
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-delivery of dihydroartemisinin and docetaxel in pH-sensitive nanoparticles for treating metastatic breast cancer via the NF-κB/MMP-2 signal pathway†
Jin Tao a,
Zeng Tana,
Lu Diaoab,
Zhonghua Jia,
Jiahuan Zhua,
Wei Chena and
Ying Hu*ab
a,
Zeng Tana,
Lu Diaoab,
Zhonghua Jia,
Jiahuan Zhua,
Wei Chena and
Ying Hu*ab
aZhejiang Pharmaceutical College, No. 888, East Section, Yinxian Main Road, The Zone of Higher Education, Ningbo, Zhejiang, China. E-mail: pharmhawk@126.com
bSchool of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang, China
First published on 13th June 2018
Abstract
Metastasis is a major barrier in cancer chemotherapy. Prolonged circulation and rapid, specific intracellular drug release are two main goals in the development of nanoscale drug delivery systems to treat metastatic breast cancer. In this study, we investigated the anti-metastasis effect of docetaxel (DTX) in combination with dihydroartemisinin (DHA) in metastatic breast cancer 4T1 cells. We synthesized a pH-sensitive material 4-arm-PEG-DTX with a hydrazone bond and used it to construct nanoparticles that co-deliver DTX and DHA (D/D NPs). The D/D NPs had a mean size of 142.5 nm and approximately neutral zeta potential. The pH-sensitive nanoparticles allowed acid-triggered drug release at the tumor site, showing excellent cytotoxicity (IC50 = 7.0 μg mL−1), cell cycle arrest and suppression of cell migration and invasion. The mechanisms underlying the anti-metastasis effect of the D/D NPs involved downregulation of the expression of p-AKT, NF-κB and MMP-2. Therefore, D/D NPs represent a new nanoscale drug delivery system for treating metastatic breast cancer, responding to the acidic tumor microenvironment to release the chemotherapeutic drugs.
Introduction
Breast cancer is one of the greatest threats to women's health.1 20–50% of women with breast cancer finally develop metastasised cancer via lung metastasis.2,3 At present, clinical treatment of early breast cancer still relies heavily on surgical and radiation therapies.4 However, those treatments cannot target metastatic tumors in the distal organs. The first-line treatment of metastatic breast cancer is chemotherapy, but it is always associated with disadvantages such as systemic side effects and low efficiency, giving poor patient outcomes.5,6One of the most established technologies of drug delivery is the use of nanoscale delivery vehicles such as liposomes and nanoparticles for cancer therapy, which afford efficient accumulation of drugs at the tumor sites by exploiting the pathological characteristics of tumors like enhanced permeability and retention (EPR).7 However, the therapeutic efficacy of some nanoscale delivery vehicles is suboptimal, mainly because of inefficient drug loading, short lifetime and inability to release drugs at specific sites.8 Therefore, a drug delivery system that allows high drug loading, long lifetime and site-specific drug release in the tumor environment is highly desirable.
To improve drug loading efficiency, a combination of physical encapsulation and chemical conjugation is often used.9 Polyethylene glycol (PEG) is widely used in drug delivery, surface modification of biomaterials and other biomedical applications owing to its hydrophilicity, biocompatibility and low toxicity. 4-Arm-PEG is a polymer commonly used in chemical conjugation of drugs to form nanoscale vehicles with a high drug loading and enhanced stability.10 In particular, PEG modification on the surface of nanoparticles could bypass the reticuloendothelial system and enhance exposure in blood because hindered nanoparticles interact with biomolecules, thereby prolonging the circulating time11 and promoting passive targeting of the tumor environment.12 Therefore, we aimed to develop a 4-arm-PEG based drug delivery system to increase drug loading via drug conjugation to the polymer and encapsulation.
To ensure specific drug release at the tumor site, specific characteristics of the tumor environment could be exploited,13,14 such as pH,15 enzymes16 and redox potential.17 The pH value of the endosomes or lysosomes in tumor cells is lower (pH = 5–6) than that in normal tissues and in blood (pH = 7.4). pH-sensitive drug delivery systems have been designed for use in tumor therapy, which allows drug release in response to the lower pH in tumor cells but not in normal cells.18 Thus, we aimed to develop a pH-sensitive drug delivery system based on a hydrazone linkage between the drug and the polymer, which is expected to release the drug under acidic environments.19
Docetaxel (DTX) is widely used as an effective anticancer agent for breast cancer.20 Despite the remarkable anticancer activity of DTX, its therapeutic use is limited by metastatic breast cancer. It is reported that first-line treatment of metastatic breast cancer with DTX is only 40–50% effective, with the effectiveness decreasing as the second- and third-line treatments.21 Therefore, a combination of DTX and other conventional chemotherapeutics for treating metastatic cancers is essential.22 Dihydroartemisinin (DHA), a derivative of artemisinin, presents good anticancer efficacy in vitro and in vivo.23 DHA mediates cell cycle arrest, induces apoptosis, blocks angiogenesis and inhibits tumor metastasis.24,25 Furthermore, combining DHA with other chemotherapeutics induces synergetic effects. We thus hypothesized that a combination of DTX and DHA could induce synergistic effects in inhibiting metastasis of breast cancer. In view of the poor water solubility of DTX and DHA, we need a nanovector to increase the solubility of the drugs.
Here, we developed a novel pH-sensitive drug delivery system against metastatic breast cancer (Scheme 1). We formed a DTX conjugate with 4-arm-PEG via a hydrazone bond, affording the acid sensitive material 4-arm-PEG-DTX. 4-Arm-PEG-DTX can self-assemble and encapsulate DHA forming a nanostructured system (D/D NPs). D/D NPs are covered by PEG on the surface to prolong circulation, and upon entry into tumor tissues will release DTX and DHA via acidic cleavage of the hydrazine bond and breakdown of the nanostructure. We also evaluated the effects of D/D NPs on the growth and metastasis of 4T1 tumors.
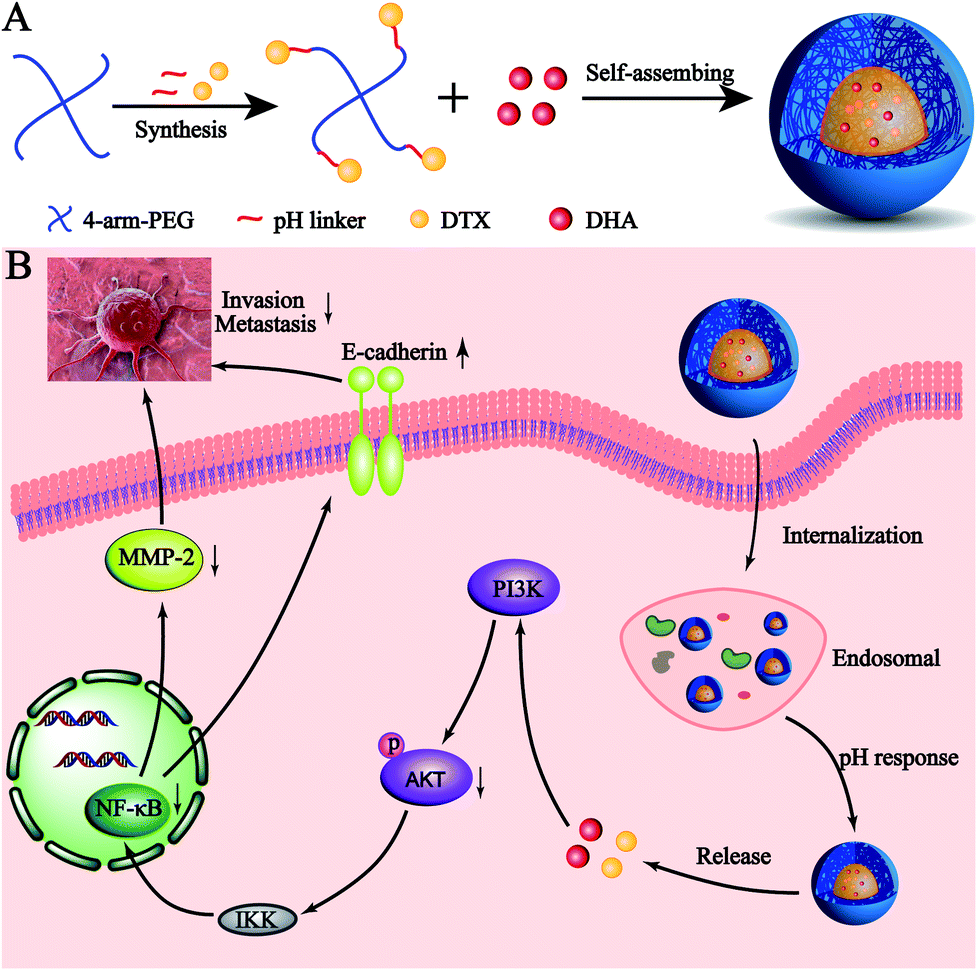 | ||
| Scheme 1 The co-delivery of DTX and DHA as acid-sensitive nanoparticles. | ||
Result
Synthesis and characterization of 4-arm-PEG-DTX
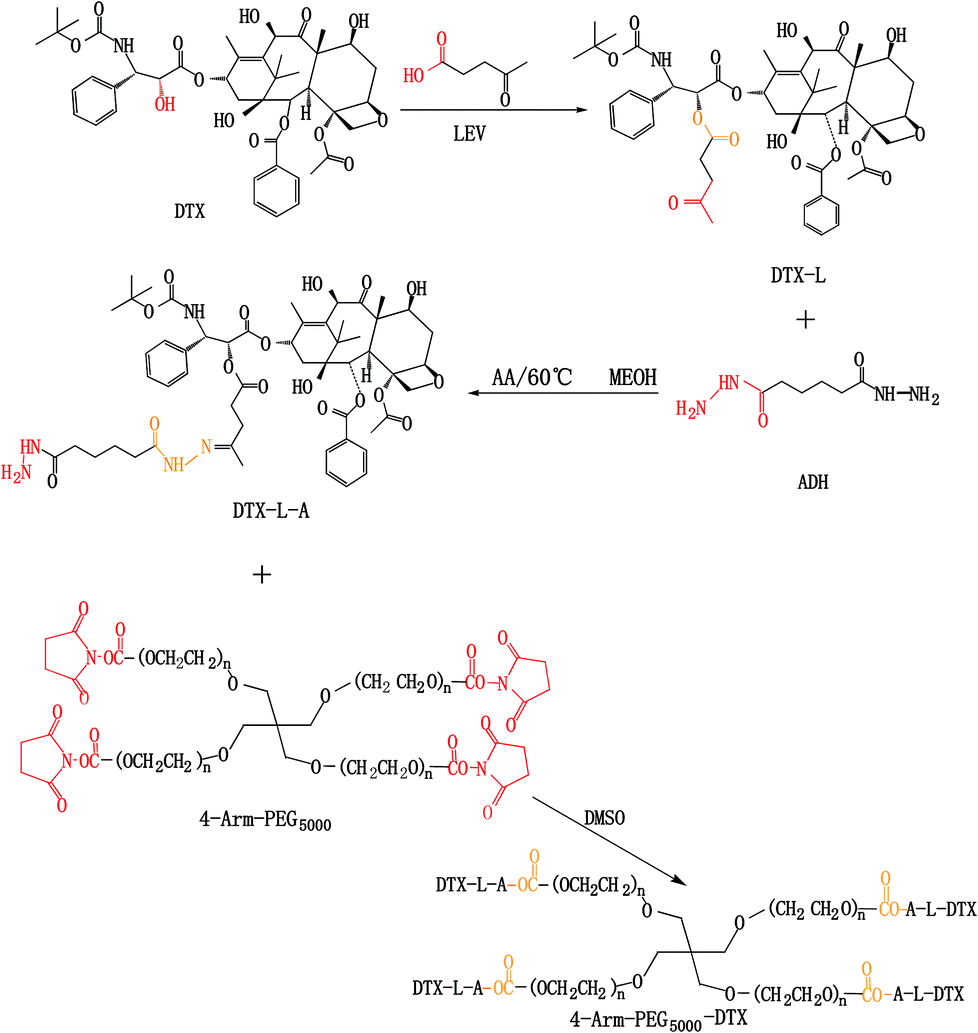
4-Arm-PEG-DTX was successfully synthesized as shown in Scheme 2. The chemical structures of the intermediates and product were confirmed by the 1H-NMR spectra shown in Fig. 1. The chemical structure of DTX-L was confirmed, with two multiplets corresponding to the two methylene groups O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH2–CH2–CO observed at 2.6 and 2.75 ppm in the 1H-NMR (Fig. 1B), like previous reported.19 DTA-L-A was evidenced by the presence of two proton peaks at 1.75 and 2.25 ppm derived from the methylenes of ADH (Fig. 1D). The –CH2–CH2– group in the 4-arm-PEG5000-DTX product occur at 3.5 ppm in the 1H-NMR (Fig. 1F).
C–CH2–CH2–CO observed at 2.6 and 2.75 ppm in the 1H-NMR (Fig. 1B), like previous reported.19 DTA-L-A was evidenced by the presence of two proton peaks at 1.75 and 2.25 ppm derived from the methylenes of ADH (Fig. 1D). The –CH2–CH2– group in the 4-arm-PEG5000-DTX product occur at 3.5 ppm in the 1H-NMR (Fig. 1F).
 | ||
| Scheme 2 Synthesis of 4-arm-PEG5000-DTX. | ||
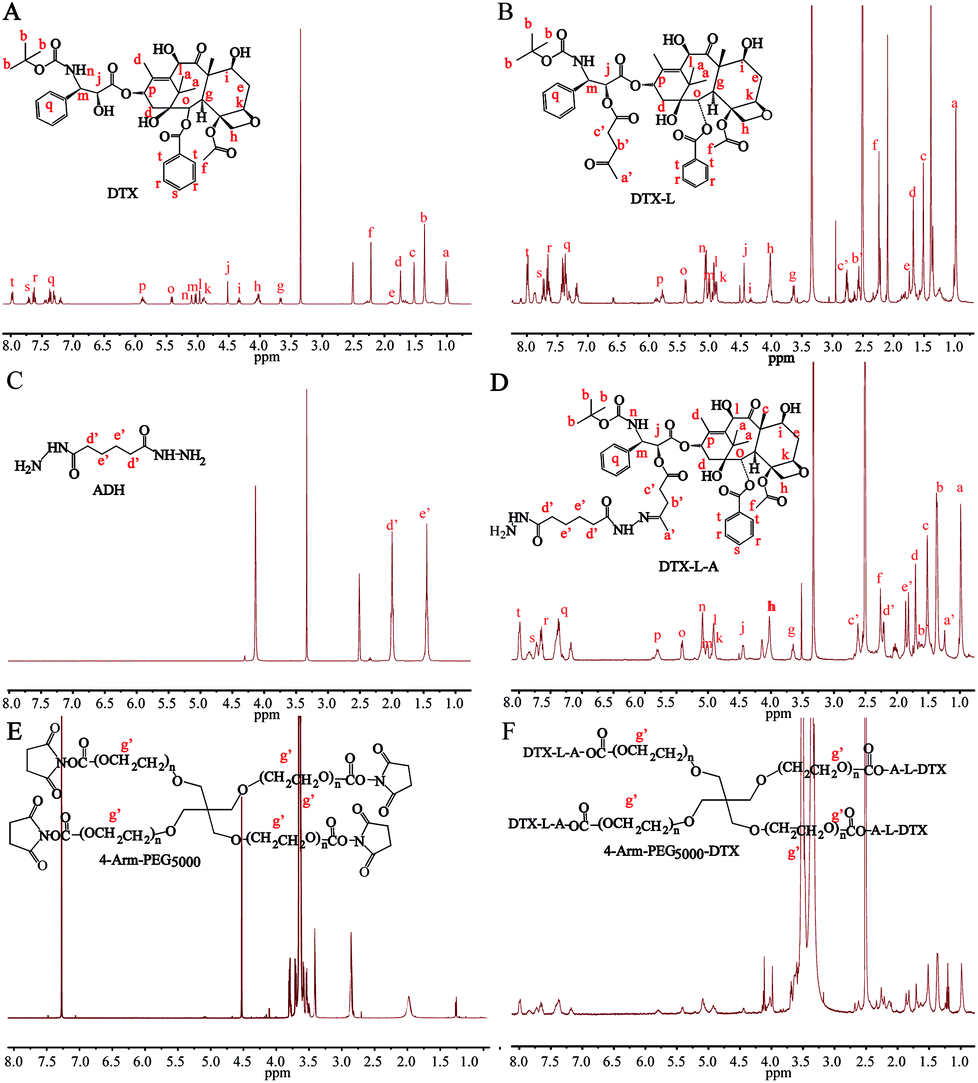 | ||
| Fig. 1 1H-NMR spectra of DTX-L, DTX-L-A and 4-arm-PEG5000-DTX. | ||
Characterization of D/D NPs
The particle size of the self-assembled DTX-NPs was 123.5 nm (PDI = 0.098) and that of D/D NPs, which encapsulated DHA, was 142.5 nm (PDI = 0.102) with the PDI values indicating good homogeneity (Fig. 2A and B). The zeta potential of DTX-NPs and D/D NPs was about −1.0 mV owing to the protective effect of PEG. From the TEM images of DTX-NPs and D/D NPs shown in Fig. 2C, the morphology of NPs was spherical and core–shell structure with a particle size smaller than the detection limit of the particle size analyzer. The drug loading and encapsulation efficiency of DHA were 7.5% ± 0.5% and 90.1 ± 4.8% (Table S1†). It is noteworthy that NPs smaller than 200 nm are associated with enhanced passive targeting by the EPR effect and enhanced drug accumulation at the tumor site. The D/D NPs remained stable after treatment with pH 7.4 PBS or contained 10% FBS PBS solution for 72 h where the particle size and PDI did not show any significant changes. However, the particle size and PDI increased at about 10 h after treatment with PBS at pH 5.0 and pH 6.2 (Fig. 2D). The D/D NPs were stable at pH 7.4 and serum-containing solution. These indicated the higher stability of D/D NPs at normal body environmental which could prolong circulation time.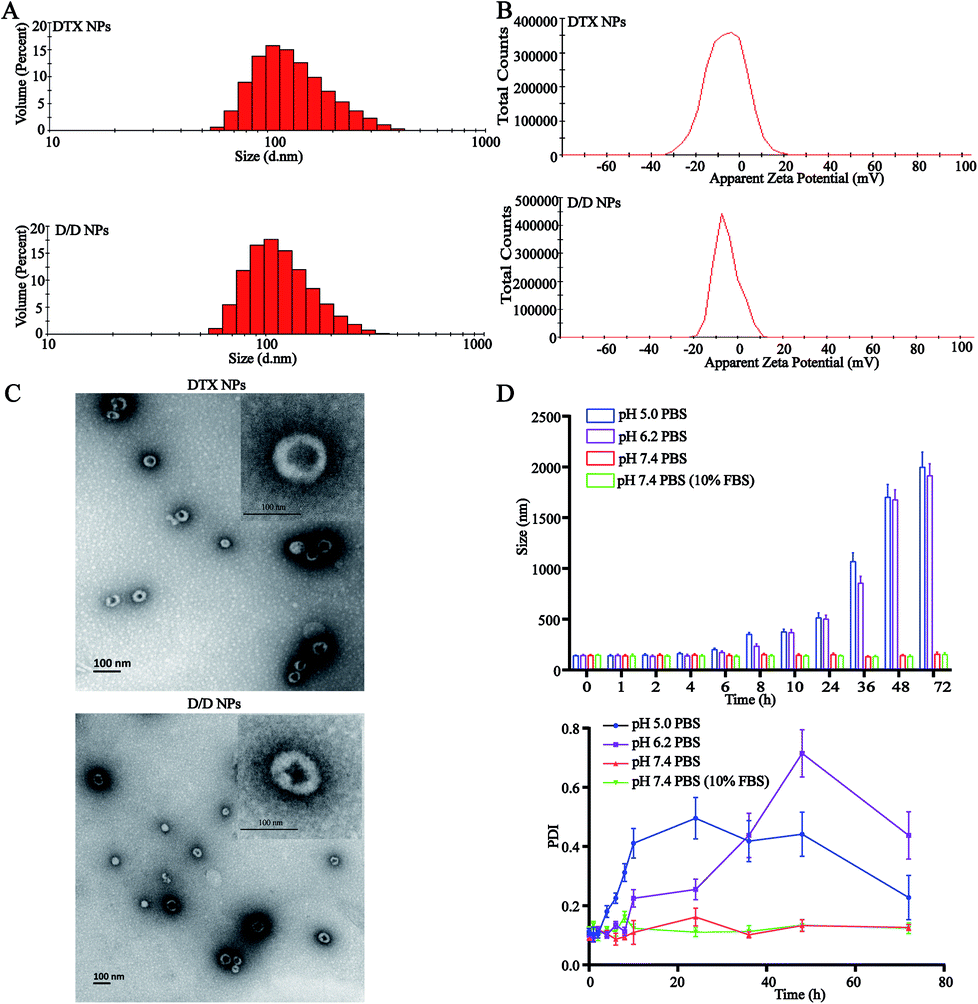 | ||
| Fig. 2 Characterization of the D/D NPs. (A) The size distribution of DTX-NPs and D/D NPs. (B) The zeta potential of DTX NPs and D/D NPs. (C) The TEM images of DTX-NPs and D/D NPs. (D) The stability of D/D-NPs in PBS buffers at different pH. | ||
Drug release from NPs
The in vitro release of DTX from the NPs in response to an acidic environment was further evaluated to confirm the pH sensitivity of the NPs. As shown in Fig. 3, the amount of DTX released from the DTX-NPs and D/D NPs in pH 7.4 PBS was about 35% over 72 h, with none detected at 10 h. These were consistent with the above stability results. The greater release of DTX from DTX-NPs and D/D NPs at pH 5.0 (about 75%) than at pH 6.2 confirmed the acid sensitivity of D/D NPs.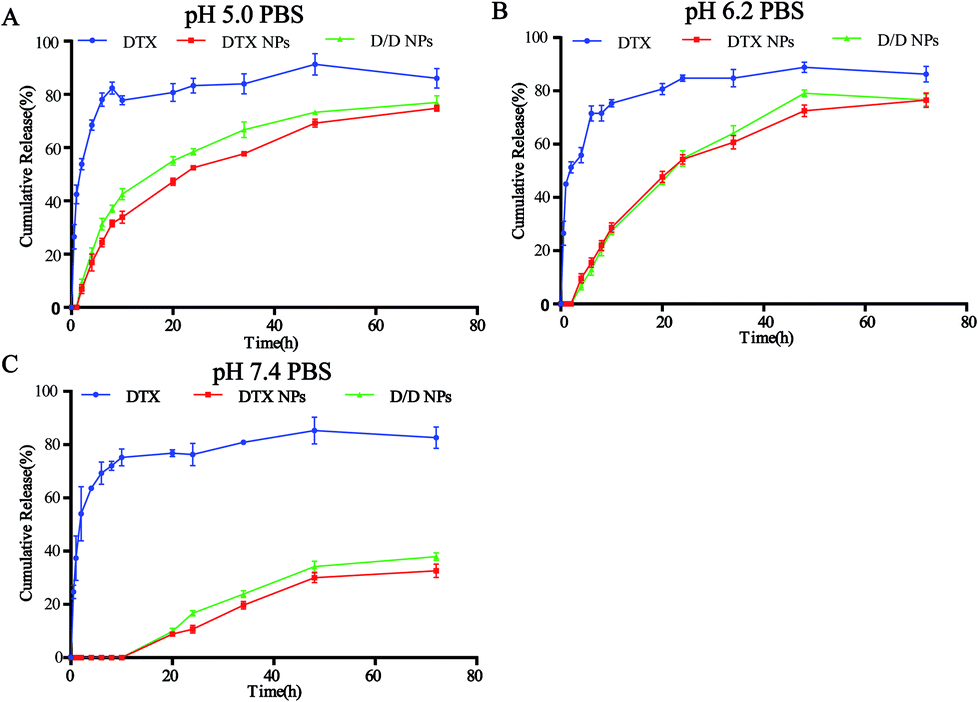 | ||
| Fig. 3 In vitro DTX release study of the NPs in PBS buffers at pH 5.0, 6.2 and 7.4. | ||
Cellular uptake of D/D NPs
The cellular uptake of D/D NPs was tracked using a fluorescence microscope. For the visualization of the loaded drugs, DHA was replaced by C6, which is commonly used as a hydrophobic dye with high fluorescence emission intensity, and C6 was encapsulated in DTX-NPs to form C6/DTX NPs. As shown in Fig. 4A, the green fluorescence of C6/DTX NPs was weaker than that of free C6 after treatment of 4T1 cells for 3 h. Also, the mean fluorescence intensity of C6/DTX NPs was lower than that of free C6 as measured by FCM, a result similar to measurement by a fluorescence microscope (Fig. 4B and C). This may be caused by the hydrophilic PEG on the surface of the NPs reducing cellular uptake.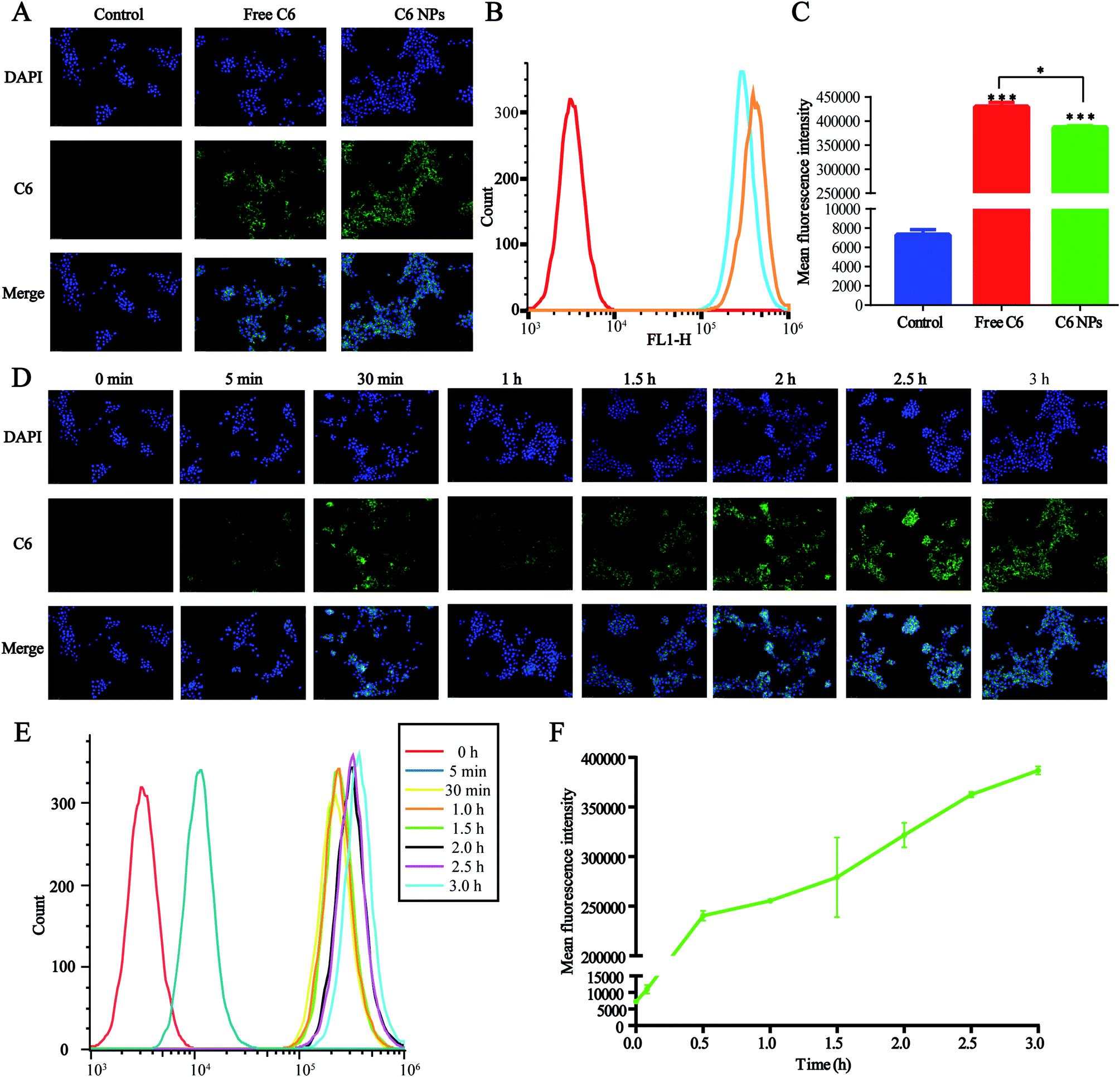 | ||
| Fig. 4 Cellular uptake of NPs in the 4T1 cells. (A) Fluorescence microscopic images of 4T1 cells after incubation for 3 h with free C6 and C6-NPs. C6 were green and the nuclei were stained with DAPI (blue). (B and C) Mean fluorescence intensity analysis by FCM. (D) Fluorescence microscopic images of the 4T1 cells for determining cellular uptake kinetics of C6-NPs at different time points. (E and F) Mean fluorescence intensity analysis of cellular uptake kinetics by FCM. | ||
We also studied the kinetics of cellular uptake of NPs. As shown in Fig. 4D–F, the green fluorescence and mean fluorescence intensity were time-dependent and after treatment of 4T1 cells with NPs for 3 h, cellular uptake and metabolism were comparable. The green fluorescence observed in the cytoplasm suggested that the NPs were accumulated in the cytoplasm.
In vitro cytotoxicity
Cell viability assays with free DTX, free D/D, DTX-NPs and D/D NPs were carried out using 4T1 cells with DTX concentrations of 0.001, 0.01, 0.1, 1, 10.0, and 100 μg mL−1 (Fig. 5A). Free DTX at below 10 μg mL−1 showed slight cytotoxic effects on 4T1 cells at 48 h after treatment, whereas free D/D showed a significant cytotoxic effect on 4T1 cells with the reduced cell viability. The co-encapsulation of DTX/DHA in D/D NPs saw a significant decrease in cell viability compared with that with free D/D and DTX NPs, by virtue of the ability of the NPs to accumulate in cells for longer time. The cytotoxic effects of different formulations were found in the following order: free DTX < free D/D < DTX-NPs < D/D NPs, with their respective IC50 values being 86.7, 56.7, 40.8 and 7.0 μg mL−1 after treatment for 48 h. These results suggested that a combination of DTX and DHA and co-encapsulation of DTX/DHA in D/D NPs induced synergistic cytotoxic effects on 4T1 cells.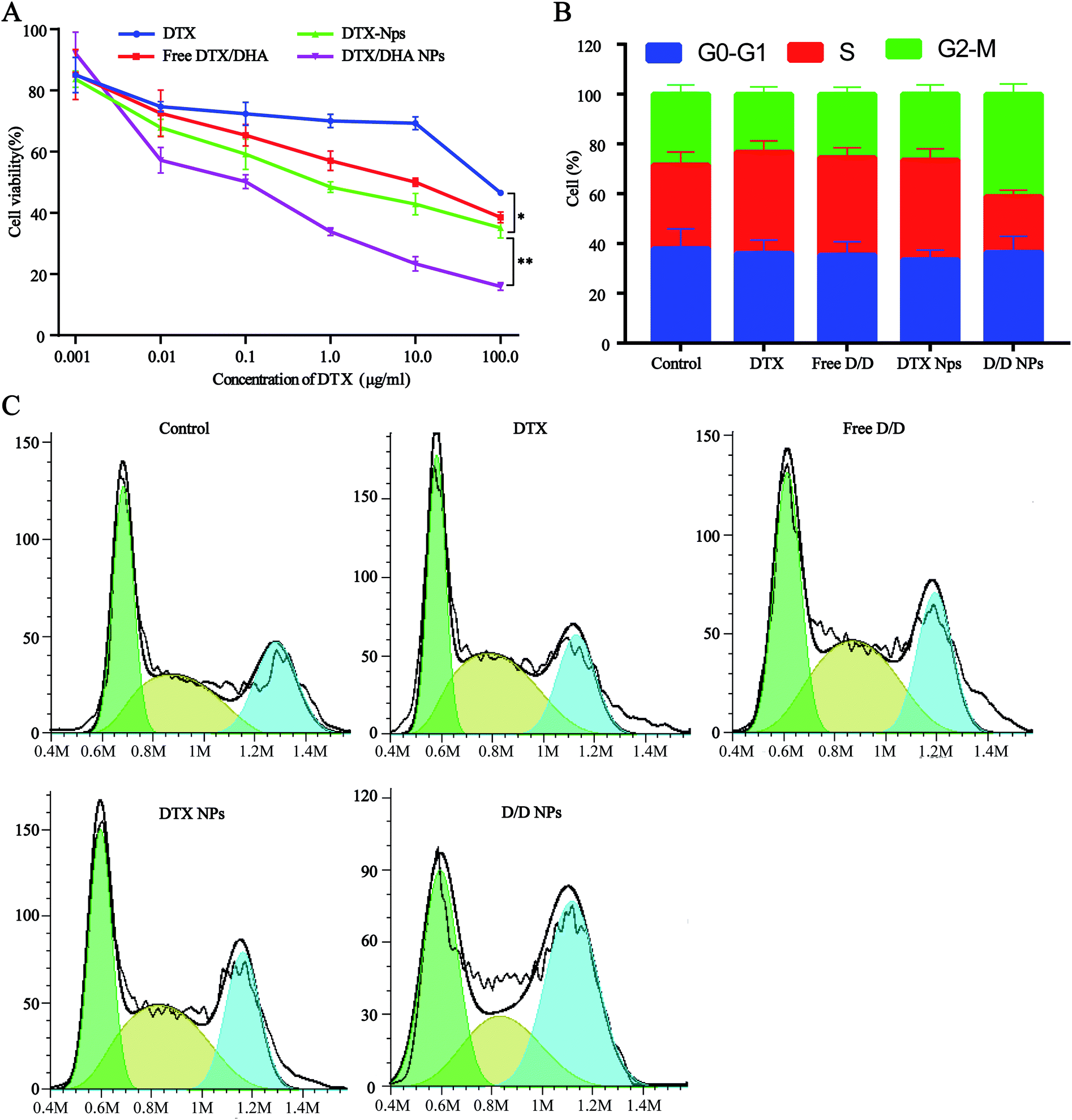 | ||
| Fig. 5 Antitumor activity assays of different formulations. (A) Cytotoxicity test in 4T1 cells. (B and C) Cell cycle rate. | ||
In vitro cell cycle study
The cell cycle study aimed to evaluate the mechanism of D/D NPs on the inhibition of cell growth, and whether it was related to cell cycle arrest. We used FCM to analyze the distribution of cells in the different phases. After treatment with DTX, free D/D and DTX-NPs at 50 ng mL−1, the percentage of cells in the S phase was 40.0%, 37.9% and 39.2% respectively, compared with 32.9% of in the control (Fig. 5B and C). Interestingly, the G2/M arrest induced by D/D NPs was significantly higher than that by other formulations, indicating that D/D NPs inhibited cell growth via G2/M arrest.In vitro migration and invasion studies
The inhibitory effect of D/D NPs on cell motility was determined by the wound healing assay. As shown in Fig. 6A and D, untreated cells exhibited strong migration healing ability with disappearance of the wound, indicating good metastasis of the 4T1 cells. D/D NPs showed a higher inhibitory effect on cell motility (28.4% wound confluence rate) than free DTX, free D/D and DTX NPs with wound confluence rates of 81.5%, 78.2% and 68.1% respectively.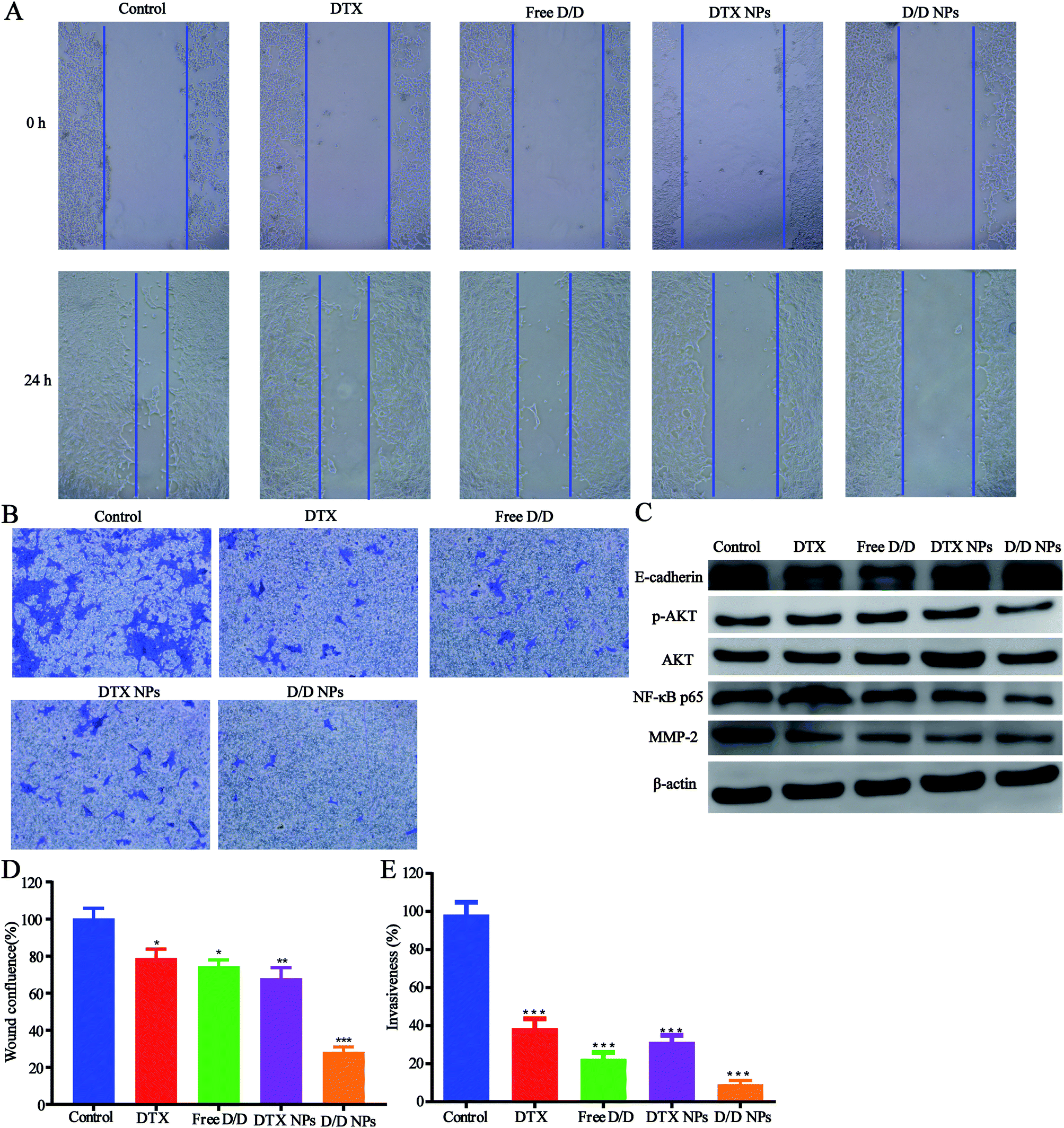 | ||
| Fig. 6 The wound healing, invasion assays and the mechanism of the anti- metastasis. (A) Images of the wound healing assays. (B) Microscopic images of the invasion assays. (C) Western blotting for determining protein expression after treatment with different formulations. (D) Quantitative analysis of the wound healing of 4T1 cells treated with different formulations for 24 h. (E) Quantitative analysis of the invasion assays of 4T1 cells with transwell. | ||
In addition, transwell assays were performed to detect the invasion of 4T1 cells after treatment with drugs. As shown in Fig. 6B and E, the invasion of 4T1 cells was significantly decreased after treatment with DTX and DHA. Moreover, D/D NPs showed higher inhibitory effects on cell invasion (9.2% invasiveness) than free DTX, free D/D and DTX NPs with invasiveness of 39.4%, 22.5% and 31.5%, respectively. These data suggested that D/D NPs could inhibit cell motility and invasion, whereas DTX and DHA induced synergistic effects on the inhibition of cell migration and invasion.
The mechanism of inhibition of 4T1 cell metastasis
We further studied the effects of D/D NPs on the expression of some proteins such as AKT, NF-κB, MMP-2 and E-cadherin, which play keys roles in the anti-metastasis pathways. As shown in Fig. 6C, the expression of E-cadherin was increased, and the expression of p-AKT, NF-κB p65 and MMP-2 was decreased. In addition, the expression of E-cadherin was higher in cells treated with D/D NPs, compared with those treated with free DTX, free D/D and DTX NPs. The expression of p-AKT, NF-κB p65 and MMP-2 was lower in cells treated with D/D NPs, compared with those treated with free DTX, free D/D and DTX NPs. These results indicated that a combination of DTX and DHA induced anti-metastasis via the NF-κB p65/MMP-2 pathways and the effect of D/D NPs was the strongest.Discussion
Breast cancer is the biggest killer that threatens women's health, and metastatic breast cancer is the most pernicious. Owing to the limited effectiveness of radiation and surgical therapies for metastatic tumors, chemotherapy remains the primary treatment. DTX, one of the taxane anticancer drugs widely used clinically, inhibits cell proliferation via cell cycle arrest.26 Its use in the treatment of metastatic breast cancer is still weakly effective, necessitating the use of a combination of drugs. In clinical practice in China, traditional Chinese medicine has been used to complement chemotherapeutic drugs in the clinical treatment of cancers.27 This implies that the active ingredients present in the Chinese medicine exert synergistic effects with the chemotherapeutic drugs used in standard cancer therapy.Artemisinin are excellent drugs in treating malaria, and they also exhibit antitumor activity.28 DHA is an important derivative of artemisinin and is the main metabolite of artemisinin drugs in vivo. DHA presents excellent antitumor activity through cell cycle arrest and induction of apoptosis.29 The Huang study suggested that the combination of DHA and doxorubicin could enhance the sensitivity of HCT8/ADR drug resistant cells to doxorubicin and showed synergistic effect against antitumor multidrug resistance.25 In line with these results, our study also supported that the combination of DHA and DTX showed synergistic effects on the inhibition of cell proliferation and cell cycle arrest.
DTX and DHA are poorly soluble anticancer drugs, and tend to accumulate in various body tissues for non-specific targets, causing serious adverse reactions and limiting their clinical applications.30 The development of new technologies in drug delivery has allowed the improvement of solubility and reduction of side effects of anticancer drugs.30,31 One kind of nanoscale delivery system is composed of a hydrophobic unit on one side and a hydrophilic unit on the other.32 Thus, hydrophilic polymers were linked with hydrophobic drugs as amphiphilic polymers and this material was used to form nanoparticles. Liang33 have used Pluronic P123 conjugated with DTX to prepare micelles, which represent useful vehicles for drug delivery. However, the drug loading efficiency of DTX in Pluronic P123-DTX was only 11.2%. Considering that nanoparticles with high drug loading are expected to be efficient drug delivery vehicles, we used PEG with 4-arm to link DTX, affording high drug loading efficiency of about 40%, and further used this material to prepare D/D NPs which encapsulated DHA.
With most nanoscale delivery systems, passive tumor targeting by the EPR effect means that nanovectors smaller than 200 nm allow drugs to accumulate at the tumor site and not in the normal tissues.34 To avoid uptake of the nanovectors by macrophages, the surfaces of the nanovectors are always modified with hydrophilic molecules like PEG and polysialic acid.35 We conjugated 4-arm-PEG with DTX to prepare the nanoparticles, which achieved long circulation time in the blood.
A large number of nanoscale delivery systems that respond to different characteristics of the tumor microenvironment, such as acid, enzymes and redox potential, have been widely studied in cancer treatments.36,37 For instance, doxorubicin has been designed to form stimuli-responsive nanoplatform using mesoporous silica nanomaterial with benzoic–imine bond, which upon cleavage in the acidic tumor environment, release DOX and other drug, which express better antitumor therapeutic efficiency in vitro and in vivo.38,39 Here, we successfully synthesized pH-responsive materials 4-arm-PEG-DTX with hydrazone bonds (Fig. 1) and used it to encapsulate DHA to prepare pH-sensitive D/D NPs (Fig. 2). The NPs released DTX at pH 6.2 and 5.0, and with about 30% of DTX released at pH 7.4 at 72 h (Fig. 3).
The treatment of metastatic breast cancer relies not only on the inhibition of tumor growth, but also the inhibition of metastasis. We studied the antitumor activity of D/D NPs in the inhibition of 4T1 cell proliferation, migration and invasion. Thus, D/D NPs are expected to effectively treat metastatic breast cancer, with evidence supporting the synergistic effects of the combination of DTX and DHA in treating metastatic breast cancer. In our study of the mechanism of D/D NPs in anti-metastasis, we identified from previous research that DTX regulates the expression of NF-κB to inhibit cancer cell proliferation,40 while DHA could also influence the expression of NF-κB, via the AKT/NF-κB or NF-κB/GLUT1 pathway.41 Our data suggested that the combination of DTX and DHA decreased the expression of NF-κB more than free DTX did. The D/D NPs were found to reduce the expression of MMP-2, which reduce cytoplasmic matrix and basement membrane to promote tumor metastasis, and the expression of p-AKT were also reduced which occur upstream of NF-κB. Therefore, our data suggested that the D/D NPs could reduce metastasis of breast cancer by acting on the AKT/NF-κB/MMP-2 pathway.
Experimental
Materials and cell lines
DTX, DHA were purchased from Meilun (Danian, China); 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma (Shanghai, China); 4-arm-PEG-SCM5000 was purchased from Xuanguang (Ningbo, China).The murine mammary carcinoma cell lines 4T1 were purchased from Cell Bank of Shanghai, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 μg mL−1 streptomycin, 100 U mL−1 penicillin, 2.5 g L−1 glucose and 0.11 g L−1 sodium pyruvate at 37 °C in a humidified atmosphere containing 5% CO2.
Synthesis of 4-arm-PEG-DTX
The derivative of DTX with levulinic acid (DTX-L) was synthesized from previously reported procedures. Briefly, DTX, lev, EDC and DMAP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:1.5:0.5, mole ratio) were dissolved in dichloromethane, and the reaction mixture was stirred at 4 °C for 12 h. Dichloromethane was then evaporated and the crude product was dissolved in ethyl acetate. The solution was washed with 1% HCl, saturated NaHCO3 and saturated NaCl solutions sequentially. The crude product was purified by preparative liquid chromatography.
:1.5:1.5:0.5, mole ratio) were dissolved in dichloromethane, and the reaction mixture was stirred at 4 °C for 12 h. Dichloromethane was then evaporated and the crude product was dissolved in ethyl acetate. The solution was washed with 1% HCl, saturated NaHCO3 and saturated NaCl solutions sequentially. The crude product was purified by preparative liquid chromatography.
The synthesis of the hydrazone compound DTX-L-A was carried out by reacting DTX-L with adipic dihydrazide (ADH). Briefly, DTX-L and ADH (1:2, mole ratio) were dissolved in methanol, acetic acid was added and the mixture was stirred at 60 °C for 3 h. After the reaction mixture was filtered and the residue was washed with water to remove excess ADH, the crude product was purified by preparative liquid chromatography.
Finally, 4-arm-PEG5000-SCM and DTX-L-A were conjugated using carboxyl-to-amine crosslinking. Briefly, 4-arm-PEG5000-SCM and DTX-L-A (1:4, mole ratio) were dissolved in methanol and stirred at room temperature for 2 h. The 4-arm-PEG-DTX product was precipitated by adding excess cold ether.
Preparation and characterization of D/D NPs
The nanoparticles D/D NPs were prepared by the dialysis method. 4-Arm-PEG-DTX and DHA at a weight ratio of 12:1 were dissolved in DMSO, dialyzed in a dialysis bag (MWCO 10 KDa) with the pH 7.40 PBS changed every 2 h for 24 h followed by filtration through a 0.22 μm filter membrane to remove free DHA. The size and zeta potential of nanoparticles were measured by a Zetasizer Nanoparticle Analyzer (Malvern, UK). The morphology of nanoparticles was captured by a transmission electron microscope.
The DHA concentration was measured using a HPLC system. The absorbance was measured at 210 nm. The drug loading and encapsulation efficiency of DHA were calculated by the equations:
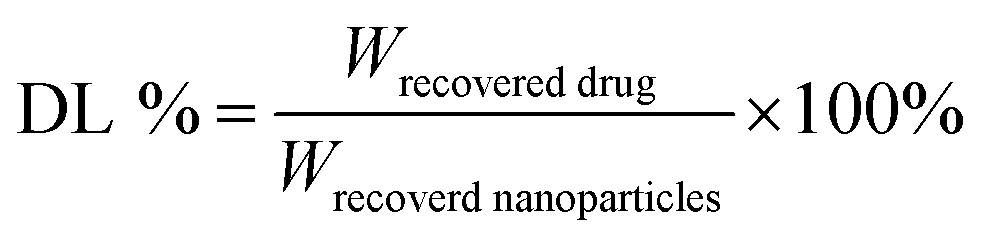
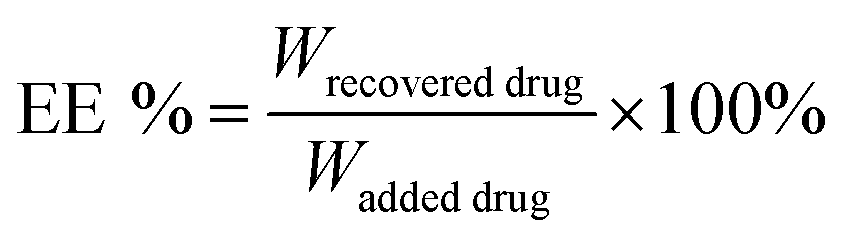
The stability of D/D NPs in 0.01 M PBS at pH 7.4, 6.3 and 5.0 was investigated. Then, the size of nanoparticles was measured at different time points.
In vitro drug release study
A dialysis method was used to study the drug release of nanoparticles in vitro. The free drug, DTX-NPs and D/D NPs were sealed in a dialysis bag and dialyzed in PBS containing 0.5% tween (pH 7.4, 6.2 and 5.0) at 37 °C under continuously shaking at 100 rpm. At the predetermined time points, the release media were removed and replenished with new release media. Drug concentrations were measured by HPLC at different time points.Cellular uptake
The rhodamine-6 (C6) loaded D/D NPs were prepared. 4T1 cells were seeded into 24-well plates at a density of 2 × 104 cells per well and cultured overnight. The cells were treated with rhodamine-6 and rhodamine-6 loaded D/D NPs for 15 min, 30 min, 1 h, 2 h, 3 h and 4 h, followed by formalin-fixation and nuclei staining with DAPI for 5 min at room temperature. Then the cells were washed three times with PBS and subjected to fluorescence microscopy. Cellular fluorescence was also quantified using a flow cytometer.In vitro cytotoxicity assay
The MTT assay was used to evaluate the cytotoxicity on 4T1 cells. 4T1 cells were seeded into 96-well plates at a density of 5 × 103 cells per well and cultured overnight. The cells were incubated with different samples of free DTX, free DTX/DHA (D/D), DTX-NPs and D/D NPs at various concentrations for 48 h. They were then incubated with MTT for 4 h and the medium was removed, 150 μL of DMSO was added to dissolve the crystals. Absorbance of the samples was measured at 490 nm with a microplate reader (Multiskan, Thermo, USA). Cell viability was calculated according to the equation: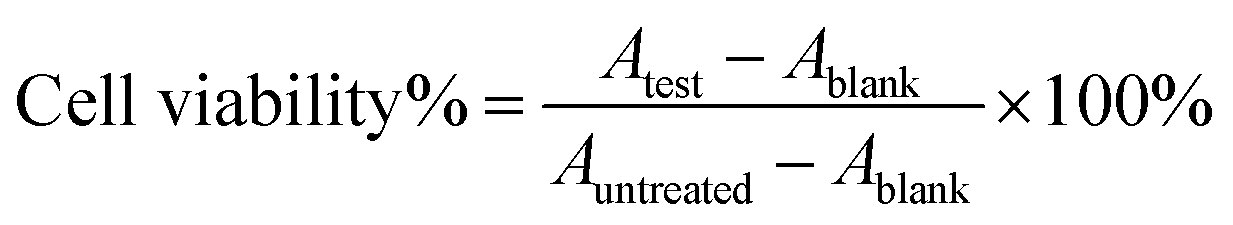
In vitro cell cycle assay
4T1 cells were seeded into 12-well plates at a density of 5 × 104 cells per well and cultured overnight. The cells were treated with free DTX, free D/D, DTX-NPs and D/D NPs at a concentration of 50 ng mL−1 of DTX. After 48 h of incubation, the cells were harvested, washed and stained with propidium iodide (PI) according to the manufacturer's instructions. The evaluation of cell cycle was performed using flow cytometry.Wound healing and invasion assay
4T1 cells were seeded into 12-well plates and cultured overnight at 90% confluency. Two hundred pipette microlitre tips were used to scratch the cells to generate the wound. The cells were then washed with PBS and incubated with free DTX, free D/D, DTX-NPs and D/D NPs at a concentration of 50 ng mL−1 of DTX. Images of wound healing were obtained at 0 h and 24 h using a microscope (DMi1, Leica, UK).The transwells were coated with Matrigel and incubated at 37 °C for 3 h. Media without FBS contained 105 number of cells were added into the upper chamber with 8 μm pore filters and 600 μL of normal medium was added into the lower chamber. After incubation for 24 h, Matrigel was removed and cells in the lower chamber were stained with 0.5% crystal violet solution for 15 min. Images of the stained cells were obtained using a microscope (DMi8, Leica, UK) and the cells were counted to calculate the migration rate.
Western blotting
4T1 cells were seeded into 12-well plates and cultured overnight, the cells were treated with free DTX, free D/D, DTX-NPs and D/D NPs at a concentration of 50 ng mL−1 of DTX. Twenty-four hours after drug treatment, the total protein was extracted from the cells and quantified by BCA analysis. We performed western blotting to examine the expression of AKT, NF-κB p65 and MMP-2 proteins.Statistical analysis
All experiments were performed in a minimum of triplicates and the data were shown as mean ± SD. Statistical analysis was conducted using the GraphPad Prism software. The significance was evaluated using Student's t-test and P < 0.05 was considered statistically significant.Conclusions
We successfully developed pH-sensitive nanoparticles based on 4-arm-PEG to co-deliver DTX and DHA to the tumor environment. The combination of DTX and DHA, as well as the rapid, pH-triggered drug release have proved to inhibit the growth and metastasis of breast cancer cells. The D/D NPs downregulated the expression of NF-κB and MMP-2, which was associated with anti-metastasis. These results highlight that D/D NPs are potentially effective nanoscale delivery vehicles which co-deliver DTX and DHA for treating metastatic breast cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (81773673 and 21502172), Science and Technology Innovation Team Project of Ningbo Science and Technology Bureau, China (No. 2015C110027), Key Laboratory of Ningbo, China (No. 2016A22002), Foundation of Zhejiang educational committee (No. Y201738467) and Scientific Research Project of Zhejiang Pharmaceutical College (No. ZPCSR2017004).Notes and references
- S. Tang, Q. Meng, H. Sun, J. Su, Q. Yin, Z. Zhang, H. Yu, L. Chen, Y. Chen and W. Gu, Adv. Funct. Mater., 2016, 26, 6033–6046 CrossRef.
- C. Fontanella, V. Fanotto, K. Rihawi, G. Aprile and F. Puglisi, Clin. Exp. Metastasis, 2015, 32, 819–833 CrossRef PubMed.
- V. Angeloni, N. Contessi, M. C. De, S. Bertoldi, M. C. Tanzi, M. G. Daidone and S. Farè, Acta Biomater., 2017, 63, 306–316 CrossRef PubMed.
- A. Ring, M. Reed, R. Leonard, I. Kunkler, H. Muss, H. Wildiers, L. Fallowfield, A. Jones and R. Coleman, Br. J. Cancer, 2011, 105, 189–193 CrossRef PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef PubMed.
- N. M. Gerhards and S. Rottenberg, Drug Resist. Updates, 2018, 36, 30–46 CrossRef PubMed.
- X. Wan, Y. Min, H. Bludau, A. Keith, S. S. Sheiko, R. Jordan, A. Z. Wang, M. Sokolsky-Papkov and A. V. Kabanov, ACS Nano, 2018, 12, 2426–2439 CrossRef PubMed.
- S. Shen, J. Xia and J. Wang, Biomaterials, 2016, 74, 1–18 CrossRef PubMed.
- Y. Liu, J. Peng, S. Wang, M. Xu, M. Gao, T. Xia, J. Weng, A. Xu and S. Liu, NPG Asia Mater., 2018, 10, e458 CrossRef.
- Y. Su, P. Burnouf, K. Chuang, B. Chen, T. Cheng and S. R. Roffler, Nat. Commun., 2017, 8, 15507 CrossRef PubMed.
- W. Tao, X. Zhu, X. Yu, X. Zeng, Q. Xiao, X. Zhang, X. Ji, X. Wang, J. Shi, H. Zhang and L. Mei, Adv. Mater., 2017, 29, 1603276 CrossRef PubMed.
- J. Sun, L. Jiang, Y. Lin, E. M. Gerhard, X. Jiang, L. Li, J. Yang and Z. Gu, Int. J. Nanomed., 2017, 12, 1517–1537 CrossRef PubMed.
- X. Chen, L. Liu and C. Jiang, Acta Pharm. Sin. B, 2016, 6, 261–267 CrossRef PubMed.
- K. Zhang, P. Yang, J. Zhang, L. Wang and H. Wang, Chin. Chem. Lett., 2017, 28, 1808–1816 CrossRef.
- C. Xu, P. Wang, J. Zhang, H. Tian, K. Park and X. Chen, Small, 2015, 11, 4321–4333 CrossRef PubMed.
- N. Li, H. Cai, L. Jiang, J. Hu, A. Bains, J. Hu, Q. Gong, K. Luo and Z. Gu, ACS Appl. Mater. Interfaces, 2017, 9, 6865–6877 Search PubMed.
- C. T. Nguyen, T. Huyen Tran, M. Amiji, X. Lu and R. Kasi, Nanomedicine, 2015, 11, 2071–2082 CrossRef PubMed.
- T. H. Tran, T. Ramasamy, J. Yeon Choi, H. T. Nguyen, T. Pham Thanh, J.-H. Jeong, K. Sae, H.-G. Choi, C. Soon and J. Kim, Asian J. Pharm. Sci., 2015, 10, 5249–5262 Search PubMed.
- Q. Yang, L. Li, W. Sun, Z. Zhou and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 13251–13261 Search PubMed.
- M. Martin, T. Pienkowski, J. Mackey, M. Pawlicki, J.-P. Guastalla, C. Weaver, E. Tomiak, T. Al-Tweigeri, L. Chap, E. Juhos, R. Guevin, A. Howell, T. Fornander, J. Hainsworth, R. Coleman, J. Vinholes, M. Modiano, T. Pinter, S. C. Tang and C. Vogel, N. Engl. J. Med., 2005, 352, 2302–2313 CrossRef PubMed.
- L. Tiainen, M. Tanner, O. Lahdenperä, P. Vihinen, A. Jukkola, P. Karihtala, N. Paunu, T. Huttunen and P. L. Kellokumpu-Lehtinen, Anticancer Res., 2016, 36, 6431 CrossRef PubMed.
- S. M. Swain, J. Baselga, S. B. Kim, J. Ro, V. Semiglazov, M. Campone, E. Ciruelos, J. M. Ferrero, A. Schneeweiss and S. Heeson, N. Engl. J. Med., 2015, 372, 724–734 CrossRef PubMed.
- Y. Qin, G. Yang, M. Li, H. Liu, W. Zhong, X. Yan, K. Qiao, J. Yang, D. Zhai and W. Yang, Oncotarget, 2017, 8, 103815 Search PubMed.
- J. Zhao, Y. Pan, X. Li, X. Zhang, Y. Xue, T. Wang, S. Zhao and Y. Hou, Cell. Physiol. Biochem., 2017, 43, 589–601 CrossRef PubMed.
- X. Kang, H. Wang, H. Peng, B. Chen, W. Zhang, A. Wu, Q. Xu and Y. Huang, Acta Pharmacol. Sin., 2017, 38, 885–896 CrossRef PubMed.
- J. Tao, J. Xu, F. Chen, B. Xu, J. Gao and Y. Hu, Eur. J. Pharm. Sci., 2017, 111, 540–548 CrossRef PubMed.
- F. Qi, L. Zhao, A. Zhou, B. Zhang, A. Li, Z. Wang and J. Han, BioSci. Trends, 2015, 9, 16–34 CrossRef PubMed.
- S. Roy, R. He, A. Kapoor, M. Forman, J. R. Mazzone, G. H. Posner and R. Arav-Boger, Antimicrob. Agents Chemother., 2015, 59, 3870–3879 CrossRef PubMed.
- C. Hu, L. Zhou and Y. Cai, Cancer Biol. Ther., 2014, 15, 279–288 CrossRef PubMed.
- A. A. Date, J. Hanes and L. M. Ensign, J. Controlled Release, 2016, 240, 504–526 CrossRef PubMed.
- T. A. Ahmed and B. M. Aljaeid, Drug Des., Dev. Ther., 2016, 10, 483–507 CrossRef PubMed.
- H. Wang, P. Agarwal, S. Zhao, R. X. Xu, J. Yu, X. Lu and X. He, Biomaterials, 2015, 72, 74–89 CrossRef PubMed.
- Y. Liang, Z. Su, Y. Yao and N. Zhang, Materials, 2015, 8, 379–391 CrossRef PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef PubMed.
- P. R. Wardwell, M. B. Forstner and R. A. Bader, Arthritis Res. Ther., 2015, 17, 1–11 Search PubMed.
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538–558 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- X. Zeng, G. Liu, W. Tao, Y. Ma, X. Zhang, F. He, J. Pan, L. Mei and G. Pan, Adv. Funct. Mater., 2017, 27, 1605985 CrossRef.
- W. Cheng, J. Nie, N. Gao, G. Liu, W. Tao, X. Xiao, L. Jiang, Z. Liu, X. Zeng and L. Mei, Adv. Funct. Mater., 2017, 27, 1704135 CrossRef.
- E. Kim, M. Matsuse, V. Saenko, K. Suzuki, A. Ohtsuru, N. Mitsutake and S. Yamashita, Thyroid, 2012, 22, 717–724 CrossRef PubMed.
- J. Jiang, G. Geng, X. Yu, H. Liu, J. Gao, H. An, C. Cai, N. Li, D. Shen, X. Wu, L. Zheng, Y. Mi and S. Yang, Oncotarget, 2016, 7, 87271–87283 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: 10.1039/c8ra02833h |
| This journal is © The Royal Society of Chemistry 2018 |