut from the center of the healing defect from each paraffin-embedded tissue block. The sections were stained with haematoxylin–eosin, Verhoeff's haematoxylin with green trichrome19 and picrosirius red (Direct Red 80, Sigma Aldrich, Munich, Germany) diluted in saturated picric acid solution was used to visualize the type I collagen (Rich and Whittaker, 2005). We used a circular polarizing filter (Hama, Monheim, Germany) crossed with a quarterwave λ/4 filter below the analyzer filter (U-GAN, Olympus, Tokyo, Japan) mounted on the Olympus CX41 microscope (Olympus, Tokyo, Japan). Methodology of the histological evaluation was described in detail elsewhere.20,212.15 Statistical analysis
Quantitative data are presented as mean values ± standard deviation (SD). The averaged values were determined from at least 3 independently prepared samples. Results were evaluated statistically using one-way analysis of variance and the Student–Newman–Keuls test (SigmaStat 12.0, Systat). The data from the histological analysis were processed with Statistica Base 9.1 (Stat-Soft, Inc., Tulsa, OK, USA). Testing of the randomness of bone distribution among compartments was based on the chi-squared distribution. The Mann–Whitney U test was used to test the quality of population medians among the groups in the study. All results were considered statistically significant if p was <0.05.3 Results
3.1 Scaffold development and characterization
Scaffolds containing osteogenic supplements (OS) were prepared using blend centrifugal spinning technology. The technology is based on mixing a polymer solution with OS prior to the spinning process. Due to the substances mixing in a liquid state, the OS are distributed along the fibers. The polymer selected for fiber development was PCL dissolved in acetic acid and formic acid. The acidic environment enabled the combination with high quantities of β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone. The solutions were prepared containing different amounts of β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone. Centrifugal spinning was performed using spinneret with G30 orifices and a rotation speed of 10000 rpm.
The centrifugal spinning process resulted in the formation of a 3D fibrous mesh. The mesh was formed by fibers forming a porous structure with a thickness of about 3 mm. An analysis by SEM confirmed the fibrous character of all scaffolds (Fig. 1). A stereological analysis (Table 2) showed that with increase of OS concentration, the mean fiber diameter increased.
 | ||
| Fig. 1 Images from scanning electron microscopy of scaffolds. Micrograph of scaffold morphology with lower magnification (scale bar 20 μm) (A–E), detailed micrograph of fibers showing phosphate microdomains (scale bar 5 μm) (F–J), detailed fiber surface after 31 days washing in PBS (scale bar 5 μm) (K–O). | ||
| OS1 | OS2 | OS5 | OS10 | PCL | |
|---|---|---|---|---|---|
| a asc-2-P, ascorbate -2-phosphate; β-GP, β-glycerol phosphate; dex, dexamethasone; OS, osteogenic supplement; PCL, poly-ε-caprolactone; SD, standard deviation. | |||||
| Fiber diameter (nm) | 739.7 | 824.9 | 1569.7 | 697.8 | 974.6 |
| Fiber diameter − SD (nm) | 159.3 | 271.2 | 1067.7 | 527.7 | 299.1 |
| Maximum pore size (μm2) | 205.0 | 157.9 | 204.5 | 33.3 | 326.0 |
| Percentage of pores > 5 μm2 (%) | 51.1 | 43.3 | 58.0 | 39.3 | 46.6 |
| Mean pore size (μm2) | 11.7 | 10.1 | 16.3 | 2.6 | 13.7 |
| Mean pore size − SD (μm2) | 20.5 | 18.9 | 25.9 | 5.9 | 29.8 |
For the control PCL, the mean fiber diameter was 974.6 ± 299.1 nm (Table 2). Mean pore size was about 13.7 ± 29.8 μm2 and maximum pore size was 326 μm2. The scaffold contained polydisperse pores and the percentage of pores bigger than 5 μm2 was 46.6%. For the OS1 sample with the lowest concentration of OS, the mean fiber thickness decreased to 739.7 ± 159.3 nm. The mean pore size decreased to 11.7 ± 20.5 μm2. In case of OS2 group the fiber size further increased to 824.9 ± 271.2 nm. Similarly, in the group OS5 the mean fiber diameter increased to 1569.7 ± 1067.7 nm. The group showed high heterogeneity in fiber diameter, which may be related to change of solution properties. In case of OS10, the mean fiber diameter was 697.8 ± 527.7. Diameter of OS10 was lower compared to OS5, however the mesh contained higher presence of defects with non-fibrous morphology (Fig. 1N).
The encapsulation of OS was verified by FTIR-ATR spectroscopy (Fig. 2). The measurement showed the incorporation of β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone into the scaffolds. The spectra of PCL without modification mainly contained a sharp peak corresponding to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups at 1700 cm−1 and C–H groups at 2800–2950 cm−1. The addition of OS resulted in an increase of –O–H group resonance at 3200–3500 cm−1. The hydroxyl group is present in all added supplements. The encapsulation of β-glycerol phosphate and ascorbate-2-phosphate was indicated by an increase in resonance of –PO4 groups at 1000 cm−1. The presence of dexamethasone was indicated by an increase of CC resonance at 1600 cm−1.
O groups at 1700 cm−1 and C–H groups at 2800–2950 cm−1. The addition of OS resulted in an increase of –O–H group resonance at 3200–3500 cm−1. The hydroxyl group is present in all added supplements. The encapsulation of β-glycerol phosphate and ascorbate-2-phosphate was indicated by an increase in resonance of –PO4 groups at 1000 cm−1. The presence of dexamethasone was indicated by an increase of CC resonance at 1600 cm−1.
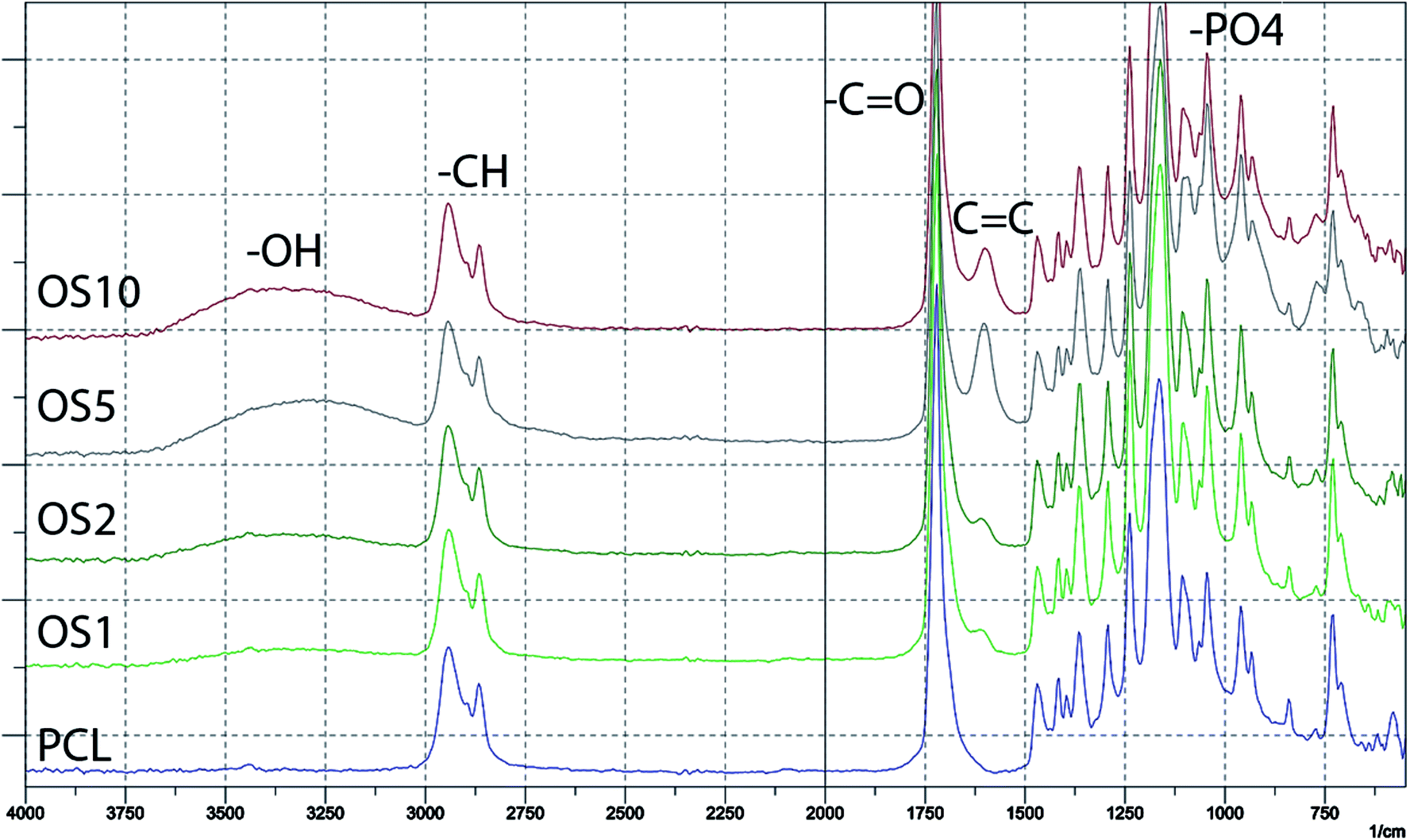 | ||
| Fig. 2 Spectra of prepared scaffolds measured by FTIR-ATR spectroscopy. | ||
In addition, a high resolution-SEM (HR-SEM) with EDS detector was used to analyze the distribution of phosphate in the samples (Fig. 3). Phosphate was detected in all samples. The signal is distributed across all fibers. However, the phosphate was located in clusters, indicating the presence of β-glycerol phosphate and ascorbate-2-phosphate signals. In addition, in OS5 and OS10 samples, the phosphate and oxygen weight percentage (Table 3) was higher, indicating successful encapsulation of β-glycerol phosphate and ascorbate-2-phosphate.
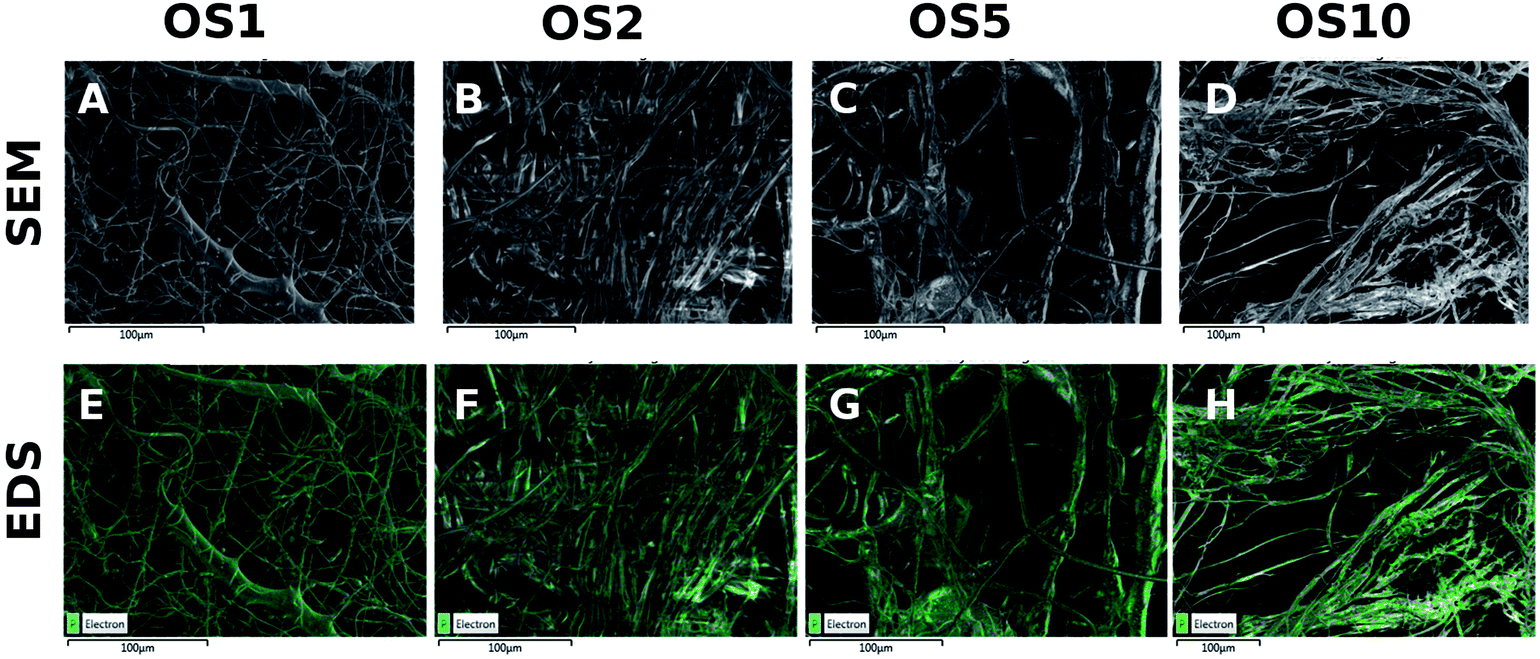 | ||
| Fig. 3 HR-SEM analysis of PCL scaffolds. Images from scanning electron microscopy (A–D). Distribution of phosphate in PCL scaffolds with encapsulated OS, measured by HR-SEM with EDS detector (E–H). | ||
| Sample | C | P | O | Sample | C | P | O | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt% | SD | wt% | SD | wt% | SD | wt% | SD | wt% | SD | wt% | SD | ||
| OS1 | 78.75 | 0.06 | 0.9 | 0.03 | 18.64 | 0.04 | OS5 | 68.76 | 0.07 | 2.8 | 0.04 | 23.82 | 0.05 |
| OS2 | 78.28 | 0.06 | 0.65 | 0.03 | 19.77 | 0.05 | OS10 | 68.05 | 0.07 | 3.73 | 0.04 | 22.89 | 0.05 |
Nevertheless, the release of phosphate contained in β-glycerol phosphate and ascorbate-2-phosphate was performed to analyze the bioavailability. The release was performed for 31 days (Fig. 4). The absolute release showed that the highest absolute amount of phosphate was released from the OS10 scaffold. A dose dependent total release was observed. The lowest amount was released from the OS1 and OS2 samples. The amount in OS5 was two times lower than in the OS10 samples. Interestingly, the cumulative release profile shows, that from the OS1 and OS2 samples, the release was more rapid than from the OS5 and OS10 samples. This indicates that the OS were located near the surface of the fibers, and after 10 days the depletion of OS leads to slower release. In the case of OS5 and OS10, the release was sustained and showed near zero order kinetics. The half-time of release was fastest for the OS2 sample (8 days). OS1 showed a half time of 14.1 days and for the samples with the highest concentration of OS, OS5 and OS10, the half time of release was 23.8 and 23.2 days, respectively. The encapsulation efficiency calculated from total phosphate samples was highest for OS1 and reached 98.2 ± 62.1%. OS2 had lowest encapsulation efficiency reaching only 24.6 ± 10.8%. Samples OS5 and OS10 had 40.1 ± 22.4% and 35.9 ± 0.9% encapsulation efficiency.
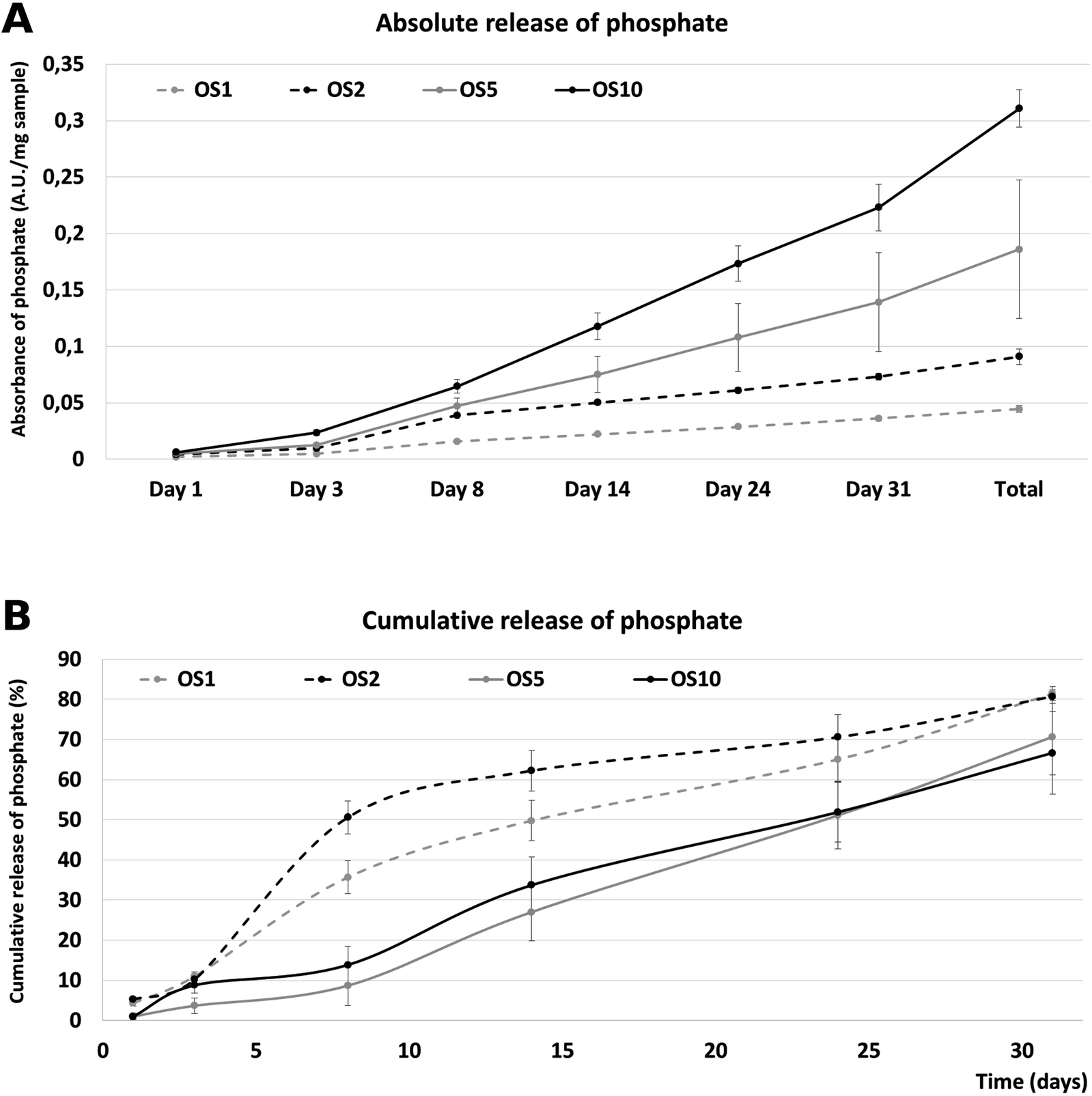 | ||
| Fig. 4 Release of phosphate from fibers. Absolute release of phosphate over time (A). Cumulative release of phosphate from fibers (B). | ||
Evaluation of dexamethasone release was problematic due to low concentration of dexamethasone in scaffolds. The maximal concentration of released dexamethasone was between 3.9 (OS1) and 39 ng (OS10). These values are under detection limit of both HPLC22 and UV-Vis methods.23 However, the effect of osteogenic supplements released from scaffolds was tested on model on Saos2 and hMSCs.
The results showed the successful encapsulation of OS to the fibrous structure and sustained release of factors for more than 10 days for OS1 and OS2 and for more than 30 days for OS5 and OS10.
3.2 Saos2 adhesion, proliferation and metabolic activity on scaffolds
In this study we tested fibers with 4 different concentrations of OS: once concentrated OS marked as OS1, two times concentrated (OS2), five times concentrated (OS5) and ten times concentrated (OS10). Two control samples were chosen, the cells seeded on plain PCL and cultivated in growth medium (CGM) and the cells seeded on plain PCL and cultivated in growth medium with OS (COM). To examine the effect of diverse concentrations of OS we tested the metabolic activity, proliferation and osteogenic differentiation of seeded cells.The amount of DNA on scaffolds seeded with Saos2 was evaluated for DNA content (Fig. 5A), cells proliferated on all scaffolds as the amount of DNA was increasing. From day 7 we observed a huge growth of cells on the OS1–10 samples and CGM but on the COM samples cell ingrowth was slower. As Saos2 are normally cultured without OS, it possibly indicates that higher doses of OS work as inhibition agents on the Saos2 osteosarcoma cell line. The results are in agreement with confocal microscopy observation (data not shown).
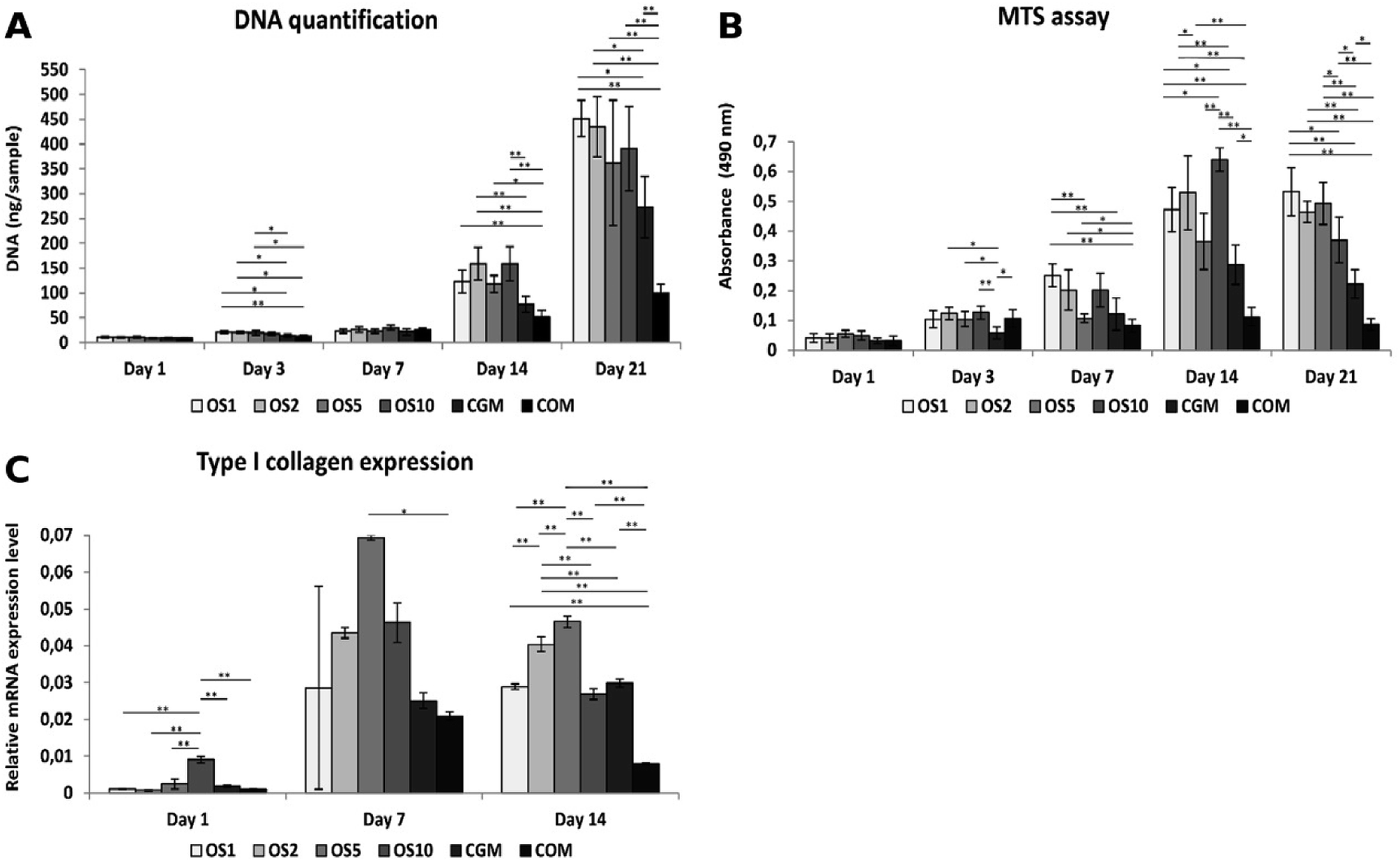 | ||
| Fig. 5 Saos2 adhesion, proliferation, metabolic activity and type I collagen expression on scaffolds. Cell adhesion and proliferation was measured using quantification of DNA (A). Metabolic activity was determined from MTS assay (B). Relative expression of type I collagen was analyzed by qPCR (C). Statistical significance is shown above the columns (*p < 0.05; **p < 0.01). | ||
Metabolic activity measured using an MTS metabolic assay (Fig. 5B) revealed a constant value of absorbance on the COM sample from day 3, although the gradual proliferation of cells was detected. Moreover, only on the OS1 and OS5 samples was the continual increase in absorbance measured. On day 7, we detected the highest absorbance on the OS1 sample. On day 14, statistically higher absorbance was observed on the OS1–2 and –10 samples compared to the CGM and COM. Cells on the OS5 and CGM samples were more metabolically active compared to cells on the COM sample. On day 21, the absorbance detected on the OS1–10 samples were in all cases higher than the rate of absorbance measured on the CGM and COM samples.
3.3 Osteogenic marker expression of Saos2
Osteocalcin is an ECM protein associated with osteogenic differentiation. Indirect immunofluorescence staining was undertaken to visualize this protein (Fig. 6A–F). Clearly, on all samples on day 21 we observed osteocalcin, which was the most visible on the OS1–10 samples. However, even on both control groups we were able to detect high doses of osteocalcin as this protein is typically expressed by Saos2 cells.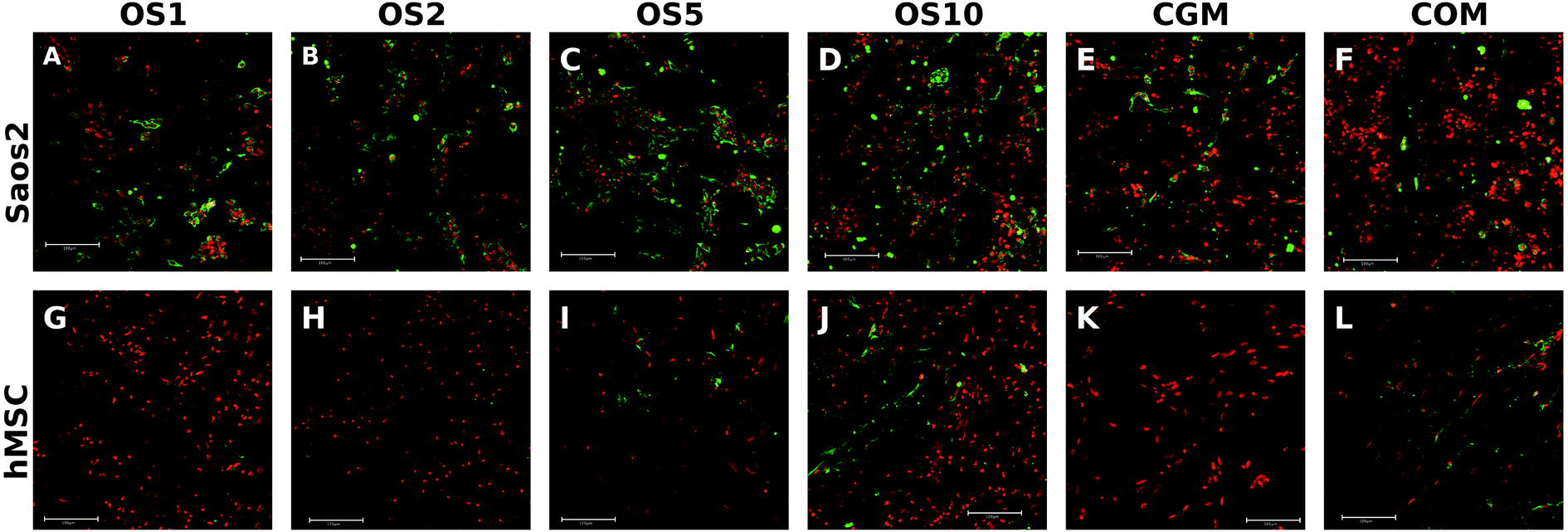 | ||
| Fig. 6 Immunofluorescence staining of osteocalcin. Saos2 stained on day 21 (A–F) and hMSCs stained on day 14 (G–L). Osteocalcin was stained using indirect fluorescence (green color) and cell nuclei using propidium iodide (red color) and visualized using a confocal microscope. Magnification 200×, scale 100 μm. | ||
Relative expression of type I collagen, an osteogenic marker, was evaluated on days 1, 7 and 14 (Fig. 5C). On day 1, cells on the OS10 sample expressed type I collagen statistically the most of all tested samples. On day 7 the normality test failed, thus one-way Anova on ranks revealed only that type I collagen expression on the OS5 sample was statistically significant in comparison to the COM sample. However, we can observe a trend that not only the OS5 samples induced higher expression but also the OS2 and OS10 samples promoted the expression of type I collagen in comparison to both control groups. On day 14, the highest expression of type I collagen was detected on the OS5 sample, followed by OS2 sample.
3.4 hMSCs adhesion, proliferation and metabolic activity on scaffolds
Cell proliferation was measured as the amount of DNA present on the scaffolds on days 1, 3, 7, 14, and 21 (Fig. 7A). On day 1, cells adhered similarly on all scaffolds. On the following days 7 and 14, the largest number of cells was seen on the OS10 sample. Moreover, on day 7, comparable amount of DNA was shown on COM. However, amount of cells decreased on COM on day 14. On day 21, the cell number decreased on all samples and was comparable to each other except for OS5, where the DNA amount was the lowest.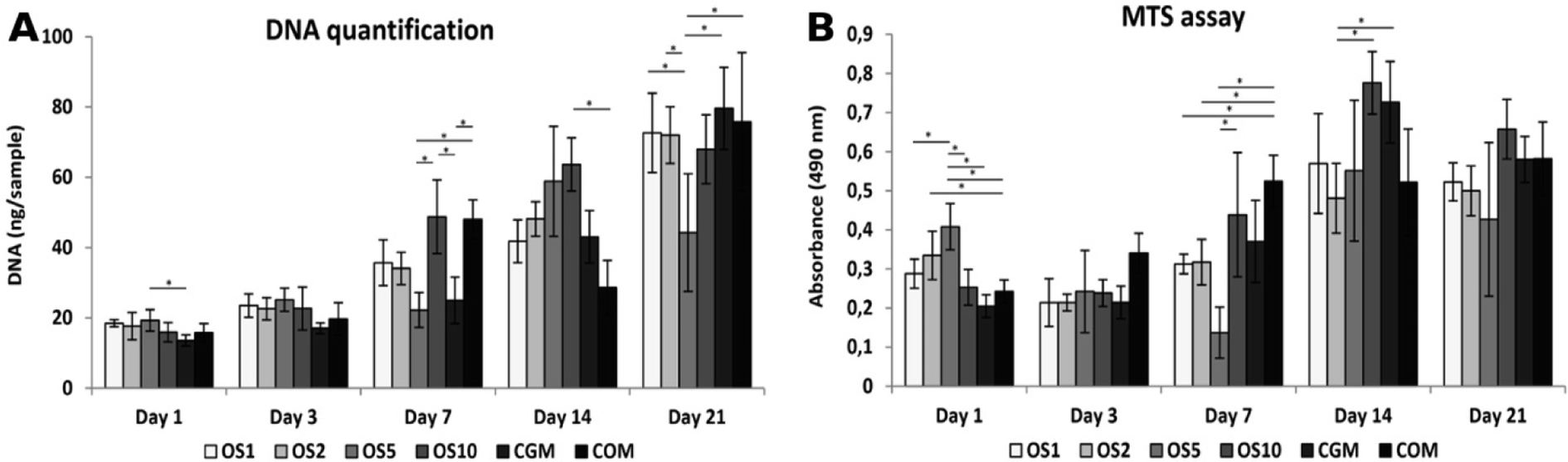 | ||
| Fig. 7 hMSCs adhesion, proliferation and metabolic activity on scaffolds. Cell adhesion and proliferation was measured using quantification of DNA (A). Metabolic activity was determined from MTS assay (B). Statistical significance is shown above the columns (*p < 0.05; **p < 0.01). | ||
The metabolic activity of the cells was measured using an MTS assay (Fig. 7B). The highest absorbance was measured on OS5 on day 1. On the OS2 sample, measured absorbance was higher compared to the CGM. On day 3, metabolic activity was comparable on all samples. On day 7, absorbance was the highest on the COM. On day 14, the highest metabolic activity was measured on the OS10 and CGM samples. There was no difference measured in metabolic activity between the samples on day 21. As OS are standardly added into the hMSC's culture media for induction of osteogenic differentiation we observed no detrimental or strong positive effect of OS, released from scaffolds, on hMSCs proliferation and metabolic activity.
To visualize the adhesion and distribution of hMSC on scaffolds, cells were labeled using fluorescence staining and detected using confocal microscopy and also detected using SEM. As it is visible from Fig. 8, cells adhered on all scaffolds, but the best adhesion was shown on OS5. In the following days of the experiment, cell's density continuously increased. Results correlated well with data from DNA quantification.
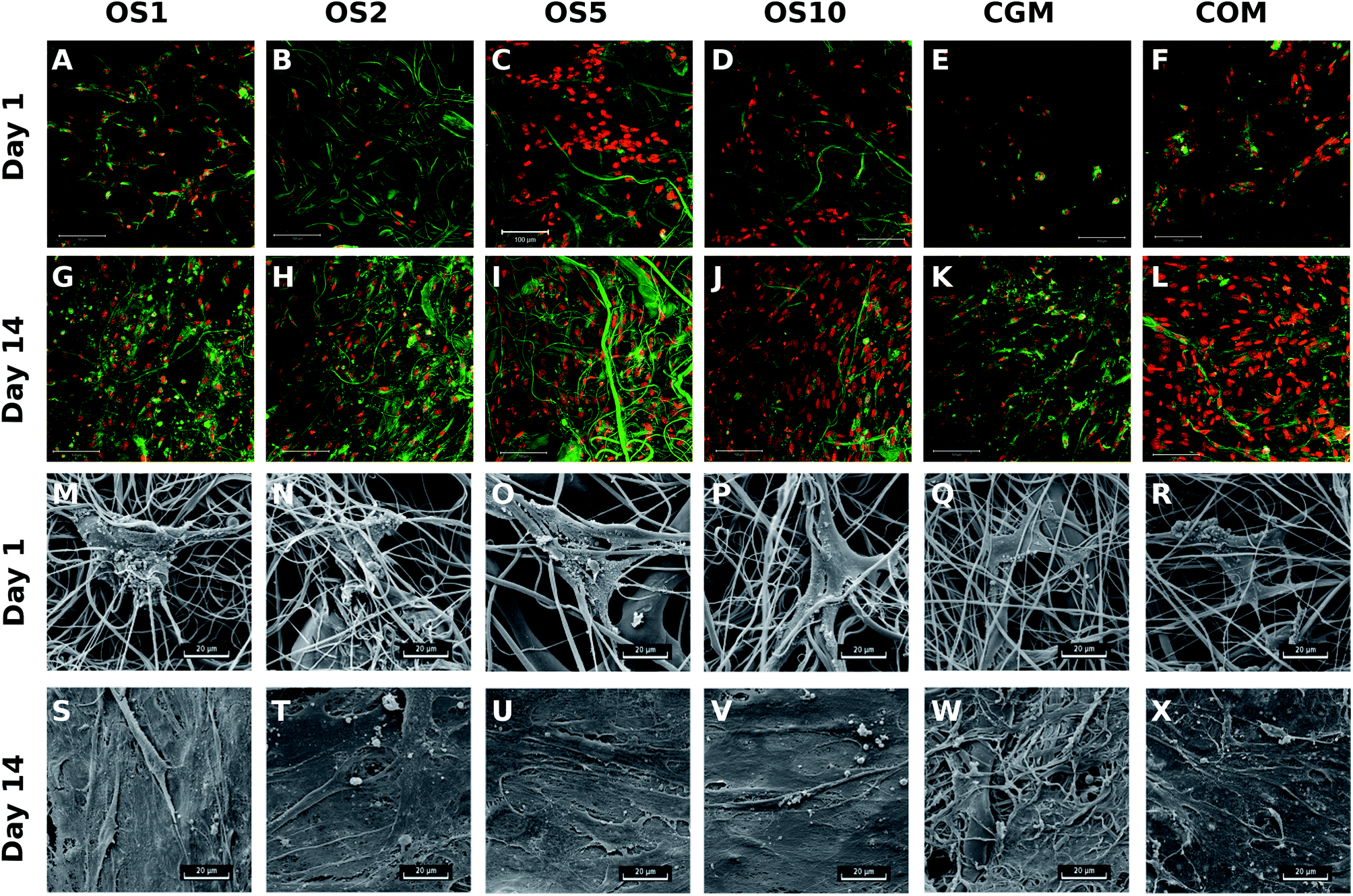 | ||
| Fig. 8 Visualization of hMSCs adhesion and distribution on scaffolds using a confocal microscope. Cell nuclei were stained using propidium iodide (red color) and cell internal membranes using DiOC3 (green color). Magnification 200×, scale 100 μm (A–L). hMSCs adhesion and interaction on fibrous scaffolds visualized using scanning electron microscopy. Magnification 3000×, scale 20 μm (M–X). | ||
3.5 Osteogenic differentiation of hMSCs
Osteogenic differentiation of hMSC was detected as an activity of ALP and qPCR analysis of osteogenic markers. ALP activity was similar in all samples on day 1 (Fig. 9A). On day 7, the highest activity was detected on the COM sample. The activity was higher on OS10 compared to OS1. On day 14 and 21, ALP activity was higher on the COM sample compared to CGM and OS2, but with other samples it was on the same statistical level.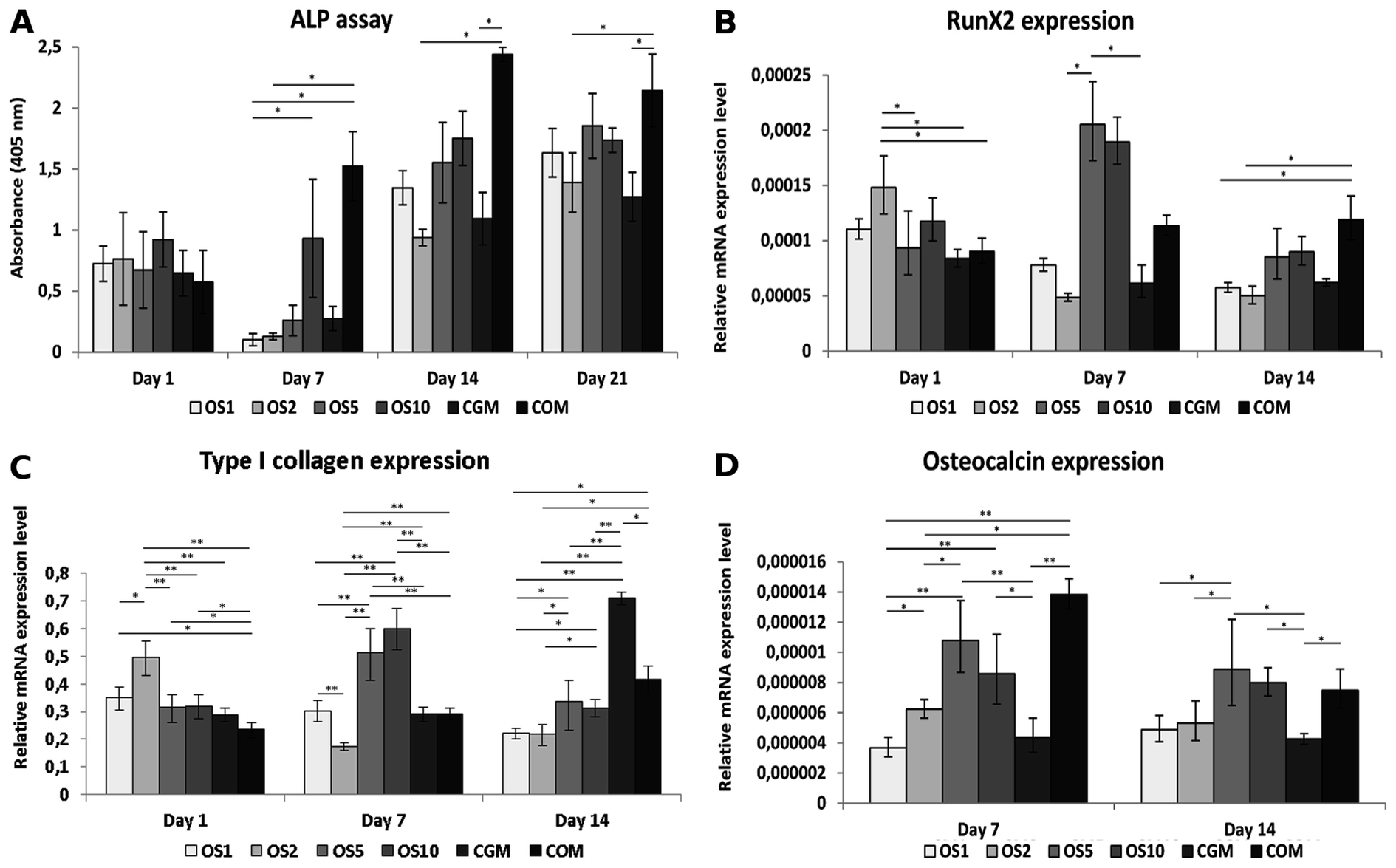 | ||
| Fig. 9 Osteogenic differentiation of hMSCs was detected using ALP activity measurement and qPCR analysis of osteogenic markers. ALP activity of hMSCs (A), relative expression of RunX2 (B), type I collagen (C) and osteocalcin (D) was determined by qPCR analysis. Statistical significance is shown above the columns (*p < 0.05; **p < 0.01). | ||
qPCR was used to detect the expression of proteins typically associated with osteogenic differentiation. RunX2 was chosen as an early osteogenic marker (Fig. 8B). The highest expression was detected on the OS2 sample on day 1. On day 7, the highest expression was seen on the OS5 and OS10 samples. Last day, day 14, on the COM sample we observed highest expression of mRNA for RunX2. Similar to RunX2, type I collagen was the most expressed on OS2 on day 1 (Fig. 9C). On day 7, the highest expression was seen on the OS5 and OS10 samples. The expression of type I collagen decreased on these samples on day 14, when the most expressed protein was detected on the CGM sample. The expression of osteocalcin (Fig. 8D), the late osteogenic marker, was low, but there were still significant differences between the samples. The highest expression of osteocalcin was detected on the OS5, OS10 and COM samples respectively on day 7 and 14.
To visualize expressed osteocalcin on the samples, protein was stained using immunofluorescence staining and observed using confocal microscopy. Osteocalcin production was very low on day 7 (data not shown) as it is a late marker of osteogenic differentiation. On day 14, osteocalcin was visible mainly on the OS5, OS10 and COM samples (Fig. 6G–L).
3.6 In vivo regeneration of critical size bone defect using fibrous scaffold with OS
Scaffold OS5 (with 200 μg mL−1 ascorbate-2-phosphate, 50 mM β-glycerol phosphate, 500 nM dexamethasone) was further tested in vivo on a rabbit model. Bone regeneration was tested in a critical sized defect in femur condyle for 12 weeks. The scaffold showed good manipulation during the surgical procedure. There was no evidence of any inflammation or any complication of healing. All defects in both groups were completely filled with newly formed tissue. Joint cavities were without any sign of inflammation. All animals survived to the date of scheduled euthanasia.3.7 Histological evaluation of regenerated bone defects
Defects of control group were filled mainly with adipose tissue (Fig. 10). In defects filled with fibrous scaffolds, the newly formed bone trabecules were found preferentially in the outer compartment. Some bone trabecules were found also in the middle compartment, but none in the inner compartment. In group II, the total bone volume within the whole defect was greater than in group I (p < 0.05). There was no difference between bone volume within the whole defect in group I and group III (p = 1). There was no significant difference between bone volume within the whole defect in group II and group III (p = 0.064). Differences of bone volume in the outer, middle and central compartments were no significant. There were not found inflammation reaction for the scaffolds and their components. Occasionally, small deposit of inflammation white cells and multinucleated giant cells from foreign bodies were found.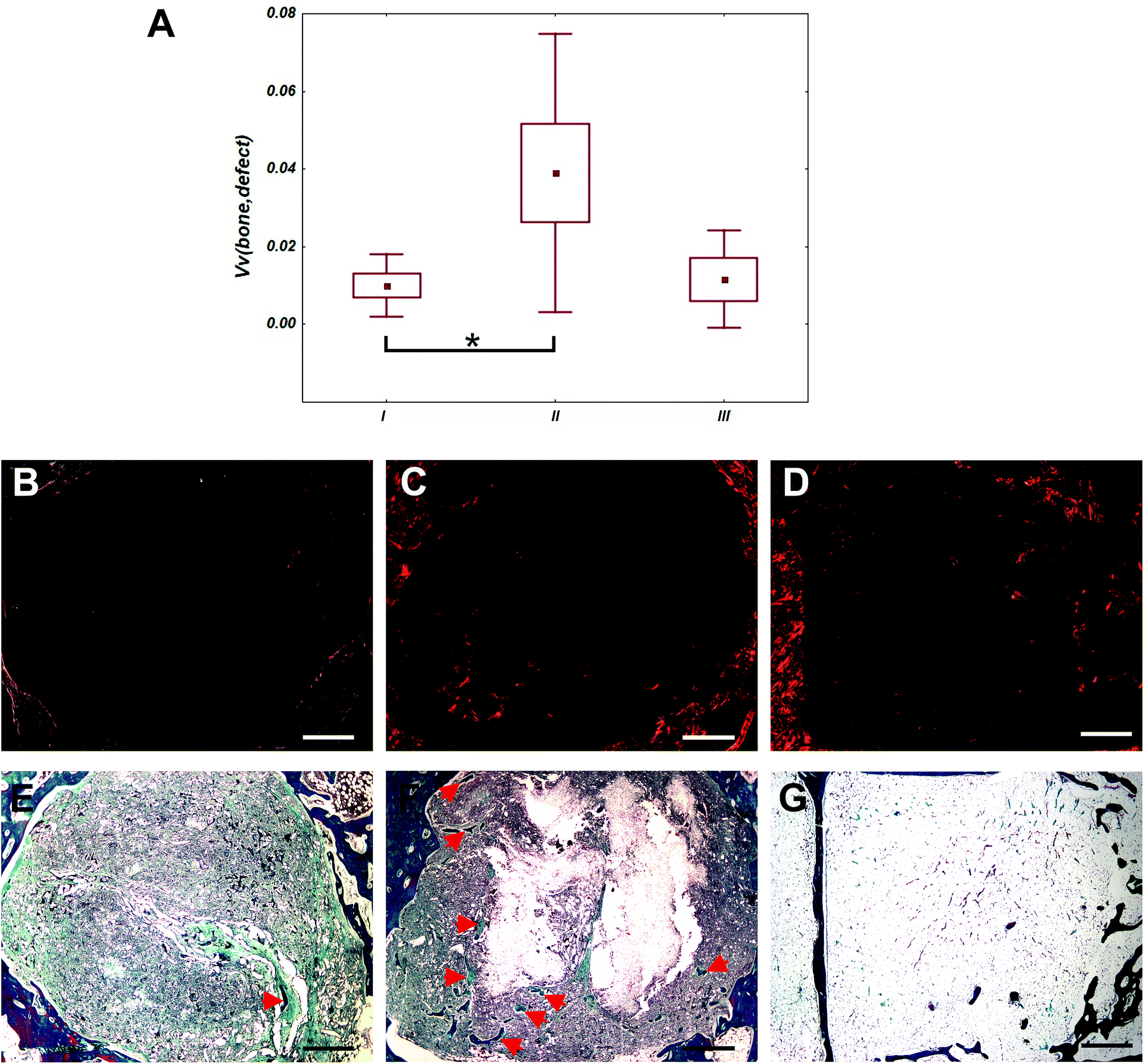 | ||
| Fig. 10 Between-group comparison of bone volume. Bone quantity was expressed as volume fraction (Vv) of bone tissue within the whole reference volume of the defect (Box and Whisker plot showing mean, standard error, and standard deviation. The differences were considered statistically significant as *p < 0.05). Bone tissue and collagen connective tissue contain type I collagen were stained using picrosirius red (A). In group I (B) and II (C), newly formed bone trabecules were found in outer compartment of bone defect. In group III (D), there was mainly adipose tissue and small vessels in bone defect. Newly formed bone trabecules were stained using Verhoeff's haematoxylin with green trichrome. In group I (E) and II (F), a few newly formed bone trabecules (red arrows) were found in outer compartment of bone defect. In group III (G), there was mainly adipose tissue in bone defect. Scale 1000 μm. | ||
4 Discussion
Centrifugal spinning technology enables the formation of nanostructured 3D scaffolds with fibrous morphology. As we have shown in our previous works, 3D nanofibers prepared by centrifugal spinning technology are beneficial for the stimulation of cell adhesion and proliferation.18 The fibrous meshes were prepared from PCL – biocompatible and biodegradable polyester widely used in tissue engineering applications. PCL fibrous scaffolds were tested in vitro in various settings.17,24–30 These studies demonstrate the potential of fibrous scaffolds for bone tissue engineering.However, despite the bio mimicking properties of fibrous scaffolds, the improved osteoinductive properties could be achieved by release of active molecules. Fibrous scaffolds serve as drug delivery systems for encapsulated bioactive compounds and enable prolongation of their bioavailability. A variety of combinations of fibrous scaffolds with bioactive compounds like BMP-2, vascular growth factor or basic fibroblast growth factor was reported.18,31–38 However, use of growth factors is problematic due to their price, stability and lack of medical approval (i.e. by the EMA or FDA). For instance, for bone tissue engineering only two growth factor formulations are approved to date – BMP-2 (Infuse) and BMP-7 (Osigraft).39 Therefore, alternative osteoinductive supplements are applied in bone tissue engineering. Platelets are valuable sources of osteoinductive growth factors and their use as a natural product enable efficient combination with scaffolds for application in bone tissue engineering.18,21,37 In addition, OS were shown to efficiently stimulate osteogenic differentiation of MSCs and stimulate osteogenic cells. Among these molecules, dexamethasone, ascorbic acid, vitamin D3, β-glycerol phosphate and L-proline are the most efficient molecules.
In the current work, we prepared novel 3D scaffolds by centrifugal spinning of a blend containing PCL as a matrix forming polymer and β-glycerol phosphate, ascorbate-2-phosphate and dexamethasone as pro-osteogenic drugs. The technology of blend centrifugal spinning was reported by Mary et al.40 for short term delivery of tetracycline. When used as systems for bone tissue engineering, the scaffolds should be able to release the content for prolonged periods (weeks to months). The centrifugal spinning process enabled the formation of scaffolds with high loading of active molecules. The β-glycerol phosphate was dissolved in concentrations from 40 mg g−1 of PCL to 400 mg g−1 of PCL, ascorbate-2-phosphate in concentrations from 750 μg g−1 of PCL to 7500 μg g−1 of PCL. Dexamethasone was present in low concentrations due to the strong stimulation properties of transcription and potential toxic effect at higher concentration.41 OS were dissolved in PCL solution and processed via centrifugal spinning. The loading of high concentrations of OS shows the low affection of centrifugal spinning via the presence of OS. The mean fiber size for low concentrations was smaller than in the case of plain PCL. This could be connected with the increased conductivity of the polymeric solution. However, in the case of the OS5 sample, the size was significantly higher due to the presence of microfibers. The increase is connected with the increased internal viscosity of the sample caused by the interaction of PCL chains and β-glycerol phosphate. Similarly, the number of pores bigger than 5 μm2 was highest in the OS5 sample. Interestingly, sample OS10 showed a decreased mean fiber diameter and lower presence of pores bigger than 5 μm2. However, the sample contained higher presence of defects indicating that increase of osteogenic additive concentration in OS10 group results in aberrant fiber formation. This could be caused by increased cohesion of polymeric solution and thus lower elongation of emitted polymeric droplet resulting in formation of non-fibrous defects. The results indicated that the blend centrifugal spinning process enables the incorporation of high concentrations of OS without the loss of the fibrous properties of the scaffolds, however the diameter and quality of fibers is decreasing with OS supplement concentration increase.
The presence of OS in the samples was confirmed by FTIR-ATR spectroscopy and RTG analysis of samples in HR-SEM. The infrared spectroscopy identified an increase in bands corresponding to OS, indicating the successful encapsulation of OS. The signal showed dose dependent behavior and with higher concentrations of added OS the signal was stronger. The RTG analysis of elements on the surface of fibers showed that with increased concentrations of drugs in solution, the amount of C decrease and amount of P and O increase. This indicates that O and P from β-glycerol phosphate, dexamethasone and ascorbate-2-phosphate are present in the structure of the scaffold. Nevertheless, the map of spatial distribution of phosphate shows distribution to distinct spots on the fibers. The spots are probably crystals of β-glycerol phosphate formed between PCL chains. The number of spots increases with the concentration of OS and leads to the formation of a connected network of active molecule aggregates. The structure impacts the scaffold's release properties. The internal morphology of the fibers is among the most crucial variables affecting the release kinetics from emulsion nanofibers. As in other nanofiber types, release from osteogenic molecule loaded nanofibers is governed by diffusion or degradation mechanisms.42 The degradation mechanism is dominant in fibers from degradable materials. Upon degradation, the drugs are liberated from the polymeric structure depending on the dissolution of the polymeric matrix.43,44 Since PCL is a slowly degradable material, this type of release is less dominant in the case of our fibers. The release from materials that are non-degradable in the timeframe of drug release is governed by diffusion rate through the polymeric matrix.45 In the case of the proposed scaffolds, the rate limiting factor is the diffusion rate of water and drugs through the polymeric matrix polymer. The rate is dependent on the internal structure of the fibers and the number of contact points available for the solvent to dissolve the core polymer. In the case of fibers with non-continuous islands of active molecule, the release depends on the interconnection of such aggregates.37 The solvent dissolves first, and the aggregates on the surface of the fibers result in rapid release of their cargo. If the droplets are not interconnected or the solvent diffusion is slowed down, the availability of the drug decreases. The lower loading of OS in the OS1 and OS2 samples resulted in a lower number of aggregates along the fibers. However, the release data indicate that most of the drugs containing phosphate, β-glycerol phosphate and ascorbate-2-phosphate, were able to diffuse out of the fiber and 80% of the drug was released on day 31. On the contrary, the structure of the fibers with higher loading (OS5 and OS10) showed numerous contact points on the surface. In the case of aggregate interconnection, the solvent could reach aggregates deeper in the fibers and result in sustained release of high concentrations of OS. The release of OS5 and OS10 fibers was continuous over the release period and a higher total amount of OS was released, as illustrated by the model of measured phosphate release. The fibers were able to release high concentrations of OS over the 31 day period. Nevertheless, the release at day 31 was about 60% and the release rate was constant, without slowing down and achieving release plateau. Therefore, the scaffold seemed to further release the OS even after the release monitoring period. Such long-term releasing system is advantageous for bone tissue engineering applications. A similar microparticle-based system for the delivery of OS was shown to release β-glycerol phosphate for 25 days.11 However, upon incomplete encapsulation, the small molecules due to their high solubility in aqueous solvents often show burst release.12,46 For instance, dexamethasone loaded electrospun PCL scaffolds were able to release dexamethasone for 6 days, however, more than 50% of the drug was released during the first 24 hours. Despite fast release, system was able to stimulate osteogenic differentiation of hMSCs.47 Lower encapsulation rate may be attributed to loss during centrifugal spinning process, where with increased concentration of active molecules, the number of defects and fiber morphology was decreased. Therefore, the non-fibrous defects had different trajectory after emission from spinerete resulting in lower accumulation on collector. This resulted in decreased encapsulation efficiency. However, the non-linearity of encapsulation efficiency still resulted in scaffold formation with different active molecule content.
The fibrous scaffolds with OS were further tested on relevant cell types – the Saos2 osteosarcoma cell line and hMSCs. The test on the Saos2 cell line showed improved proliferation and viability on OS loaded scaffolds compared to control scaffolds both in osteogenic and non-osteogenic medium. Similarly, the scaffolds with OS showed increased production of osteocalcin (protein production increase) and type I collagen (mRNA increase). The indirect immunostaining method of osteocalcin was performed on day 21 on cultivated Saos2 cells. We observed higher doses of osteocalcin on all samples with incorporated OS in comparison to both control groups. Type I collagen showed the highest mRNA expression on samples OS2–OS10, compared to both control groups in the case of cultivated Saos2 cells, meaning that higher doses of OS on the COM sample had a negative effect on the expression of type I collagen in cultured Saos2. The results clearly demonstrated the positive effect of released OS on Saos2 cells.
hMSCs that were seeded on the same scaffolds showed different trends during the culture period. The amount of DNA as well as metabolic activity were comparable for the whole time of the culture period, meaning that the incorporation of OS showed no positive as well as no detrimental effect on cell culture. Those results are in agreement with results obtained by confocal microscopy and SEM observation. Similar results were obtained by Shi et al. who tested the effect of released OS from microspheres on hMSC proliferation. The best cell proliferation was observed on the control group cultivated in growth medium.11 Peter et al.48 confirmed that after rat MSC reached confluency the ongoing exposure of cells to OS had no further positive effect on cell proliferation. However, experiments that study the effect of OS on hMSCs, seeded in lower cell density in comparison to our cell seeding density, indicate the positive effect of OS in culture medium on cell proliferation or metabolic activity,49,50 meaning that in confluent cell layers there are other important factors that mainly affect cell differentiation. However, the results of the study show the osteoinductive properties of the prepared scaffolds. ALP activity was measured on the scaffold cultured with hMSCs to study the effect of released OS on the osteogenic differentiation of hMSCs. The trend in ALP activity of cells cultivated on scaffolds with incorporated OS was slightly lower in comparison to the COM samples, but higher compared to cell cultured in growth medium. Many studies have tested ALP activity in diverse cultivation conditions. However, the complete differentiation medium reaches the best ALP activity in cultured hMSCs.48–50 Expression of mRNA for key osteogenic transcription factor – RunX2 was observed on the OS1–10 scaffolds. hMSCs cultivated on samples marked as OS5 and OS10 reached the peak in expression of mRNA for RunX2 on day 7. A higher amount of expressed mRNA for type I collagen was observed on day 7 on the OS5 and OS10 samples and on day 14 on the CGM sample. In addition, the differentiation into osteogenic lineage was confirmed by the indirect immunostaining method of osteocalcin. On day 14 we observed stained osteocalcin on the OS5, OS10 and COM samples. Thus, we conclude that the presence of OS in higher doses influenced the osteogenic differentiation. Osteocalcin visualization on hMSCs is in agreement with the results obtained by qPCR. These findings again indicated that the OS5, OS10 and COM samples promoted the osteogenic differentiation the most of all tested groups. This means that the incorporation of OS in samples marked as OS5 and OS10 showed a comparable effect to OS freely added into growth medium at the concentration standardly used for the induction of osteogenic differentiation in vitro. Those results are in agreement with the study of Shi et al.11 who observed the highest expression of mRNA for RunX2, type I collagen and osteocalcin on scaffolds releasing OS.
Osteoinductive properties of the centrifugal spun scaffold releasing OS were confirmed in in vivo study. While defects of the control group were filled dominantly with the adipose tissue, cells from surrounding tissues infiltrated the fibrous scaffolds and started to form bone trabecules. Fibers served to cells as a scaffolding material helping them to settle inside the defect and synthetized bone ECM. Bone trabecules were formed preferentially from the side of the defect, what is typical for cell-free scaffolds. OS released from the scaffolds induced osteoinduction, the amount of the bone trabecules was higher compared to PCL fibers without the OS.
The proposed scaffolds showed the osteoinductive properties and show high potential for application in cell-free tissue engineering. The use of the cell-free scaffolds possesses several advantages compared to scaffolds that are seeded with cells prior to implantation into the defect site. The aspiration of cells is connected with an extra surgical procedure that could lead on the donor side to morbidity, pain or inflammation. Moreover, during the ex vivo expansion and cultivation the cells can undergo unwanted phenotypic changes. Thus, cell-free scaffolds designated as drug delivery carriers can induce in the defect cell migration, proliferation and finally differentiation into the desired cell type.16 Besides osteoinductive properties, the scaffolds based on centrifugal spinning technology are highly advantageous in the formation of the real 3D structure of the scaffolds. Classical electrospun scaffolds have limited thickness and pore size, which does not enable rapid cell penetration into the scaffolds.17,51 Centrifugal spinning technology enables the penetration of cells thorough the fibrous scaffold without the need for additional components (i.e. like salt crystals).51 Scaffold porosity is an important property in bone tissue engineering. Generally, pores of a size of less than 100 μm do not favor the ingrowth of mineralized extracellular matrix and also do not support cell migration.52 Optimal pore size for bone tissue engineering ranges from 100–400 μm.53 However, in vivo study where PCL scaffolds with the range of pore diameters from 350–800 μm were tested did not reveal differences in new bone formation.54 In our case, the maximal pore size of the scaffolds was about 200 μm2, which facilitated cell penetration thorough the scaffolds. It was proved in our previous study, where cells cultured in static conditions penetrated into the depth of 80–100 μm.18 In the case of the OS10 sample, the pore size was lower, which may hamper in vivo bone formation using these scaffolds.
5 Conclusion
The above-mentioned properties of prepared OS releasing scaffolds clearly shows that the fibrous scaffolds prepared by blend centrifugal spinning technology are promising for application in bone tissue engineering. The materials prepared of PCL are biodegradable, biocompatible and the polymer is approved by the FDA for medical use. In addition, the utilized OS could be obtained in qualities applicable in medicine. Compared to growth factors, OS are longer lasting molecules with prolonged activity in solutions11 and could be obtained at a lower price. Combined with the results of the in vitro study, the prepared scaffolds served as drug delivery systems releasing OS for a period of 31 days and were shown to have favorable morphological and osteoinductive properties for use in bone tissue engineering. These scaffolds, with incorporated OS, showed enhanced metabolic activity and cell proliferation while Saos2 cells were cultivated. According to the differentiation assays from the Saos2 and hMSCs cultivation, we conclude that the sample marked OS5 was the most potent one to induce osteogenic differentiation in vitro for both cell types. The obtained results were comparable or better than the sample, where the cells were cultivated in medium with added OS. Osteoinductive properties of OS5 scaffold were further confirmed in in vivo study on critical sized bone defect of rabbit. This scaffold, serving as drug delivery system, has great potential for bone tissue engineering as it is osteoinductive, porous, 3D and able to allow the sustained release of OS lasting for up to one month.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study has been supported by the Czech Science Foundation (grants no. 15-15697S, 16-14758S), the Ministry of Education, Youth, and Sports of the Czech Republic (Research Programs NPU I:LO1309, NPU I:LO1508) and the Internal Grant Agency of the Ministry of Health of the Czech Republic (MZ-VES project no. 16-29680A, 16-28637A, 17-32285A and 17-31276A) and MV projects no. VI20152018010, MOTOL 9775.References
- R. Lucas, A. Martins, M. Severo, P. Silva, T. Monjardino, A. R. Gaio, C. Cooper and H. Barros, Sci. Rep., 2017, 7, 3754 CrossRef PubMed.
- M. Mehta, K. Schmidt-Bleek, G. N. Duda and D. J. Mooney, Adv. Drug Delivery Rev., 2012, 64, 1257–1276 CrossRef PubMed.
- J. Wiltfang, F. R. Kloss, P. Kessler, E. Nkenke, S. Schultze-Mosgau, R. Zimmermann and K. A. Schlegel, Clin. Oral Implants Res., 2004, 15, 187–193 CrossRef PubMed.
- J. C. Banwart, M. A. Asher and R. S. Hassanein, Spine, 1995, 20, 1055–1060 CrossRef PubMed.
- R. Dimitriou, G. I. Mataliotakis, A. G. Angoules, N. K. Kanakaris and P. V. Giannoudis, Injury, 2011, 42(suppl 2), S3–S15 CrossRef PubMed.
- M. Saini, Y. Singh, P. Arora, V. Arora and K. Jain, World J. Clin. Cases, 2015, 3, 52–57 CrossRef PubMed.
- M. A. Velasco, C. A. Narváez-Tovar and D. A. Garzón-Alvarado, BioMed Res. Int., 2015, 2015, 729076 Search PubMed.
- P. V. Giannoudis, H. Dinopoulos and E. Tsiridis, Injury, 2005, 36(suppl 3), S20–S27 CrossRef PubMed.
- J. R. Woodard, A. J. Hilldore, S. K. Lan, C. J. Park, A. W. Morgan, J. A. Eurell, S. G. Clark, M. B. Wheeler, R. D. Jamison and A. J. Wagoner Johnson, Biomaterials, 2007, 28, 45–54 CrossRef PubMed.
- E. Filova, M. Rampichova, A. Litvinec, M. Drzik, A. Mickova, M. Buzgo, E. Kostakova, L. Martinova, D. Usvald, E. Prosecka, J. Uhlik, J. Motlik, L. Vajner and E. Amler, Int. J. Pharm., 2013, 447, 139–149 CrossRef PubMed.
- X. Shi, Y. Wang, R. R. Varshney, L. Ren, Y. Gong and D.-A. Wang, Eur. J. Pharm. Sci., 2010, 39, 59–67 CrossRef PubMed.
- H. Kim, H. Suh, S. A. Jo, H. W. Kim, J. M. Lee, E. H. Kim, Y. Reinwald, S.-H. Park, B.-H. Min and I. Jo, Biochem. Biophys. Res. Commun., 2005, 332, 1053–1060 CrossRef PubMed.
- S. Srouji, I. Blumenfeld, A. Rachmiel and E. Livne, Cell Tissue Banking, 2004, 5, 223–230 CrossRef PubMed.
- X. Niu, Q. Feng, M. Wang, X. Guo and Q. Zheng, J. Controlled Release, 2009, 134, 111–117 CrossRef PubMed.
- P. Yilgor, K. Tuzlakoglu, R. L. Reis, N. Hasirci and V. Hasirci, Biomaterials, 2009, 30, 3551–3559 CrossRef PubMed.
- E. Amler, E. Filova, M. Buzgo, E. Prosecka, M. Rampichova, A. Necas, P. Nooeaid and A. R. Boccaccini, Nanomedicine, 2014, 9, 1083–1094 CrossRef PubMed.
- M. Rampichova, J. Chvojka, M. Buzgo, E. Prosecka, P. Mikes, L. Vyslouzilova, D. Tvrdik, P. Kochova, T. Gregor, D. Lukas and E. Amler, Cell Proliferation, 2013, 46, 23–37 CrossRef PubMed.
- M. Rampichová, M. Buzgo, A. Míčková, K. Vocetková, V. Sovková, V. Lukášová, E. Filová, F. Rustichelli and E. Amler, Int. J. Nanomed., 2017, 12, 347–361 CrossRef PubMed.
- J. Kocova, Folia Morphol., 1970, 15, 293–295 Search PubMed.
- M. Rampichova, J. Chvojka, V. Jencova, T. Kubikova, Z. Tonar, j. Erben, M. Buzgo, j. Dankova, A. Litvinec, K. Vocetkova, M. Plencner, E. Prosecka, V. Sovkova, V. Lukasova, M. Kralickova, D. Lukas and E. Amler, Biomed. Mater., 2018, 13(2), 025004 CrossRef PubMed.
- E. Prosecká, M. Rampichová, A. Litvinec, Z. Tonar, M. Králíčková, L. Vojtová, P. Kochová, M. Plencner, M. Buzgo, A. Míčková, J. Jančář and E. Amler, J. Biomed. Mater. Res., Part A, 2015, 103, 671–682 CrossRef PubMed.
- Q. Chen, D. Zielinski, J. Chen, A. Koski, D. Werst and S. Nowak, J. Pharm. Biomed. Anal., 2008, 48, 732–738 CrossRef PubMed.
- R. B. Friedrich, A. Ravanello, L. C. Cichota, C. M. B. Rolim and R. C. R. Beck, Quim. Nova, 2009, 32, 1052–1054 CrossRef.
- K. Vocetkova, M. Buzgo, V. Sovkova, D. Bezdekova, P. Kneppo and E. Amler, Cell Proliferation, 2016, 49, 568–578 CrossRef PubMed.
- J. Venugopal, L. L. Ma, T. Yong and S. Ramakrishna, Cell Biol. Int., 2005, 29, 861–867 CrossRef PubMed.
- Y. M. Ko, D. Y. Choi, S. C. Jung and B. H. Kim, J. Nanosci. Nanotechnol., 2015, 15, 192–195 CrossRef PubMed.
- L. Koepsell, L. F. Zhang, D. Neufeld, H. Fong and Y. Deng, Macromol. Biosci., 2011, 11, 391–399 CrossRef PubMed.
- H. Yoshimoto, Y. M. Shin, H. Terai and J. P. Vacanti, Biomaterials, 2003, 24, 2077–2082 CrossRef PubMed.
- R. A. Thibault, L. Scott Baggett, A. G. Mikos and F. K. Kasper, Tissue Eng., Part A, 2010, 16, 431–440 CrossRef PubMed.
- R. Kang, Y. Luo, L. Zou, L. Xie, H. Lysdahl, X. Jiang, C. Chen, L. Bolund, M. Chen, F. Besenbacher and C. Bunger, RSC Adv., 2014, 4, 5734–5739 RSC.
- X. L. Jia, C. G. Zhao, P. Li, H. Zhang, Y. Huang, H. Li, J. Fan, W. Feng, X. Y. Yuan and Y. B. Fan, J. Biomater. Sci., Polym. Ed., 2011, 22, 1811–1827 CrossRef PubMed.
- W. X. Cao, H. Zhang and X. Y. Yuan, Chem. J. Chin. Univ., 2011, 32, 1396–1400 Search PubMed.
- Y. Su, Q. Q. Su, W. Liu, J. R. Venugopal, X. M. Mo, S. Ramakrisha and C. T. Donghua Univ, BMP2/Dexamethasone Loaded and Release from Core-shell Nanofibers for Bone Tissue Engineering, Donghua Univ Press, Shanghai, 2011 Search PubMed.
- L. H. Yin, S. H. Yang, M. M. He, Y. C. Chang, K. J. Wang, Y. D. Zhu, Y. H. Liu, Y. R. Chang and Z. H. Yu, J. Mater. Sci.: Mater. Med., 2017, 28, 15 CrossRef PubMed.
- H. Zhang, J. Liang, Y. Ding and P. Li, Mater. Lett., 2016, 181, 119–122 CrossRef.
- M. Rubert, J. Dehli, Y. F. Li, M. B. Taskin, R. D. Xu, F. Besenbacher and M. L. Chen, J. Mater. Chem. B, 2014, 2, 8538–8546 RSC.
- M. Buzgo, M. Rampichova, K. Vocetkova, V. Sovkova, V. Lukasova, M. Doupnik, A. Mickova, F. Rustichelli and E. Amler, RSC Adv., 2017, 7, 1215–1228 RSC.
- M. D. Schofer, S. Fuchs-Winkelmann, C. Gräbedünkel, C. Wack, R. Dersch, M. Rudisile, J. H. Wendorff, A. Greiner, J. R. J. Paletta and U. Boudriot, Sci. World J., 2008, 8 CrossRef PubMed.
- M. P. G. Bostrom and D. A. Seigerman, HSS J., 2005, 1, 9–18 CrossRef PubMed.
- L. Amalorpava Mary, T. Senthilram, S. Suganya, L. Nagarajan, J. Venugopal, S. Ramakrishna and V. R. Giri Dev, eXPRESS Polym. Lett., 2013, 7, 238–248 CrossRef.
- C. K. Yeung, K. P. Chan, C. K. M. Chan, C. P. Pang and D. S. C. Lam, Jpn. J. Ophthalmol., 2004, 48, 236–242 CrossRef PubMed.
- R. Srikar, A. L. Yarin, C. M. Megaridis, A. V. Bazilevsky and E. Kelley, Langmuir, 2008, 24, 965–974 CrossRef PubMed.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef PubMed.
- J. Zeng, L. Yang, Q. Liang, X. Zhang, H. Guan, X. Xu, X. Chen and X. Jing, J. Controlled Release, 2005, 105, 43–51 CrossRef PubMed.
- M. Gandhi, R. Srikar, A. L. Yarin, C. M. Megaridis and R. A. Gemeinhart, Mol. Pharm., 2009, 6, 641–647 CrossRef PubMed.
- H. Kim, H. W. Kim and H. Suh, Biomaterials, 2003, 24, 4671–4679 CrossRef PubMed.
- A. Martins, A. R. C. Duarte, S. Faria, A. P. Marques, R. L. Reis and N. M. Neves, Biomaterials, 2010, 31, 5875–5885 CrossRef PubMed.
- S. J. Peter, C. R. Liang, D. J. Kim, M. S. Widmer and A. G. Mikos, J. Cell. Biochem., 1998, 71, 55–62 CrossRef PubMed.
- M. J. Coelho and M. H. Fernandes, Biomaterials, 2000, 21, 1095–1102 CrossRef PubMed.
- N. Jaiswal, S. E. Haynesworth, A. I. Caplan and S. P. Bruder, J. Cell. Biochem., 1997, 64, 295–312 CrossRef PubMed.
- M. Rampichova, M. Buzgo, J. Chvojka, E. Prosecka, O. Kofronova and E. Amler, Cell Adhes. Migr., 2014, 8, 36–41 CrossRef PubMed.
- S. F. Hulbert, F. A. Young, R. S. Mathews, J. J. Klawitter, C. D. Talbert and F. H. Stelling, J. Biomed. Mater. Res., 1970, 4, 433–456 CrossRef PubMed.
- S. H. Oh, I. K. Park, J. M. Kim and J. H. Lee, Biomaterials, 2007, 28, 1664–1671 CrossRef PubMed.
- S. M. M. Roosa, J. M. Kemppainen, E. N. Moffitt, P. H. Krebsbach and S. J. Hollister, J. Biomed. Mater. Res., Part A, 2010, 92A, 359–368 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |