
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative functionalization of aliphatic and aromatic amino acid derivatives with H2O2 catalyzed by a nonheme imine based iron complex†
Barbara Ticconia,
Arianna Colcerasaa,
Stefano Di Stefano a,
Osvaldo Lanzalunga*a,
Andrea Lapia,
Marco Mazzonnaa and
Giorgio Olivob
a,
Osvaldo Lanzalunga*a,
Andrea Lapia,
Marco Mazzonnaa and
Giorgio Olivob
aDipartimento di Chimica, Università degli Studi di Roma “La Sapienza” and Istituto CNR di Metodologie Chimiche (IMC-CNR), Sezione Meccanismi di Reazione, c/o Dipartimento di Chimica, Università degli Studi di Roma “La Sapienza”, P.le A. Moro 5, I-00185 Rome, Italy. E-mail: osvaldo.lanzalunga@uniroma1.it
bInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Spain
First published on 23rd May 2018
Abstract
The oxidation of a series of N-acetyl amino acid methyl esters with H2O2 catalyzed by a very simple iminopyridine iron(II) complex 1 easily obtainable in situ by self-assembly of 2-picolylaldehyde, 2-picolylamine, and Fe(OTf)2 was investigated. Oxidation of protected aliphatic amino acids occurs at the α-C–H bond exclusively (N-AcAlaOMe) or in competition with the side-chain functionalization (N-AcValOMe and N-AcLeuOMe). N-AcProOMe is smoothly and cleanly oxidized with high regioselectivity affording exclusively C-5 oxidation products. Remarkably, complex 1 is also able to catalyze the oxidation of the aromatic N-AcPheOMe. A marked preference for the aromatic ring hydroxylation over Cα–H and benzylic C–H oxidation was observed, leading to the clean formation of tyrosine and its phenolic isomers.
Introduction
Oxidative functionalization of amino acids has received a great deal of attention in recent years. This interest has its roots in the great significance for human health of the reactions of amino acids and peptides with reactive oxygen species (ROS), which are implicated in various disease states and aging processes.1 Reactions of ROS with amino acids are also important in structural analysis and protein targeting.2 Finally, oxidations of amino acid derivatives and oxidative postsynthetic modifications of peptides have found interesting applications in the production of useful synthetic intermediates for the preparation of medicinal agents3 and chiral auxiliaries in asymmetric reactions.4Product analysis of the oxidation of amino acids and peptides have been carried out by several research groups using both metal based or organic oxidants.5 Such studies clearly showed that these reactions may lead to oxidation of amino acid side chains, backbone cleavage, cross-linking, unfolding and loss of enzymatic activity.
A growing interest has been recently devoted to sustainable oxidations catalyzed by nonheme iron complexes and environmentally friendly H2O2 as the terminal oxidant. These species can be considered simple models of iron oxygenases, and were found to efficiently catalyze aliphatic C–H hydroxylation with high selectivity.6 Thus, they represent good candidates as catalysts for the side-chain functionalization of amino acid residues in proteins. A significant advantage lies in the possibility of finely tuning their oxidation by varying the nature of the ligand. For example, the two aminopyridine nonheme Fe(PDP) and Fe(CF3PDP) complexes (PDP = [N,N′-bis(2-pyridylmethyl)]-2,2′-bipyrrolidine) catalyze the oxidative modification of aliphatic amino acids proline, leucine, valine, and norvaline triggered by C–H oxidation with preservation of the stereocenters.3e,7
Along with aminopyridine iron catalysts, an interesting yet underdeveloped class of nonheme models is represented by imine iron complexes where the central iron metal is coordinated to an imine based ligand.8 These catalysts have received a minor attention with respect to their amine analogues since they are considered less robust and stable under the oxidizing conditions employed, yet some of these complexes proved particularly efficient as oxidation catalysts.
For example, we found that the nonheme iron complex 1, easily and quantitatively obtained by spontaneous self-assembly of cheap and commercially available 2-picolylaldehyde, 2-picolylamine, and Fe(OTf)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio (Fig. 1) is able to catalyze aliphatic C–H hydroxylation with H2O2 9,10 with yields and turnover numbers comparable to those reported for far more sophisticated nonheme aminopyridine iron-complexes. Remarkably, we found clear evidence that no free radical processes are involved in the C–H hydroxylation process and that a metal based oxidant is responsible for the C–H functionalization.10
:2:1 ratio (Fig. 1) is able to catalyze aliphatic C–H hydroxylation with H2O2 9,10 with yields and turnover numbers comparable to those reported for far more sophisticated nonheme aminopyridine iron-complexes. Remarkably, we found clear evidence that no free radical processes are involved in the C–H hydroxylation process and that a metal based oxidant is responsible for the C–H functionalization.10
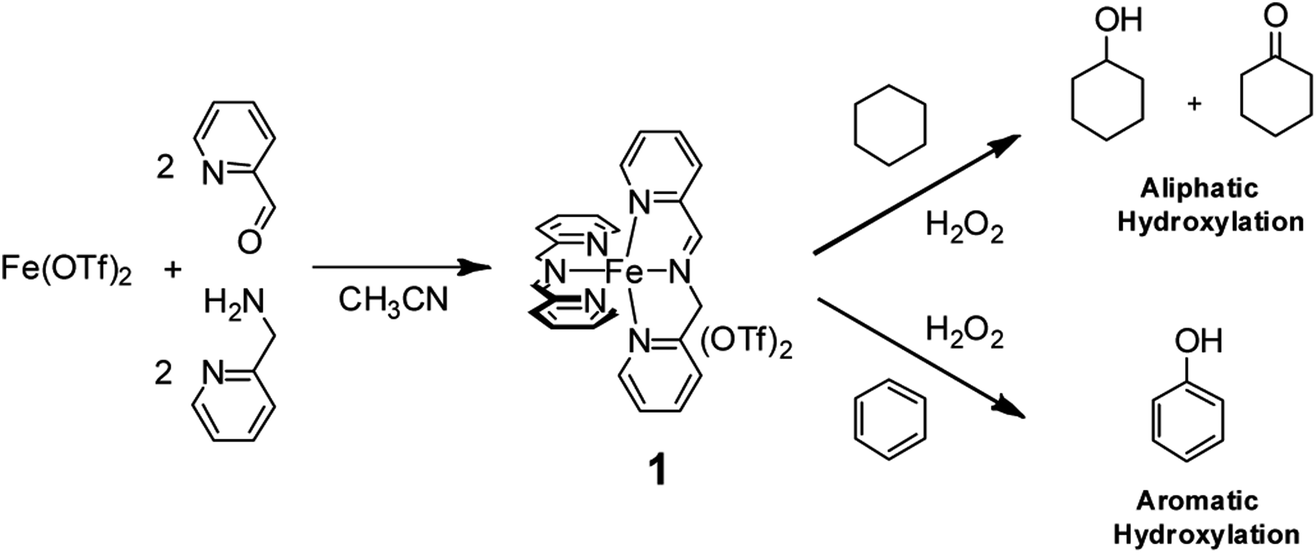 | ||
| Fig. 1 Oxidation of aliphatic and aromatic hydrocarbons with H2O2 promoted by the nonheme imine-based iron complex 1. | ||
Moreover, we recently found that the imine complex 1 also catalyzes the hydroxylation of aromatic rings with H2O2 under mild conditions with good efficiency.11 It has to be remarked that in the oxidation of alkylaromatics and benzylic alcohols, the 1/H2O2 system has a striking preference for aromatic hydroxylation over side-chain oxidation.11,12 The above results obtained in the oxidation of aliphatic and aromatic hydrocarbons catalyzed by imine complex 1 prompted us to investigate the oxidation of α-amino acids containing both aliphatic and aromatic side-chains in order to obtain information on the reactivity and selectivity patterns in these oxidative processes. The results are reported herewith.
Results and discussion
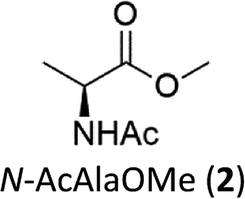
A series of methyl esters of N-acetylated amino acids (2–8, Fig. 2) were prepared to model individual residues within a polypeptide chain. Both the carboxylic acid and the amino function have been protected in order to avoid their coordination to the iron ion, which could affect the reactivity of the oxidizing species.6k,7 Substrates 2–8 were synthesized from the corresponding L-amino acids according to a literature procedure13 (see Experimental section).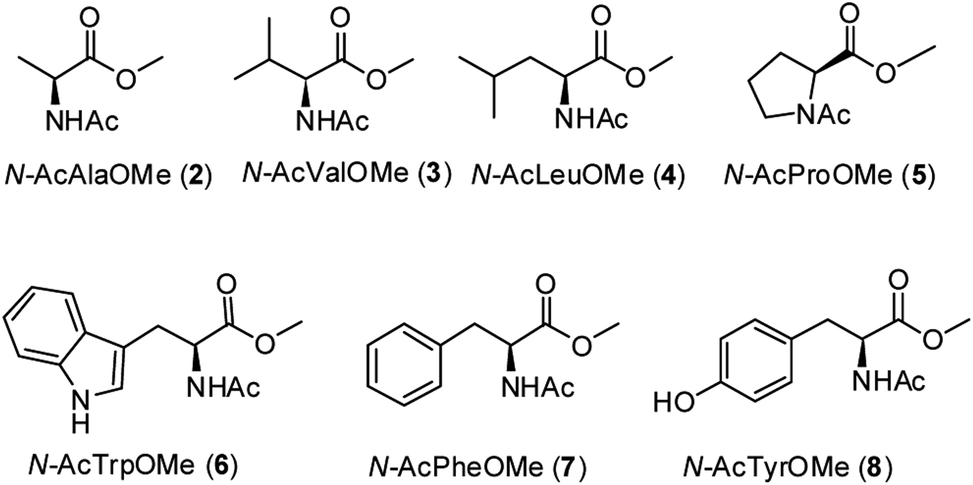 | ||
| Fig. 2 N-acetyl amino acids methyl esters (2–8) investigated in this work. | ||
Oxidations with the 1/H2O2 system were carried out in acetonitrile in the conditions previously adopted for aliphatic and aromatic substrates9–11 at 25 °C for 30–90 min in the presence of 1 mol% catalyst and 2.5 molar equivalents of H2O2 slowly added over 30 minutes through a syringe pump. The results are shown in Table 1. Reaction products were identified by GC-MS and 1H NMR analyses (see ESI†). In all cases the mass balance was satisfactory (>80%).
| Substrate | Unreacted substrateb | Productsb (Yields%) | ||
|---|---|---|---|---|
| a Reaction conditions: 250 mol% H2O2, 1 mol% catalyst 1, CH3CN at 25 °C, reaction time 30–90 minutes, oxidant added by syringe pump (30 min).b Recovered substrates and product yields (%), determined by GC and 1H NMR analysis using bibenzyl as an internal standard, are referred to the initial amount of substrate. Average of at least two runs (error ± 5%). | ||||
 |
72 | 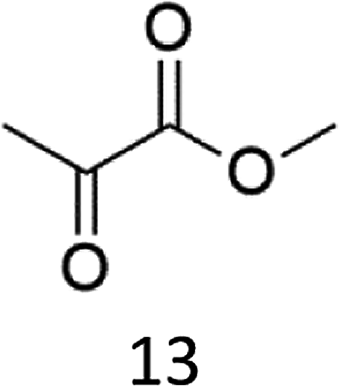 |
||
 |
70 | 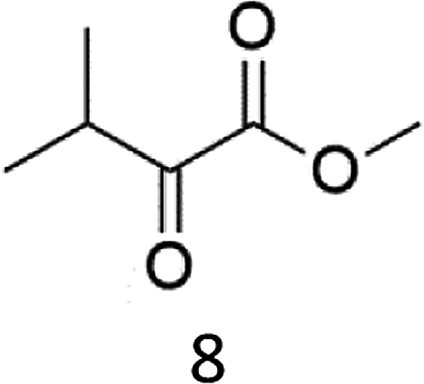 |
 |
|
 |
73 | 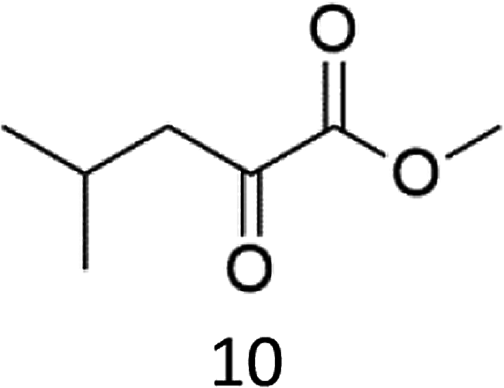 |
 |
|
 |
64 | 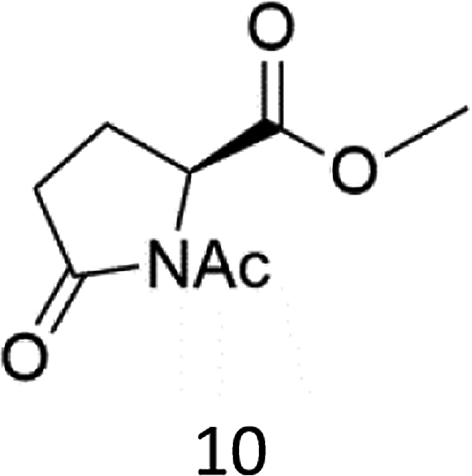 |
 |
|
 |
73 | 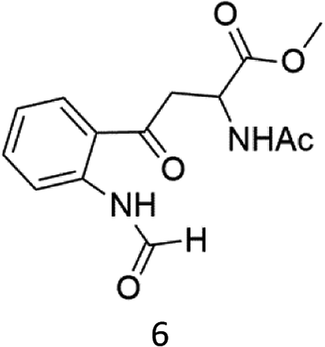 |
||
 |
65 | 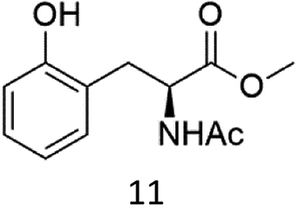 |
 |
 |
 |
98 | |||
Control experiments showed that no oxidation of amino acids occurred in the absence of iron catalyst, nor when using H2O2 and Fe(OTf)2 as catalyst. Moreover similar product yields were observed in the reactions carried out under air or nitrogen, thus reinforcing the idea that autoxidation processes are not responsible of products formation.
The oxidation of the alanine substrate 2 leads to the formation of methyl piruvate as the exclusive reaction product in 13% yield. Methyl piruvate likely derives from decomposition of α-hydroxyalanine produced after initial hydroxylation of the Cα–H bond by the iron based oxidant (Scheme 1). No competitive side chain oxidation is observed due to the low reactivity of the strong methyl C–H bonds.
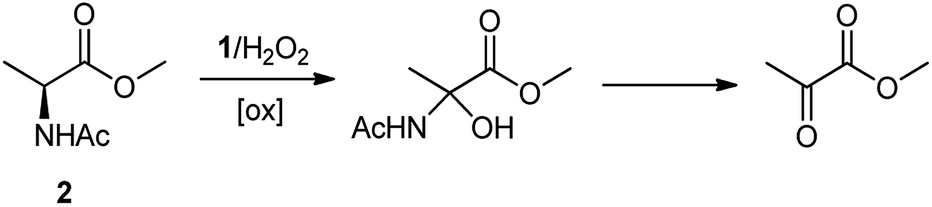 | ||
| Scheme 1 Formation of methyl piruvate in the oxidation of N-AcAlaOMe (2) with the 1/H2O2 system. | ||
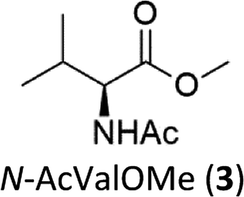
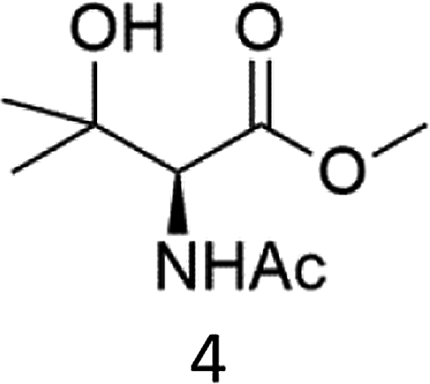
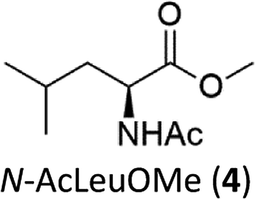
A competition between the side-chain and Cα–H oxidation is instead observed with other aliphatic amino acids 3–5 containing more reactive secondary and tertiary C–H bonds. Oxidation of N-AcValOMe (3) furnished methyl 3-methyl-2-oxobutanoate as the major product (8%) via initial Cα–H hydroxylation accompanied by N-acetyl-β-hydroxyvaline methyl ester (4%) which in turn derives from hydroxylation of the tertiary C–H bond. Cα–H hydroxylation is also the preferred reaction pathway in the oxidation of N-AcLeuOMe (4) with 4-methyl-2-oxo-pentanoic acid methyl ester and 2-acetylamino-4,4-dimethyl-4-butanolide formed in 10% and 5% yields, respectively. The latter is obtained after lactonization of the first formed N-acetyl-γ-hydroxyleucine methyl ester (Scheme 2).
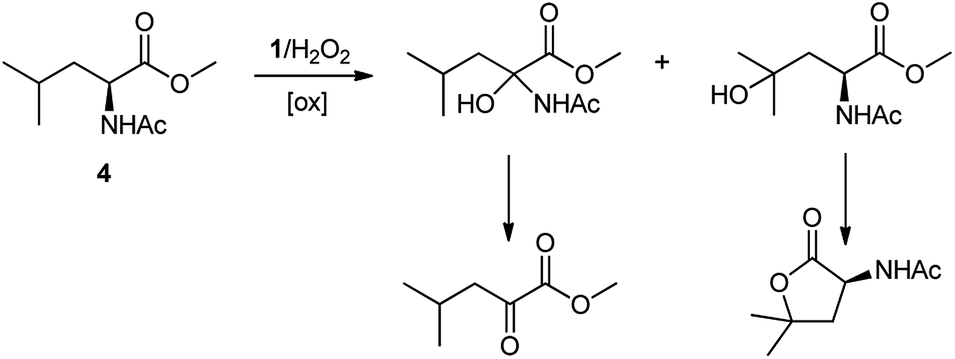 | ||
| Scheme 2 Formation of Cα–H and side-chain oxidation products in the reaction of N-AcLeuOMe (4) with the 1/H2O2 system. | ||
In the likely hypothesis of a hydrogen atom transfer (HAT) mechanism from the substrate to the oxidizing species, ‡ the observed preference for Cα–H hydroxylation is in accordance with a relatively late transition state structure where the stability of the incipient α-carbon centered radical plays a major role even though in the case of substituted amino acids such as Ala, Val and Leu, a nonbonding interaction between the amide carbonyl and side chain R group destabilizes the coplanar conformation of the radical formed which does not benefit of captodative stabilization.14
Thus, the catalytic behavior of the 1/H2O2 system is different from what observed in the oxidations of aliphatic amino acids promoted by aminopyridine nonheme iron catalysts. Exclusive side-chain functionalization was observed as in the oxidation of leucine and valine promoted by Fe(PDP) and Fe(CF3PDP) complexes.3e On the other hand, no products have been observed in the oxidation of amino acids Ala, Val, Leu with KHSO5 promoted by the pentadentate nonheme complex [FeII(N4Py)]2+ with the active species iron(IV)-oxo complex [FeIV(O)(N4Py)]2+ decaying at a rate faster than the hydrogen abstraction from these substrates.15
The selectivity pattern observed with the 1/H2O2 system resembles that of the cumyloxyl radical (CumO˙) where exclusive HAT from α-C–H bond with Ala and a competition between side-chain vs. Cα–H bond cleavage for N-AcLeuOMe (4) were observed.16 However, no HAT reaction from the tertiary β-C-H bond of N-AcValOMe (3) has been observed with CumO˙, thus indicating that our system is less sensitive to steric effects in the HAT process.
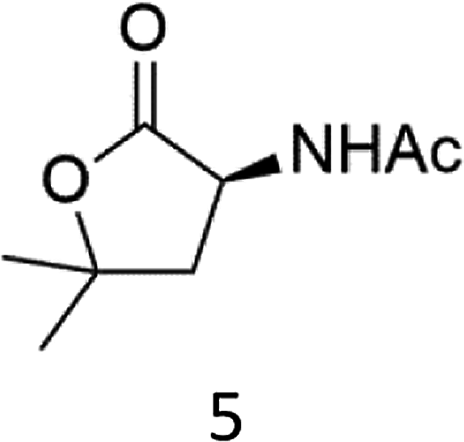
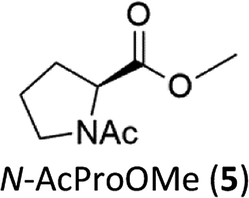
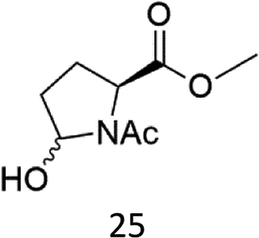
N-AcProOMe (5) was the most reactive substrate with the 1/H2O2 system. N-Ac-5-hydroxyproline methyl esters (mixture of diastereomers) and N-Ac-5-oxoproline methyl ester were formed as oxidation products in a combined 35% yield (36% conversion). Selective C-5 functionalization well matches the expected high reactivity of the C5–H bonds on the basis of the intrinsically high HAT reactivity due to polar effects of the C–H bonds in the δ-carbon atom.3e,5c,14,16a, Similar results were found in the oxidations promoted by aminopyridine nonheme Fe(PDP) and Fe(CF3PDP) complexes.3e However, it has to be remarked that the lower yields of C-5 proline oxidation with the 1/H2O2 system when compared to those obtained with the Fe(PDP) system (77%) are obtained at a considerably lower catalyst loading (1% vs. 25%).
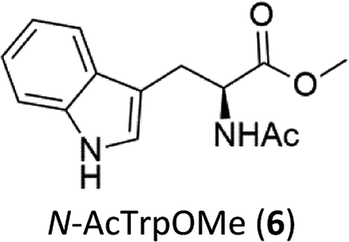
C–H functionalization of phenylalanine and other aromatic amino acids are particularly attractive transformations and have been exploited for the preparation of unnatural amino acids and structural modification of small peptides.3c,17 Oxidation of N-AcTrpOMe (6) with the 1/H2O2 system led to small amounts of side chain functionalization products (see ESI† for details). The major one, formed in 6% yield, was N-formyl-N-acetylkynurenine methyl ester deriving from the oxidative cleavage of the indole ring. The same product had been observed in the oxidation of N-AcTrpOMe (6) with singlet oxygen or with N3˙ in γ-radiolysis experiments18 and its formation may be rationalized according to the mechanism reported in Scheme 3 where the first step involves the abstraction of hydrogen from the indole N–H bond.
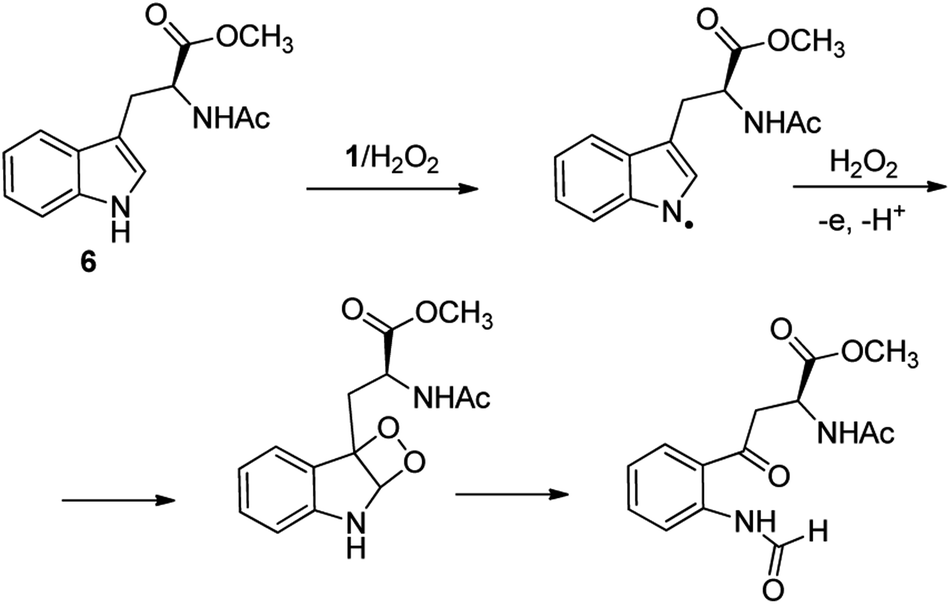 | ||
| Scheme 3 Formation of N-formyl-N-acetylkynurenine methyl ester in the oxidation of N-AcTrpOMe (6) with the 1/H2O2 system. | ||
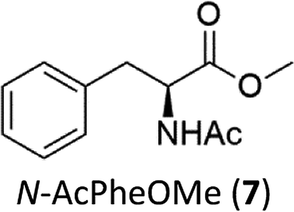
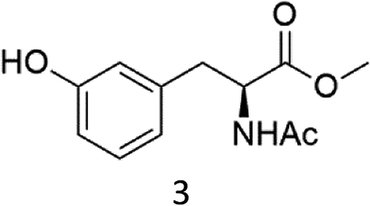
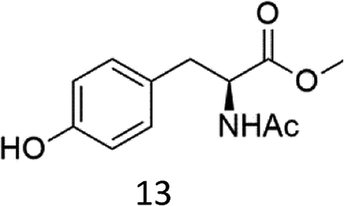
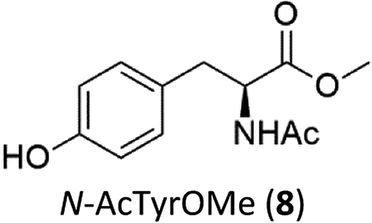
Oxidation of N-AcPheOMe (7) with the 1/H2O2 system leads to the formation of N-AcTyrOMe (8) as the main reaction product (14%) accompanied by the two isomeric o-OH and m-OH phenolic derivatives. No products deriving from Cα–H or benzylic hydroxylation have been observed.
The high chemoselectivity for the aromatic hydroxylation with respect to the oxidation of α-CH and β-CH bonds is a noteworthy feature of this transformation and is in line with the preference for aromatic over aliphatic alcohol oxidation previously reported.11,12 The 1/H2O2 system indeed revealed especially suitable for the hydroxylation of aromatic rings11 and it does not suffer the irreversible phenol binding to the iron center that generally prevents catalytic turnover with nonheme iron catalysts.19 Moreover with this system the oxidation tends to stop at the phenol stage without quinone formation. The latter is an undesired overoxidation product that flaws other iron-catalyzed oxidations20 as reported for example in the oxidation of phenylalanine with the iron(IV)-oxo complex [FeIV(O)(N4Py)]2+ where orthoquinone was the main product.21
It is worth to note that while direct phenylalanine to tyrosine oxidation can be easily accomplished by enzymes (in particular the nonheme iron enzyme phenylalanine hydroxylase22), few chemical oxidants are competent for this transformation. For example, the oxidation of Phe by hydroxyl radicals produced by Fenton reactions or γ-radiolysis also leads to a mixture of o-, m-, p-hydroxyphenylalanines but suffers the limitation of a low positional selectivity and partial overoxidation to 2,3- and 3,4-dihydroxyphenylalanines.23 The higher o-, m-, p- positional selectivity observed with our system is in accordance with a metal based SEAr mechanism proposed in a previous study for the oxidation of aromatic compounds.11
In accordance with the high selectivity for p-hydroxyphenylalanine, no products were observed in the 1/H2O2 promoted oxidation of N-AcTyrOMe (8), with the substrate being recovered almost quantitatively. This result is somewhat surprising since phenolic O–H bond is particularly activated towards HAT reagents, including high-valent iron–oxo species. For instance, [FeIV(O)(N4Py)]2+ reacts much faster with the electron-rich phenol moiety of N-AcTyrNHtBu than with the aromatic ring of N-AcPheNHtBu.15
In view of the relevance of the Phe to Tyr transformation, reaction conditions were subsequently screened varying the amounts of catalyst and oxidant in order to optimize the Tyr yields. The results are shown in Table 2.
| Entry | Cat. (mol%) | H2O2 (eq) | Cosolv H2O | Conv. (%) | Yields of hydroxyphenylalanines | |||
|---|---|---|---|---|---|---|---|---|
| o | m | p | ||||||
| a Yields (%) are referred to the initial amount of substrate. Reaction time 75 minutes.b Reaction time 120 min. | ||||||||
| 1 | 1 | 2.5 | — | 35 | 11 | 3 | 13 | |
| 2 | 3 | 2.5 | — | 47 | 13 | 9 | 24 | |
| 3 | 5 | 2.5 | — | 36 | 9 | 6 | 16 | |
| 4 | 3 | 3.5 | — | 45 | 11 | 9 | 24 | |
| 5 | 3 | 5 | — | 37 | 4 | 6 | 9 | |
| 6b | 1 | 2.5 | — | 69 | 9 | 2 | 20 | |
| 7 | 3 | 2.5 | 7% | 45 | 6 | 10 | 24 | |
| 8 | 3 | 2.5 | 14% | 42 | 6 | 10 | 20 | |
| 9 | 3 | 2.5 | 21% | 40 | 7 | 10 | 20 | |
A pleasant improvement in Tyr yield was observed on increasing catalyst loading from 1 to 3% (entry 2). A further increase in catalyst loading led to a lower substrate conversion (entry 3). With the optimal amount of catalyst (3%) the efficiency decreased by increasing the added oxidant to 3.5 and 5 molar equiv. (entries 4 and 5). By increasing reaction time from 75 to 120 min lower yields of phenolic products and mass balance were observed likely due to formation of overoxidation products (entry 6). Interestingly, the catalyst activity and selectivity are maintained in the presence of water as cosolvent. Substrate conversions and Tyr yields are almost unaffected up to 20% (v/v) water content (entries 7–9).
The high chemoselectivity displayed by the 1/H2O2 system for the aromatic hydroxylation in Phe prompted us to investigate further the high preference for aromatic hydroxylation through intermolecular competitive oxidation experiments of N-AcPheOMe (7) and other aliphatic amino acids. To this purpose, equimolar mixtures of N-AcPheOMe (7) and N-AcValOMe (3), N-AcLeuOMe (4) or N-AcProOMe (5) were subjected to the standard oxidation conditions (see Table 1), and the results are reported in Fig. 3. Quite surprisingly, in the competitive oxidation experiments of N-AcPheOMe (7) with N-AcValOMe (3) or N-AcLeuOMe (4) formation of a mixture of o-, m-, p-hydroxyphenylalanines was accompanied by trace amounts of products deriving from the oxidation of aliphatic amino acids. Phe aryl oxidation also well competes with the C-5 oxidation of reactive N-AcProOMe (5), with product yields comparable with those observed in the oxidation of N-AcPheOMe (7) alone. These results confirm the marked preference for the electrophilic aromatic substitution over the HAT process displayed by the active species formed in the 1/H2O2 system, a property that makes this catalyst unique and particularly attractive in comparison with other nonheme iron complexes.
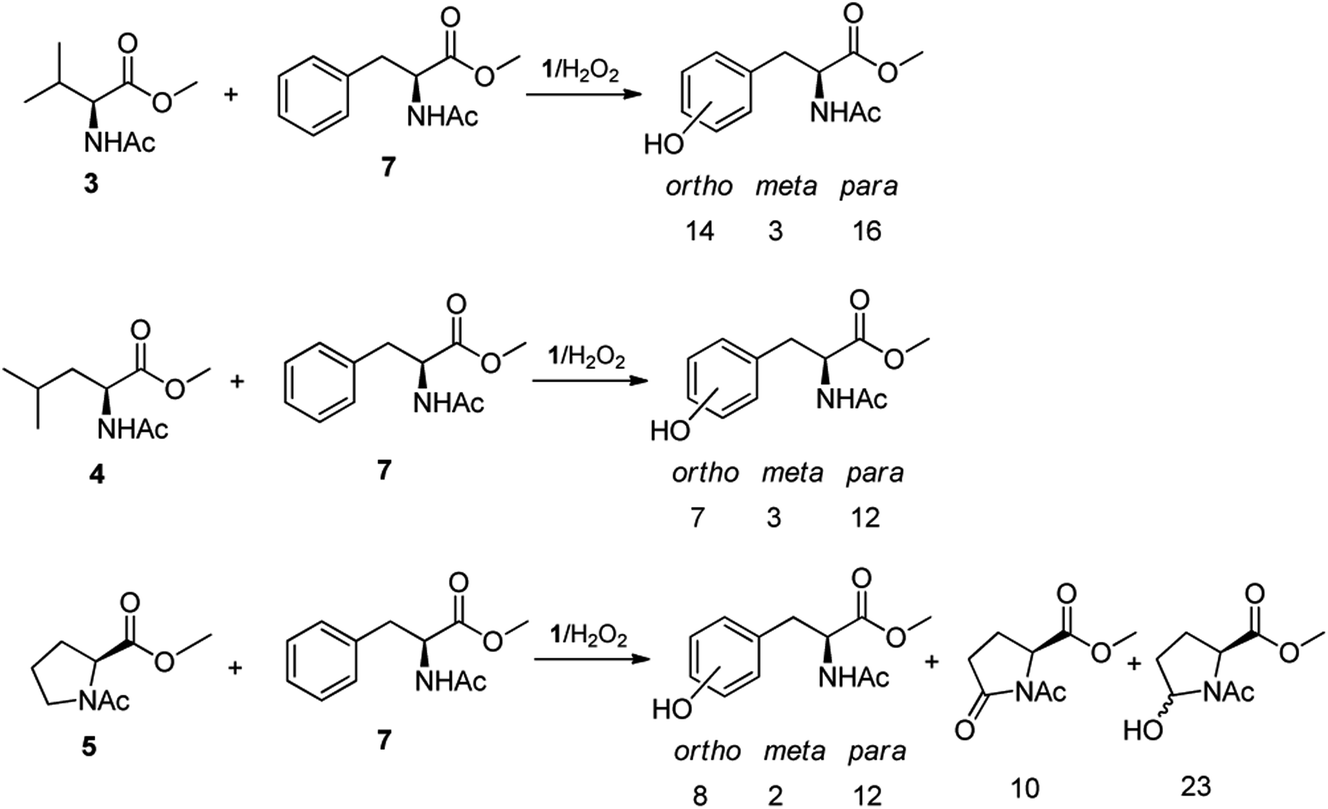 | ||
| Fig. 3 Oxidation products and yields in the competitive experiments of N-AcPheOMe (7) with N-AcValOMe (3), N-AcLeuOMe (4) and N-AcProOMe (5). | ||
Such a clear preference for the aromatic hydroxylation pave the way to the possible selective oxidative functionalization of the Phe aryl rings in polypeptide chains, which is currently under investigation in our laboratories.
Conclusion
The readily available iminopyridine iron(II) complex 1 was found to promote the α-C–H or side-chain oxidation of aliphatic amino acids with H2O2 and a regioselectivity dictated by the substrate structure. In the oxidation of N-AcPheOMe (7) it showed a marked preference for aromatic hydroxylation over Cα–H and benzylic C–H oxidation unlocking the key conversion of Phe to Tyr with a sustainable oxidant and a cheap and easily obtainable iron catalyst. Extension of this methodology to the site-specific modification and/or cleavage of peptides and proteins is current underway in our laboratory.Experimental
Instruments and general methods
GC analyses were carried out on a gas chromatograph equipped with a capillary methylsilicone column (30 m × 0.25 mm × 25 μm). GC-MS analyses were performed with a mass detector (EI at 70 eV) coupled with a gas chromatograph equipped with a melted silica capillary column (30 m × 0.2 mm × 25 μm) covered with a methylsilicone film (5% phenylsilicone, OV5). NMR spectra were recorded on a 300 MHz spectrometer and were internally referenced to the residual proton solvent signal.Materials
All reagents and solvents employed were of the highest purity available and used without further purification. Methyl esters of N-acetylated amino acids 2–8 were prepared as reported in the literature.13 In a typical procedure L-amino acid (17 mmol) was dissolved in acetic anhydride (44 mmol) in methanol (7.6 mL). The mixture was stirred under reflux for 6 h, after cooling to room temperature the solvent was evaporated under reduced pressure and the residue was triturated with ethyl acetate, filtrated and dried in vacuo. With this procedure it was possible to isolate directly the methyl esters 5–8 and the N-acetylated amino acids N-Ac-Ala, N-Ac-Val and N-Ac-Leu. Methyl esters 2–4 were prepared by adding potassium carbonate (30 mmol) to a solution of N-acetylated amino acids (10 mmol) in DMF (36 mL). The reaction was kept at room temperature for 24 h. After this time, iodomethane (30 mmol) was added and stirring was continued for additional 24 h. The solvent was evaporated under reduced pressure and the reaction mixture was extracted with ethyl acetate (2 × 50 mL). The combined organic layers were successively washed with H2O (60 mL), 5% aqueous Na2S2O3 (3 × 60 mL), and brine (3 × 60 mL), dried over anhydrous Na2SO4 and filtered. Substrates 2–8 were purified by recrystallization or by column chromatography. Spectral data are in accordance with those reported in the literature (see ESI†).Product analysis of the oxidation of 2–8 with the 1/H2O2 system
In a typical oxidation experiment Fe(CF3SO3)2(CH3CN)2 (1.09 mg, 2.50 μmol), picolylamine (50 μL of a solution 0.1 M in CH3CN 5.0 μmol) and picolylaldehyde (50 μL of a solution 0.1 M in CH3CN, 5.0 μmol) were mixed in a vial at 25 °C. Substrate (250 μmol) and CH3CN were then added up to a total volume of 700 μL. A solution of H2O2 in CH3CN (1.74 M diluted from a 30% w/w H2O2) was added over 30 minutes by syringe pump under vigorous stirring, and left reacting for additional 5 minutes (Pro), 45 minutes (Phe), 60 minutes (Ala, Val, Leu, Trp). At this point an internal standard was added (bibenzyl, 25 μmol) and the reaction mixture was filtered over a short pad of SiO2 with 10 mL of AcOEt. The filtered solution was subjected to GC and GC-MS analysis or evaporated to furnish the product mixture for the 1H NMR analysis. The following oxidation products were identified by comparison with authentic specimens or by comparison of their spectral data with those reported in the literature: methyl piruvate,24 methyl 3-methyl-2-ketobutanoate,25 (3S)-N-acetyl-hydroxyvaline methyl ester,5j 4-methyl-2-oxo-pentanoic acid methyl ester,26 (2S)-2-acetylamino-4,4-dimethyl-4-butanolide,5j (2S,5S)- and (2S,5R)-N-Ac-5-hydroxyproline methyl esters (mixture of diastereomers),27 (2S)–N-acetyl-5-oxoproline methyl ester,28 (2S)–N-acetyl-m-hydroxyphenylalanine methyl ester,29 N-formyl-N-acetylkynurenine methyl ester.18 (2S)–N-acetyl-o-hydroxyphenylalanine methyl ester was isolated from the crude reaction mixture by column chromatography and characterized as follows:1H NMR (300 MHz, CDCl3): δ = 7.17–7.11 (dt, 1H), 7.00–6.97 (dd, 1H), 6.86–6.78 (m, 2H), 6.62–6.60 (m, 1H), 4.65–4.59 (m, 1H), 3.73 (s, 3H), 3.21–3.15 (dd, 1H), 2.99–2.92 (dd, 1H), 2.04 (s, 3H). 13C NMR (75 MHz, CDCl3): δ = 172.6, 171.0, 154.9, 131.1, 128.6, 122.3, 119.9, 115.9, 53.6, 52.4, 33.3, 22.8. HR-MS (ESI-TOF): m/z = 260.0897 [M + Na]+ calcd. for C12H15NO4Na, found 260.0899.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are due to the Ministero dell’Istruzione, dell'Università e della Ricerca (MIUR), for financial support and to the CIRCC, Interuniversity Consortium of Chemical Catalysis and Reactivity.References
- (a) W. M. Garrison, Chem. Rev., 1987, 87, 381–398 CrossRef; (b) E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef PubMed; (c) C. J. Easton, Chem. Rev., 1997, 97, 53–82 CrossRef PubMed.
- (a) A. Schepartz and B. Cuenoud, J. Am. Chem. Soc., 1990, 112, 3247–3249 CrossRef; (b) D. Hoyer, H. Cho and P. G. Schultz, J. Am. Chem. Soc., 1990, 112, 3249–3250 CrossRef; (c) M. Zhong, L. Lin and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2111–2115 CrossRef PubMed; (d) J. Gallagher, O. Zelenko, A. D. Walts and D. S. Sigman, Biochemistry, 1998, 37, 2096–2104 CrossRef PubMed; (e) G. Xu and M. R. Chance, Chem. Rev., 2007, 107, 3514–3543 CrossRef PubMed; (f) S. D. Maleknia and K. M. Downard, Chem. Soc. Rev., 2014, 43, 3244–3258 RSC.
- (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef PubMed; (b) C. Annese, I. Fanizza, C. D. Calvano, L. D'Accolti, C. Fusco, R. Curci and P. G. Williard, Org. Lett., 2011, 13, 5096–5099 CrossRef PubMed; (c) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef PubMed; (d) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef PubMed; (e) T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214–219 CrossRef PubMed.
- C. J. Easton, Pure Appl. Chem., 1997, 69, 489–494 CrossRef.
- (a) B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef PubMed; (b) C. L. Hawkins and M. J. Davies, J. Chem. Soc., Perkin Trans. 2, 1998, 2617–2622 RSC; (c) R. Saladino, M. Mezzetti, E. Mincione, I. Torrini, M. Paglialunga Paradisi and G. Mastropietro, J. Org. Chem., 1999, 64, 8468–8474 CrossRef; (d) M. B. Goshe, Y. H. Chen and V. E. Anderson, Biochemistry, 2000, 39, 1761–1770 CrossRef PubMed; (e) C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, 2001, 1504, 196–219 CrossRef PubMed; (f) B. N. Nukuna, M. B. Goshe and V. E. Anderson, J. Am. Chem. Soc., 2001, 123, 1208–1214 CrossRef PubMed; (g) B. D. Dangel, J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2001, 123, 8149–8150 CrossRef PubMed; (h) A. K. Croft, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2003, 125, 4119–4124 CrossRef PubMed; (i) M. J. Davies, Biochim. Biophys. Acta, 2005, 1703, 93–109 CrossRef PubMed; (j) M. R. Rella and P. G. Williard, J. Org. Chem., 2007, 72, 525–531 CrossRef PubMed; (k) Z. I. Watts and C. J. Easton, J. Am. Chem. Soc., 2009, 131, 11323–11325 CrossRef PubMed; (l) Q. Raffy, D.-A. Buisson, J.-C. Cintrat, B. Rousseau, S. Pin and J. P. Renault, Angew. Chem., Int. Ed., 2012, 51, 2960–2963 CrossRef PubMed; (m) P. E. Morgan, D. I. Pattison and M. J. Davies, Free Radical Biol. Med., 2012, 52, 328–339 CrossRef PubMed.
- (a) K. Chen and L. Que Jr, J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef PubMed; (b) K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que Jr, J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef PubMed; (c) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef PubMed; (d) M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef PubMed; (e) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (f) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef; (g) M. S. Chen and M. C. White, Science, 2012, 335, 807–809 CrossRef PubMed; (h) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef PubMed; (i) O. Cussò, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285–14298 RSC; (j) W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef PubMed; (k) G. Olivo, O. Cussò and M. Costas, Chem. - Asian J., 2016, 11, 3148–3158 CrossRef PubMed; (l) G. Olivo, O. Cussò, M. Borrell and M. Costas, J. Biol. Inorg Chem., 2017, 22, 425–452 CrossRef PubMed.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef PubMed.
- (a) A. C. Lindhorst, S. Haslinger and F. E. Kühn, Chem. Commun., 2015, 51, 17193 RSC; (b) G. Olivo, O. Lanzalunga and S. Di Stefano, Adv. Synth. Catal., 2016, 358, 843–863 CrossRef.
- G. Olivo, G. Arancio, L. Mandolini, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2014, 4, 2900–2904 Search PubMed.
- G. Olivo, M. Nardi, D. Vìdal, A. Barbieri, A. Lapi, L. Gómez, O. Lanzalunga, M. Costas and S. Di Stefano, Inorg. Chem., 2015, 54, 10141–10152 CrossRef PubMed.
- G. Capocasa, G. Olivo, A. Barbieri, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2017, 7, 5677–56864 Search PubMed.
- G. Olivo, S. Giosia, A. Barbieri, O. Lanzalunga and S. Di Stefano, Org. Biomol. Chem., 2016, 14, 10630–10635 Search PubMed.
- (a) R. C. Wende, A. Seitz, D. Niedek, S. M. M. Schuler, C. Hofmann, J. Becker and P. R. Schreiner, Angew. Chem., Int. Ed., 2016, 55, 2719–2723 CrossRef PubMed; (b) P. Fatus, J. Bachl, S. Oehm, A. I. JimØnez, C. Cativiela and D. Díaz Díaz, Chem.–Eur. J., 2013, 19, 8861–8874 CrossRef PubMed.
- V. A. Burgess, C. J. Easton and M. P. Hay, J. Am. Chem. Soc., 1989, 111, 1047–1052 CrossRef.
- A. I. Abouelatta, A. A. Campanali, A. R. Ekkati, M. Shamoun, S. Kalapugama and J. J. Kodanko, Inorg. Chem., 2009, 48, 7729–7739 CrossRef PubMed.
- (a) M. Salamone, F. Basili and M. Bietti, J. Org. Chem., 2015, 80, 3643–3650 CrossRef PubMed; (b) L. M. Pipitone, G. Carboni, D. Sorrentino, M. Galeotti, M. Salamone and M. Bietti, Org. Lett., 2018, 20, 808–811 CrossRef PubMed.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, J. M. Alvarez-Gutiérrez, A. Ballesteros and J. M. . González, Angew. Chem., Int. Ed., 2004, 43, 325–329 CrossRef PubMed.
- X. Fang, F. Jin, H. Jin and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1998, 259–263 RSC.
- (a) O. V. Makhlynets and E. V. Rybak-Akimova, Chem.–Eur. J., 2010, 16, 13995–14006 CrossRef PubMed; (b) A. Thibon, V. Jollet, C. Dibal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem.–Eur. J., 2012, 18, 2715–2724 CrossRef PubMed.
- (a) P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC; (b) A. C. Lindhorst, J. Schutz, T. Netscher, W. Bonrath and F. E. Kühn, Catal. Sci. Technol., 2017, 7, 1902–1911 RSC.
- A. R. Ekkati and J. J. Kodanko, J. Am. Chem. Soc., 2007, 129, 12390–12391 CrossRef PubMed.
- T. Flatmark and R. C. Stevens, Chem. Rev., 1999, 99, 2137–2160 CrossRef PubMed and references therein.
- Z. Maskos, J. D. Rush and W. H. Koppenol, Arch. Biochem. Biophys., 1992, 296, 521–529 CrossRef PubMed.
- Y. Si, J. Chen, F. Li, J. Li, Y. Qin and B. Jiang, Adv. Synth. Catal., 2006, 348, 898–904 CrossRef.
- H. Poisel, Chem. Ber., 1978, 111, 3136–3139 CrossRef.
- S. Thiede, P. R. Wosniok, D. Herkommer, A. Schulz-Fincke, M. Gütschow and D. Menche, Org. Lett., 2016, 18, 3964–3967 CrossRef PubMed.
- K. Irie, A. Ishida, T. Nakamura and T. Oh-Ishi, Chem. Pharm. Bull., 1984, 32, 2126–2139 CrossRef.
- S. Yoshifuji, K. Tanaka, T. Kawai and Y. Nitta, Chem. Pharm. Bull., 1985, 33, 5515–5521 CrossRef.
- C. E. Humphrey, M. Furegati, K. Laumen, L. La Vecchia, T. Leutert, J. C. D. Müller-Hartwieg and M. Vögtle, Org. Process Res. Dev., 2007, 11, 1069–1075 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Other experimental details, selected GC and GC-MS chromatograms and characterization of the products of some oxidation reactions. See DOI: 10.1039/c8ra02879f |
| ‡ The structure of the active species is still unknown, the hypothetical formation of an imine based nonheme iron-oxo complex (see ref. 10) is currently under investigation. |
| This journal is © The Royal Society of Chemistry 2018 |