
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen deficient Pr6O11 nanorod supported palladium nanoparticles: highly active nanocatalysts for styrene and 4-nitrophenol hydrogenation reactions†
Nan Jiang,
Xiao Zhou,
Yi-Fan Jiang,
Zhi-Wei Zhao,
Liu-Bo Ma,
Cong-Cong Shen,
Ya-Nan Liu,
Cheng-Zong Yuan,
Shafaq Sahar and
An-Wu Xu *
*
Division of Nanomaterials and Chemistry, Hefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China, Hefei, Anhui 230026, People's Republic of China. E-mail: anwuxu@ustc.edu.cn
First published on 14th May 2018
Abstract
The design and development of highly efficient and long lifetime Pd-based catalysts for hydrogenation reactions have attracted significant research interest over the past few decades. Rational selection of supports for Pd loadings with strong metal-support interaction (SMSI) is beneficial for boosting catalytic activity and stability. In this context, we have developed a facile approach for uniformly immobilizing ultra-small Pd nanoparticles (NPs) with a clean surface on a Pr6O11 support by a hydrogen thermal reduction method. The hydrogenations of p-nitrophenol and styrene are used as model reactions to evaluate the catalytic efficiency. The results show highly efficient styrene hydrogenation performance under 1 atm H2 at room temperature with a TOF value as high as 8957.7 h−1, and the rate constant value of p-nitrophenol reduction is 0.0191 s−1. Strong metal-support interaction and good dispersion of Pd nanoparticles, as demonstrated by XPS and HRTEM results, contribute to the excellent hydrogenation performance. Electron paramagnetic resonance (EPR) results suggest the presence of oxygen vacancies in the support, which serve as electron donors and enhance the adsorption and activation of reactants and subsequent conversion into products. Moreover, the catalyst can be recovered and reused up to 10 consecutive cycles without marked loss of activity. Overall, our results indicate that oxygen-deficient Pr6O11 nanorods (NRs) not only play a role as support but also work as the promoter to substantially boost the catalytic activities for organic transformations, therefore, providing a novel strategy to develop other high-performance nanostructured catalysts for environmental sustainability.
Introduction
Metal-catalyzed hydrogenation reactions are important for large-scale production of pharmaceuticals, polymers, fine chemicals, and so on.1,2 Palladium nanoparticles (Pd NPs), as one of the most active noble metals, has attracted increasing attention in these reactions. Unfortunately, Pd-based catalyst methods have significant drawbacks, including high cost, easy aggregation due to high surface energy and poor recycling performance.3,4 Immobilizing Pd NPs on various supports, such as metallic oxides,5 organic polymers,6 carbon7 or biomaterials,8 has shown enhanced catalytic performance and stability in organic transformation reactions. In particular, anchoring noble metal NPs on metal oxide with oxygen vacancies not only exhibits good dispersion but also activates the reactants and accelerates the reactions.9 On the other hand, the interaction between noble metals and supports (known as strong metal-support interaction, SMSI) will influence both the activity and selectivity during catalytic processes.10,11 Therefore, rational selection of supports should be taken into consideration in the development of highly efficient and stable catalysts.The oxides of rare earth elements have been widely used in high-performance luminescent devices, catalysts, promoters, and many others on account of their unique electronic, optical, magnetic and chemical properties.12–14 Praseodymium oxide, an important rare earth material, has been studied as an adsorbent for dye removal,15 Au-catalyzed support for CO oxidation,16 catalysts for ethane and ethylene synthesis,17 ceramic pigments18 and other functional materials. Praseodymium oxides have various kinds of stoichiometrical oxides, for instance, Pr2O3, PrO2, Pr4O7 and so on, among these oxides, Pr6O11 is a stable phase in air at ambient temperature,16,19 and it is an n-type semiconductor with high electrical conductivity involving the 4f shell of their ions,20 which could trigger electronic interactions with loaded noble metals through SMSI, thus is in favor of adsorption and activation of reactants and subsequent conversion into final products.21 Bearing these in mind, and taking the advantages of oxygen-deficient oxide support, in this work, Pr6O11 nanorods are prepared by thermal treatment of Pr(OH)3 precursor in air and used as support for loading Pd NPs.
In this context, we develop a hydrogen thermal reduction method to load Pd NPs on oxygen-deficient Pr6O11 support, and study the catalytic performances as heterogeneous catalyst for hydrogenation of styrene and 4-nitrophenol. X-ray powder diffraction (XRD), high-resolution transmission electron microscopy (HRTEM), X-ray photoemission spectroscopy (XPS), electron paramagnetic resonance (EPR) were carried out to characterize Pd/Pr6O11 nanocatalysts. The results demonstrate that obtained Pd/Pr6O11 nanocatalysts exhibit superior catalytic activities in hydrogenation reactions of styrene and 4-nitrophenol. When the actual loading weight content of Pd NPs is 1.42 wt%, the turnover frequency (TOF) value of styrene hydrogenation is as high as 8957.7 h−1 at room temperature, which is comparable to the highest value (8973 h−1) for hydrogenation of styrene that has been reported to date.5 Excellent catalytic activity is also observed in 4-nitrophenol hydrogenation reaction, which rate constant value is measured to be 0.0191 s−1. The unique electron structure of rare earth metal oxides, the oxygen-deficient Pr6O11 support, and the existing SMSI facilitate the adsorption and activation of reactants, thus resulting in highly efficient activities in hydrogenation reactions. We anticipate that this study will help to encourage a further research and application of other rare earth metal oxides as supports for catalytic reactions and enhance large-scale production of chemicals with advanced catalytic performances and increased environmental sustainability.
Experimental section
Chemicals
Praseodymium nitrate hexahydrate (Pr(NO3)3·6H2O, 99.9%) was purchased from Aladdin Ltd. (Shanghai, China). Sodium tetrachloropalladate (II) (Na2PdCl4, 98%) was purchased from Sigma-Aldrich. Ammonium hydroxide (NH3·H2O, 25%), styrene, absolute ethanol (EtOH, 99.5%), sodium borohydride (NaBH4) and 4-nitrophenol (reagent grade) were obtained from Sinopharm Chemical Reagent Co. Ltd. All chemicals reagents were used without further purification.Synthesis of Pr(OH)3 and Pr6O11 NRs
In a typical synthesis, 800 mg of Pr(NO3)·6H2O was dissolved in 50 mL of distilled water at room temperature followed by adding ammonium hydroxide dropwise into Pr(NO3)3 aqueous solution under violent magnetic stirring until the pH value reached to 8. A light green colloidal precipitate of Pr(OH)3 appeared immediately. The solution was transferred into stainless steel Teflon lined autoclave after stirring another 20 min and heated at 120 °C for 8 h. After naturally cooling to room temperature, the resulting products were collected by centrifugation and washed several times using distilled water and ethanol, finally dried 12 h at 60 °C under vacuum oven. Pr6O11 NRs were synthesized by thermal treatment of as-obtained Pr(OH)3 NRs at 600 °C in air for 3 h.Synthesis of Pd/Pr6O11 nanocatalyst
The Pd NPs were loading on praseodymium oxide nanorods by wet impregnation and H2 reduction method. In a detail, 1410 μL of Na2PdCl4 (10 mM) aqueous solution (the nominal weight content was 1.5 wt%) was dispersed into 20 mL of distilled water and then 100 mg of Pr6O11 sample was added. After ultrasonic treatment for several minutes, then the mixture was stirred vigorously for 12 h at room temperature for palladium ion adsorption. Subsequently, the solid was recovered, rinsed with distilled water and ethanol, and dried at 60 °C overnight. Finally, the as-prepared solid was calcined at 350 °C (5 °C min−1) for 2 h in 5% H2/Ar gas, and Pd/Pr6O11 sample was obtained.Characterizations
X-ray powder diffraction (XRD) patterns of samples were carried out by Philips X'Pert Pro Super diffractometer, which was radiated by graphite monochromatized Cu Kα (λ = 1.54178 Å). The operating voltage was maintained at 40 kV, the current was maintained at 200 mA and analyzed in the range of 10° ≤ 2θ ≤ 70°. Scanning electron microscopy (SEM) images were performed on a field-emission scanning electron microanalyzer (JEOL JSM-6700F, 15 kV). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM), scanning transmission electron microscopy (STEM) images and energy dispersive X-ray spectroscopy (EDX) analysis were measured on FEI Talos F200X operated at 200 kV. The X-ray photoemission spectroscopy (XPS) was performed on Perkin-Elmer RBD upgraded PHI-5000C ESCA system. JEOL JES-FA200 EPR spectrometer was used here to examine the electron paramagnetic resonance (EPR) spectra with the operating parameters 140 K, 9064 MHz, 0.998 mW, X-band. The actual concentration of Pd was analyzed by inductively coupled plasma-mass (ICP-MS) optical emission spectroscopy on the optima 7300 DV, Perkin-Elmer. The reaction conversion measurements were carried out using gas chromatography (GC) (Agilent 7890B).General experimental procedure for catalytic hydrogenation of styrene
The catalytic activity measurements of Pd/Pr6O11 catalysts for styrene hydrogenation reaction were carried out by using H2 (1 atm) as the reductant at room temperature. Typically, Pd/Pr6O11 (5 mg), styrene (570 μL, 5 mmol) and 1,3,5-trimethylbenzene (internal standard, 500 μL, 3.6 mmol) were added into 10 mL of absolute ethanol under constant stirring. The progress of the reaction was detected by gas chromatography (GC) equipped with a flame ionization detector (FID) at different reaction time intervals, and the conversion ratio was calculated by the area ratio of substrate and product according to the internal standard method. The recyclability of Pd/Pr6O11 catalysts was evaluated as follows: the reaction was performed under the same conditions as described above, and a successive of operations was carried out by centrifugation and washing the solid with water and ethanol thoroughly after the end of the reaction, followed by drying at 60 °C in a vacuum oven overnight. Then the recycled Pd/Pr6O11 catalysts were re-dispersed into a new reaction system under the same conditions for the next run.General experimental procedure for 4-nitrophenol reduction reaction
In a typical procedure, 4-nitrophenol (100 μL, 10 mM) was added into NaBH4 aqueous solution (8 mL, 0.1 M) under constant stirring for about 10 min. Then Pd/Pr6O11 aqueous suspension (100 μL, 1.4 mg mL−1) was quickly added into above solution under continuous stirring until the solution turned colorless. The reaction progress of 4-nitrophenol reduction was monitored at different time intervals by the UV-visible absorption spectra of the filtrated solutions.Results and discussion
The oxygen-deficient Pr6O11 NRs supported Pd NPs with clean surface were fabricated via a simple and surfactant-free thermal reduction method. Pr(OH)3 NRs were obtained by hydrothermal treatment at 120 °C for 12 h, followed by annealing at 600 °C for 3 h to obtain Pr6O11 NRs, and the color of the products turned from light green to brown. Pd NPs were immobilized on the Pr6O11 support by thermal reduction method at 350 °C for 2 h in 5% H2/Ar (see Experimental section for details) with the color of the sample further changed to grey. As a result, the ultra-small Pd NPs were successfully loaded on oxygen-deficient Pr6O11 nanorod support.The crystal structure and phase purity of the as-prepared samples were tested by X-ray diffraction (XRD) patterns and shown in Fig. 1. The obtained Pr(OH)3 NRs adopt a hexagonal structure and the corresponding lattice parameters are a = 0.6456 nm and c = 0.3755 nm, which is consistent with the standard powder diffraction file of Pr(OH)3 (JCPDS no. 83-2304). All detectable diffraction peaks in Fig. 1b can be readily indexed to a face-centered cubic structure of Pr6O11, which are in good agreement with the standard powder diffraction file of Pr6O11 (JCPDS no. 42-1121) with lattice constant a = 0.5468 nm. No peaks from other phases can be detected, indicating the high purity of the as-made Pr6O11 NRs. All the peaks in Fig. 1b are strong and sharp, indicating the high crystallinity of Pr6O11 sample. After loading Pd NPs on the Pr6O11 support, it can be clearly seen that the diffraction peaks become broader and weaker, no peaks of Pd can be found, suggesting the ultra-low loading content of Pd and well dispersity of Pd NPs on the support (actual loading weight content was 1.42 wt% determined by ICP-MS analysis). Compared with the pristine Pr6O11 NRs, the diffraction intensities of Pd/Pr6O11 sample after loading Pd NPs decrease, which means the generation of surface disorder produced from Pd-catalyzed instant hydrogenation of Pr6O11. The H2 thermal reduction process can be expressed as adsorbed Pd(II) was first reduced to Pd(0) by H2, and then H2 molecules spontaneously dissociate on the surface of Pd(0) to produce highly active atomic hydrogen species, which could diffuse into and interact with Pr6O11 lattices, thus leading to the surface disorder and oxygen vacancies of hydrogenated Pr6O11 NRs.22
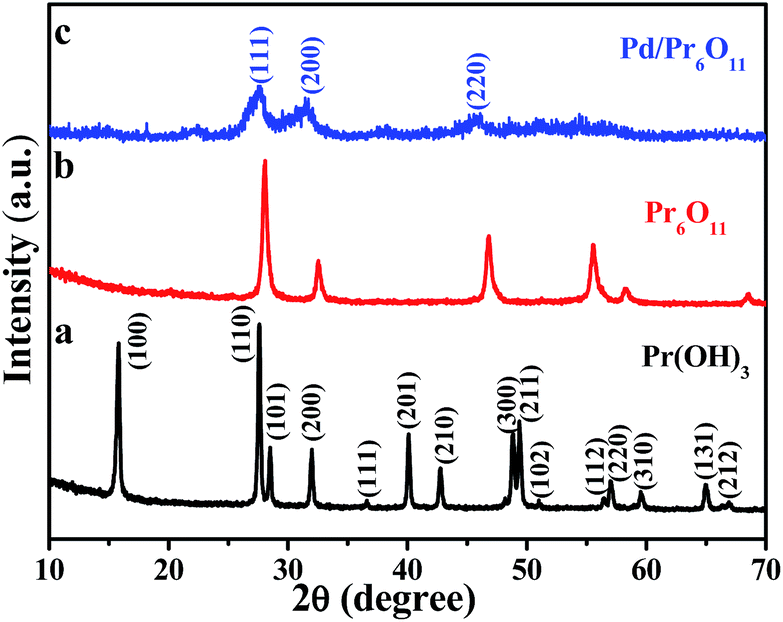 | ||
| Fig. 1 XRD patterns of (a) Pr(OH)3 precursor, (b) Pr6O11 NRs and (c) Pd/Pr6O11 nanocatalysts (1.42 wt% Pd). | ||
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) measurements were performed to characterize the morphology and structure features of as-synthesized products and the images are shown in Fig. 2. It can be seen that Pr(OH)3 NRs have straight and smooth surfaces with an average diameter of 30 nm (Fig. 2a). It is worth noting that the morphology of Pr6O11 NRs can be well maintained after annealing Pr(OH)3 NRs, but the surfaces of Pr6O11 NRs become a little coarse, which is likely due to dehydration of Pr(OH)3 during calcination process (Fig. 2b). As shown in Fig. 2c and d, the lattice fringes of Pr6O11 NRs after loading Pd NPs are assigned to the (200) plane with a spacing of 0.284 nm. Obviously, Pd NPs are uniformly deposited on the surface of Pr6O11 NRs, and the average crystallite size of Pd NPs is about 2.60 nm (Fig. 2e). The lattice spacing of Pd NPs is about 0.224 nm, which is consistent with the (111) plane of metallic Pd. Moreover, after Pd loadings, the surfaces of Pr6O11 NRs become porous and rough, which is caused by Pd-catalyzed instant hydrogenation of Pr6O11, in agreement with XRD results. Consequently, confirming the existence of strong metal-support interaction (SMSI). This phenomenon has been proved to accelerate catalytic performance and boost the stability of as-obtained catalysts.22,23 The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) measurements were adopted to further confirm the uniform distribution of Pd NPs on the Pr6O11 support. The elemental mapping images (Fig. 2f–i) clearly reveal the uniform distribution of Pr, O and Pd elements throughout the whole Pr6O11 NRs.
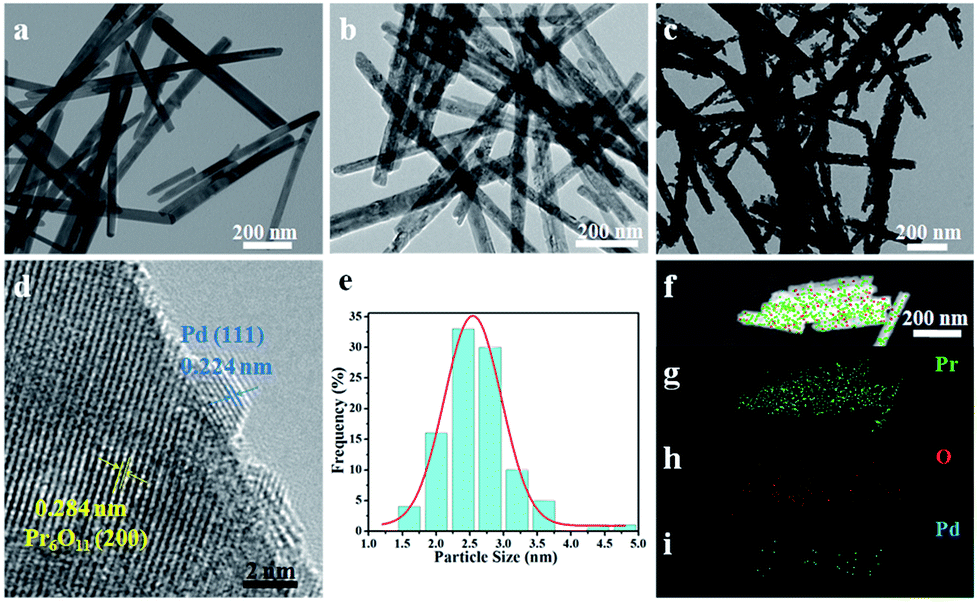 | ||
| Fig. 2 TEM images of (a) Pr(OH)3 NRs, (b) Pr6O11 NRs and (c) 1.42 wt% Pd/Pr6O11 nanocatalysts, (d) HRTEM image of Pd/Pr6O11 nanocatalysts, (e) size distribution of Pd NPs in Pd/Pr6O11 nanocatalysts, (f–i) HAADF-STEM images and corresponding elemental mappings of Pd/Pr6O11 for (g) Pr, (h) O and (i) Pd. | ||
In order to determine the elemental composition and chemical state of various elements in Pd/Pr6O11 NRs, X-ray photoelectron spectroscopy (XPS) measurements were carried out. The survey spectrum proves the existence of Pr, O and Pd elements (Fig. 3a). It can be seen that Pr 3d exhibits two strong peaks at around 933 eV and 954 eV (Fig. 3b), which correspond to Pr 3d5/2 and Pr 3d3/2 energy levels, respectively. According to the previous report, the XPS spectra of Pr 3d can be deconvoluted into three peaks, the strong peaks with binding energies of 933 eV for Pr 3d5/2 correspond to Pr3+ and other two peaks are attributed to Pr4+ (935 eV) with a shake-off satellite (930 eV).24–26 From XPS data, the atomic ratio of Pr4+ to Pr3+ was estimated to be about 0.168, and the average valence of praseodymium atoms in this sample was calculated to be ca. 3.17, which is a little lower to the expected value of 3.66 for Pr6O11, namely, the chemical formula of this sample is Pr6O9.5, therefore, XPS results confirm that oxygen vacancies indeed exist in oxide support. The corresponding O 1s XPS spectrum can be fitted into two components (Fig. 3c), the lower binding energy at 528.95 eV, accounting for about 11.98% of the O 1s spectrum, is the typical metal–oxygen bonds of Pr–O; the higher binding energy at 531.45 eV, which accounts for ca. 88.02% of the O 1s spectrum, can be readily assigned to adsorbed oxygen on the surface of catalysts.27,28 Pd 3d spectrum of Pd/Pr6O11 (Fig. 3d) can be deconvoluted into two sub-peaks as 3d5/2 and 3d3/2 levels of Pd. The spectrum of Pd 3d5/2 exhibits two different chemical states of Pd. The main peak at 337.6 eV could be ascribed to Pd(II), and the other peak centered at 335.9 eV proves the presence of metallic Pd(0). As compared to unsupported Pd NPs, the position of these two peaks shifts to higher binding energy values,24,28 thus suggesting the electronic communication and strong metal-support interaction (SMSI) occur between Pd NPs and Pr6O11 support.22
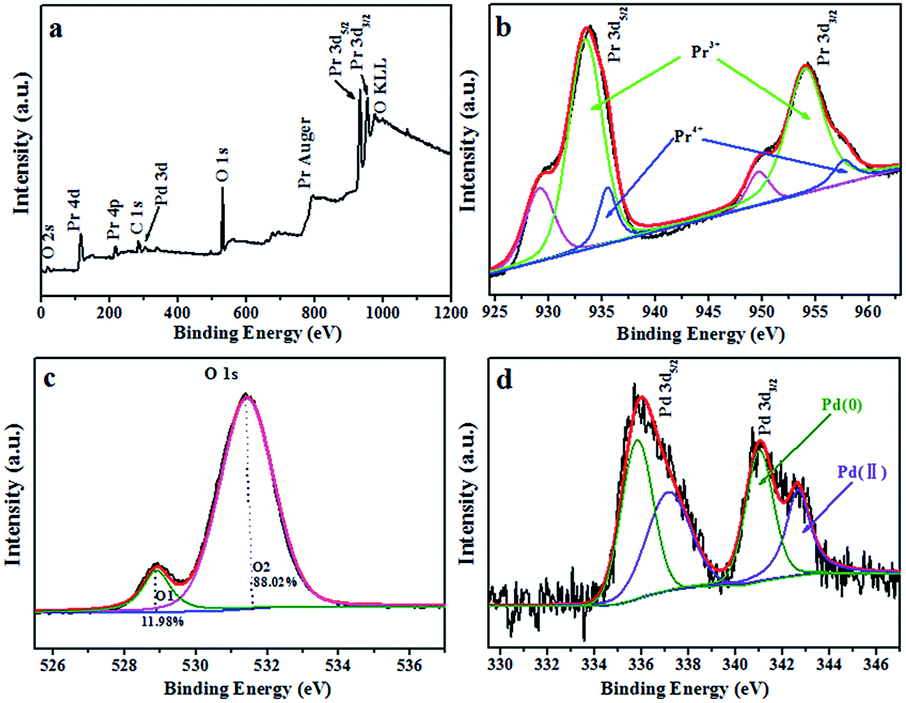 | ||
| Fig. 3 XPS analysis of Pd/Pr6O11 samples (a) survey XPS spectrum, (b) Pr 3d; (c) O 1s and (d) Pd 3d. | ||
Electron paramagnetic resonance (EPR) measurements were performed to further reveal the formation of oxygen vacancies in as-obtained Pd/Pr6O11 samples (Fig. 4). The pristine Pr6O11 NRs show no EPR signal due to its antiferromagnetic characteristics. However, after H2 thermal reduction treatment for Pd loadings, a sharp and strong signal with the g value of 2.002 appears, which has previously been assigned to the paramagnetic state of the oxygen vacancies 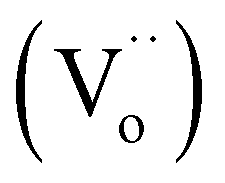 .29 Oxygen vacancies are in favor of adsorbing atmospheric oxygen molecules and O2 was reduced to ·O2−, thus producing a strong signal in EPR spectrum.30 The EPR result further confirms the presence of abundant oxygen vacancies, which could serve as free electron donors and increase both the electrical conductivity of samples and the electron density of Pd surface, consequently improving the catalytic activities.
.29 Oxygen vacancies are in favor of adsorbing atmospheric oxygen molecules and O2 was reduced to ·O2−, thus producing a strong signal in EPR spectrum.30 The EPR result further confirms the presence of abundant oxygen vacancies, which could serve as free electron donors and increase both the electrical conductivity of samples and the electron density of Pd surface, consequently improving the catalytic activities.
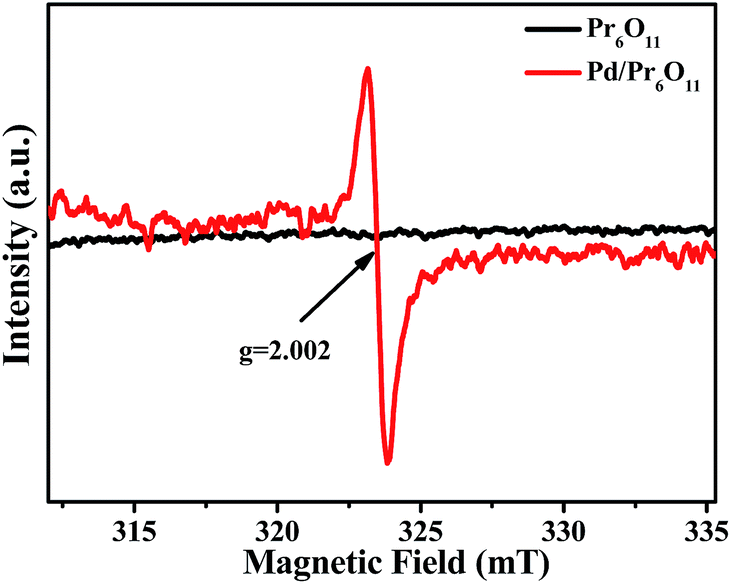 | ||
| Fig. 4 The X-band EPR spectra of Pr6O11 and Pd/Pr6O11 samples recorded at 140 K. | ||
Olefin hydrogenation reactions are frequently used in the industrial process, and has also been important reactions used in the environmentally friendly chemical process.31,32 Therefore, the catalytic activity of Pd/Pr6O11 nanocomposites was investigated for the styrene hydrogenation by using 1,3,5-trimethylbenzene as internal standard and ethanol as solvent at room temperature under 1 atm H2, as elucidated in Scheme 1. The reaction progress was monitored by gas chromatography (GC) equipped with a flame ionization detector (FID) at different reaction time intervals. Gratifyingly, we observed a highly efficient catalytic activity of the catalyst in the hydrogenation reaction. As shown in Fig. 5a, when the actual Pd loading amount is 1.42 wt% (determined by ICP-MS), the styrene was converted into ethylbenzene completely with 40 min, and the corresponding turnover frequency (TOF, calculated by moles of substrate converted per mole of Pd per hour) value reaches as high as 8957.7 h−1. To the best of our knowledge, this TOF value for styrene hydrogenation is comparable to the highest value (8973 h−1) of Pd single atoms supported on TiO2 that has ever been reported to date.5 The comparison for the catalytic activity of styrene hydrogenation is listed in Table 1, obviously, our Pd/Pr6O11 catalyst is one of the best catalysts reported so far,5,33–40 and comparable to Pd single atoms supported on TiO2 catalyst.5 It should be noted that there is no obvious activity of pure Pr6O11 support, this implies that Pr6O11 NRs only play a role as support and promoter for highly efficient catalytic performance.
 | ||
| Scheme 1 The equation of styrene hydrogenation reaction. | ||
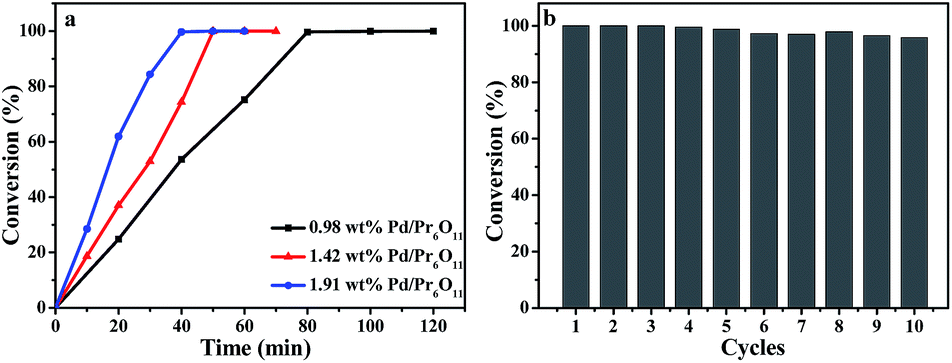 | ||
| Fig. 5 (a) Kinetics of styrene hydrogenation catalyzed by Pd/Pr6O11 samples. (b) Recycling curves of Pd/Pr6O11 catalysts for hydrogenation reaction of styrene for ten times. | ||
| Entry | Catalyst | Solvent | Conditions H2 (MPa)/T (°C) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|
| a TOF: turnover frequency calculated as the number of moles of product per mol Pd per h. | |||||
| 1 | Pd/Pr6O11 | Ethanol | 0.1/25 | 8957.7 | Our study |
| 2 | Pd/PEG | Ethanol | 0.1/25 | 660 | 33 |
| 3 | Fe3O4-NC-PZS-Pd | Ethanol | 0.1/25 | 1792 | 34 |
| 4 | Pd/C | DMF | 0.1/25 | 377.4 | 35 |
| 5 | Polymer-anchored Pd(II) schiff base catalyst | DMF | 0.1/25 | 765.6 | 35 |
| 6 | Pd single atom/TiO2 | Ethanol | 0.1/30 | 8973 | 5 |
| 7 | Pd/SiO2 | Ethanol | 2/25 | 5181 | 36 |
| 8 | Pd in microreactor | Ethanol | 0.1/35 | 1449 | 37 |
| 9 | Pd/Tm-MOF | No | 0.1/35 | 703 | 38 |
| 10 | Pd/ZIF-8 | No | 0.1/35 | 307 | 38 |
| 11 | Pd/MOF-5 | No | 0.1/35 | 682 | 39 |
| 12 | Pd/C | Ethyl acetate | 0.3/25 | 163 | 40 |
The recyclability of the catalyst for styrene hydrogenation was performed by filtering and drying of the samples at the end of the reaction, and reused in a new reaction system under the same condition and the results are displayed in Fig. 5b. It can be seen that the catalyst could be reused for up to ten successive reactions with only about 4% activity loss. Such good recycling performance can be attributed to the effective stabilization of active Pd NPs by Pr6O11 NR support, which also confirms the SMSI takes place between Pd NPs and Pr6O11 support.
As well-known, 4-NP is an organic pollutant and by-product of some industrial reactions.41,42 Therefore, it is necessary to convert 4-NP into environmentally friendly chemicals. Herein, p-nitrophenol (4-NP) hydrogenation was also chosen as a model reaction to further evaluate the catalytic activity of as-synthesized Pd/Pr6O11 catalyst, where NaBH4 was used as the reducing agent. The catalytic reduction of 4-NP was performed at room temperature by adding 100 μL (1.4 mg mL−1) Pd/Pr6O11 nanocatalysts into mixed solution of 4-NP (100 μL, 10 mM) and NaBH4 (8 mL, 0.1 M). The reaction progress was monitored by UV-vis absorption spectroscopy, as shown in Fig. 6. As the amount of NaBH4 is in excess compared to 4-NP, therefore the concentration of BH4− can be considered as constant throughout the reaction, indicating only 4-NP and p-aminophenol (4-AP) influence the reaction kinetics.43,44 The intensity of absorption peak at 400 nm is related to the 4-NP concentration, while the intensity of absorption peak at 300 nm corresponds to the 4-AP concentration (reaction product).45–47 From Fig. 6a, it can be seen that after the introduction of the as-synthesized catalyst, the absorption peak at 400 nm dropped quickly in intensity, along with the increased intensity of the peaks at 300 nm. Moreover, the color of the solution changed from bright yellow to colorless. This phenomenon suggests that the 4-NP was efficiently reduced to 4-AP and proving the excellent catalytic performance of the as-obtained nanocatalysts. The hydrogenation reaction completed very fast, as evidenced by the fact that the absorption peak at 400 nm nearly decreased to zero within 180 s. Fig. 6b exhibits the relationship between ln(Ct/C0) and reaction time (t), where C0 and Ct are the concentrations of 4-NP at time 0 and t, respectively. The apparent rate constant k was calculated by fitting with a model function of the form ln(Ct/C0) = −kt, and the results are shown in the ESI Table S1,† together with the reported data for comparison. It can be seen that the apparent rate constant value for our Pd/Pr6O11 catalyst is 0.0191 s−1, which is much higher than many of previously reported catalysts for 4-NP reduction.48–51
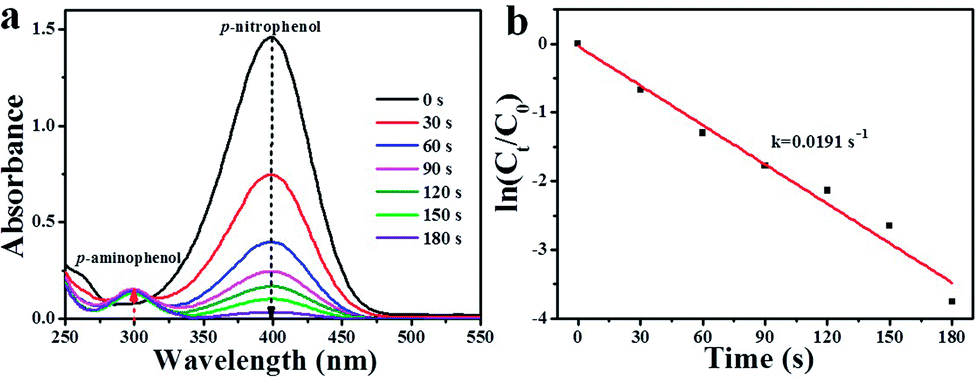 | ||
| Fig. 6 (a) Successive UV-vis spectra of 4-NP reduction reaction over Pd/Pr6O11 catalyst. (b) Kinetic curves for the reduction of 4-NP catalyzed by Pd/Pr6O11. Ct and C0 are 4-NP concentrations at time t and 0, respectively. The actual weight content of Pd was 1.42 wt%. | ||
Above results demonstrate that our developed Pd/Pr6O11 nanocomposites exhibit highly efficient styrene and 4-NP hydrogenation performance, the reasons for such enhanced catalytic performance may be rationalized by the following factors. (1) As confirmed by TEM and HRTEM analysis, Pd NPs have clean surface without any other organic groups and ultra-small particle size, what's more, they are uniformly anchored on the Pr6O11 support, this of very importance to high-performance Pd-based catalyst.52,53 (2) Both the praseodymium (Pr) and palladium (Pd) elements exist in two kinds of valence states (Pr3+/Pr4+ and Pd0/Pd2+), indicating the SMSI and electronic communication occur between Pd NPs and Pr6O11 support.22,28 (3) The XPS and EPR results reveal the existence of SMSI and oxygen vacancies in Pr6O11 NRs. It has been reported that oxygen vacancies carry free two electrons, which can serve as electron donors,54 namely, the electrons can transfer from support to Pd NPs, increasing the electrical conductivity and the adsorption of reactants. The energy barrier required to cleave H–H bond could be decreased with the help of oxygen vacancies, thus facilitating the dissociation of H2,55,56 this is favorable for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond activation of styrene and accelerating the rate of styrene hydrogenation.9,57,58 Taken together, the emerging SMSI and oxygen vacancies play important roles in promoted catalytic activity.59,60
C bond activation of styrene and accelerating the rate of styrene hydrogenation.9,57,58 Taken together, the emerging SMSI and oxygen vacancies play important roles in promoted catalytic activity.59,60
Conclusions
In summary, we have combined a simple and surfactant-free thermal reduction method with rare earth metal oxide support to fabricate oxygen-deficient and surface-clean Pd/Pr6O11 nanocatalysts. The styrene and 4-NP hydrogenation reactions are tested as model reactions to evaluate the catalytic performances of as-obtained nanocomposites, the results show that Pd/Pr6O11 nanorods indeed display highly efficient catalytic activity and superior stability. Specifically, the TOF value for styrene hydrogenation is as high as 8957.7 h−1, which is comparable to the highest TOF value that has been reported so far. And the rate constant value for 4-NP hydrogenation reaction is measured to be 0.0191 s−1. The as-synthesized nanocatalyst exhibits excellent catalytic activity and stability up to 10 consecutive cycles. It is found that Pd nanoparticles have ultra-small particle size and are evenly distributed on the support, and Pd-catalyzed instant hydrogenation of Pr6O11 leads to oxygen vacancies and rough surface of Pr6O11 nanorods, consequently inducing strong metal-support interaction, this could accelerate the reactions and improve the stability. Moreover, the oxygen vacancies in Pr6O11 support can serve as electron donors and enable the high electron density of Pd species, thus promoting the activation of CC bond, which boosts the catalytic activity toward styrene and 4-NP hydrogenation reactions. We anticipate that this work not only opens a new possibility for designing highly efficient catalysts for hydrogenation reactions, but also helps to trigger the potential utilization of rare earth elements and their oxides in other catalytic systems, which is beneficial for the large-scale production of chemicals with advanced catalytic performance and environmental sustainability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The special funding support from the National Natural Science Foundation of China (51572253, 21271165), Scientific Research Grant of Hefei Science Center of CAS (2015SRG-HSC048), and Cooperation between NSFC and Netherlands Organization for Scientific Research (51561135011) is acknowledged.References
- T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636–2654 RSC.
- S. D. Jackson and L. A. Shaw, Appl. Catal., A, 1996, 134, 91–99 CrossRef.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 8572–8580 CrossRef.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef PubMed.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef PubMed.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef PubMed.
- M. J. Gronnow, R. Luque, D. J. Macquarrie and J. H. Clark, Green Chem., 2005, 7, 552–557 RSC.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef PubMed.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef.
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef PubMed.
- G. Y. Adachi and N. Imanaka, Chem. Rev., 1998, 98, 1479–1514 CrossRef.
- A.-W. Xu, Y.-P. Fang, L.-P. You and H.-Q. Liu, J. Am. Chem. Soc., 2003, 125, 1494–1495 CrossRef PubMed.
- A.-W. Xu, Y. Gao and H.-Q. Liu, J. Catal., 2002, 207, 151–157 CrossRef.
- T. Zhai, S. Xie, X. Lu, L. Xiang, M. Yu, W. Li, C. Liang, C. Mo, F. Zeng, T. Luan and Y. Tong, Langmuir, 2012, 28, 11078–11085 CrossRef PubMed.
- P. X. Huang, F. Wu, B. L. Zhu and G. R. Li, J. Phys. Chem. B, 2006, 110, 1614–1620 CrossRef PubMed.
- K. Asami, K.-i. Kusakabe, N. Ashi and Y. Ohtsuka, Appl. Catal., A, 1997, 156, 43–56 CrossRef.
- C.-W. Nahm, J. Am. Ceram. Soc., 2010, 93, 3056–3059 CrossRef.
- M. Fernandez-Garcia, A. Martinez-Arias, J. C. Hanson and J. A. Rodriguez, Chem. Rev., 2004, 104, 4063–4104 CrossRef PubMed.
- O. Hirsch, K. Kvashnina, C. Willa and D. Koziej, Chem. Mater., 2017, 29, 1461–1466 CrossRef.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- M. Imran, A. B. Yousaf, X. Zhou, Y.-F. Jiang, C.-Z. Yuan, A. Zeb, N. Jiang and A.-W. Xu, J. Phys. Chem. C, 2017, 121, 1162–1170 Search PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2016, 9, 120–127 CrossRef PubMed.
- G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, 1979, p. 64 Search PubMed.
- S. Balachandran, K. Thirumalai and M. Swaminathan, RSC Adv., 2014, 4, 27642–27653 RSC.
- C. K. Narula, L. P. Haack, W. Chun, H.-W. Jen and G. W. Graham, J. Phys. Chem. B, 1999, 103, 3634–3639 CrossRef.
- H. He, H. X. Dai, K. W. Wong and C. T. Au, Appl. Catal., A, 2003, 251, 61–74 CrossRef.
- Y. F. Jiang, C. Z. Yuan, X. Xie, X. Zhou, N. Jiang, X. Wang, M. Imran and A. W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 9756–9762 Search PubMed.
- H. L. Guo, Q. Zhu, X. L. Wu, Y. F. Jiang, X. Xie and A. W. Xu, Nanoscale, 2015, 7, 7216–7223 RSC.
- Q. Zhu, Y. Peng, L. Lin, C. M. Fan, G. Q. Gao, R. X. Wang and A. W. Xu, J. Mater. Chem. A, 2014, 2, 4429–4437 Search PubMed.
- Y.-M. Cheng, J.-R. Chang and J.-C. Wu, Appl. Catal., 1986, 24, 273–285 CrossRef.
- A. Tahara, H. Tanaka, Y. Sunada, Y. Shiota, K. Yoshizawa and H. Nagashima, J. Org. Chem., 2016, 81, 10900–10911 CrossRef PubMed.
- F. A. Harraz, S. E. El-Hout, H. M. Killa and I. A. Ibrahim, J. Catal., 2012, 286, 184–192 CrossRef.
- S. Yang, C. Cao, Y. Sun, P. Huang, F. Wei and W. Song, Angew. Chem., Int. Ed., 2015, 54, 2661–2664 CrossRef PubMed.
- S. M. Islam, A. S. Roy, P. Mondal and N. Salam, Appl. Organomet. Chem., 2012, 26, 625–634 CrossRef.
- Y. Wang, A. V. Biradar, C. T. Duncan and T. Asefa, J. Mater. Chem., 2010, 20, 7834–7841 RSC.
- Y. Lan, M. Zhang, W. Zhang and L. Yang, Chem.–Eur. J., 2009, 15, 3670–3673 CrossRef PubMed.
- Y. Pan, D. Ma, H. Liu, H. Wu, D. He and Y. Li, J. Mater. Chem., 2012, 22, 10834–10839 RSC.
- M. Sabo, A. Henschel, H. Fröde, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827–3832 RSC.
- C.-B. Hwang, Y.-S. Fu, Y.-L. Lu, S.-W. Jang, P.-T. Chou, C. C. Wang and S. J. Yu, J. Catal., 2000, 195, 336–341 CrossRef.
- K. B. Narayanan and N. Sakthivel, J. Hazard. Mater., 2011, 189, 519–525 CrossRef PubMed.
- H. Terada, Biochim. Biophys. Acta, Rev. Bioenerg., 1981, 639, 225–242 CrossRef.
- J. Zhang, G. Chen, D. Guay, M. Chaker and D. Ma, Nanoscale, 2014, 6, 2125–2130 RSC.
- N. Xue, R.-J. Yu, C.-Z. Yuan, X. Xie, Y.-F. Jiang, H.-Y. Zhou, T.-Y. Cheang and A.-W. Xu, RSC Adv., 2017, 7, 2351–2357 RSC.
- Z. Dong, X. Le, C. Dong, W. Zhang, X. Li and J. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef.
- Z. Dong, X. Le, Y. Liu, C. Dong and J. Ma, J. Mater. Chem. A, 2014, 2, 18775–18785 Search PubMed.
- Z. Dong, X. Le, X. Li, W. Zhang, C. Dong and J. Ma, Appl. Catal., B, 2014, 158, 129–135 CrossRef.
- X. Lu, X. Bian, G. Nie, C. Zhang, C. Wang and Y. Wei, J. Mater. Chem., 2012, 22, 12723–12730 RSC.
- Z. Jin, M. Xiao, Z. Bao, P. Wang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 6406–6410 CrossRef PubMed.
- Y. Mei, Y. Lu, F. Polzer and M. Ballauff, Chem. Mater., 2007, 19, 1062–1069 CrossRef.
- P. Zhang, R. Li, Y. Huang and Q. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 2671–2678 Search PubMed.
- A. Aijaz, Q. L. Zhu, N. Tsumori, T. Akita and Q. Xu, Chem. Commun., 2015, 51, 2577–2580 RSC.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef PubMed.
- J. Guzman, S. Carrettin and A. Corma, J. Am. Chem. Soc., 2005, 127, 3286–3287 CrossRef PubMed.
- C. M. Fan, L. F. Zhang, S. S. Wang, D. H. Wang, L. Q. Lu and A. W. Xu, Nanoscale, 2012, 4, 6835–6840 RSC.
- R. J. Madix, Science, 1986, 233, 1159–1166 Search PubMed.
- J. Horiuti and M. Polanyi, Nature, 1933, 132, 819 CrossRef.
- J. Horiuti and M. Polanyi, Nature, 1934, 134, 377 CrossRef.
- Y. Liu, C. Xiao, Z. Li and Y. Xie, Adv. Energy Mater., 2016, 6, 1600436 CrossRef.
- S. Zhang, J. Li, W. Gao and Y. Qu, Nanoscale, 2015, 7, 3016–3021 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02831a |
| This journal is © The Royal Society of Chemistry 2018 |