
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular networking prospection and characterization of terpenoids and C15-acetogenins in Brazilian seaweed extracts†
Ana Cláudia Philippus‡
 ,
Gabriele A. Zatelli‡,
Tauana Wanke,
Maria Gabriela de A. Barros,
Satomy A. Kami,
Cintia Lhullier,
Lorene Armstrong,
Louis P. Sandjo and
Miriam Falkenberg*
,
Gabriele A. Zatelli‡,
Tauana Wanke,
Maria Gabriela de A. Barros,
Satomy A. Kami,
Cintia Lhullier,
Lorene Armstrong,
Louis P. Sandjo and
Miriam Falkenberg*
Federal University of Santa Catarina (UFSC), Postgraduate Program in Pharmacy, Health Sciences Center, Florianópolis, 88040-900, Brazil. E-mail: miriam.falkenberg@gmail.com
First published on 21st August 2018
Abstract
Molecular networking (MN) can efficiently dereplicate extracts and pure compounds. Red algae of the genus Laurencia are rich in halogenated secondary metabolites, mainly sesquiterpenes and C15-acetogenins. Brown algae of the genus Dictyopteris produce mainly C11-hydrocarbons, sesquiterpenes and sulfur-containing compounds, while Dictyota and Canistrocarpus are reported to contain mainly diterpenes. This study performs an exploratory MN analysis of 14 extracts from algae collected in Brazil (including the oceanic islands) and characterizes the secondary metabolites from the analyzed species. The extracts and some isolated metabolites were analyzed by LC-MS using the FastDDA algorithm, and the MS/MS spectra were submitted to GNPS and displayed in Cytoscape 3.5.1. The GNPS platform generated 68 individual nodes and nine family networks. The MN exploratory analysis indicated chemical differences among species, and also in sampling sites for the same species. For some extracts, it was possible to identify mass values that could correspond to terpenoids and C15-acetogenins that have already been isolated from those or related species. An interesting chemodiversity was highlighted between Laurencia catarinensis from two nearby islands, and this was revealed and was also suggested by the family networks. Many nodes in the MN could not be characterized, and these metabolites can be used as targets for isolation in future works.
Introduction
Recently, new approaches have been developed to guide the isolation of new metabolites, with reduced costs and effort. MS/MS-based Global Natural Products Social (GNPS) Molecular Networking (MN) is an example of a platform that can be easily integrated into natural product workflows, to efficiently dereplicate extracts and pure compounds.1,2 Using a special computational algorithm to compare the degree of spectral similarity between the MS/MS spectra of different compounds, this technique provides a visual overview of all the ions that were detected and fragmented in the MS experiment.3 The key is based on structurally related natural products that share similar tandem mass fragmentation patterns, and molecular families that tend to cluster together and can be visualized as a network.2,4 This dereplication strategy has been widely used for the identification of marine and microorganism-derived natural products.4,5The marine environment represents approximately 70% of the earth's surface, and has a huge biodiversity, characterized by a wide chemical diversity of natural products.6,7 The Brazilian maritime area, known as the “Blue Amazon”, has one of the longest coastlines in the world, and five oceanic islands.8 However, it is still under-explored as a source of natural products. This marine area is largely located in tropical zone, contributing to the development of a rich and diverse marine biota. Many marine algae species of the genera Laurencia, Dictyota, Canistrocarpus and Dictyopteris, can be found in this area.
While red algae of the genus Laurencia J. V. Lamouroux are rich in halogenated secondary metabolites, belonging mainly to the sesquiterpenes, diterpenes, triterpenes, and C15-acetogenins classes,9 brown alga of the genus Dictyopteris produce mainly C11-hydrocarbons, sesquiterpenes and sulfur-containing compounds.10 In addition, Dictyota and Canistrocarpus are chemically composed of diterpenes.11–16 The metabolites produced by algae mainly serve as chemical defenses against herbivorous or pathogenic micro-organisms.16 Many of these metabolites have been confirmed as presenting anticoagulant, antifouling, antimicrobial, antioxidant, herbivore-deterrent and cytotoxic activities.9, 10, 17–20
This study reports an exploratory MN analysis of fourteen algae extracts of algae collected mostly from Brazilian oceanic islands and the characterization of terpenoids and C15-acetogenins from some of the analyzed species.
Results and discussion
Samples were collected in 14 different locations. These samples were identified as belonging to seven species (Table 1). Analysis of the MN data revealed that 10 of the collected samples were distributed in nine family networks (F1–F9) of which: F1 presented 19 nodes, F2 showed 12 nodes, F3–F6 contained three nodes, and F7–F9 comprised two nodes (Fig. 1). Metabolites from extracts of red and brown algae clustered in different family networks, as was expected. Moreover, two extracts of brown (DJ1, DP2) and two of red algae (LD3 and LI4) were not included in any family network. The entire network was formed by 117 nodes, including 68 individual nodes.| Species | Code | Collection site | Date | ||||
|---|---|---|---|---|---|---|---|
| a Collection performed in different points. | |||||||
| Canistrocarpus cervicornis | CC1 | Rocas Atoll, Rio Grande do Norte/Brazil | 03/2015a | ||||
| CC2 | Rocas Atoll, Rio Grande do Norte/Brazil | 03/2015a | |||||
| CC3 | Rocas Atoll, Rio Grande do Norte/Brazil | 03/2015a | |||||
| Dictyopteris jolyana | DJ1 | Fernando de Noronha Archipelago, Pernambuco/Brazil | 10/2014 | ||||
| Dictyopteris plagiogramma | DP1 | Fernando de Noronha Archipelago, Pernambuco/Brazil | 11/2014 | ||||
| DP2 | Fernando de Noronha Archipelago, Pernambuco/Brazil | 11/2014 | |||||
| DP3 | Rocas Atoll, Rio Grande do Norte/Brazil | 05/2016 | |||||
| Dicyota mertensii | DM1 | Trindade Island, Espírito Santo/Brazil | 08/2014 | ||||
| DM2 | Fernando de Noronha Archipelago, Pernambuco/Brazil | 10/2016 | |||||
| DM3 | São Pedro and São Paulo Archipelago, Pernambuco/Brazil | 10/2016 | |||||
| Laurencia catarinensis | LC1 | Xavier Island, Santa Catarina/Brazil | 06/2015 | ||||
| LC2 | Arvoredo Island Santa Catarina/Brazil | 02/2008 | |||||
| Laurencia dendroidea | LD3 | Bombinhas, Santa Catarina/Brazil | 11/2015 | ||||
| Laurencia intricata | LI4 | Rocas Atoll Rio Grande do Norte/Brazil | 03/2016 | ||||
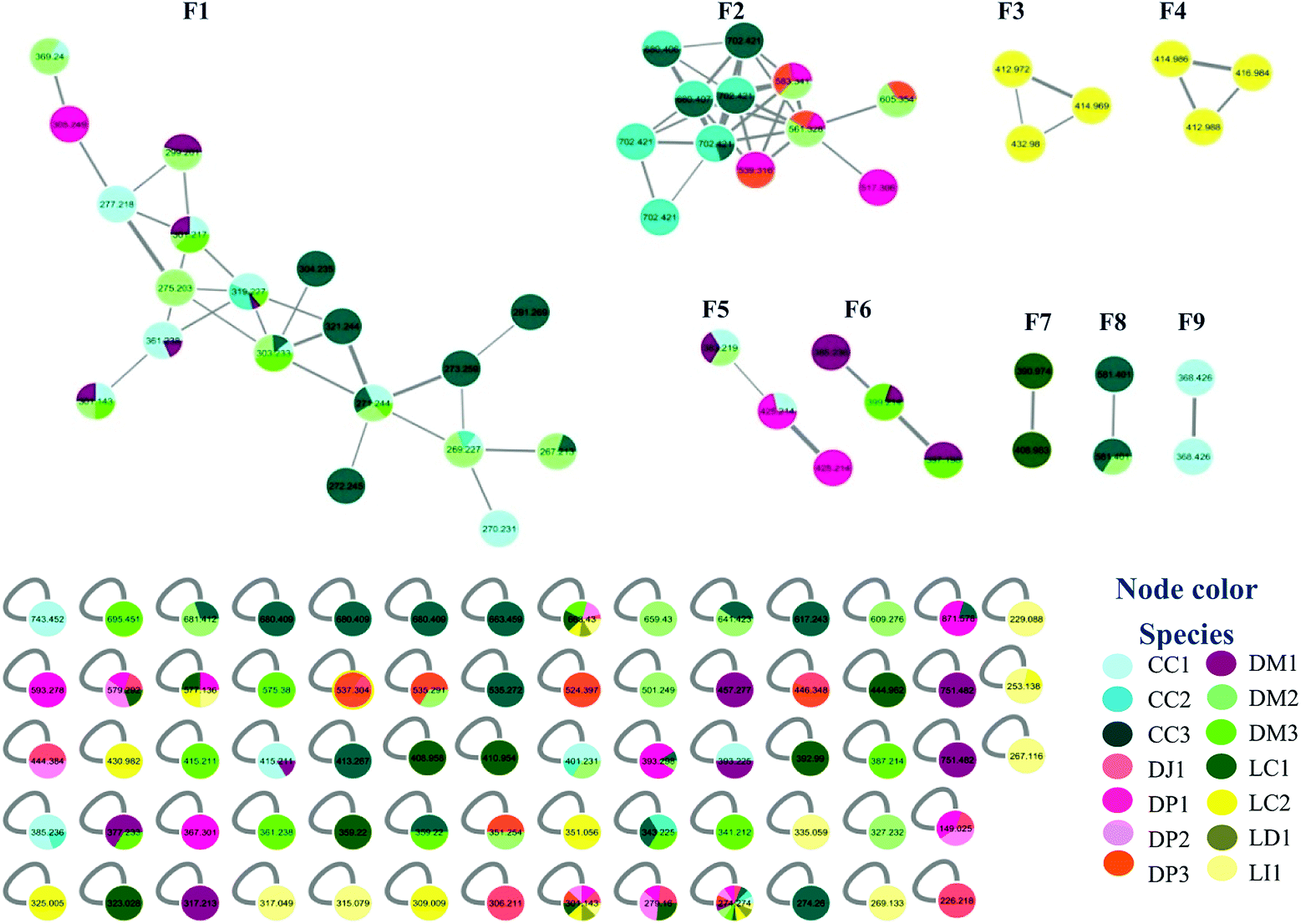 | ||
| Fig. 1 Molecular networking of Brazilian algae extracts. | ||
F1 (Fig. 2A) showed a node with m/z 319.2274 [C20H30O3 + H]+ (calcd 319.2273) corresponding to the peak at 8.96 min in the LC-MS data for the CC1, CC2, DM1 and DM3 extracts. Fragments were also observed resulting from loss of H2O (18 Da), and two H2O molecules (36 Da) at m/z 301.2172 [C20H28O2 + H]+ (calcd 301.2167) and m/z 283.2065 [C20H26O2 + H]+ (calcd 283.2062). Its MS/MS data matches those of the xeniane diterpene 4β-hydroxydictyodial (1).15 Similar MS2 behavior was observed for the node at m/z 361.2383 [C22H32O4 + H]+ (calcd 361.2379) detected at 10.48 min in the CC1 and DM1 LC-MS data. This metabolite differs from 1 by 42 Da, suggesting that it is an acetylated derivative. The above data, together with the data reported in the literature, suggest the structure of 4β-acetoxydictyodial A (2), another xeniane diterpene.14
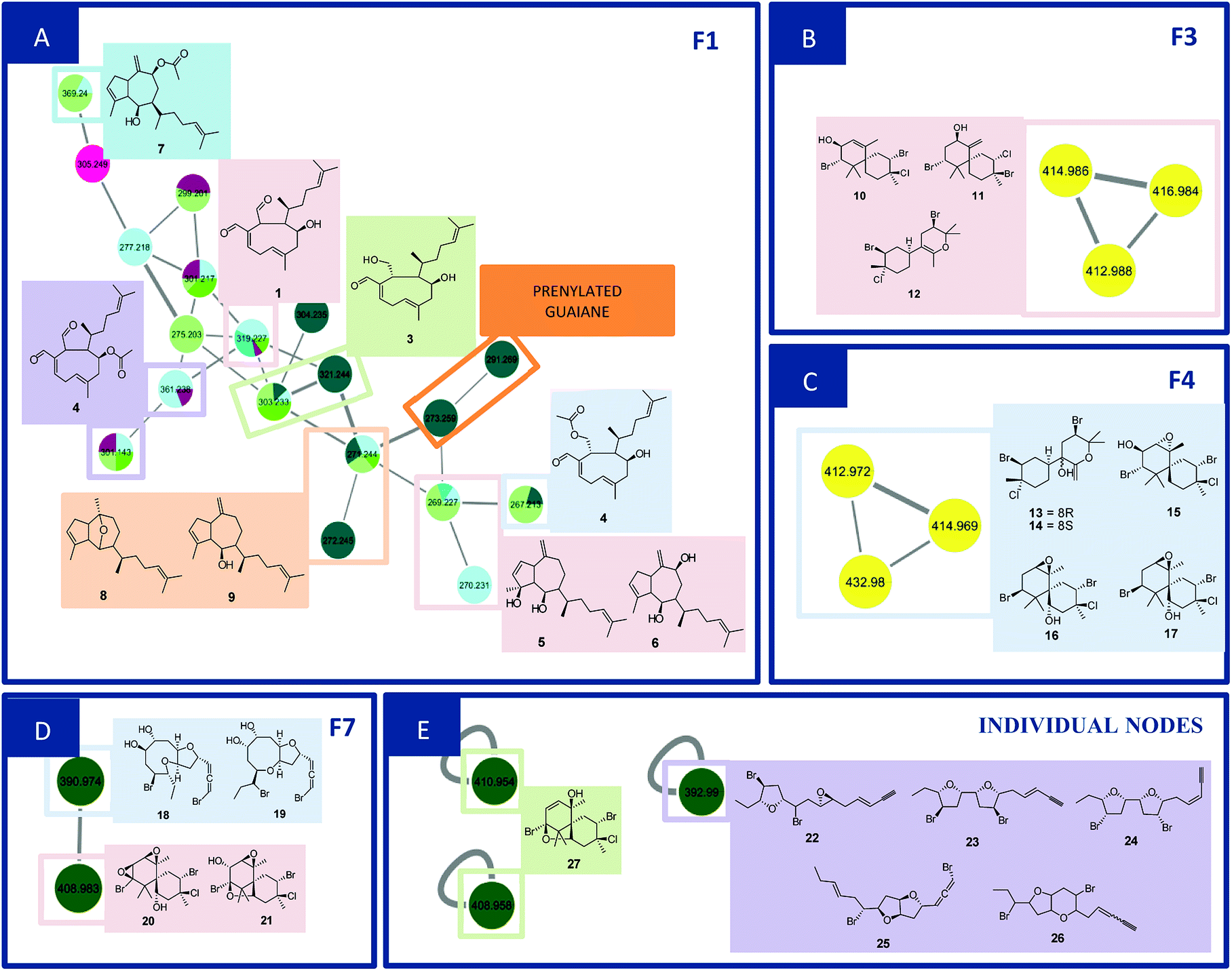 | ||
| Fig. 2 Family 1 (A), 3 (B), 4 (C), 7 (D) and individual nodes (E) and related compounds. | ||
The third node m/z 321.2441 [C20H32O3 + H]+ (calcd 321.2429) (RT 7.74 min) lost H2O (18 Da) to afford m/z 303.2332 [C20H30O2 + H]+ (calcd 303.2324). This precursor was found in the CC1, CC3, DM2 and DM3 LC-MS data. Moreover, daughter ions were observed at m/z 285.2236 [C20H28O + H]+ (calcd 285.2218) and m/z 267.2100 [C20H26 + H]+ (calcd 267.2113). Both ions were produced from the loss of 2 (36 Da) and 3 (54 Da) H2O molecules, respectively. These data suggest another xeniane diterpene 18,4-dihydroxydictyo-19-al A (3).15
The node m/z 267.2134, detected at 7.32 min in the CC3 and DM1 LC-MS spectra, turned out to be a base peak ion, which was the fragment of the precursor m/z 385.2359 [C22H34O4 + Na]+ (calcd 385.2355) as observed in the node mass spectrum. Fragments resulting from the loss of NaOH (40 Da), [NaOH (40 Da) and H2O (18 Da)], [NaOH (40 Da), H2O (18 Da) and ketene (42 Da)], and [NaOH (40 Da), 2H2O (36 Da) and ketene (42 Da)], were obtained at m/z 345.2441 [C22H33O3]+ (calcd 345.2424), m/z 327.2324 [C22H31O2]+ (cald 327.2319), m/z 285.2236 [C22H29O]+ (calcd 285.2213) and m/z 267.2134 [C22H27]+ (calcd 267.2107), respectively. The literature data suggested another xeniane congener, and the structure of 18-acetoxy-4-hydroxydictyo-19-al A (4) was considered a possibility.15
Another F1 node at m/z 269.2265 (detected at 11.84 min in CC1, CC2, and DM2) is also a base peak ion of the precursor at m/z 327.2287 [C20H32O2 + Na]+ (calcd 327.2300). This precursor produced m/z 287.2375 [C20H31O]+ (calcd 287.2369) and m/z 269.2265 [C20H28 + H]+ (calcd 269.2264) after losing NaOH (40 Da), [NaOH (40 Da) and H2O (18 Da)]. The information outlined above, in conjunction with the literature search led to two previously reported compounds. Since the completed structure is not always possible using tandem mass data alone, dictyotadiol (5) and dictyol B (6) were suggested as potential structures.14,16 Similar MS/MS behavior was observed for the node at m/z 369.2395 [C22H34O3 + Na]+ (calcd 369.2406) that corresponded to the peak at 12.00 min in the CC1 and DM2 LC-MS data. This metabolite differs from 5 and 6 by 42 Da suggesting that it is an acetylated derivative. The above data, together with the data reported in the literature led to the structure of dictyol B acetate (7), another guaiane prenylated diterpene.16
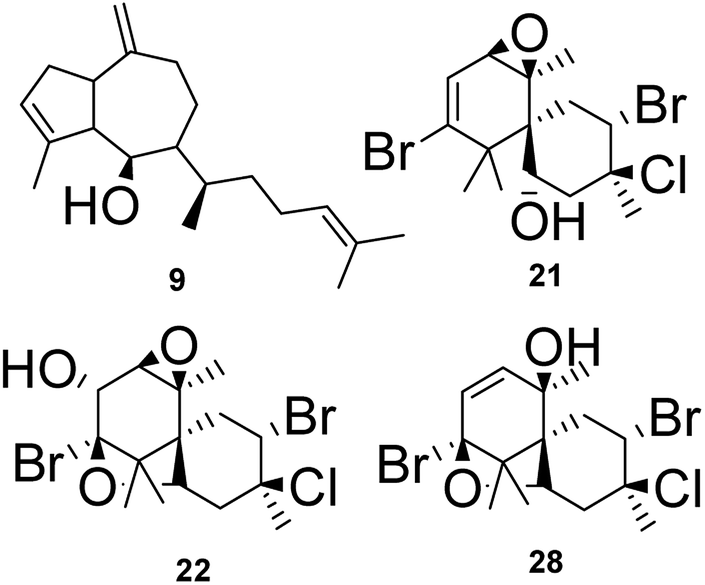
The node at m/z 271.2439 [C20H31O + H]+ (13.09 min in CC1, CC2, DM2 and DM3) represents a base peak ion formed when the precursor at m/z 289.2550 [C20H32O + H]+ (calcd 289.2531) lost H2O (18 Da). The literature search, together with the above information, led to the hypotheses of two prenylated guaiane diterpenes, namely, dictyoxide (8) and pachydictyol A (9).13 Although two metabolites already reported in the literature were suggested, the node could refer to 9, since this compound was later isolated from the extract DM2.
The node m/z 291.2691 [C20H34O + H]+ (RT 4.15 min in CC3) afforded m/z 273.2586 [C20H32 + H]+ (calcd 273.2582) as a fragment ion after losing H2O (18 Da). The literature search resulted in a no-hit compound as chemotaxonomy was considered. However, the mass spectrum of this node gave the precursor ion with 2 Da lighter than 8 and 9 suggesting a prenylated guaiane diterpene analog.
In general, MN clustered in F1 metabolites are mainly from the brown algae Canistrocarpus cervicornis and Dictyota mertensii (Fig. 1), with similar fragmentation patterns to the diterpenes already reported for the Dictyota species. Although the literature for C. cervicornis regards the presence of dolastane and secodolastane, curiously only the guaiane prenylated and xeniane types were observed in the nodes of F1. This suggest that C. cervicornis from Rocas Atoll is chemically diverse from other specimens collected on the Brazilian Coast. Compounds 8 and 9 were previously identified in D. mertensii,12,13 while 1–4 and 6 were obtained from D. crenulata14,15 and 5 and 7 were identified in D. dichotoma.21
The nodes with m/z 270.2309, m/z 272.2452 and m/z 304.2349 were found in LC-MS, but these peaks correspond to isotopic patterns of the nodes with m/z 269.2265, m/z 271.2439 and m/z 303.2332, respectively.
F2 was characterized by heavier mass values for the nodes, however none of the structures was established.
Families F3 (Fig. 2B) and F4 (Fig. 2C) are formed by three nodes each and composed only of metabolites produced by L. catarinensis from Arvoredo Island (LC2). F3 contains the nodes m/z 412.9883, m/z 414.9859 and m/z 416.9840. These three mass values were found in the same spectrum, and belong to the same metabolite. This mass spectrum showed m/z 412.9883, 414.9859, 416.9840 and 418.9828 at a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:5:1, corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms and one chlorine atom. Two metabolites were observed at 7.74 and 8.11 min with this particular isotopic pattern and the mass value of m/z 412.9883. The elemental composition [C15H23Br2ClO + H]+ (calcd m/z 412.9882) is compatible with halogenated marine sesquiterpenes. Its fragmentation spectrum revealed ions at m/z 333.0627/335.0609/337.0566 (4:5:1), m/z 253.1281/255.0815 (3:1), m/z 217.1458; the first fragment was formed after the loss of HBr (80 Da), while the second fragment resulted from the elimination of 2 HBr (160 Da); the last fragment was produced after the elimination of 2 HBr and HCl (196 Da). The foregoing data in conjunction to the data reported in the literature led to the structure of some chamigrane-type sesquiterpenes, such as 9-hydroxy-4,10-dibromo-3-chloro-α-chamigrene (10)22–24 and 2,10-dibromo-3-chloro-8-hydroxy-β-chamigrene (11),25 along to the irregular rearranged bisabolane-related sesquiterpene laucapyranoid A (12).26 The metabolite detected at 7.74 and 8.11 min might be related, due to similar fragmentation behavior. Compound 10 was reported for many Laurencia species, such as L. nidifica,22 L. nipponica23 and L. pacifica,24 while compound 11 was reported only for L. nipponica.25
:7:5:1, corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms and one chlorine atom. Two metabolites were observed at 7.74 and 8.11 min with this particular isotopic pattern and the mass value of m/z 412.9883. The elemental composition [C15H23Br2ClO + H]+ (calcd m/z 412.9882) is compatible with halogenated marine sesquiterpenes. Its fragmentation spectrum revealed ions at m/z 333.0627/335.0609/337.0566 (4:5:1), m/z 253.1281/255.0815 (3:1), m/z 217.1458; the first fragment was formed after the loss of HBr (80 Da), while the second fragment resulted from the elimination of 2 HBr (160 Da); the last fragment was produced after the elimination of 2 HBr and HCl (196 Da). The foregoing data in conjunction to the data reported in the literature led to the structure of some chamigrane-type sesquiterpenes, such as 9-hydroxy-4,10-dibromo-3-chloro-α-chamigrene (10)22–24 and 2,10-dibromo-3-chloro-8-hydroxy-β-chamigrene (11),25 along to the irregular rearranged bisabolane-related sesquiterpene laucapyranoid A (12).26 The metabolite detected at 7.74 and 8.11 min might be related, due to similar fragmentation behavior. Compound 10 was reported for many Laurencia species, such as L. nidifica,22 L. nipponica23 and L. pacifica,24 while compound 11 was reported only for L. nipponica.25
F4 presented similar feature to F3, showing three nodes at m/z 412.9717, m/z 414.9692 and m/z 432.9804. These three mass values were found in the same spectrum and belong to the same metabolite. They originated from the precursor at m/z 428.9848, which appeared alongside m/z 430.9824, 432.9804 and 434.9787 at a ratio of 3:7:5:1, corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms and one chlorine atom. This precursor ion lost H2O (18 Da) and produced the fragment at m/z 410.9749, with the same isotopic pattern. Three isomeric metabolites were found with this particular isotopic pattern and the mass value of m/z 428.9831 at 7.04, 7.45 and 9.99 min. Their elemental composition [C15H23Br2ClO2 + H]+ (calcd m/z 428.9831) supported the presence of halogens in these marine sesquiterpenes. A literature search led to the structure of sesquiterpenes related to laucapyranoids B (13), C (14),26 4,10-dibromo-3-chloro-7,8-epoxy-9-hydroxychamigrane (15),26 4,10-dibromo-3-chloro-7,8-epoxychamigrane (16)27 and 4,10-dibromo-3-chloro-7,8-epoxy-5-hydroxychamigrane (17).28 Compounds 13 and 14 were already reported for L. caespitosa,26 while compound 15 was found in L. glomerata28 and L. flagellifera,29 and 17 in L. scoparia.28
None of the structure was established for F5 and F6.
F7 (Fig. 2D) contained only two nodes at m/z 390.9736 and m/z 408.9829. The first ion was generated from the precursor m/z 424.9951 (RT 5.46 min in LC-MS of LC1), which appeared alongside m/z 426.9961 and 428.9933 at a ratio of 1:2:1. This isotopic proportion indicated the presence of 2 bromine atoms in this compound. Its elemental composition [C15H22Br2O4 + H]+ (calcd m/z 424.9963) supported the brominated nature of this metabolite. The MS spectrum revealed ions at m/z 406.9875/408.9829/410.9832 (1:2:1) and m/z 388.9743/390.9736/392.9700 (1:2:1), furnished respectively by a successive loss of H2O (18 Da), and 2H2O (36 Da). Moreover, further fragment ions were observed in the MS2 spectrum at m/z 327.0549 [M + H–H2O–Br]+, m/z 309.0506 [M + H–2H2O–Br]+, m/z 271.0301 [M + H–2H2O–(1-bromopropa-1,2-diene)]+ and m/z 240.0135 [M + H–2H2O–(1-bromopropa-1,2-diene)–C2H6]+. The foregoing data in conjunction to those reported in the literature led to the structural hypotheses of two brominated C15-acetogenins namely laurendecumallenes A (18) and B (19), which has already been reported in the chemical study of L. decumbens.30
Node m/z 408.9829, in turn, was a fragment ion of the precursor m/z 442.9620 (RT 7.93 min), which appeared in the mass spectrum alongside m/z 444.9616, 446.9571 and 448.9570 at a ratio of 3:7:5:1, corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms and one chlorine atom. Its elemental composition [C15H21Br2ClO3+H]+ (calcd m/z 442.9624) supported the halogenated nature of this marine sesquiterpene. The MS spectrum showed a fragment ion at m/z 406.9875/408.9829/410.9832 (1:2:1) after loss of HCl (36 Da). Furthermore, the MS2 spectrum gave a fragment ion at m/z 309.0487 after eliminating 2H2O (36 Da), HBr (80 Da) and HCl (36 Da). As reported for F3 and F4, these findings indicated a chamigrane sesquiterpene type, which was compatible with the structures of prepacifenol epoxide (20)39 and johnstonol (21).22 Since 20 and 21 were isolated from LC1 extract and presented the same retention times and fragmentation patterns, it is possible that this node corresponds to both compounds. Nevertheless, according to the literature, prepacifenol epoxide may be converted into johnstonol.22 Compound 20 was also isolated from L. composita,31 L. johnstonii,20 L. nidifica,22 L. okamurai,32 while compound 21 was reported for L. nidifica,22 L. okamurai32 and L. pacifica.33
For F8 and F9, it was also not possible to establish any structure.
The individual node with m/z 392.9902 also corresponded to a metabolite produced by L. catarinensis from Xavier Island (LC1 extract). This mass spectrum showed m/z 390.9898, 392.9902 and 394.9876 at ratio of 1:2:1, corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms. Two isomeric metabolites were observed at 4.96 and 6.39 min with this particular isotopic pattern and the mass value of m/z 390.9898. Its elemental composition [C15H20Br2O2 + H]+ (calcd m/z 392.9888) confirmed a dibrominated metabolite. The literature search led to the hypotheses of some C15-acetogenins containing five-membered cyclic ethers ring, such as laureepoxide (22) (one tetrahydrofuran ring),34 (3E)-elatenyne (23) and elatenyne (24) (two isolated tetrahydrofuran rings),30,35 and kumausallene (25) (two fused tetrahydrofuran rings),25 along with laurobtusin (26), an acetogenin-containing six-membered cyclic ether ring.36 Compound 22 was reported to L. nipponica,34 23 to L. majuscula,35 24 to L. decumbens30 and L. elata,37 25 to L. nipponica25 and 26 to L. obtusa.36
The individual nodes at m/z 408.9581 and 410.9542 correspond to the metabolite from LC1 extract with a retention time at 9.80 min. These mass values were found in the same spectrum alongside m/z 412.9509 and m/z 414.9525 at a ratio of 3:7:5:1 corresponding to the isotopic pattern of a metabolite containing 2 bromine atoms and one chlorine atom. This ion was produced by the precursor at m/z as observed in the mass spectrum. Loss of H2O [C15H21Br2ClO2 − H2O + H]+ (calcd m/z 428.9654). These data were also indicative of a chamigrane-type sesquiterpene, and this compound was suggested to be pacifenol (27), since MS data and RT were in agreement with those from pacifenol isolated from LC1 (L. catarinensis from Xavier Island). This metabolite was reported for some Laurencia species, such as L. filiformis,38 L. majuscula39 and L. pacifica.40
Overall, the diagnosis of the above dereplication data reveals how the marine ecosystem can vary according to geographical location. Marine chemistry is strongly based on the interaction and diversity of animal, plant and microorganism species.40 LC1 and LC2, two extracts obtained from the same species of algae collected in relatively close islands from the Southern Brazilian Coast, interestingly supported this fact. For LC1, L. catarinensis collected in Xavier Island, three chamigrane-type sesquiterpenes (20, 21 and 27) were characterized; alongside with seven C15-acetogenins (18, 19, 22, 23, 24, 25, 26), while for LC2, L. catarinensis collected in Arvoredo Island, three compounds related to caespitol (12, 13 and 14) were suggested, together with four chamigrene-type sesquiterpenes (10, 11, 16, 17). Previous chemical work with L. catarinensis collected in Arvoredo Island41 found mostly caespitol-related metabolites, but no C15-acetogenins. Meanwhile, from the ongoing investigation of L. catarinensis from Xavier Island, the three chamigranes characterized in the extract LC1 have been isolated and structurally identified and some fractions presented NMR data suggestive of C15-acetogenins. It was observed that Canistrocarpus cervicornis from Rocas Atoll (CC1-3) clustered mostly with Dictyota mertensii from three different oceanic islands (DM1-3), while extracts from Dictyopteris spp. presented mostly isolated nodes. Nevertheless, the isolation of further metabolites is needed, in order to allow a better evaluation of those hypotheses. It is also possible that some of the discussed nodes correspond to new metabolites with the same molecular formula structures and similar fragmentation patterns to compounds already reported in the literature.
Previous studies on the Laurencia species also showed a different chemical composition in relation to the collection sites.42,43 Prevezols A and B, alongside acetogenins were identified from L. obtusa collected in Greece,44,45 while the same species collected on the Italian coast produced obtusalenes V, VI, VII and IX.45 Furthermore, L. microcladia from some Greek islands showed different chemical profiles according to the collection sites.46–49
No compound could be identified in the extracts of Dictyopteris spp., Laurencia dendroidea and L. intricata.
Experimental
Chemicals and materials
Acetonitrile (HPLC grade) and formic acid were purchased from Tedia (São Paulo, Brazil), while ethyl acetate, dichloromethane, hexane and methanol were provided by Vetec Química Fina Ltda (São Paulo, Brazil). Deuterated chloroform was purchased from Cambridge Isotope Laboratories, Inc (Massachusetts, EUA) and silica gel (40–63 μm) from Sigma-Aldrich (Missouri, EUA). Milli-Q system (18.2 MΩ, Milipore, Simplipak. France) was used to produce ultrapure water for aqueous solutions. Filtration of the samples was performed with syringe filter nylon (13 MM; 0.22 μM) from Allcrom (São Paulo, Brazil). Precoated TLC plates (200 mm thickness, silica gel 60 F254, Silicycle, Inc.) were used for thin-layer chromatography.General experimental procedures
1D and 2D NMR spectra for isolated metabolites were obtained on Bruker DRX 400 MHz spectrometer (Bruker, Massachusetts). HRESIMS data were recorded with Xevo G2-Xs QTof mass spectrometer (Waters Corporation, Milford, MA). Vacuum liquid chromatography (VLC) was performed on silica gel column (40–63 μm; 5 mm × 5.5 mm). Medium pressure liquid chromatography (MPLC) was carried out on Sepacore system (Buchi, Switzerland) using silica gel column (40–63 μm; 15 mm × 920 mm). Solid Phase Extractions (SPE) were performed using Supelclean™ LC-Si SPE cartridges. Semipreparative HPLC was carried out on a Pharmacia LKB 2252 liquid chromatography system equipped with refractive index detector Shodex RI-102, using a Supelcosil column (10 mm × 250 mm × 5 μm, silica).Organism collection and extraction
Ten brown (Dictyopteris plagiogramma, Dictyopteris jolyana, Dictyota mertensii and Canistrocarpus cervicornis) and four red algae (Laurencia catarinensis, L. dendroidea, L. intricata) samples were obtained from different sites and in different seasons (Table 1), and dried under cold air. The dried material was exhaustively extracted with dichloromethane and methanol (2:1) at room temperature and the crude extracts were dried under vacuum.
LC-MS analysis
The fourteen crude extracts and the isolated metabolites were dissolved in acetonitrile with a final concentration at 2.0 mg mL−1, and then filtered over nylon filters. The samples and the blank were injected in an aliquot of 2.0 μL to an Acquity UPLC system equipped with a BEH C18 column (2.1 mm × 50 mm; 1.7 μm), PDA detector and a quaternary bomb. Temperatures of the sample tray and column were set at 20 °C e 40 °C. The flow rate was 0.3 mL min−1 of a gradient mobile phase of CH3CN/H2O (0.1% formic acid): from 80% to 70% of H2O in 1 min and then decreasing until 15% of CH3CN in 11 min. This proportion lasted 2 min; from 15% to 80% in 1 min; 5 min of 80% of H2O were used to return to the initial condition.The mass spectrometer was set to observe m/z in a range of 100–1500 in positive ESI mode using the FastDDA algorithm to acquire MS/MS spectra. The capillary voltage was set at 4.0 kV and the cone voltage at 40 V. The cone gas flow was 200 L h−1 with an ion source temperature of 80 °C. The desolvation temperature was 300 °C, and the desolvation gas flow was 900 L h−1. The analyses were accurate, and leucine enkephalin was used as reference (lock mass m/z 556.2771). The collision energy was between 25 and 35 eV and argon was used as collision gas for the tandem mass. The software MassLynx V4.1 (Waters Corporation, Milford, USA) was used to process the LC-MS data.
After preliminary MN analysis of the extracts, the same LC-MS method was applied to isolated metabolites for comparison of retention times and MS/MS data.
Molecular networking
All chromatograms and MS/MS spectra obtained for the 14 crude extracts were digitally converted into .mzML files using MSConvert software (http://www.proteowizard.sourceforge.net), and then submitted to the Global Natural Product Social MN (GNPS) platform (http://gnps.ucsd.edu). The MN was generated by interconnecting MS/MS spectra with precursor ion mass tolerance of 0.002 Da, fragment ion mass tolerance of 0.02 Da, minimum peak intensity equal to 50, a minimum matched of fragment ions and library search minimum matched peaks equal to four. The remaining parameters were kept the same as suggested by the GNPS platform. The MN generated was displayed on Cytoscape 3.6.0.Isolation of compounds
The extracts DM2 and LC2 were subjected to vacuum column chromatography on silica gel, using hexane with increasing amounts of EtOAc, followed by EtOAc with increasing amounts of MeOH as the mobile phase, to yield 16 fractions (DA1–DA16) and 20 fractions (LA1–LA20), respectively. Fraction DA2 (10% EtOAc in hexane, 48.9 mg) was fractionated by normal-phase classic column, using hexane with increasing amounts of EtOAc as eluents, to yield 6 fractions (DB1–DB6). Fraction DB3 (17.5 mg) was purified by normal-phase HPLC, using cyclohexane/EtOAc (8:2) as eluents, to yield 9 (DC3, 2.2 mg). Fractions LA3 and LA4 (5% and 10% EtOAc in hexane, 345.0 mg) were fractionated by normal-phase MPLC, using hexane with increasing amounts of EtOAc as eluent, to yield 48 fractions (LB1–LB48). Fractions LB20 and LB26 (155.0 mg) were purified by normal-phase MPLC, using hexane with increasing amounts of EtOAc as eluent, to yield 27 (C44–45, 14.2 mg) and 21 (C48–50, 17.4 mg). Fraction LA5 (15% EtOAc in hexane, 114.0 mg) was fractionated by normal-phase MPLC using hexane with increasing amounts of EtOAc as the mobile phase, to yield 75 fractions (LD1–LD75). Fractions LD15, LD16 and D17 (79.0 mg) were purified by normal-phase HPLC, using cyclohexane/EtOAc (95:5) as eluents, to yield 20 (LE2, 36.0 mg).
Data of isolated metabolites
:1.5); 1H NMR (CDCl3, 300 MHz) and 13C NMR (CDCl3, 75 MHz) data were in agreement to the literature.13:1); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 75 MHz) data were in agreement to the literature.39 Positive ESIMS [M + H]+ m/z 442.9628, 444.9620, 446.9571, 448.9523 (calc. for C15H22Br2ClO3, 442.9624). RT 7.93 min.:1); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 75 MHz) data were in agreement to the literature.50 Positive ESIMS [M + H]+ m/z 442.9620, 444.9616, 446.9571, 448.9570 (calc. for C15H22Br2ClO3, 442.9624). RT 7.93 min.:1); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 75 MHz) data were in agreement to the literature.51 Positive ESIMS [M − HCl + H]+ m/z 408.9576, 410.9532, 412.9537, 414.9507 (calc. for C15H19Br2ClO, 408.9569). RT 9.80 min.Conclusions
The MN exploratory analyses indicated chemical differences among the extracts from different brown and red algae species, and among different collection sites for the same species, including sites closer to the coast (LC1 and LC2) and on the oceanic islands. For some extracts it was possible to identify mass values compatible with metabolites already reported in the literature, such as terpenoids (F1, F3, F4 and F7 besides individual nodes) and C15-acetogenins (F7 and individual nodes) already isolated from those or related species. Furthermore, one diterpene and three sesquiterpenes were isolated from Dictyota mertensii (DM2) and Laurencia catarinensis (LC2) extracts, respectively. Due to the limitations of mass spectrometry for structure assignment, the metabolites reported here could also have an isomeric skeleton of the proposed structure. Many nodes in the MN could not be characterized, and these metabolites could be used as targets for isolation in a future work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Profs. Vassilios Roussis and Efstathia Ioannou (University of Athens) for the support by isolation and identification of metabolites from L. catarinensis, to team of SISBIOTA-Mar and PROSPECMar (USP, UFF, UFSC, UFRJ, UA, UCSD, UFBA, FTC, UFRN, UNESP) projects for collecting the algal material, to Prof. Paulo Horta (UFSC) for the identification of algae, and to CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for the financial support to PROSPECMAR (grant number 458548/2013-8). ACP, GAZ, TW and MGAB thank Capes for their fellowships.References
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. Liaw, Y. Yang, H. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. Allard, P. Phapale, L. Nothias, T. Alexandrov, M. Litaudon, J. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef PubMed.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef PubMed.
- R. A. Quinn, L. Nothias, O. Vining, M. Meehan, E. Esquenazi and P. C. Dorrestein, Trends Pharmacol. Sci., 2017, 38, 143–154 CrossRef PubMed.
- C. B. Naman, R. Rattan, S. E. Nikoulina, J. Lee, B. W. Miller, N. A. Moss, L. Armstrong, P. D. Boudreau, H. M. Debonsi, F. A. Valeriote, P. C. Dorrestein and W. H. Gerwick, J. Nat. Prod., 2017, 80, 625–633 CrossRef PubMed.
- L. V. Costa-Lotufo, F. Carnevale-Neto, A. E. Trindade-Silva, R. R. Silva, G. G. Z. Silva, D. V. Wiilke, F. C. L. Pinto, B. D. B. Sahm, P. C. Jimenez, J. N. Mendonça, T. M. C. Lotufo, O. D. L. Pessoa and N. P. Lopes, Chem. Commun., 2017, 16, 1952–1955 Search PubMed.
- V. Ruiz-Torres, J. A. Encinar, M. Herranz-López, A. Pérez-Sánchez, V. Galiano, E. Barrajón-Catalán and V. Micol, Molecules, 2017, 22, E1037 CrossRef PubMed.
- C. B. B. Matta, É. T. Souza, A. C. Queiroz, D. P. Lira, M. V. Araújo, L. H. A. Cavalcante-Silva, G. E. C. Miranda, J. X. Araújo-Júnior, J. M. Barbosa-Filho, B. V. O. Santos and M. S. Alexandre-Moreira, Mar. Drugs, 2011, 9, 307–318 CrossRef PubMed.
- T. Z. Serafini, G. B. França and J. Andriguetto-Filho, journal of integrated Coastal Zone Management, 2010, 10, 281–301 Search PubMed.
- M. Harizani, E. Ioannou and V. Roussis, Prog. Chem. Org. Nat. Prod., 2016, 102, 91–252 Search PubMed.
- G. A. Zatelli, A. C. Philippus and M. B. Falkenberg, Rev. Bras. Farmacogn., 2018, 28, 243–260 CrossRef.
- V. Teixeira and A. Kelecom, Sci. Total Environ., 1988, 75, 271–283 CrossRef.
- A. B. Alvarado and W. H. Gerwick, J. Nat. Prod., 1985, 48, 132–134 CrossRef.
- S. P. Freitas, A. S. Oliveira, J. C. De-Paula, R. C. Pereira, D. N. Cavalcanti and V. L. Teixeira, Nat. Prod. Commun., 2007, 2, 13–15 Search PubMed.
- J. C. De-Paula, L. B. Bueno, D. N. Cavalcanti, Y. Yoneshigue-Valentin and V. L. Teixeira, Molecules, 2008, 13, 1253–1262 CrossRef PubMed.
- M. P. Kirkup and R. E. Moore, Phytochemistry, 1983, 22, 2539–2541 CrossRef.
- D. J. Faulkner, B. N. Ravi, J. Finer and J. Clardy, Phytochemistry, 1977, 16, 991–993 CrossRef.
- L. V. Costa-Lotufo, D. V. Wilke, P. C. Jimenez and R. A. Epifanio, Quim. Nova, 2009, 32, 703–716 CrossRef.
- E. M. Bianco, V. L. Teixeira and R. C. Pereira, Braz. J. Oceanogr., 2010, 58, 213–218 CrossRef.
- R. B. G. Camara, L. S. Costa, G. P. Fidelis, L. T. D. B. Nobre, N. Dantas-Santos, S. L. Cordeiro, M. S. S. P. Costa, L. G. Alves and H. A. O. Rocha, Mar. Drugs, 2011, 9, 124–138 CrossRef PubMed.
- Y. N. Nariyoshi, E. A. C. Guedes, H. A. O. Rocha and L. M. Pinotti, J. Pharm. Pharm. Sci., 2013, 5, 815–818 Search PubMed.
- D. J. Faulkner, M. O. Stallard and C. Ireland, Tetrahedron Lett., 1974, 15, 3571 CrossRef.
- J. Kimura, N. Kamada and Y. Tsujimoto, Bull. Chem. Soc. Jpn., 1999, 72, 289–292 CrossRef.
- M. Suzuki, E. Kurosawa and A. Furusaki, Bull. Chem. Soc. Jpn., 1988, 61, 3371–3373 CrossRef.
- W. Fenical, Phytochemistry, 1976, 15, 511–512 CrossRef.
- M. Suzuki, M. Segawa, T. Suzuki and E. Kurosawa, Bull. Chem. Soc. Jpn., 1983, 56, 3824–3826 CrossRef.
- M. Chang, J. T. Vazquez, K. Nakanishi, F. Cataldo, D. M. Estrada, J. Fernandez, A. GAllardo, J. D. Martin, M. Norte and R. Perez, Phytochemistry, 1989, 28, 1417–1424 CrossRef.
- J. F. Elsworth and R. H. Thomson, J. Nat. Prod., 1989, 52, 893–895 CrossRef.
- D. J. Kennedy, I. A. Selby and R. H. Thomson, Phytochemistry, 1988, 27, 1761–1766 CrossRef.
- M. L. Bittner, M. Silva, V. J. Paul and W. Fenical, Phytochemistry, 1985, 24, 987–989 CrossRef.
- N.-Y. Ji, X.-M. Li, K. Li and B.-G. Wang, J. Nat. Prod., 2007, 70, 1499–1502 CrossRef PubMed.
- M. Masuda, T. Abe, T. Suzuki and M. Suzuki, Phycologia, 1996, 35, 550–562 CrossRef.
- M. Ojika, Y. Shizuri and K. Yamada, Phytochemistry, 1982, 21, 2410–2411 CrossRef.
- J. J. Sims, W. Fenical, R. M. Wing and P. Radlick, Tetrahedron Lett., 1972, 13, 195 CrossRef.
- A. Fukuzawa, M. Aye, Y. Takaya, T. Masamune and A. Murai, Phytochemistry, 1990, 29, 2337–2339 CrossRef.
- K. Kim, M. R. Brennan and K. L. Erickson, Tetrahedron Lett., 1989, 30, 1757–1760 CrossRef.
- S. Imre, A. Öztunç and S. Islimyeli, Doga: Turk Kim. Derg., 1987, 11, 119 Search PubMed.
- J. G. Hall and J. A. Reiss, Aust. J. Chem., 1986, 39, 1401–1409 CrossRef.
- J. Jongaramruong, A. J. Blackman, B. W. Skelton and A. H. White, Aust. J. Chem., 2002, 55, 275 CrossRef.
- S. Caccamese, A. Compagnini and R. M. Toscano, J. Nat. Prod., 1986, 49, 173 CrossRef.
- B. M. Howard, A. M. Nonomura and W. Fenical, Biochem. Syst. Ecol., 1980, 8, 329 CrossRef.
- M. P. Puglisi, J. M. Sneed, K. H. Sharp, R. Ritson-Williams and V. J. Paul, Nat. Prod. Rep., 2014, 31, 1510–1553 RSC.
- E. G. Lyakhova, A. I. Kalinovsky, S. A. Kolesnikova, V. E. Vaskovsky and V. A. H. Stonik, Phytochemistry, 2004, 65, 2527–2532 CrossRef PubMed.
- V. Cassano, J. C. De-Paula, M. T. Fujii, B. A. P. Gama and V. L. Teixeira, Biochem. Syst. Ecol., 2008, 36, 223–226 CrossRef.
- N. Mihopoulos, C. Vagias, E. Mikros, M. Scoullos and V. Roussis, Tetrahedron Lett., 2001, 42, 3749–3752 CrossRef.
- D. Iliopoulou, C. Vagias, C. Harvala and V. Roussis, Phytochemistry, 2002, 59, 111–116 CrossRef PubMed.
- G. Guella and F. A. Pietra, Helv. Chim. Acta, 2000, 83, 2946–2952 CrossRef.
- M. Kladi, C. Vagias, G. Furnari and D. Moreau, Tetrahedron Lett., 2005, 46, 5723–5726 CrossRef.
- M. Kladi, H. Xenaki, C. Vagias, P. Papazafiri and V. Roussis, Tetrahedron, 2006, 62, 182–189 CrossRef.
- M. Kladi, C. Vagias, P. Papazafiri, G. Furnari, D. Serio and V. Roussis, Tetrahedron, 2007, 63, 7606–7611 CrossRef.
- C. R. Kaiser, Quim. Nova, 2000, 23, 231–236 CrossRef.
- C. R. Kaiser, L. F. Pitombo and A. C. Pinto, Spectrosc. Lett., 1998, 31, 573–585 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra, IR of the four isolated compounds. See DOI: 10.1039/c8ra02802h |
| ‡ Ana Cláudia Philippus and Gabriele Andressa Zatelli contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |