DOI:
10.1039/C8RA02939C
(Paper)
RSC Adv., 2018,
8, 17183-17190
Design, synthesis and biological evaluation of N-arylsulfonyl carbazoles as novel anticancer agents†
Received
5th April 2018
, Accepted 30th April 2018
First published on 10th May 2018
Abstract
In this work, a set of structurally diverse synthetic carbazoles was screened for their anticancer activities. According to structure–activity relationship studies, carbazoles with an N-substituted sulfonyl group exhibited better anticancer activity. Moreover, compound 8h was discovered to show the most potent anticancer effects on Capan-2 cells by inducing apoptosis and cell cycle arrest in G2/M phase. Finally, the in vivo study demonstrated that 8h prevented the tumor growth in PANC-1 and Capan-2 xenograft models without apparent toxicity.
1. Introduction
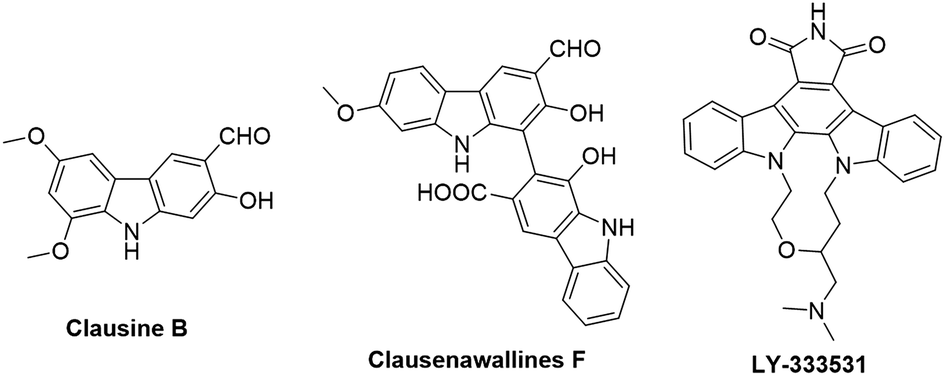
Carbazoles and their derivatives are of great interest in the research fields of synthetic chemistry and medicinal chemistry, due to their tricyclic structure and widespread pharmacological properties.1 In general, carbazoles constitute a useful class of polycyclic aromatic drugs displaying diverse activities including antibacterial,2 neuroprotective,3 anti-inflammatory,4 antioxidant,5 and anti-HIV functions.6 More importantly, carbazoles are found to have effective antiproliferative activity on tumor cells (Fig. 1). Clausine B, a naturally occurring carbazole alkaloid, was found to be active (IC50 < 30 μg mL−1) against four human cancer cell lines including MDA-MB-231 (breast cancer), HeLa (cervical cancer), CAOV3 (ovarian cancer) and HepG2 (hepatic cancer).7 Clausenawallines F showed cytotoxicity against oral cavity cancer (KB) and small-cell lung cancer (NCI-H187) with IC50 values of 10.2 μM and 4.5 μM, respectively.8 Due to the important pharmacological activities of the reported natural occurring carbazoles, an array of compounds containing carbazole motifs has been designed and synthesized for biological evaluation. For example, LY-333531 was reported as an anticancer agent by inhibiting Pim-1 kinase with IC50 value of 0.2 μM.9,10
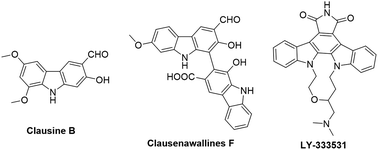 |
| | Fig. 1 Representative carbazoles with anticancer activities. | |
In the recent years, other research groups and ours have demonstrated that cyclic diphenyliodoniums (CDIs) are useful synthetic synthons for complex polycyclic arenes.11–20 Due to the structural similarity of carbazoles and CDIs, it was envisioned that the insertion of nitrogen into the CDIs would be able to afford carbazoles. Indeed, we previously developed a synthetic strategy for carbazoles via the reaction of CDIs with various amines which was mediated by Cu(OAc)2 at low cost.11 With this strategy, an in-house chemical library of carbazoles with structural diversity and complexity was successfully built. The previous biological neuroprotective screening of the carbazole library found that some N-phenyl carbazoles showed good neuroprotection of neuronal cells HT22 against cell injury induced by glutamate or homocysteic acid.12 Pancreatic cancer is one leading cause of cancer-related death and remains a serious clinical problem due to poor prognosis and the limited drug options.21 In the continuation of our efforts to discover novel and potent anticancer agents, our in-house synthetic carbazoles were further screened for their potential antiproliferation of pancreatic cancer cells. The screening assay provided a structural starting point for further structure–activity study. In this work, we report the design and synthesis of structurally diversified carbazoles and their biological evaluation, leading to the discovery of novel N-arylsulfonyl carbazoles with both in vitro and in vivo antitumor therapeutic activities.
2. Results and discussion
2.1 Chemistry
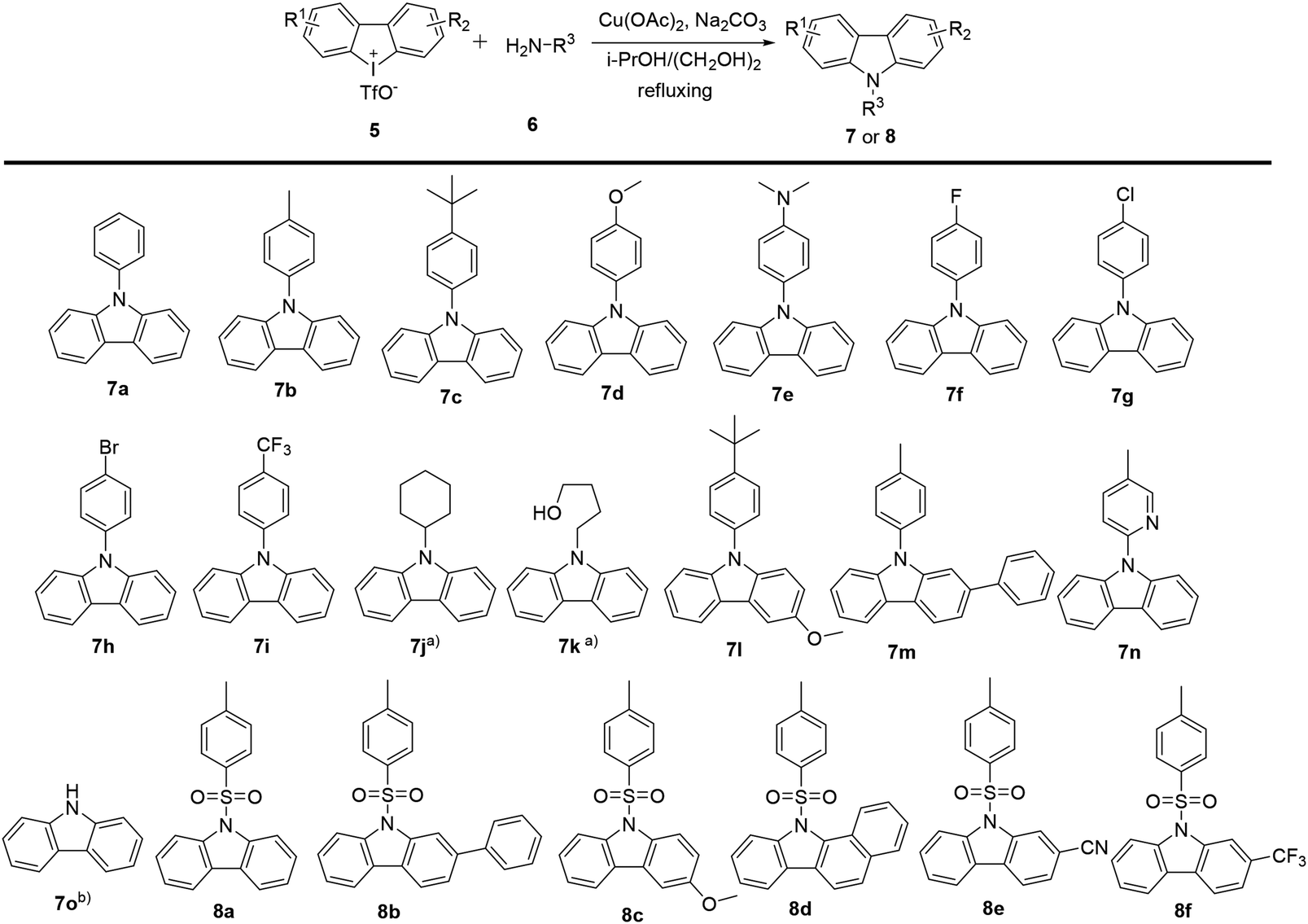
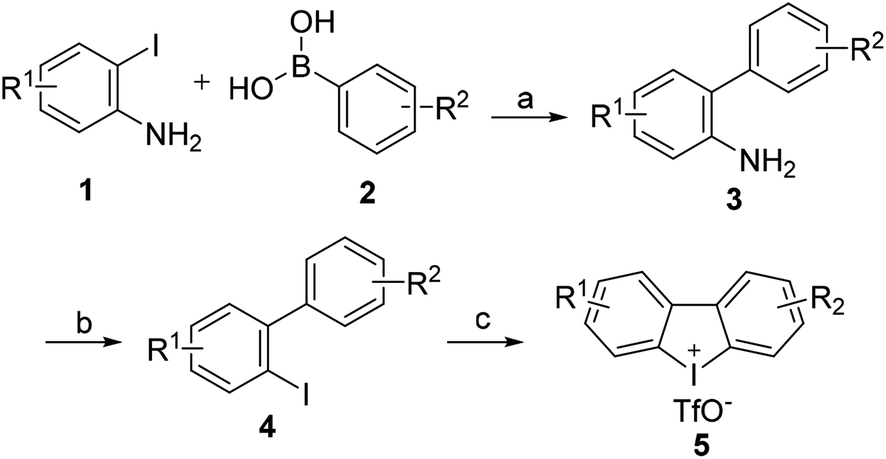
2.1.1 General procedures for the synthesis of carbazoles with cyclic diphenyliodoniums. In previous work, we have demonstrated the construction of N-substituted carbazoles starting from cyclic diphenyl iodoniums.11 The procedures for the synthesis of carbazoles were depicted in Scheme 1. The reactions of cyclic diphenyl iodoniums 5 with amines 6 were performed in a refluxing isopropanol/ethylene glycol (9/1) mixed solvent at argon atmosphere for 16 h, with Cu(OAc)2 as the catalyst and Na2CO3 as the base. Thus, the reactions effectively afforded a series of N-aryl (7a–7i and 7l–7m), N-pyridinyl (7n), N-aliphatic (7j, 7k) and N-arylsulfonyl (8a–8f) substituted carbazoles at moderate to good yields. Besides, carbazole (7o) was also accessible after desulfonylation of 8a. A mechanism study for this conversion was reported in our previous work.11,12
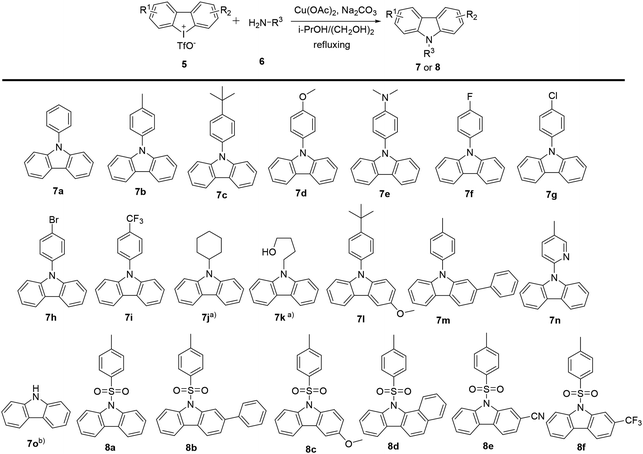 |
| | Scheme 1 Synthesis of carbazoles. Reaction conditions: diphenyleniodonium 5 (0.1 mmol), amine 6 (4 equiv.), Na2CO3 (3 equiv.), Cu(OAc)2 (0.2 equiv.), 16 h, in refluxing i-PrOH/(CH2OH)2 (1.8/0.2 mL), argon atmosphere. (a) Additional CuI (0.2 equiv.) was added. (b) 7o was obtained from the desulfonylation of 8a. | |
2.2 Biology
2.2.1 In vitro anticancer screening. The in vitro antitumor activities of the synthesized carbazole derivatives against human cancer cell lines, including two pancreatic cancer cell lines (PANC-1 and Capan-2), were assayed by MTS method. A summary of these results was shown in Table 1. N-Phenyl substituted carbazoles were found to display moderate to good neuroprotective activity by mediating ROS decrease in the neuronal cells HT22.12 However, in this context, it was observed that the treatment with most of N-phenyl (7a–7i and 7l, 7m), N-pyridinyl (7n), N-aliphatic substituted carbazoles (7j, 7k) and carbazole 7o led to an inhibition of cell viability less than 50% at the tested concentrations. Thus, compounds 7a–7o were excluded in further study, which was based on criterion of NCI-60 program.22 Meanwhile, most of the N-arylsulfonyl substituted carbazoles (8a–8f) exhibited potent cytotoxic activities against PANC-1 and Capan-2 cell lines. Furthermore, among these effective carbazoles, different substituents on the carbazole motif and the N-sulfonyl group played a crucial role in gaining antiproliferative activities. Carbazole with a trifluoromethyl (8f) was the best compared to the ones with alternative functional groups such as 8a, 8c (OMe), 8b and 8d (phenyl) and 8e (CN).
Table 1 The primary antiproliferative screening of 7a–7o and 8a–8f on PANC-1 and Capan-2 pancreatic cancer cell lines
| Compound |
Inhibitory rate of PANC-1 viability (%) |
Inhibitory rate of Capan-2 viability (%) |
| 30 μM |
10 μM |
30 μM |
10 μM |
| 7a |
25.02 |
20.38 |
31.48 |
25.25 |
| 7b |
12.59 |
16.42 |
21.01 |
23.82 |
| 7c |
22.8 |
18.61 |
20.65 |
15.85 |
| 7d |
25.9 |
17.65 |
34.8 |
12.91 |
| 7e |
43.5 |
24.4 |
49.29 |
20.56 |
| 7f |
11.75 |
34.27 |
22.55 |
28.82 |
| 7g |
3.8 |
3.81 |
20.32 |
11.75 |
| 7h |
71.1 |
19.05 |
38.12 |
18.58 |
| 7i |
54.31 |
25.61 |
14.53 |
20.05 |
| 7j |
19.78 |
18.18 |
15.93 |
32.61 |
| 7k |
44.34 |
36.68 |
35.65 |
33.72 |
| 7l |
5.4 |
23.76 |
35.98 |
30.36 |
| 7m |
9.61 |
13.16 |
42.79 |
38.41 |
| 7n |
34.73 |
33.84 |
24.94 |
20.91 |
| 7o |
35.9 |
36.27 |
ND |
27.03 |
| 8a |
43.66 |
41.55 |
67.81 |
61.57 |
| 8b |
59.8 |
63.98 |
70.54 |
66.79 |
| 8c |
54.17 |
54.71 |
69.39 |
57.72 |
| 8d |
53.92 |
34.94 |
72.62 |
72.04 |
| 8e |
58.6 |
61.2 |
28.46 |
28.39 |
| 8f |
56.67 |
48.05 |
82.2 |
66.6 |
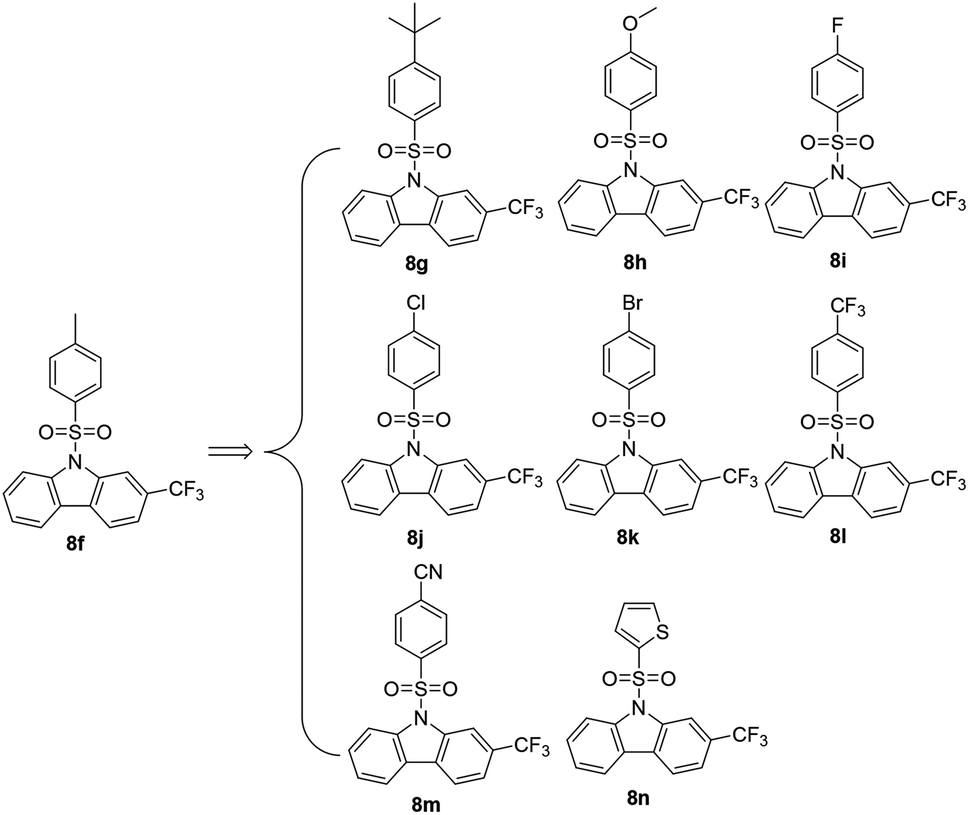
2.2.2 Optimization of compound 8f. Based on the structure of 8f (Scheme 2), a preliminary structure–activity relationship (SAR) study was conducted in order to find more potent agents, as shown in Table 2. In terms of substituents on the arylsulfonyl, tert-butyl (t-butyl, 8g), methoxy (OMe, 8h), and cyano (CN, 8m) enhanced the anticancer efficiency. On the contrary, the carbazoles with an N-arylsulfonyl substituted by fluorine (F, 8i), chlorine (Cl, 8j) and trifluoromethyl (CF3, 8l) lost their anticancer efficacy drastically. Interestingly, the carbazole with an N-arylsulfonyl substituted by bromine (Br, 8k) showed more effective cytotoxicity than its counterparts 8i (–F) and 8j (–Cl). A thiophenyl group to replace phenyl of arylsulfonyls did not help to improve the inhibitory effect of 8n compared to the leading compound 8f. It is worth mentioning that 8g–8n were characterized by NMR and HRMS. Finally, the most potent compound 8h was taken for further investigation for its antiproliferation against four different types of human cancer cell lines PANC-1 (pancreatic cancer), Capan-2 (pancreatic cancer), SGC-7901 (gastric cancer), HCT-116 (colon cancer), and HL-60 (leukemia), while 5-fluorouracil (5-FU) was selected as a positive control. As shown in the Table 3, 8h demonstrated a broad spectrum of antitumor activities. Under our experiment conditions, the antiproliferative activities of 8h were found to be more potent than 5-FU. Moreover, compound 8h showed much lower toxicity (IC50, 20.04 μM) to normal HFL-1 fibroblast cell line, indicating a good selectivity between human cancer and normal cells.
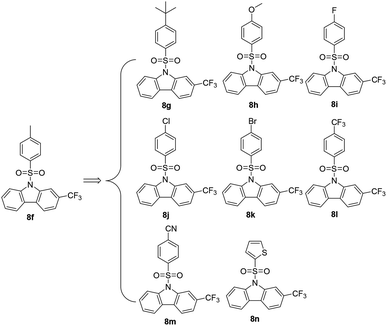 |
| | Scheme 2 Newly synthetic arylsulfonyl N-substituted carbazoles for more potent anticancer agents. | |
Table 2 The antiproliferative evaluation of 8g–8n on PANC-1 and Capan-2 pancreatic cancer cell lines
| Compound |
Inhibitory rate of PANC-1 viability (%) |
Inhibitory rate of Capan-2 viability (%) |
| 30 μM |
10 μM |
30 μM |
10 μM |
| 8g |
35.5 |
25.1 |
36.1 |
44.1 |
| 8h |
72.2 |
68.2 |
86.8 |
89.4 |
| 8i |
5.6 |
13.42 |
19.55 |
14.4 |
| 8j |
36.8 |
16.5 |
58.7 |
52.4 |
| 8k |
53.9 |
54.8 |
84.9 |
73.3 |
| 8l |
43.3 |
8.49 |
29.7 |
31.41 |
| 8m |
76.9 |
59.9 |
80.2 |
64.0 |
| 8n |
61.6 |
57.3 |
40.4 |
49.1 |
Table 3 In vitro IC50 value (μM) of 8h against tumor cell lines and normal cell line using MTS assaya
| Compound |
Cell line (IC50 in μM) |
| PANC-1 |
Capan-2 |
SGC-7901 |
HCT-116 |
HL-60 |
HFL-1 |
| NT: not tested. Values are expressed as mean ± standard deviation (SD) of three independent duplicate experiments. |
| 8h |
3.69 ± 0.30 |
1.30 ± 0.28 |
2.25 ± 0.21 |
1.90 ± 0.14 |
2.20 ± 0.70 |
20.04 ± 2.48 |
| 5-Fu |
19.31 ± 2.76 |
32.40 ± 3.70 |
16.56 ± 1.32 |
10.03 ± 0.88 |
NT |
NT |
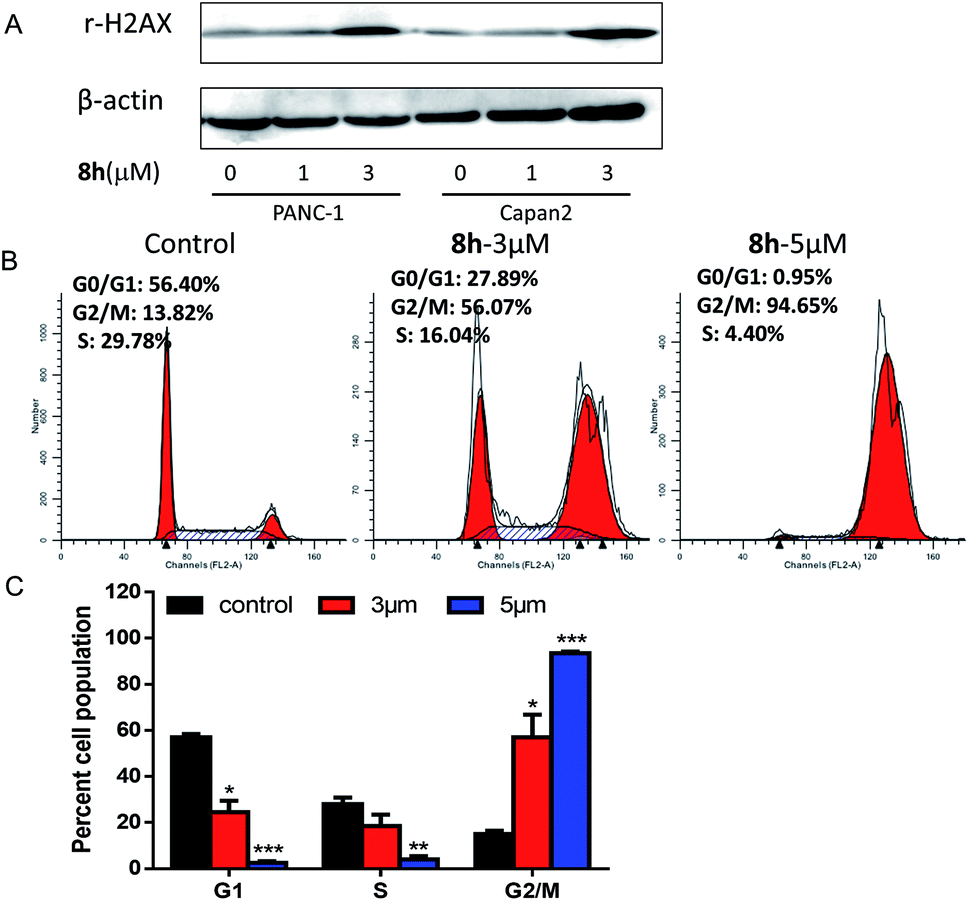
2.2.3 The effect of 8h on cell cycle of Capan-2 cancer cells. As a planar and aromatic polycyclic compound, carbazole derivative 8h was reasoned to interact with DNA and induce a DNA damage.23 Indeed, the treatment with 8h in PANC-1 and Capan-2 cells increased the level of γ-H2AX protein, which is a sensitive molecular marker of DNA damage (Fig. 2A). Considering that DNA damage has been reported to induce cell cycle arrest,24–26 the effect of 8h on cell cycle distribution was also studied at different concentrations on Capan-2 cells. As shown in Fig. 2B–C, the treatment of Capan-2 cells at 5 μM resulted in more than 90% of the cells arrested in G2/M phase. The substantial accumulation of cancer cells in G2/M phase induced by 8h might contribute to the antiproliferation of the compound.
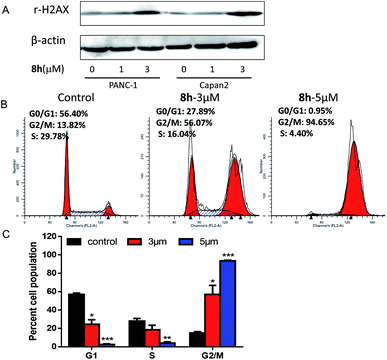 |
| | Fig. 2 Compound 8h induced DNA damage and cell cycle arrest in G2/M phase. (A) Protein γ-H2AX was analyzed by western blot. PANC-1 and Capan-2 cells were treated with 1 or 3 μM of compound 8h for 24 h. (B) Effect of compound 8h at 3 μM or 5 μM on the cell cycle of Capan-2 for 24 h. Cellular DNA contents were measured by PI staining using flow cytometry. The numbers indicate the normalization of each phase of cell cycle. (C) Histograms demonstrated the percentage of Capan-2 at different phases of the cell cycle. Error bars represent mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. | |
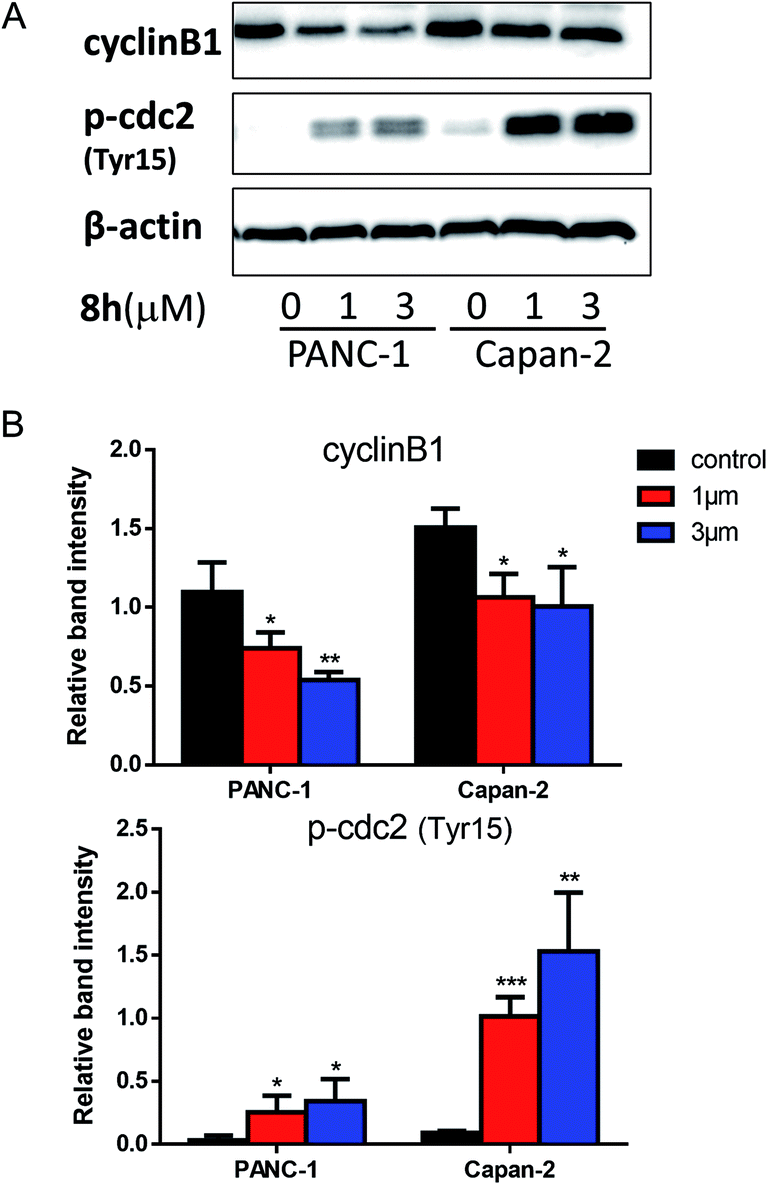
2.2.4 The effect of 8h on the levels of cell-cycle regulatory proteins. To explore the potential molecular mechanism for 8h-induced cell cycle arrest at the G2/M phase, we examined the protein expression of cyclin B1 and phospho-Tyr15-Cdc2 which are crucial regulators in G2/M phase transition.27,28 It is well known that the cell cycle is primarily regulated by CDK-cyclin complexes containing cyclin-dependent kinases (CDKs) and their cofactor cyclins.29 The progression from G2 to mitosis is driven by the formation and activation of cdc2 (CDK1)-cyclin B1 complex.30 As one of cdc2 negative regulator sites, Tyr15 phosphorylation inhibits cdc2 activity.27 In PANC-1 and Capan-2 cells, 8h significantly decreased the expression level of cyclin B1 and increased the expression level of phospho-Tyr15-cdc2 (Fig. 3A–B), thus favoring cell cycle arrest at G2/M phase.
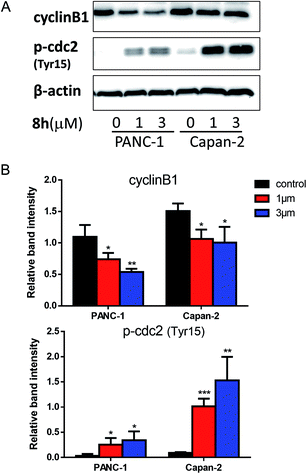 |
| | Fig. 3 The effect of compound 8h on the level of cyclin B1, p-Tyr15-cdc2. (A) Western blot was used to determine the expression of cyclin B1, p-Tyr15-cdc2 in PANC-1 and Capan-2 cells treated in the absence or presence (1 or 3 μM) of 8h for 24 h. (B) Densitometric analysis of cyclin B1, p-Tyr15-cdc2 proteins normalized with β-actin. The expression ratio of related proteins was quantified against β-actin using ImageJ software. Data were derived from three independent replicates. Bars means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. | |
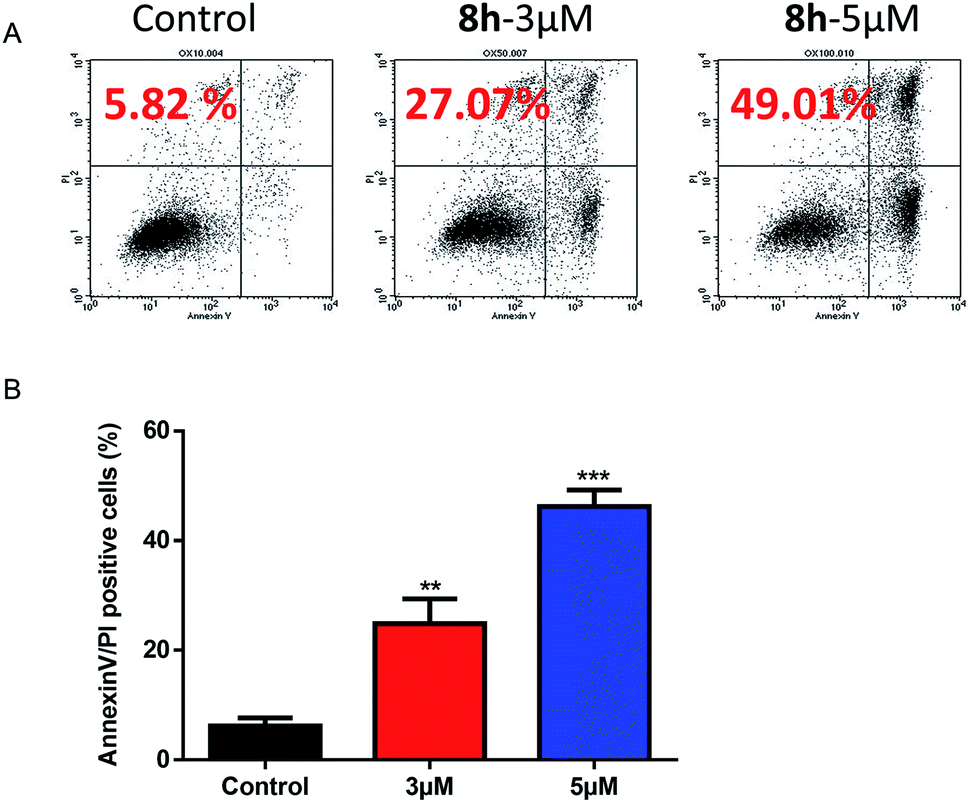
2.2.5 The effect of 8h on the apoptosis induction of Capan-2 cancer cells. We further investigated whether an induced apoptosis contributed to 8h's lethality by Annexin V-FITC and PI staining using flow cytometer. The apoptotic effect of compound 8h (3 μM and 5 μM) was evaluated on Capan-2 cells. Data indicated that 8h significantly increased the percentage of apoptotic cells in a dose-dependent manner compared to the control (Fig. 4A–B).
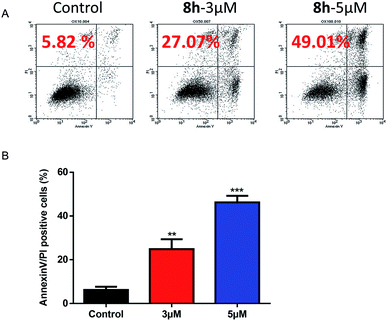 |
| | Fig. 4 Flow cytometric analysis of 8h induced apoptosis in Capan-2 cells using Annexin V-FITC and PI double staining. (A) Apoptosis rates were determined after Capan-2 cells were treated with 8h at 3 μM or 5 μM for 48 h. Percentage numbers indicate cell death population. (B) Percentage of Annexin V/PI positive cells were derived from three independent replicates. Data shown are mean values ± SD (error bar). **p < 0.01; ***p < 0.001. | |
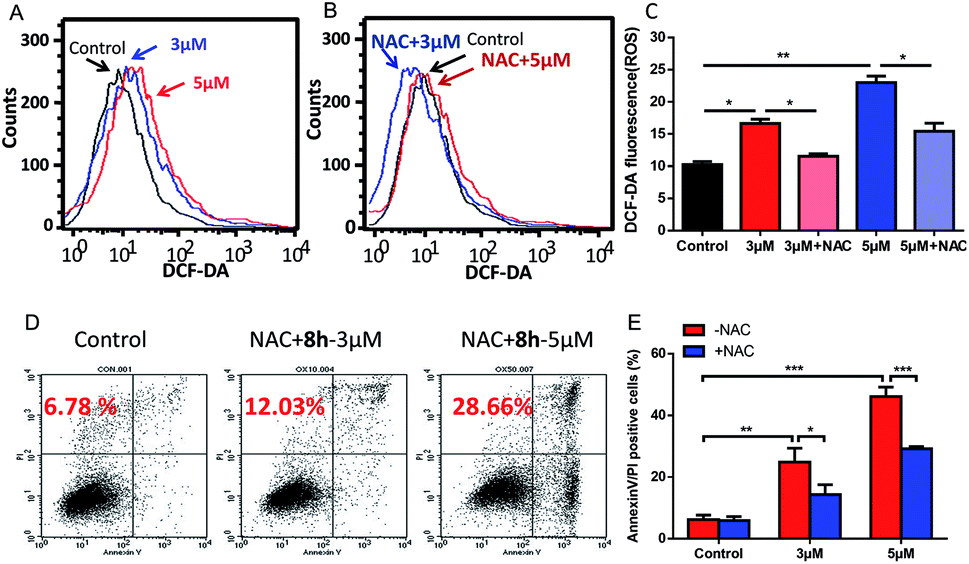
2.2.6 The role of ROS in mediating 8h-induced apoptosis. Reactive oxygen species (ROS) are well known to be a major player in redox signaling and a critical mediator of apoptosis.31,32 The cancer cells in advanced stage tumors often exhibit high oxidative stress, suggesting that it might be possible to preferentially kill these cancer cells by pharmacologically increasing the ROS level transiently.33 In previous work, we demonstrated that such strategy was effective to eliminate cancer cells by mediating intracellular ROS increase.34,35 Therefore, it was of our interest whether such N-arylsulfonyl carbazole 8h could elevate the intracellular ROS level in cancer cells. To test this possibility, the ROS level was measured when Capan-2 cells were subjected to 8h. As shown in Fig. 5A–C, 8h caused a concentration-dependent increase of cellular ROS, which could be partially reversed by pretreatment with an antioxidant N-acetyl-L-cysteine (NAC), a scavenger of ROS. Importantly, pretreatment with NAC also resulted in marked protection against 8h-induced apoptosis (Fig. 5D–E), suggesting that the ROS generation induced by 8h might play an important role in the cell apoptosis.
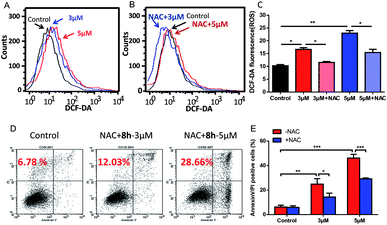 |
| | Fig. 5 Induction of ROS increase by compound 8h and its partial reversion by an antioxidant NAC. (A) DCF-DA staining showed the effect of 3 μM (blue line) and 5 μM (red line) of 8h on cellular ROS levels after 24 h treatment. (B and C) Pretreatment with NAC effectively reversed the ROS accumulation. Capan-2 cells were pre-treated with 3 mM NAC for 1 h and then incubated with compound 8h (3 or 5 μM) for 24 h. Error bars represent mean ± SD. (D and E) Compound 8h-induced apoptosis in Capan-2 cells was partially prevented by pre-treatment with NAC compared to Fig. 4. Capan-2 cells were pre-treated with 3 mM NAC for 1 h, and then incubated with the indicated concentrations (3 or 5 μM) of 8h for 24 h. Error bars represent mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. | |
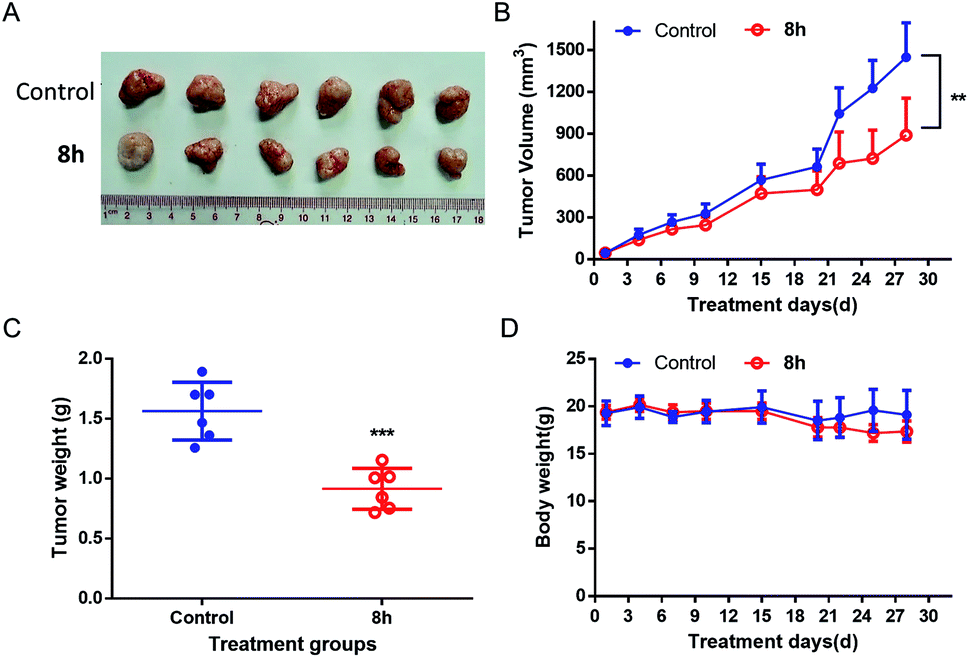
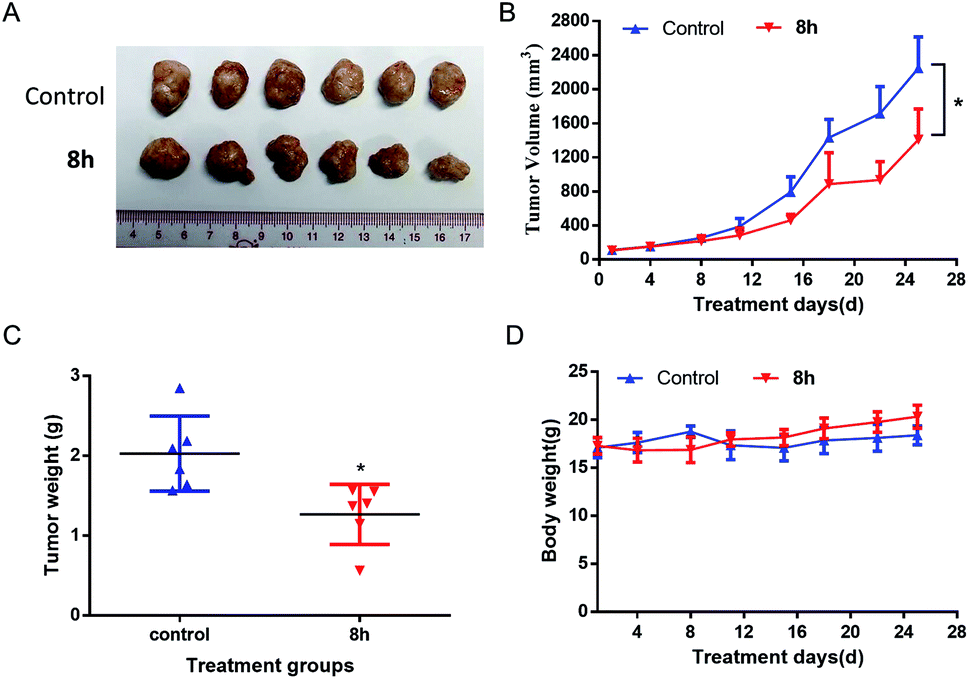
2.2.7 In vivo anticancer activity of 8h. Based on the promising in vitro anticancer activity of 8h toward various cell lines, especially pancreatic cancer cell lines, we next evaluated in vivo anti-cancer activity of 8h in PANC-1 and Capan-2 xenograft mice. Compared with the control group, PANC-1 tumor growth was substantially reduced in the mice treated with 8h intraperitoneally three times weekly at a 40 mg kg−1 dosage, with approximately 40% volume inhibition (Fig. 6A–B). Furthermore, the treatment with 8h for 28 days significantly diminished tumor weight (Fig. 6C). Meanwhile, no significant weight loss or illness was observed in the treatment group (Fig. 6D). Moreover, the anticancer effect of 8h was also observed in another pancreatic cancer cell line Capan-2 xenograft mice (Fig. 7). Taken together, 8h exhibited significant in vivo antitumor efficacy at a safe dose.
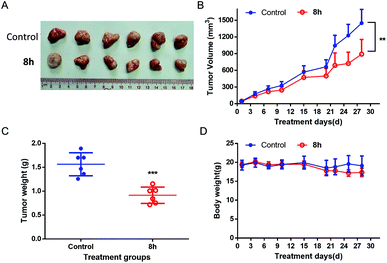 |
| | Fig. 6 In vivo antitumor effect of compound 8h in PANC-1 xenograft model. 8h was administered three times per week intraperitoneally at 40 mg kg−1 dosage. (A) Image of all dissected tumors. (B) The effect of 8h on the PANC-1 tumor growth. (C) Tumor weight at day 28 after 4 weeks of treatment. (D) Mice weights were measured twice a week. Bars means ± SD. **p < 0.01; ***p < 0.001. | |
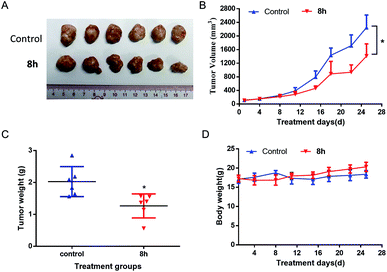 |
| | Fig. 7 In vivo antitumor effect of compound 8h in Capan-2 xenograft model. 8h was administered three times per week intraperitoneally at 40 mg kg−1 dosage. (A) Image of all dissected tumors. (B) The effect of 8 h on the Capan-2 tumor growth. (C) Tumor weight after 25 days of treatment. (D) Mice weights were measured twice a week. Bars means ± SD. *p < 0.05. | |
3. Conclusions
In conclusion, a chemical library of N-substituted carbazoles was synthesized conveniently, and all the compounds were screened for a potential anticancer activity. Results demonstrated that the N-arylsulfonyl substituted carbazoles showed significant antiproliferative ability. A preliminary SAR study found that the synthetic compound 8h displayed the most potent cytotoxic activity against PANC-1, Capan-2, SGC-7901, HCT-116, HL-60 cancer cells. Moreover, compound 8h exhibited promising in vivo anticancer therapeutic efficacy in PANC-1 and Capan-2 xenograft mice models without an apparent side effect. The following preliminary investigation of its action model mechanism implied that the ROS mediated-apoptosis as well as the G2/M cell cycle arrest might contribute to the anticancer activity of 8h. Environmentally benign and easily accessible synthesis and the potent anticancer activity may make these N-arylsulfonyl substituted carbazoles to be promising drug candidates to kill cancer cells.
4. Experiments
4.1 Chemistry
4.1.1 General information. All solvents were commercially available and were used without a further purification unless stated. The chemicals used were either purchased from commercial sources or prepared according to literature procedures. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance spectrometer 400 at 400 and 100 MHz respectively. Chemical shifts are given in ppm (δ) referenced to CDCl3 with 7.26 for 1H and 77.10 for 13C, and to d6-DMSO with 2.50 for 1H and 39.52 for 13C. In the case of multiplet, the signals are reported as intervals. Signals are abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants are expressed in hertz. The progress of the reactions was monitored by thin-layer chromatography on a glass plate coated with silica gel with fluorescent indicator (GF254). Column chromatography was performed on silica gel (200–300 mesh).
4.1.2 General procedures for the synthesis of cyclic diphenyleneiodoniums. The procedures11 for the synthesis of cyclic diphenyliodoniums 5 were depicted in Scheme 3. Catalyzed by Pd(PPh3)4, substituted 2-iodoaniline 1 reacted with functionalized phenlyboronic acid 2 yielded areneamine 3 at a good yield. 3 was readily transformed into iodoarene 4 via a Sandermeyer reaction, and then cyclic diphenyliodoniums 5 could be obtained after 4 was treated with mCPBA and triflic acid (TfOH) for 2 h at room temperature.
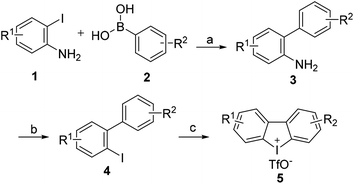 |
| | Scheme 3 Synthesis of cyclic diphenyliodoniums (5). Reaction conditions: (a) Pd(PPh3)4, K3PO4, EtOH, reflux, 6 h. (b) (i). NaNO2, 0 °C THF, 4 M HCl, (ii). KI, rt. (c) mCPBA (85%), TfOH, CH2Cl2, rt, 2 h. | |
4.1.3 Synthesis and characterization of the carbazole derivatives.
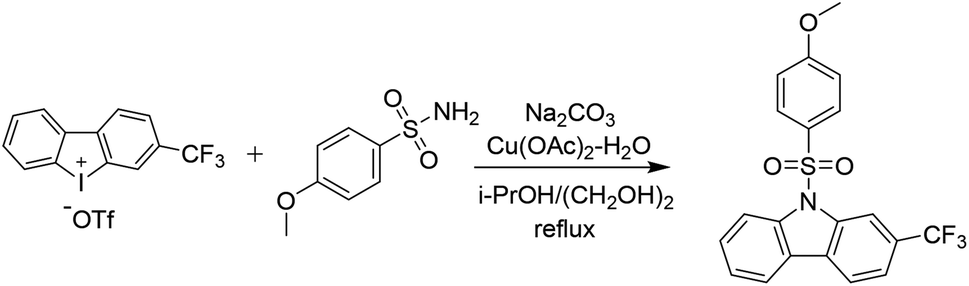
The general procedure for the preparation of these carbazoles was exemplified by the synthesis of 8h:To a stirred solution of 3-(trifluoromethyl)dibenzo[b,d]iodol-5-ium trifluoromethanesulfonate (100 mg, 0.2 mmol, 1 eq.) in i-PrOH (1.8 mL) and ethylene glycol (0.2 mL), was added 4-methoxybenzenesulfonamide (151 mg, 0.8 mmol, 4 equiv.), sodium carbonate (64 mg, 0.6 mmol, 3 equiv.), Cu(OAc)2 (8 mg, 0.04 mmol, 0.2 equiv.). The reaction proceeded at a reflux for 16 h under argon atmosphere before i-PrOH was removed by a rotary evaporation. The remained mixture was partitioned between water and EtOAc, and the aqueous phase was extracted with EtOAc. The combined organic phases were washed with H2O and brine, dried over anhydrous Na2SO4, evaporated in a vacuo. The residue was purified by column chromatography on a silica gel (PE/EtOAc = 15/1–5/1) to provide carbazole 8h.
4.1.3.1 9-((4-(tert-Butyl)phenyl)sulfonyl)-2-(trifluoromethyl)-9H-carbazole (8g). A white solid, 63% yield. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.37 (d, J = 8.4 Hz, 1H), 8.00 (dd, J = 16.8, 7.9 Hz, 2H), 7.77 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.37 (d, J = 8.7 Hz, 2H), 1.21 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 158.3, 139.3, 137.9, 134.9, 128.8, 126.6, 126.5, 125.2, 124.3, 120.8, 120.5, 115.3, 112.5, 35.4, 31.0 ppm. HRMS calcd for C23H20F3NO2S [M + H]+: 432.1245, found: 432.1239.
4.1.3.2 9-((4-Methoxyphenyl)sulfonyl)-2-(trifluoromethyl)-9H-carbazole (8h). A white solid, 69% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.36 (d, J = 8.5 Hz, 1H), 7.98 (dd, J = 17.1, 8.0 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 6.79 (d, J = 8.9 Hz, 2H), 3.74 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 164.2, 139.3, 137.9, 129.4, 129.2, 128.8, 128.7, 125.9, 125.2, 124.3, 123.1, 120.8, 120.5, 115.3, 114.5, 112.6, 112.5, 55.7 ppm. HRMS calcd for C20H14F3NO3S [M + H]+: 406.0724, found: 406.0721.
4.1.3.3 9-((4-Fluorophenyl)sulfonyl)-2-(trifluoromethyl)-9H-carbazole (8i). A white solid, 46% yield. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.34 (d, J = 8.3 Hz, 1H), 7.99 (dd, J = 17.7, 7.9 Hz, 2H), 7.89–7.78 (m, 2H), 7.65 (d, J = 7.9 Hz, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.44 (t, J = 7.3 Hz, 1H), 7.03 (t, J = 7.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.3, 164.8, 139.2, 137.8, 133.6, 133.6, 129.7, 129.5, 129.4, 129.0, 125.8, 125.4, 124.7, 123.1, 120.9, 120.6, 116.9, 116.7, 115.3, 112.6, 112.5 ppm. HRMS calcd for C19H11F4NO2S [M − H]−: 392.0369, found: 392.0371.
4.1.3.4 9-((4-Chlorophenyl)sulfonyl)-2-(trifluoromethyl)-9H-carbazole (8j). A white solid, 52% yield. 1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 7.99 (dd, J = 18.1, 8.0 Hz, 2H), 7.74 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.32 (d, J = 8.6 Hz, 2H). 13CNMR (100 MHz, CDCl3) δ 141.1, 139.2, 137.8, 136.0, 129.7, 129.4, 129.0, 128.0, 125.4, 124.8, 121.3, 121.3, 121.0, 120.7, 115.3, 112.6, 112.6 ppm. HRMS calcd for C19H11ClF3NO2S [M + H]+: 410.0029, found: 410.0028.
4.1.3.5 9-((4-Bromophenyl)sulfonyl)-2-(trifluoromethyl)-9H-carbazole (8k). A white solid, 49% yield. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.33 (d, J = 8.5 Hz, 1H), 7.99 (dd, J = 17.9, 8.0 Hz, 2H), 7.66 (d, J = 8.7 Hz, 3H), 7.58 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 8.7 Hz, 2H), 7.44 (t, J = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 139.2, 137.8, 136.5, 132.7, 129.7, 129.5, 129.0, 128.0, 125.5, 124.8, 121.3, 121.0, 120.7, 115.4, 112.6 ppm. HRMS calcd for C19H11BrF3NO2S [M + H]+: 453.9724, found: 453.9718.
4.1.3.6 2-(Trifluoromethyl)-9-((4-(trifluoromethyl)phenyl)sulfonyl)-9H-carbazole (8l). A white solid, 57% yield. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.00 (dd, J = 18.5, 7.9 Hz, 2H), 7.93 (d, J = 8.1 Hz, 2H), 7.67 (d, J = 8.1 Hz, 1H), 7.64–7.57 (m, 3H), 7.45 (t, J = 7.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 140.9, 139.1, 137.7, 136.0, 135.7, 129.9, 129.5, 129.1, 127.2, 126.6, 126.6, 125.7, 125.5, 125.0, 124.3, 123.0, 121.5, 121.0, 120.8, 115.3, 112.6, 112.5 ppm. HRMS calcd for C20H11F6NO2S [M − H]−: 442.0337, found: 442.0335.
4.1.3.7 4-((2-(Trifluoromethyl)-9H-carbazol-9-yl)sulfonyl)benzonitrile (8m). A white solid, 51% yield. 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.00 (dd, J = 18.6, 7.9 Hz, 2H), 7.89 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.2 Hz, 1H), 7.63 (dd, J = 7.6, 5.0 Hz, 2H), 7.62–7.57 (m, 1H), 7.46 (t, J = 7.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 141.2, 138.9, 137.6, 133.2, 130.0, 129.6, 129.2, 127.2, 125.6, 125.2, 121.7, 121.7, 121.1, 120.8, 118.1, 116.8, 115.3, 112.6, 112.6 ppm. HRMS calcd for C20H11F3N2O2S [M − H]−: 399.0415, found: 399.0415.
4.1.3.8 9-(Thiophen-2-ylsulfonyl)-2-(trifluoromethyl)-9H-carbazole (8n). A white solid, 72% yield. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.00 (dd, J = 16.7, 7.8 Hz, 2H), 7.66 (d, J = 8.1 Hz, 1H), 7.63–7.56 (m, 2H), 7.48–7.42 (m, 2H), 6.92 (dd, J = 5.0, 3.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 139.0, 137.6, 136.9, 133.4, 132.9, 129.5, 129.2, 128.8, 127.5, 125.7, 125.4, 124.7, 123.0, 121.1, 120.7, 120.4, 115.5, 112.8 ppm. HRMS calcd for C17H10F3NO2S2 [M + H]+: 382.0183, found: 382.0181.
4.2 Biological assay
4.2.1 Cell lines and cell culture. Human cell lines Capan-2 (pancreatic cancer) and HCT-116 (colon cancer) were purchased from American Tissue Culture Collection (Manassas, VA, USA). Cell lines PANC-1 (pancreatic cancer), SGC-7901 (gastric cancer), HL-60 (leukemia) and HFL-1 (normal human lung fibroblast) were purchased from the cell bank, Chinese Academy of Sciences (Shanghai, China). The medium for cell culture were specified by the instructions of cell repository banks. All medium were obtained from Invitrogen Life Technologies (Carlsbad, CA, USA) and supplemented with 10% fetal bovine serum (GIBCO, Carlsbad, CA, USA). All cell lines were cultured at 37 °C in a humidified incubator with 5% CO2 atmosphere.
4.2.2 MTS assay. Cell viability was determined by MTS assay (Promega, Madison, WI, USA).36 Cells were seeded in a 96-well plate at a density of 3000 cells per well. We simultaneously prepared blank wells containing media only. The stock solutions of test compounds were dissolved in appropriate volume so that the final DMSO concentration during treatment in cells was not more than 0.1%. Treated cells were cultured with indicated compounds while control cells were treated with 0.1% (v/v) DMSO. After 72 h of treatment, 20 μL MTS reagent was added to each well and incubated for an additional 4 hours at 37 °C. The optical density (OD) was determined by microplate reader (Thermo, Helsinki, Finland) at 490 nm. The experiment was repeated for three independent times. The percentages of cell viability and inhibitory rate of cell viability were calculated using formulas:
| Inhibitory rate of cell viability (%) = 100 − cell viability (%) |
4.2.3 Cell cycle analysis. Cells were seeded in 6-well plates with a density of 2 × 105 cells per well and incubated with different concentrations of drugs for 24 h. The cells were harvested and fixed with 70% alcohol at 4 °C for 2 h and then incubated with RNase at 37 °C for 30 min followed by addition of PI. Finally, cell cycle distribution was detected using FACS Calibur flow cytometer (BD Biosciences).
4.2.4 Apoptosis assay. Apoptosis was detected by an Annexin-V/PI assay kit (KeyGEN, Nanjing, China). After the drug treatment, cells were harvested and washed. The cells were then stained with Annexin-V-FITC and propidium iodide (PI) in binding buffer for 15 min and apoptotic rates were analyzed using FACS flow cytometer (BD Biosciences, San Diego, CA, USA). Comparisons between groups were tested by one-way ANOVA analysis.
4.2.5 Analysis of ROS. Cells were seeded in six-well plates at 2 × 105 cells per well overnight and treated with compound 8h at indicated concentrations for 24 h. Then the cells were collected and stained with CMH2DCF-DA (Sigma-Aldrich) followed by flow cytometry analysis using FACS Calibur flow cytometer (BD Biosciences). In some experiments of ROS analysis, cells were pre-treated with 3 mM NAC (Sigma-Aldrich, St. Louis, MO, USA) for 1 h prior to the exposure to compound 8h. Statistical comparisons were analyzed by one-way ANOVA.
4.2.6 Western blot analysis. Collected cells were homogenized in lysis buffer (5% SDS, 10 mM EDTA, 50 mM NaCl, 10 mM Tris–HCl). Protein concentrations were measured using pierce BCA protein assay (Thermo Fisher, Rockford, IL, USA). Proteins were run on a standard SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in 5% milk, incubated with primary antibody at a concentration of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, then incubated with secondary antibody at a concentration of 1:10000 and read using ECL detection system (Bio-Rad, Richmond, CA). β-Actin was used as a loading control. Anti-γ-H2AX (phospho-Ser139), anti-cyclinB1, anti-phospho-cdc2 (Tyr15), and anti-β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). The expression ratio of cyclinB1 and p-cdc2 (Tyr15) proteins was quantified against β-actin using ImageJ software. One-way ANOVA was used for statistical analysis.
:1000, then incubated with secondary antibody at a concentration of 1:10000 and read using ECL detection system (Bio-Rad, Richmond, CA). β-Actin was used as a loading control. Anti-γ-H2AX (phospho-Ser139), anti-cyclinB1, anti-phospho-cdc2 (Tyr15), and anti-β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). The expression ratio of cyclinB1 and p-cdc2 (Tyr15) proteins was quantified against β-actin using ImageJ software. One-way ANOVA was used for statistical analysis.
4.2.7 Animal study. The exponentially growing PANC-1 and Capan-2 cells were collected and injected subcutaneously into the flanks of 5 week-old female immune-deficient BALB/c nude mice (Vital River, Beijing, China) at 2 × 106 cells or 3 × 106 cells per injection site respectively. After the formation of palpable tumors, the nude mice were randomly assigned into control and treatment groups (six mice per group). Compound 8h was dissolved in 80% PBS, 10% DMSO and 10% cremophor and administered intraperitoneally three times weekly at a dose of 40 mg kg−1. Control mice were treated with equal volume of vehicle (80% PBS, 10% DMSO and 10% cremophor). The mice body weight and tumor volume, which was calculated by the formula 0.5 × length × width2 were measured twice a week. At the end of the experiment, the mice were sacrificed, their tumors were dissected, and the tumor weights were measured. p values were calculated using the two-tailed Student t-test. The animal study was conducted in accordance with a protocol approved by the Institutional Animals Care and Use Committee of Sun Yat-sen University Cancer Center. The code for IACUC approval was L102012016020G.
4.2.8 Statistical analysis. Data were presented as mean ± SD. The statistical significance of differences was determined using the Student's t-test or one-way ANOVA. p value < 0.05 was regarded as statistically significant.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (81672952, 81430060), and Guangdong Science and Technology Program (2017A020215198).
References
- A. W. Schmidt, K. R. Reddy and H. J. Knolker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- P. Y. Wang, H. S. Fang, W. B. Shao, J. Zhou, Z. Chen, B. A. Song and S. Yang, Bioorg. Med. Chem. Lett., 2017, 27, 4294–4297 CrossRef CAS PubMed.
- A. A. Pieper, S. Xie, E. Capota, S. J. Estill, J. Zhong, J. M. Long, G. L. Becker, P. Huntington, S. E. Goldman, C. H. Shen, M. Capota, J. K. Britt, T. Kotti, K. Ure, D. J. Brat, N. S. Williams, K. S. MacMillan, J. Naidoo, L. Melito, J. Hsieh, J. De Brabander, J. M. Ready and S. L. McKnight, Cell, 2010, 142, 39–51 CrossRef CAS PubMed.
- A. D. Favia, D. Habrant, R. Scarpelli, M. Migliore, C. Albani, S. M. Bertozzi, M. Dionisi, G. Tarozzo, D. Piomelli, A. Cavalli and M. De Vivo, J. Med. Chem., 2012, 55, 8807–8826 CrossRef CAS PubMed.
- D. Pratico, Trends Pharmacol. Sci., 2008, 29, 609–615 CrossRef CAS PubMed.
- K. M. Meragelman, T. C. McKee and M. R. Boyd, J. Nat. Prod., 2000, 63, 427–428 CrossRef CAS.
- W. N. Wan Mohd Zain, A. Rahmat, F. Othman and T. Y. Yap, Malays J Med Sci, 2009, 16, 29–34 Search PubMed.
- W. Maneerat, T. Ritthiwigrom, S. Cheenpracha, T. Promgool, K. Yossathera, S. Deachathai, W. Phakhodee and S. Laphookhieo, J. Nat. Prod., 2012, 75, 741–746 CrossRef CAS PubMed.
- R. Akue-Gedu, E. Rossignol, S. Azzaro, S. Knapp, P. Filippakopoulos, A. N. Bullock, J. Bain, P. Cohen, M. Prudhomme, F. Anizon and P. Moreau, J. Med. Chem., 2009, 52, 6369–6381 CrossRef CAS PubMed.
- D. Drygin, M. Haddach, F. Pierre and D. M. Ryckman, J. Med. Chem., 2012, 55, 8199–8208 CrossRef CAS PubMed.
- D. Zhu, Q. Liu, B. Luo, M. Chen, R. Pi, P. Huang and S. Wen, Adv. Synth. Catal., 2013, 355, 2172–2178 CrossRef CAS.
- D. Zhu, M. Chen, M. Li, B. Luo, Y. Zhao, P. Huang, F. Xue, S. Rapposelli, R. Pi and S. Wen, Eur. J. Med. Chem., 2013, 68, 81–88 CrossRef CAS PubMed.
- D. Zhu, Y. Wu, B. Wu, B. Luo, A. Ganesan, F. H. Wu, R. Pi, P. Huang and S. Wen, Org. Lett., 2014, 16, 2350–2353 CrossRef CAS PubMed.
- Z. Liu, D. Zhu, B. Luo, N. Zhang, Q. Liu, Y. Hu, R. Pi, P. Huang and S. Wen, Org. Lett., 2014, 16, 5600–5603 CrossRef CAS PubMed.
- M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916–4919 CrossRef CAS PubMed.
- M. Wang, Q. Fan and X. Jiang, Org. Lett., 2018, 20, 216–219 CrossRef CAS PubMed.
- M. Wang, J. Wei, Q. Fan and X. Jiang, Chem. Commun., 2017, 53, 2918–2921 RSC.
- Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279–10282 RSC.
- S. Riedmuller and B. J. Nachtsheim, Beilstein J. Org. Chem., 2013, 9, 1202–1209 CrossRef PubMed.
- H. Xie, M. Ding, M. Liu, T. Hu and F. Zhang, Org. Lett., 2017, 19, 2600–2603 CrossRef CAS PubMed.
- A. P. Klein, Nat. Rev. Cancer, 2013, 13, 66–74 CrossRef CAS PubMed.
- In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval, ed. B. A. Teicher and P. A. Andrews, Humana press, Totowa, NJ, 2004, pp. 41–62 Search PubMed.
- C. Asche and M. Demeunynck, Anti-Cancer Agents Med. Chem., 2007, 7, 247–267 CrossRef CAS PubMed.
- M. Laiho and L. Latonen, Ann. Med., 2003, 35, 391–397 CrossRef CAS PubMed.
- K. Ishikawa, H. Ishii and T. Saito, DNA Cell Biol., 2006, 25, 406–411 CrossRef CAS PubMed.
- I. A. Shaltiel, L. Krenning, W. Bruinsma and R. H. Medema, J. Cell Sci., 2015, 128, 607–620 CrossRef CAS PubMed.
- R. Y. Zhao and R. T. Elder, Cell Res., 2005, 15, 143–149 CrossRef CAS PubMed.
- L. Tang, Y. Zhang, H. Pan, Q. Luo, X. M. Zhu, M. Y. Dong, P. C. Leung, J. Z. Sheng and H. F. Huang, Reprod. Biol. Endocrinol., 2009, 7, 144 CrossRef PubMed.
- S. Lim and P. Kaldis, Development, 2013, 140, 3079–3093 CrossRef CAS PubMed.
- T. Miyazaki and S. Arai, Cell Cycle, 2007, 6, 1419–1425 CrossRef CAS PubMed.
- J. M. Mates, J. A. Segura, F. J. Alonso and J. Marquez, Arch. Toxicol., 2012, 86, 1649–1665 CrossRef CAS PubMed.
- K. Sinha, J. Das, P. B. Pal and P. C. Sil, Arch. Toxicol., 2013, 87, 1157–1180 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- J. Wang, B. Luo, X. Li, W. Lu, J. Yang, Y. Hu, P. Huang and S. Wen, Cell Death Dis., 2017, 8, e2887 CrossRef CAS PubMed.
- D. Trachootham, Y. Zhou, H. Zhang, Y. Demizu, Z. Chen, H. Pelicano, P. J. Chiao, G. Achanta, R. B. Arlinghaus, J. Liu and P. Huang, Cancer Cell, 2006, 10, 241–252 CrossRef CAS PubMed.
- A. M. Schrand, J. B. Lin and S. M. Hussain, Methods Mol. Biol., 2012, 906, 395–402 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02939c |
| ‡ Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab