
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeak magnetic field enhances the activation of peroxymonosulfate by ZnO@Fe3O4†
Haodi Zhao,
Jing Zhang *,
Qian Ye,
Hao Xu,
Guanyu Zhou,
Meijing Wang and
Wanning Deng
*,
Qian Ye,
Hao Xu,
Guanyu Zhou,
Meijing Wang and
Wanning Deng
College of Architecture & Environment, Sichuan University, Sichuan 610065, China. E-mail: zjing428@163.com
First published on 14th May 2018
Abstract
The effect of weak magnetic field (WMF) on Acid Orange 7 (AO7) removal by ZnO@Fe3O4/peroxymonosulphate (PMS) was investigated. The results showed that the AO7 sequestration rate enhanced progressively to 0.14183 min−1 in the presence of WMF, approximately 3 times the 0.04966 min−1 in the absence of WMF. SO4˙−/SO5˙− and O2˙− radicals were generated from the decomposition of PMS catalyzed by ZnO@Fe3O4 causing the degradation of AO7. In addition, a weak magnetic field promoted the production of O2˙− radicals and transition of Fe3+ to Fe2+. Radical-pair theory was used in this study to describe the role of WMF and a possible reaction mechanism was derived. Based on that, the influence of magnetic field flux intensity, pH and the reusability of ZnO@Fe3O4 were investigated in this paper.
1. Introduction
Nowadays, water scarcity is gradually increasing as a worldwide problem due to accelerating population growth and environmental pollution.1 Thus, efficient and cost-effective techniques for wastewater treatment have attracted a significant attention due to the increasing problem of water pollution. Heterogeneous catalysis, as an essential part of Advanced Oxidation Processes (AOPs), plays an important role in addressing the issue of water pollution. The principal mechanism of the heterogeneous catalysis function is the generation of highly reactive radicals.2 Consequently, a combination of heterogeneous catalysis and other technologies like magnetic field, electric field and ultrasonication can expectedly enhance radical generation, which eventually leads to higher oxidation rates.3–5Among them, the applied magnetic field is an efficient and effective method, and it has gradually received considerable critical attention. Most of the correlational research comes to a conclusion that a weak magnetic field (WMF) enhanced the degradation of pollutants, compared with the simple catalytic system.6,7 In addition, several theories on the role of magnetic field have been proposed. For example, in the system of Guan's research, the primary role of WMF in the process of contaminants removal by zero-valent iron (ZVI) was to enhance mass transfer and to promote ZVI corrosion.8 Moreover, Some other studies showed that the effect of magnetic fields on radicals was the reason for the acceleration of reactions, which could be explained by the radical-pair theory.9 However, researches on the magnetic field to enhance the degradation of pollutants in water have been mostly restricted to ZVI and photocatalytic system.10–13 To date, few attempts has been made to investigate the role of WMF in peroxymonosulphate (PMS) system.
Researches have shown that PMS can be effectively activated by many magnetic catalysts.14–16 Among them, ZnO@Fe3O4 is a much attractive material for the disinfection of water due to its high chemical stability and it exhibits strong catalytic efficiency and high adsorption capability. In addition, ZnO@Fe3O4 having optimal magnetic properties and essential surface functionality are most promising materials to resolve various environmental problems due to their easy and rapid separation from solution via magnetic field.17,18 More importantly, the catalytic activity of ZnO@Fe3O4 could be influenced by the magnetic field because of its magnetic properties.19 Based on that, ZnO@Fe3O4 may show a higher activation of PMS in the presence of WMF.
To sum up, the specific objective of this study was focused on the influence of weak magnetic field on the oxidation and degradation of Acid Orange 7 (AO7) by ZnO@Fe3O4/PMS system. This paper attempted to establish the mechanism model of the reaction and to explain the role of WMF in this system. So the main issues addressed in this paper are: (a) the effect of WMF in ZnO@Fe3O4/PMS system, (b) the possible reaction mechanism and the role of WMF and, (c) some other influence factors.
2. Experimental section
2.1. Materials
Zinc acetate dihydrate (≥98%), ferrous chloride tetrahydrate (≥99%), ferric chloride hexahydrate (ACS reagent, 97%) and ammonia solution (25% NH3 in H2O) were procured from Chengdu Kelong Chemical Reagent Co., Ltd. (China). AO7, PMS, furfuryl alcohol, t-butyl alcohol, methanol and benzoquinone were procured from Sigma-Aldrich Co. LLC. (USA). Deionized water was used in all experiments.2.2. Preparation of ZnO@Fe3O4
A stock solution of Zn(Ac)2 was prepared by dissolving Zn(Ac)2·2H2O into deionized water. Ammonia was slowly added to the Zn(Ac)2 solution until it is clear. Then a stock solution of FeCl2 and FeCl3 was prepared in deionized water. The two solutions were added to the above Zn(Ac)2 solution with rapid stirring. After that, the mixture was stirred for a while and then transferred to a hydrothermal reactor. After reaction, on cooling to room temperature, the precipitate was separated centrifugally, washed with twice-distilled water and absolute ethanol, and then dried. Finally, it was calcined in a muffle furnace and then ZnO@Fe3O4 was synthesized.2.3. Batch experiments and catalyst characterization
Batch experiments were carried out to investigate the feasibility of AO7 removal by ZnO@Fe3O4 in the presence of WMF. Several pieces of neodymium-iron-boron permanent magnets (the maximum magnetic field intensity of each one was 10 mT) were employed to offer magnetic field. The axis line of the reactor (a 250 mL beaker) and the center of the magnetic field coincided. This magnetic field was weak and would be referred as weak magnetic field (WMF) hereafter. Unless otherwise specified, two magnets were used at the same time in the experiments.The influence of magnetic field intensity was investigated by fixing pH at 6.0 while varying the magnetic field intensity from about 10 mT to 40 mT (by changing the number of magnets from one to four). Batch reduction tests were conducted by dosing 0.10 g ZnO@Fe3O4 to 200 mL solution containing 7 mg L−1 AO7 to investigate the effect of pH. The pH was varied from 2.0 to 10.0 for different tests. 0.1 mmol L−1 of inhibitors was used in the radicals inhibition experiment and corresponding radicals can be sufficiently inhibited.20–22 If it was not otherwise specified, the experiments were carried out open to the air. Since ZnO@Fe3O4 is ferromagnetic, the presence of WMF can induce the aggregation of ZnO@Fe3O4, which may subsequently affect AO7 removal by ZnO@Fe3O4. So the solution was mixed at 300 rpm using a mechanical stirrer (D2004W, Shanghai Sile Instrument Co., Ltd). The AO7 concentration remained in solution was determined by measuring the absorbance of the solution at 484 nm on a Cary 50 UV-vis spectrophotometer (Varian, USA). The concentrations of iron and zinc leached into reaction solutions were monitored by spectrophotometry and atomic absorption spectroscopy (AAS, Analyst 300, P. E. Inc.). All batch experiments lasted for 30 min. The experimental device was constructed as shown in Scheme S1.†
After batch tests, ZnO@Fe3O4 were collected by magnetic separation, washed with deionized water and vacuum dried at 50 °C for 1 h. Such a pre-treatment could not change the particle size and crystalline structure of ZnO@Fe3O4 samples. The crystalline structure of various samples was analysed using powder X-ray diffraction (XRD). The spectra were obtained on a Bruker D8-Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5418 A), with accelerating voltage and current of 40 kV and 40 mA, respectively. Scanning electron microscopy (SEM), performed on a FEI Inspect F50, was used to evaluate the morphology, size and textural information of the samples. The surface chemical compositions of ZnO@Fe3O4 were identified by X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi, ThermoFisher Scientific) using the Mg Kα line (ht = 1253.6 eV) as an excitation source. The pressure in the XPS analysis chamber was maintained at 109 mbar. The binding energies of all peaks were referenced to the C (1s) line (285.0 eV). After Shirley background subtraction, the spectra were analyzed by the XPSPeak 4.1 Program. The magnetic hysteresis loop measurement for the pristine ZnO@Fe3O4 was carried out using a vibrating sample magnetometer (VSM; Lake Shore 735, Lake Shore Cryotronics, Westerville, OH).
3. Results and discussion
3.1. Influence of WMF on AO7 removal by ZnO@Fe3O4/PMS system
The WMF effect on AO7 removal by ZnO@Fe3O4 was investigated at pH 6.0, as shown in Fig. 1. In control experiments, almost no disappearance of AO7 was observed when only PMS was added into the solution of AO7 both in the presence and absence of WMF (Fig. S1†). However, as shown in Fig. 1, the ZnO@Fe3O4 without PMS removed 10% of AO7 within 30 min, which stemed from the adsorption of the catalyst. Furthermore, the degradation rate was not promoted by WMF, which could indicate that the magnetic field did not affect the adsorption of the catalyst.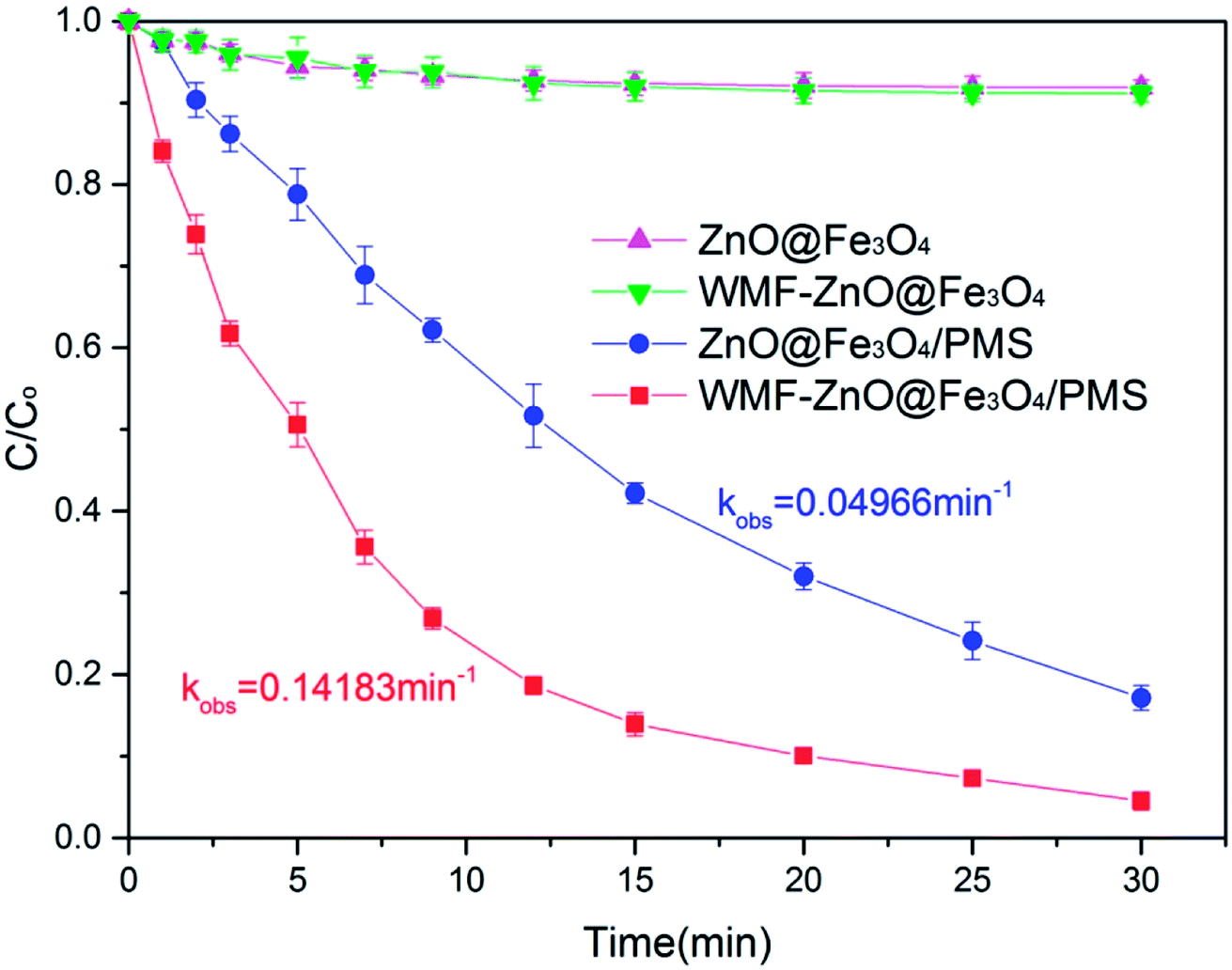 | ||
| Fig. 1 WMF accelerated AO7 removal by ZnO@Fe3O4/PMS system. Reaction conditions: [PMS]0 = 5 × 10−4 M or 0 M, [AO7]0 = 0.02 mM, [ZnO@Fe3O4]0 = 0.5 g L−1, pH = 6.0. C and C0 are AO7 concentration at time t and initial. | ||
After adding PMS, the ZnO@Fe3O4 removed 80% of AO7 within 30 min. The results indicated that ZnO@Fe3O4 may activate the PMS to remove organic pollutants. Turning now to the experimental in the presence of WMF, the ZnO@Fe3O4 exhibited much higher catalytic activity and removed 95% of AO7 within 30 min. Moreover, it removed more than 80% within only 12 min, which is a 30% improvement over the absence of WMF. Thus, the necessary reaction time to achieve complete AO7 removal could be greatly shortened by introducing WMF.
The kinetics of AO7 elimination with or without WMF could be well simulated with pseudo-first-order rate law (eqn (1))
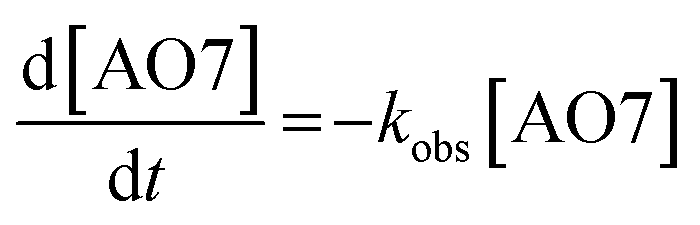 | (1) |
3.2. Influence of radical inhibitors on AO7 removal by ZnO@Fe3O4/PMS system
To speculate the role of WMF in this system, it is necessary to identify the nature of radicals generated from the decomposition of PMS catalyzed by ZnO@Fe3O4 and compare the radical production under the condition of WMF. According to relevant research,23 OH˙, SO4˙−/SO5˙− and O2˙− radicals may exist in the system. Hence, both in the presence and absence of WMF, different radical inhibitors were added to the system of ZnO@Fe3O4/PMS.When excess furfuryl alcohol was added to act as inhibitors of various radicals, the AO7 degradation was greatly suppressed and only about 10% of AO7 was removed within 30 min both in the presence and absence of WMF (Fig. 2a). It was roughly consistent with AO7 degradation by the adsorption of the catalyst which further illustrated that the degradation of AO7 mainly came from the oxidation of radicals. Besides, the AO7 degradation rate was almost the same with WMF. This was consistent with the above result that the adsorption of the ZnO@Fe3O4 was not affected by the magnetic field.
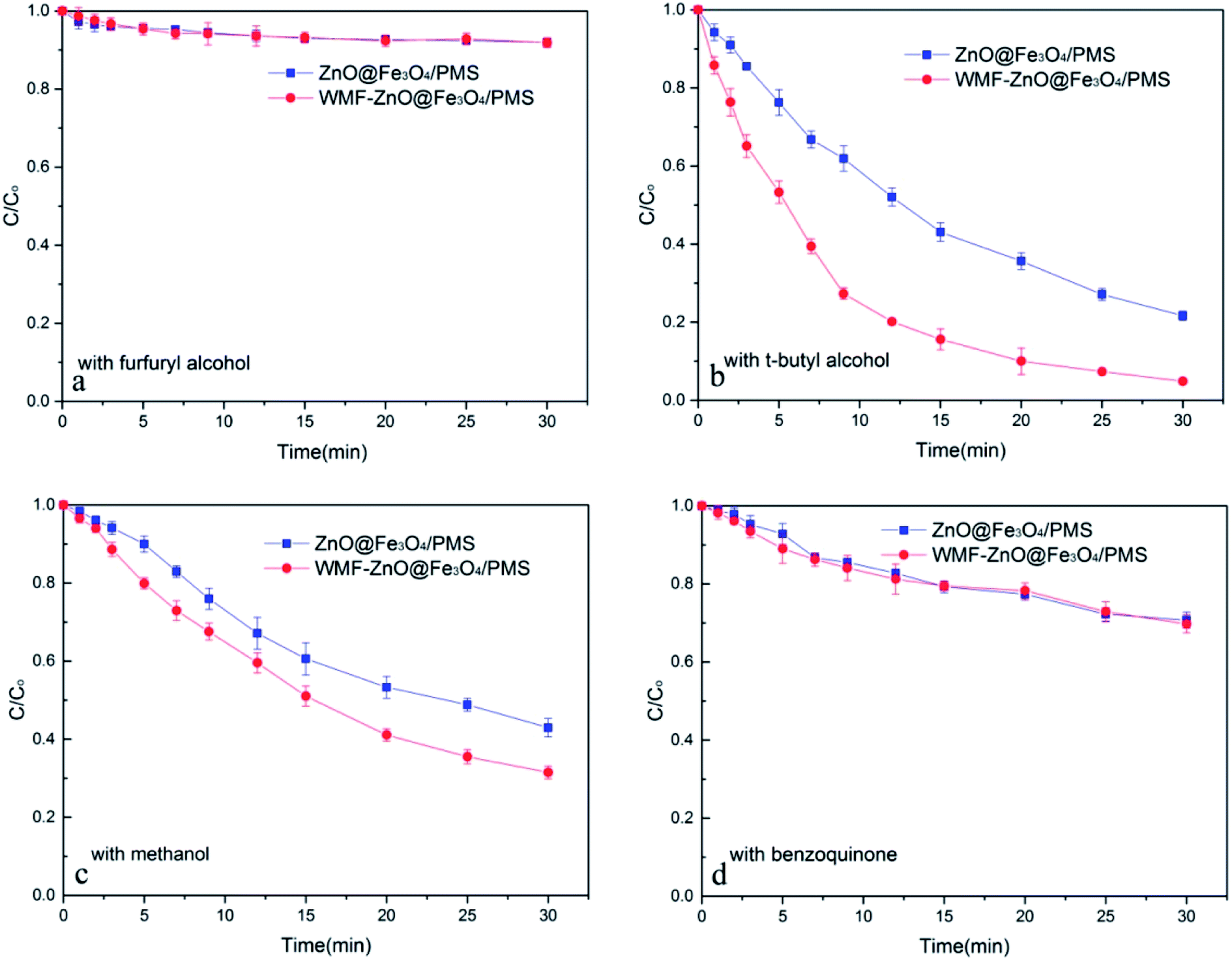 | ||
| Fig. 2 The influence of radical inhibitors on the AO7 removal by ZnO@Fe3O4/PMS system. Reaction conditions: [PMS]0 = 5 × 10−4 M, [AO7]0 = 0.02 mM, [ZnO@Fe3O4]0 = 0.5 g L−1, pH = 6.0. | ||
As shown in Fig. 2b, the addition of excess t-butyl alcohol as a well-known OH˙ radical scavenger, has little affect on the AO7 degradation. Both in the presence and absence of WMF, the degradation rates of AO7 were almost the same whether or not t-butyl alcohol added (compared with Fig. 1). Therefore, there was almost no OH˙ radical in this system. Even with WMF, the generation of OH˙ radical has not been promoted. In the control experiments, AO7 was hardly degraded by ZnO@Fe3O4/H2O2 system (Fig. S2†), which suggested that ZnO@Fe3O4 did not have the ability to activate peroxy species to produce OH˙ radicals.
In the absence of WMF, when excess of methanol was added to act as the SO4˙−/SO5˙− radicals scavenger, the AO7 degradation was suppressed by about 30% (Fig. 2c). Surprisingly, in the presence of WMF, the degradation rate of AO7 was different. As shown in Fig. 2c, the AO7 degradation was also suppressed by methanol in the presence of WMF, which showed the presence of SO4˙−/SO5˙− radicals in the system. However, comparing with no WMF system, the removal rate of AO7 was increased. It is speculate that other radicals may play an important role and be affected by WMF. According to related research,23 O2˙− radicals may exist in this system. And when excess of benzoquinone was added to act as the O2˙− radicals scavenger, the AO7 degradation was suppressed by about 60% and the degradation rate was almost the same in the presence of WMF (Fig. 2d). These evidences demonstrates that both SO4˙−/SO5˙− and O2˙− radicals were formed in the ZnO@Fe3O4-participating catalytic reaction, but O2˙− radicals exhibited a stronger effect than SO4˙−/SO5˙− radicals in the AO7 degradation by PMS catalyzed by ZnO@Fe3O4. In addition, the results illustrated that the WMF did not affect the SO4˙−/SO5˙− radicals but the O2˙− radicals in ZnO@Fe3O4/PMS system. The production rate of O2˙− radicals increased in the presence of WMF thereby enhancing the AO7 degradation in ZnO@Fe3O4/PMS system. Since O2˙− radicals were important in this system, it was necessary to check the origin of O2˙− radicals. Hence, continuous N2 purging was carry out to explore the role of O2 on the generation of O2˙− radicals. As shown in Fig. S3,† in the absence of dissolved oxygen, the AO7 degradation reduced only 6% in the absence of WMF and 7% in the presence of WMF, compared to experiments in open air (Fig. 1). It suggested that although O2 did play a role in the system, it was not a significant factor. Hence, it can be speculated that most of O2˙− radicals was generated by the reaction of ZnO@Fe3O4 with PMS.
3.3. Reaction mechanism of ZnO@Fe3O4/PMS system and the roles of WMF
In order to further reveal the reaction mechanism, it is necessary to figure out how the radicals produced and how the WMF acted on the radicals. And the generation of radicals is closely related to the surface properties of the catalyst in heterogeneous catalytic reactions.24 Therefore, XRD and SEM were used to analyze the characteristics of catalyst. X-ray powder diffraction was used to identify the components of three samples as shown in Fig. 3. The observed diffraction peaks at 2θ = 30.0°, 35.4°, 43.0°, 53.4°, 56.9° and 62.5° correspond to the (220), (311), (400), (422), (511) and (440) planes of Fe3O4 (JCPDS no. 19-0629).25–29 And it showed diffraction peaks at 2θ = 31.7°, 34.4°, 36.2°, 47.5°, 56.5°and 62.8°, which correspond to the (100), (002), (101), (102), (110) and (103) Miller indices assigned to ZnO hexagonal wurtzite lattice (JCPDS, 36-1451).30 By comparing and analyzing the XRD patterns of the three samples, the proportion of the components of the three samples was obtained, shown as Table 1. The content of ZnO decreased in the sample after use, since ZnO reacted with the PMS causing Zn2+ to dissolve.31 And Table 1 also indicated that the reaction was hardly affected by WMF (comparing with the sample 1 and sample 2). Fig. 3 also gives the SEM image of the three samples. The surface of ZnO@Fe3O4 was not corrosion after catalytic experimentation and even with WMF. We then demonstrated that the catalytic reduction of PMS was induced predominately by the heterogeneous catalysis on the surface of ZnO@Fe3O4, because the iron concentration leaching into the solutions was very low (less than 0.015 mg L−1) both in the presence and absence of WMF, which could not yield a marked activation of PMS.23 And the zinc concentration leaching into the solutions was about 5.8 mg L−1, which came from the reaction between ZnO and PMS.31 This reaction might be a small amount which would hardly cause corrosion of the catalyst. And in the control experiments, 5.8 mg l−1 Zn2+ could not activate the PMS (Fig. S4†). In addition, the concentrations of iron and zinc leaching into reaction solutions did not change in the presence of WMF. The same conclusion was drawn with the catalyst characterization. This is different from the report of Ren et al., which concluded that the corrosion of the ZVI increased with WMF.11 However, this study found that the catalytic activity of ZnO@Fe3O4 was improved by the magnetic field but the corrosion of it did not increase, which could reduce the secondary pollution caused by the dissolution of the catalyst.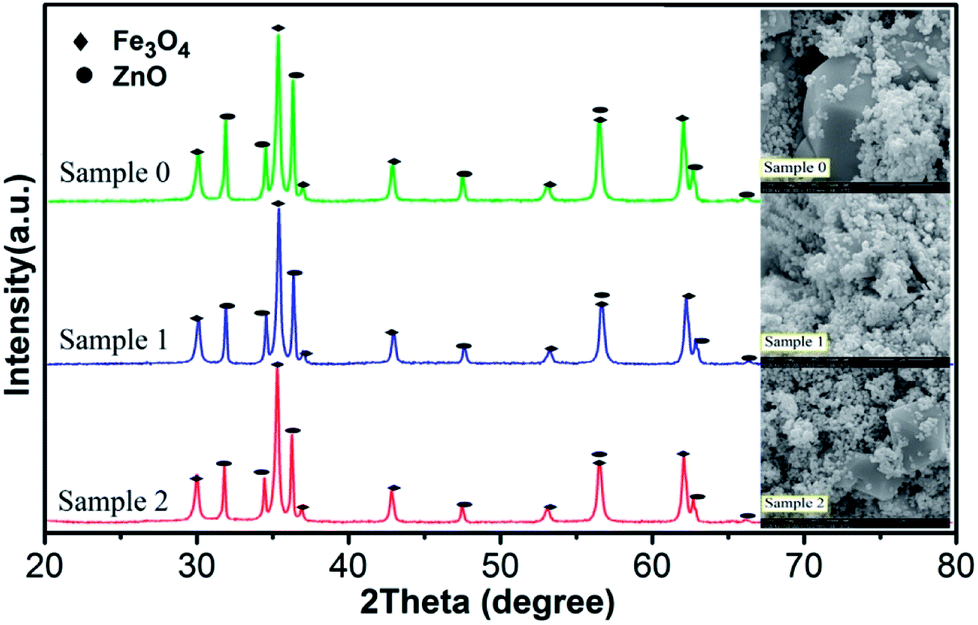 | ||
| Fig. 3 XRD patterns and SEM images of ZnO@Fe3O4: Sample 0, before used; Sample 1, after catalytic experimentation without WMF; Sample 2, after catalytic experimentation with WMF. | ||
| Fe3O4 | ZnO | |
|---|---|---|
| Sample 0 | 57.76% | 42.24% |
| Sample 1 | 63.30% | 36.70% |
| Sample 2 | 63.63% | 36.37% |
In order to analyze the effect of WMF on the catalyst, XPS was used for the analysis of the proportion of Fe3+ and Fe2+ in the catalyst. As shown in Fig. 4 and Table 2, the proportion of Fe3+ in the catalyst was increased and Fe2+ was reduced after the experiment without WMF. Nevertheless, in the presence of WMF, the proportion of Fe2+ and Fe3+ in the catalyst was almost the same as the freshly-made catalyst, which showed that WMF promoted the transition and cycle of Fe3+ and Fe2+ on catalyst surface. Further analysis showed that surface reactions between PMS and Fe3+ may make a large contribution to the PMS-activating ability of ZnO@Fe3O4 in the presence of WMF. Combined with the analysis of the radicals, presumably, O2˙− radicals might originate from the reaction of Fe3+ and PMS, which was promoted in the presence of WMF.
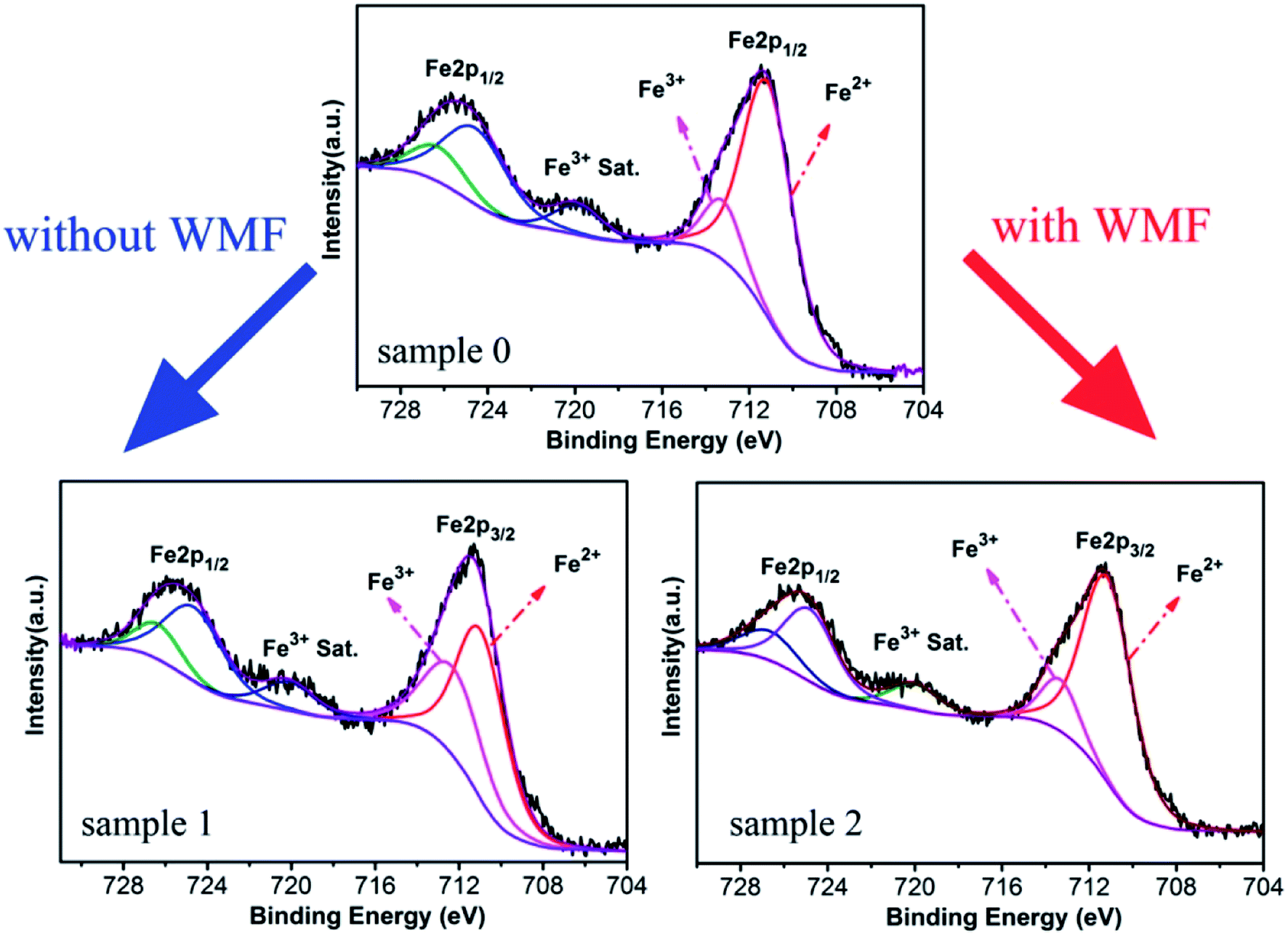 | ||
| Fig. 4 XPS spectra of ZnO@Fe3O4: Sample 1, after catalytic experimentation without WMF; Sample 2, after catalytic experimentation with WMF. | ||
| Fe3+ | Fe2+ | |
|---|---|---|
| Sample 0 | 16.24% | 83.76% |
| Sample 1 | 38.46% | 61.54% |
| Sample 2 | 18.05% | 81.95% |
Based on the above discussions and other related researches, the likely catalytic mechanism for the activation of PMS by ZnO@Fe3O4 and the role of WMF is proposed in Scheme 1. PMS molecules might be adsorbed on the surface of ZnO@Fe3O4 and then activated by the bound Fe2+ and Fe3+ to generate SO4˙−/SO5˙− and O2˙− radicals. At the same time, WMF contributed to the transition of Fe2+ and Fe3+.23,32 In addition, ZnO partially reacted with PMS to produce SO4˙−/SO5˙− radicals,31 which could cause ZnO to dissolve, but as mentioned previously, it could not be affected by the magnetic field. Besides, most of the ZnO remains in the catalyst. Because of the high oxidizing ability of radicals, the generated SO4˙−/SO5˙− and O2˙− radicals initiate the subsequent degradation of AO7. The role of WMF could be explained by the radical-pair theory of magneto chemistry.9,33,34 The radical pair may be classified as a singlet or as a triplet radical pair according to a vector model. In a reaction system, triplet–singlet (T–S) intersystem crossing involves conversion of T+, T−, or To to S. Moreover, the rate of T–S intersystem crossing in an indirect fashion depends on the magnetic field strength, since an external field caused T+ and T− to split in energy from S. As a result, conversion of T+ and T− to S was slowed down with magnetic field, which increased the reaction between radicals and AO7. Hence, the production rate of radicals was increased since the consumption of radicals has been promoted. And it was the way that WMF promoted the reaction. Moreover, at the same time, the faster consumption of radicals also promoted the reaction of Fe3+ and PMS. Consequently, the transition of Fe3+ to Fe2+ was promoted by WMF which may improve the recycling capacity of the catalyst.
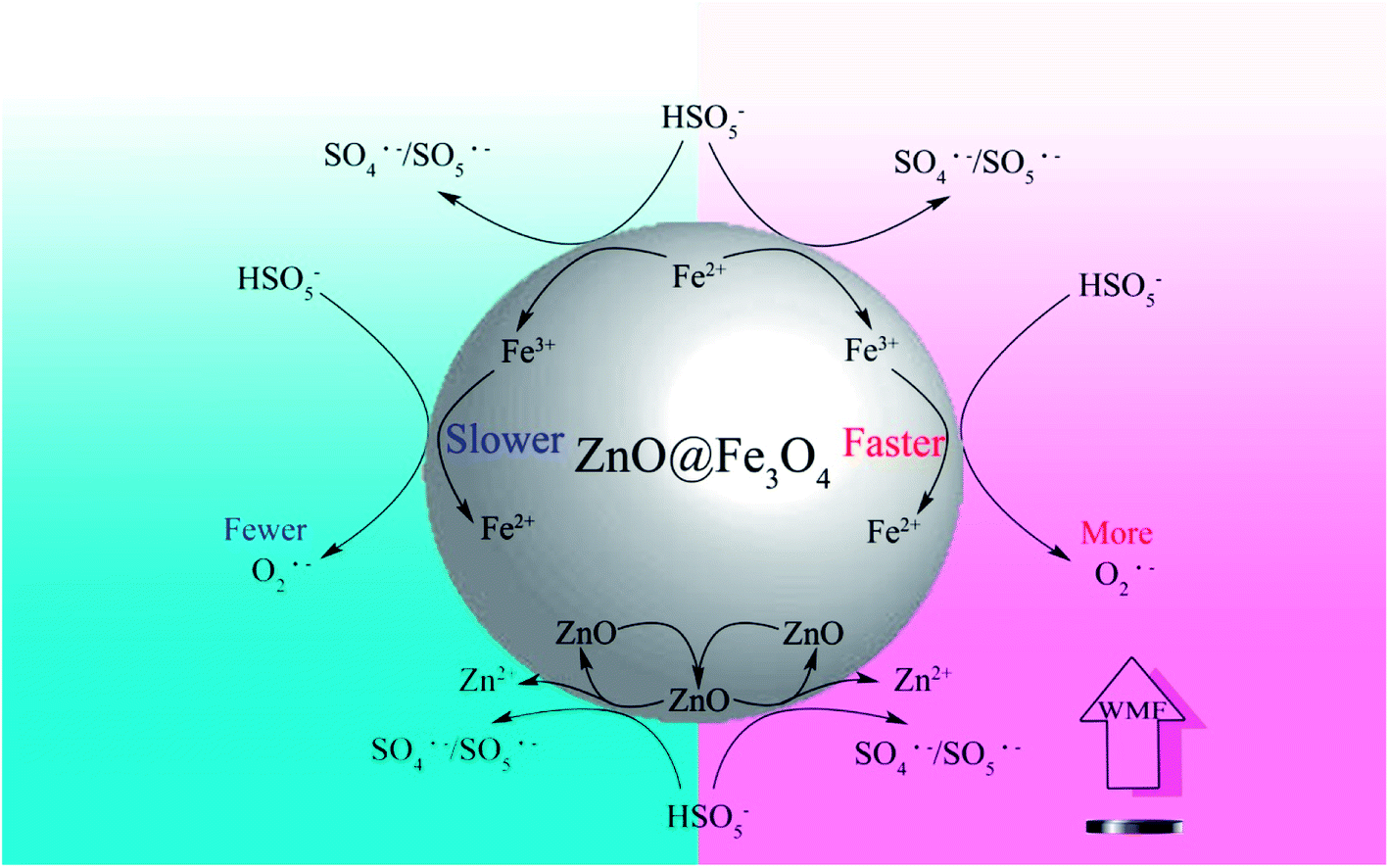 | ||
| Scheme 1 A mechanism for WMF enhancing the activation of PMS on ZnO@Fe3O4 in the degradation of organic pollutants. | ||
3.4. Influence of magnetic field intensity on AO7 removal by ZnO@Fe3O4/PMS system
The intensity of magnetic field is an important factor in the applied magnetic field, which would ultimately affect the removal effect of AO7. Therefore, the feasibility of AO7 removal by ZnO@Fe3O4 under different magnetic field intensity was investigated and shown in Fig. 5a. According to the magnetic field intensity from about 10 mT to 40 mT, the experimental conditions were divided into WMF1 to WMF4. With WMF of different intensities, the degradation rate of AO7 was all increased. The ZnO@Fe3O4 with PMS exhibited a higher catalytic activity when the magnetic field intensity was about 20 mT and removed about 95% of the added AO7 within 30 min. As the intensity of the applied magnetic field increased gradually, the auxiliary function of the WMF was also observed, but the enhancement effect gradually decreased. And it removed about 85% of the added AO7 within 30 min with the magnetic field of 40 mT, only about 5 percent higher than in the absence of WMF. The effect of WMF was similar when the magnetic field intensity was about 10 mT and 30 mT. As shown as Fig. 5b, The AO7 sequestration rate constant enhanced from 0.04966 to 0.14183 min−1 with increasing the magnetic field flux intensity from 0 to 20 mT. Although the rates of AO7 removal by ZnO@Fe3O4/PMS progressively decreased to 0.08797 and 0.07059 min−1, respectively, by increasing the flux intensity of magnetic field to 30 and 40 mT, they were larger than that by ZnO@Fe3O4/PMS without WMF. To provide a generalized basis for the comparison among different magnetic field intensity, the ratio (RWMF) of kinetic constants measured with and without the WMF was calculated, according to eqn (2).
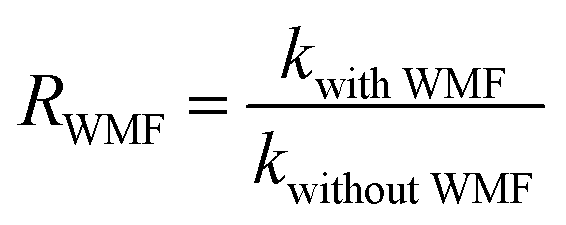 | (2) |
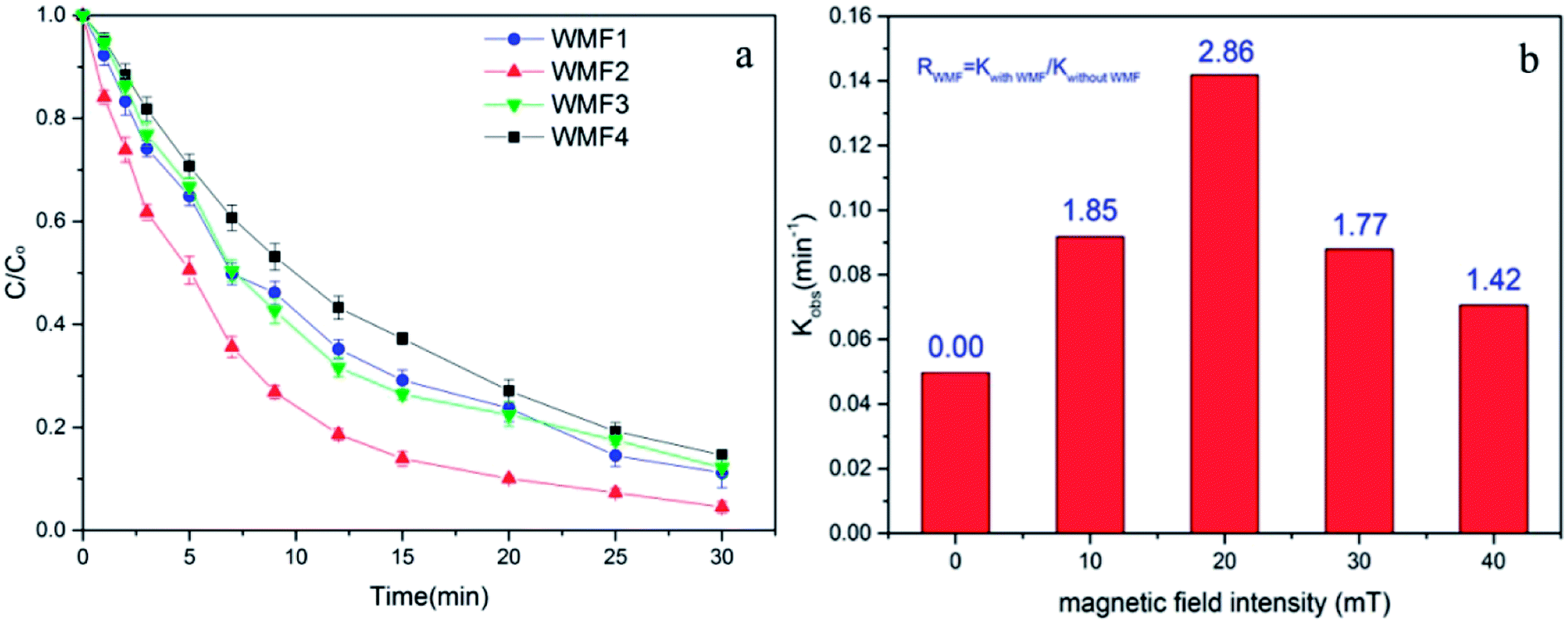 | ||
| Fig. 5 (a) Influence of WMF on AO7 removal by ZnO@Fe3O4/PMS system at different magnetic field intensity vary from 10 mT to 40 mT. (b) The pseudo-first-order rate constants of AO7 removal by ZnO@Fe3O4/PMS system and RWMF as a function of the MF flux intensity. Reaction conditions: [PMS]0 = 5 × 10−4 M, [AO7]0 = 0.02 mM, [ZnO@Fe3O4]0 = 0.5 g L−1, pH = 6.0. | ||
Explained by the radical-pair theory, the rate of T–S intersystem crossing related to the magnetic field strength.32 When the conversion is sufficiently inhibited, the role of magnetic field reaches saturation. Then the production rate of radicals would not increase with strong magnetic field. In addition, since ZnO@Fe3O4 is ferromagnetic, it might agglomerate in water. Hence the number of catalyst surface active point might reduce with a strong magnetic field, reducing the removal rate of AO7.
3.5. Influence of WMF on AO7 removal by ZnO@Fe3O4/PMS system at different pHs
In addition to the intensity of the applied magnetic field, the pH levels were also an external influence factor. The WMF effect on AO7 removal by ZnO@Fe3O4 at different pH was investigated over the pH range of 2.0–10.0 to know the WMF effects at different pH levels. Fig. 6 shows the WMF effects on AO7 removal by ZnO@Fe3O4 at various pH levels. In the absence of WMF, the AO7 removal rate increased with increasing pH from 2.0 to 8.0 but declined with a further elevation in pH. And the removal rates were very similar at pH 6.0 and 8.0. After the application of WMF, the AO7 removal rates were increased considerably over the pH range of 2.0–6.0 and declined with a further elevation in pH. The results showed that the effect of WMF related to the pH levels of the solution. This might be due to the fact that pH affected the properties and yield of radicals. In the solution with a high pH, SO4˙− radicals reacted with OH− to form OH˙ radicals.35 However OH˙ radicals would consume O2˙− radicals,36 which could slow the catalytic reaction. It is worth noting that this process was limited by yield of SO4˙− radicals so the reaction rate would not be greatly reduced. Since the consumption of O2˙− radicals, the promotion of WMF was reduced. In addition, there was an unexpected outcome at pH 2.0. This might be related to the zeta potential and electrostatic adsorption of the catalyst surface. Therefore, thezZeta potential of the catalyst was analyzed. The zeta potential of ZnO@Fe3O4 at pH 2 is +10.13, much greater than +2.88 at pH 4. Since AO7 is a negative dye, the positive charge on the surface of the catalyst will interact with AO7. It can be inferred that electrostatic adsorption of the catalyst at pH 2 leaded to rapid removal of AO7 in the first few minutes. However, in strong acidic conditions, the activation of PMS by ZnO@Fe3O4 was reduced so the degradation rate of AO7 became very slow since then. Besides, it could be seen that WMF had little effect on the electrostatic adsorption of ZnO@Fe3O4.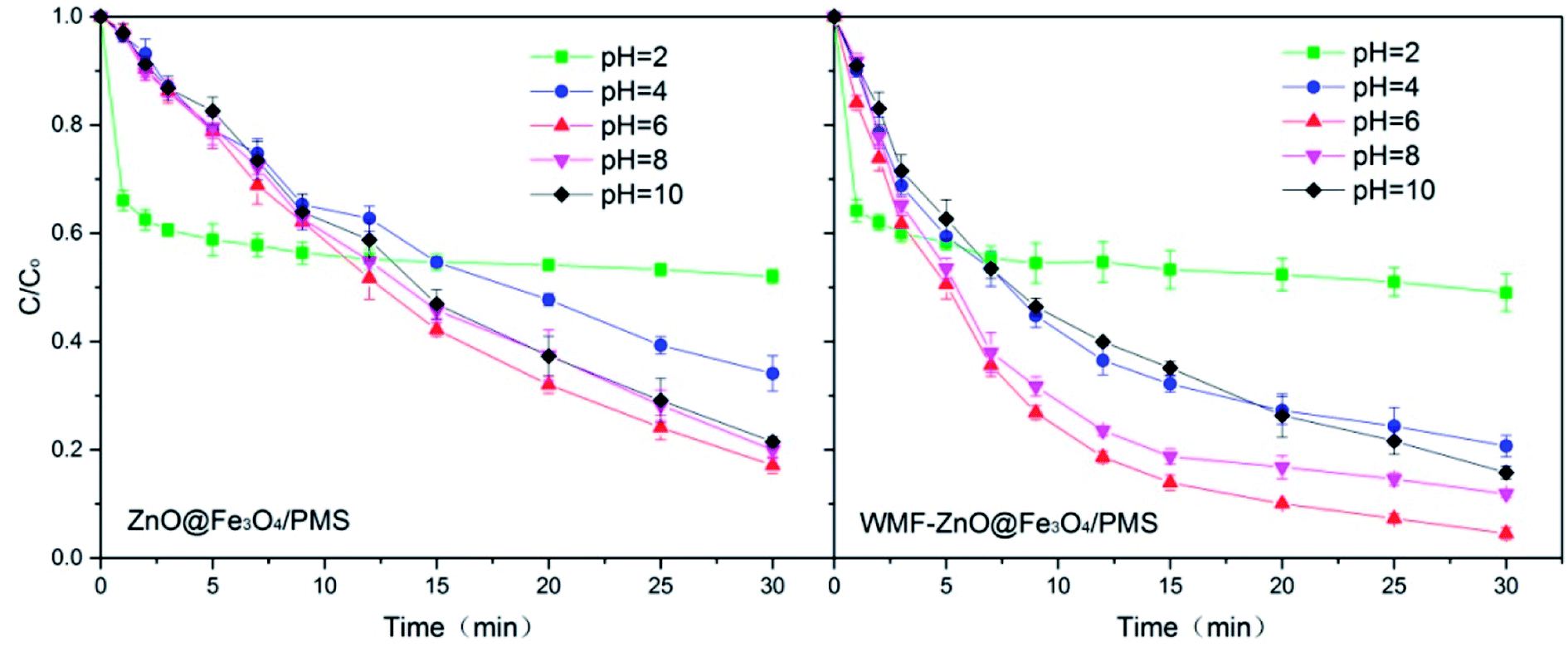 | ||
| Fig. 6 Influence of WMF on AO7 removal by ZnO@Fe3O4/PMS system at different pH levels vary from 2.0–10.0. Reaction conditions: [PMS]0 = 5 × 10−4 M, [AO7]0 = 0.02 mM, [ZnO@Fe3O4]0 = 0.5 g L−1. | ||
3.6. Magnetic properties and reusability of ZnO@Fe3O4
Since the influence of WMF is related to the magnetism of the catalyst, the magnetic properties of the film and the hysteresis loop of ZnO@Fe3O4 was investigated, as shown in Fig. 7a. The magnetization was about 6 emu g−1 when the external magnetic field reached 20 mT. In this case, the removal rate of AO7 was the highest in our research. The magnetization might be the key to the effect of the catalyst.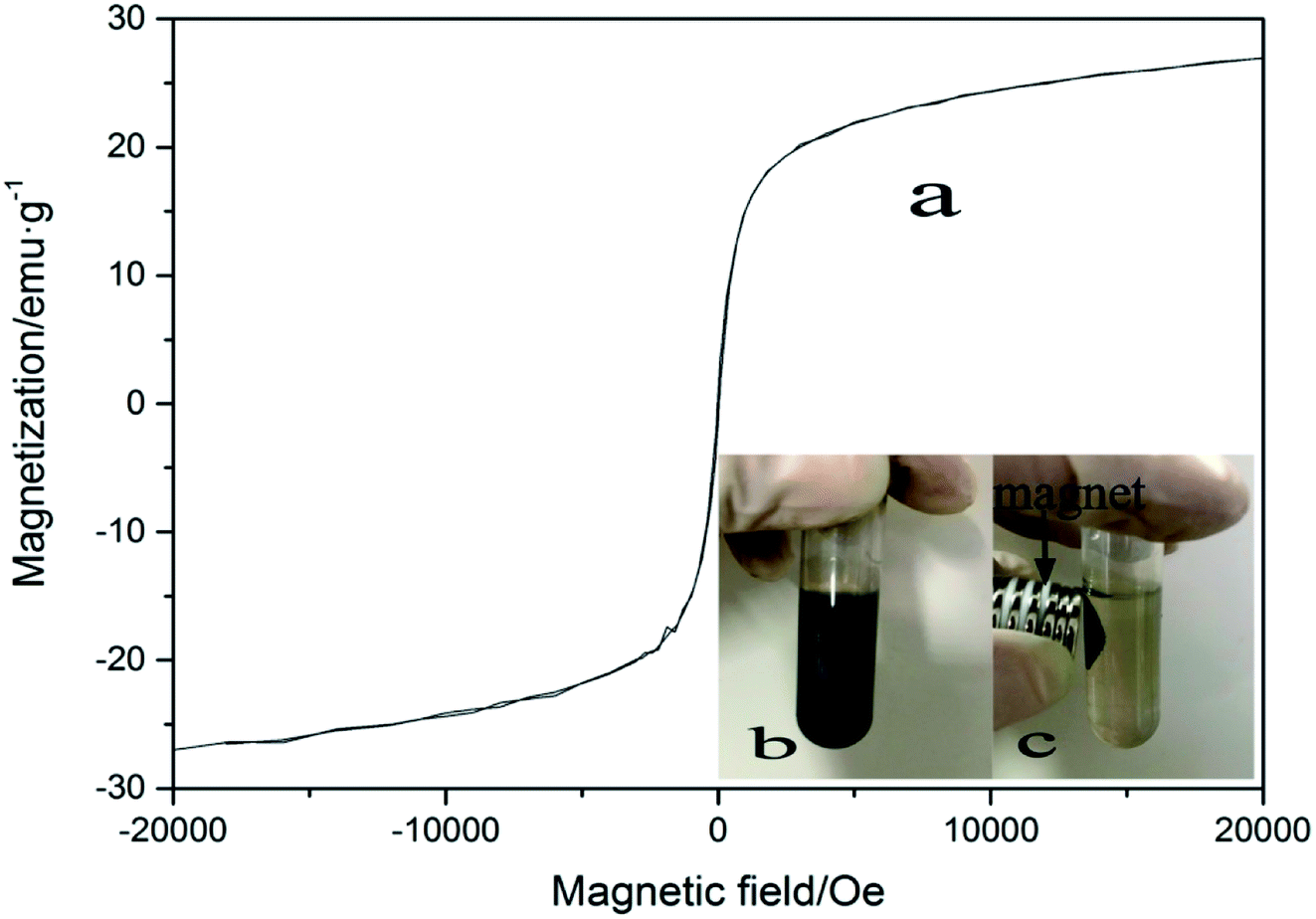 | ||
| Fig. 7 (a) VSM curves of ZnO@Fe3O4 (b) image of ZnO@Fe3O4 dispersed in pure water (c) image of ZnO@Fe3O4 external magnet. | ||
As the reusability of the catalyst is an important parameter of the degradation process, the repetitive use of ZnO@Fe3O4 was investigated in this study. A suspension of ZnO@Fe3O4 in pure water is shown in Fig. 7b. When a magnet was applied to the suspension, the magnetic catalyst immediately accumulated to the side of the centrifuge tube near the magnet, leaving the solution colourless (Fig. 7c), and demonstrating a good magnetically separating property.
AO7 degradation with recycled ZnO@Fe3O4 was investigated under the same conditions used for the freshly-made catalyst (Fig. 8). After four cycles, about 87% of the added AO7 was removed with WMF. And it still increased AO7 degradation by 13% within 30 min. The results showed the high reusability and stability of the catalyst and the effect of WMF on the recycled catalyst. And the reason for the slight decrease in degradation rate might be the reaction between ZnO and PMS, but it was a just small amount so the ZnO@Fe3O4 can maintain a high catalytic activity after repeated use.
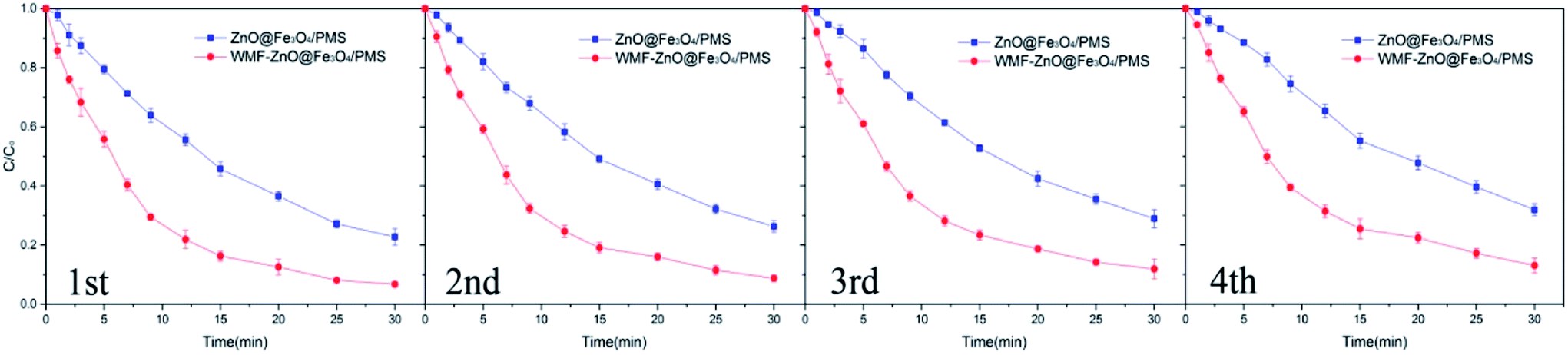 | ||
| Fig. 8 Influence of WMF on AO7 removal in four consecutive cycles of ZnO@Fe3O4. Reaction conditions: [PMS]0 = 5 × 10−4 M, [AO7]0 = 10 mg L−1, [ZnO@Fe3O4]0 = 0.5 g L−1. | ||
4. Conclusions
The results of this study revealed the enhancing effect of WMF on AO7 removal by ZnO@Fe3O4/PMS system, which was of practical and fundamental importance to the environmental decontamination by the system. Furthermore, the corrosion of the catalyst did not increase with WMF which showed the catalytic reaction was induced predominately by the heterogeneous catalysis on the surface of ZnO@Fe3O4. Besides, the results revealed that both SO4˙−/SO5˙− and O2˙− radicals are involved in the catalytic reactions, but the O2˙− radicals are the mainly radicals affected by WMF. The production rate of O2˙− radicals increased in the presence of WMF. In addition, the faster production and consumption of radicals promoted the reaction of Fe3+ and PMS which promoted the transition of Fe3+ to Fe2+ on the catalyst surface. The WMF effect observed in this study could be explained with the theory of radical-pair. Furthermore, the catalytic effect of the system was different beyond the appropriate magnetic field flux intensity and pH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Appreciation and acknowledgment are given to the National Natural Science Foundation of China (No. 51508353).References
- R. L. Mcginnis and M. Elimelech, Environ. Sci. Technol., 2008, 42, 8625–8629 CrossRef.
- T. E. Agustina, H. M. Ang and V. K. Vareek, J. Photochem. Photobiol., C, 2005, 6, 264–273 CrossRef.
- C. He, Y. Xiong and X. Zhu, Thin Solid Films, 2002, 422, 235–238 CrossRef.
- N. L. Stock, J. Peller, K. Vinodgopal and P. V. Kamat, Environ. Sci. Technol., 2000, 34, 1747–1750 CrossRef.
- X. Xiong, J. Gan, Z. Wei and S. Bo, Environ. Sci. Pollut. Res., 2016, 23, 16761–16770 CrossRef PubMed.
- L. Liang, X. Guan, Y. Huang, J. Ma, X. Sun, J. Qiao and G. Zhou, Sep. Purif. Technol., 2015, 156, 1064–1072 CrossRef.
- L. Liang, W. Sun, X. Guan, Y. Huang, W. Choi, H. Bao, L. Li and Z. Jiang, Water Res., 2014, 49, 371 CrossRef PubMed.
- J. Li, H. Qin, W.-X. Zhang, Z. Shi, D. Zhao and X. Guan, Sep. Purif. Technol., 2017, 176, 40–47 CrossRef.
- B. Brocklehurst, Chem. Soc. Rev., 2002, 33, 301–311 RSC.
- C. He, X. Liu, W. Ji and J. Zhao, Water, Air, Soil Pollut., 2016, 227, 1–12 CrossRef.
- J. Du, D. Che, X. Li, W. Guo and N. Ren, RSC Adv., 2017, 7, 18231–18237 RSC.
- W. Zhang, X.-X. Wang, H.-X. Lin and X.-Z. Fu, Acta Chim. Sin., 2005, 63, 1765–1768 Search PubMed.
- W. Zhang, X.-X. Wang, H.-X. Lin and X.-Z. Fu, Acta Chim. Sin., 2005, 63, 715–719 Search PubMed.
- F. Ji, C. Li, Y. Liu and P. Liu, Sep. Purif. Technol., 2014, 135, 1–6 CrossRef.
- S. Muhammad, P. R. Shukla, M. O. Tadé and S. Wang, J. Hazard. Mater., 2012, 215–216, 183–190 CrossRef PubMed.
- Y. Yao, Z. Yang, D. Zhang, W. Peng, H. Sun and S. Wang, Ind. Eng. Chem. Res., 2012, 51, 6044–6051 CrossRef.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef PubMed.
- S. Singh, K. C. Barick and D. Bahadur, J. Hazard. Mater., 2011, 192, 1539–1547 CrossRef PubMed.
- L. E. Bykova, V. G. Myagkov, I. A. Tambasov, O. A. Bayukov, V. S. Zhigalov, K. P. Polyakova, G. N. Bondarenko, I. V. Nemtsev, V. V. Polyakov and G. S. Patrin, Phys. Solid State, 2015, 57, 386–390 CrossRef.
- E. B. Burlakova, Russ. Chem. Rev., 1975, 44, 871 CrossRef.
- R. Palominos, J. Freer, M. A. Mondaca and H. D. Mansilla, J. Photochem. Photobiol., A, 2008, 193, 139–145 CrossRef.
- I. Gültekin, G. Tezcanligüyer and N.-H. Ince, Ultrason. Sonochem., 2009, 16, 577–581 CrossRef PubMed.
- N. Wang, L. Zhu, D. Wang, M. Wang, Z. Lin and H. Tang, Ultrason. Sonochem., 2010, 17, 526–533 CrossRef PubMed.
- Z. R. Ismagilov, S. N. Pak and V. K. Yermolaev, J. Catal., 1992, 136, 197–201 CrossRef.
- H. Yan, J. Zhang, C. You, Z. Song, B. Yu and Y. Shen, Mater. Chem. Phys., 2009, 113, 46–52 CrossRef.
- A. Demir, A. Baykal, H. Sözeri and R. Topkaya, Synth. Met., 2014, 187, 75–80 CrossRef.
- J. Wang, J. Yang, X. Li, D. Wang, B. Wei, H. Song, X. Li and S. Fu, Phys. E, 2016, 75, 66–71 CrossRef.
- Y. Mizukoshi, T. Shuto, N. Masahashi and S. Tanabe, Ultrason. Sonochem., 2009, 16, 525–531 CrossRef PubMed.
- H. Iida, K. Takayanagi, T. Nakanishi and T. Osaka, J. Colloid Interface Sci., 2007, 314, 274 CrossRef PubMed.
- J. Xia, A. Wang, X. Liu and Z. Su, Appl. Surf. Sci., 2011, 257, 9724–9732 CrossRef.
- P. Shukla, I. Fatimah, S. Wang, H. M. Ang and M. O. Tadé, Catal. Today, 2010, 157, 410–414 CrossRef.
- J. Madhavan, P. Maruthamuthu, S. Murugesan and S. Anandan, Appl. Catal., B, 2008, 83, 8–14 CrossRef.
- N. J. Turro and G. C. Weed, J. Am. Chem. Soc., 1983, 105, 1861–1868 CrossRef.
- F. S. Barnes and B. Greenebaum, Bioelectromagnetics, 2015, 36, 45–54 CrossRef PubMed.
- O. S. Furman, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef PubMed.
- C. K. Duesterberg, S. E. Mylon and T. D. Waite, Environ. Sci. Technol., 2008, 42, 8522–8527 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03108h |
| This journal is © The Royal Society of Chemistry 2018 |