
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibody-pHPMA functionalised fluorescent silica nanoparticles for colorectal carcinoma targeting†
Denisa Lizoňováa,
Monika Majerskáa,
Vlastimil Králab,
Michal Pecharc,
Robert Polac,
Marek Kovářd and
František Štěpánek *a
*a
aLaboratory of Chemical Robotics, Department of Chemical Engineering, University of Chemistry and Technology Prague, Czech Republic. E-mail: Frantisek.Stepanek@vscht.cz; Tel: +420 220 443 236
bLaboratory of Structural Biology, Institute of Molecular Genetics of the Czech Academy of Sciences, 142 20 Prague 4, Czech Republic
cLaboratory of Biomedical Polymers, Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského Nám. 2, 162 06 Prague 6, Czech Republic
dLaboratory of Tumour Immunology, Institute of Microbiology of the CAS, v.v.i., Prague, Czech Republic
First published on 13th June 2018
Abstract
The systemic application of highly potent drugs such as cytostatics poses the risks of side effects, which could be reduced by using a carrier system able to specifically deliver the encapsulated drug to the target tissue. Essential components of a nanoparticle-based drug delivery system include the drug carrier itself, a targeting moiety, and a surface coating that minimizes recognition by the immune system. The present work reports on the preparation, in vitro characterization and in vivo testing of a new delivery system consisting of fluorescent silica nanoparticles functionalised with a non-immunogenic stealth polymer poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) and a monoclonal antibody IgG M75 that specifically binds to Carbonic Anhydrase IX (CA IX). CA IX is a promising therapeutic target, as it is a hallmark of several hypoxic tumours including colorectal carcinoma. Uniquely in this work, the monoclonal antibody was covalently coupled to the surface of fluorescently labelled silica nanoparticles via a multivalent amino-reactive co-polymer rather than a traditional bivalent linker. The pHPMA-M75 functionalised SiO2 nanoparticles exhibited excellent colloidal stability in physiological media. Their in vitro characterisation by flow cytometry proved a highly specific interaction with colorectal carcinoma cells HT-29. In vivo study on athymic NU/NU nude mice revealed that the SiO2–pHPMA-M75 nanoparticles are capable of circulating in the blood after intravenous administration and accumulate in the tumour at tenfold higher concentration than nanoparticles without specific targeting, with a considerably longer retention time. Additionally, it was found that by reducing the dose administered in vivo, the selectivity of the nanoparticle biodistribution could be further enhanced in favour of the tumour.
Introduction
It is known that intravenously (i.v.) administered nanoparticles without specific surface coating are quickly removed from the bloodstream because they are recognized by the immune system.1–3 Nanoparticle administration into the bloodstream is usually followed by the adsorption of plasma proteins to the nanoparticle surface (opsonization),1,4,5 which induces macrophage phagocytosis.6,7 The phagocytosed nanoparticles are then decomposed or stored in the liver, spleen or other organs of the Reticulo-Endothelial System (RES).8 Therefore, it is necessary to minimize the protein adsorption if a prolonged nanoparticle circulation is required (e.g. vascular drug delivery, site-specific targeting).9 Several materials have been explored in order to prevent protein adsorption and prepare so called “stealth nanoparticles”.Coating by hydrophilic polymers that act as a steric barrier appears to be the most efficient approach (e.g. polyethylene glycol, polyvinyl pyrrolidone, polyvinyl alcohol etc.).5,10,11 The most commonly used polymer from this group is polyethylene glycol (PEG), which is considered as a “gold standard” for stealth polymers. However, it offers not only advantages but also certain risks (hypersensitivity reactions, anaphylaxis).12–16 Therefore, alternatives to PEG have been intensively explored recently. Co-polymers based on poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) seem very promising in the field of drug–polymer conjugate delivery and a few candidates have already been tested in clinical trials.12,17 pHPMA sufficiently prolongs the blood circulation of such conjugates and thus enables passive tumour accumulation.18–20 Other studies confirmed that pHPMA can create a stealth hydrophilic layer or even enhance the blood circulation in the case of DNA complexes,21 self-assembled vesicles,22 polymeric particles23 or viral vectors.24 However, pHPMA has not been previously combined with silica based drug carriers.
Apart from prolonging blood circulation, accumulation in the tumour can be further enhanced by active targeting to tumour-specific molecules. One such molecule is Carbonic Anhydrase IX (CA IX), a transmembrane protein overexpressed in several commonly occurring carcinomas.25 Thanks to its low expression in the normal tissue,26 it is a promising target for the delivery of anticancer drugs.27 CA IX can be targeted by various moieties such as enzyme inhibitors,47 but in order to achieve a truly specific binding, it has been shown that the monoclonal antibody IgG M75 binds CA IX28 and specifically accumulates in HT-29 human colorectal carcinoma xenografts in vivo (HT-29 cell line expressing CA IX).29 In our previous work it was confirmed that silica nanoparticles bearing IgG M75 antibody can specifically adhere to CA IX antigen under in vitro conditions.30 Therefore, the objective of the present was to prepare silica nanoparticles suitable for in vivo experiments by the covalent coupling of IgG M75 in combination with a stealth polymer pHPMA to the nanoparticle surface, and to prove that such nanoparticles are capable of specific accumulation in tumours expressing CA IX.
Beyond the factors mentioned above, particle size is another important parameter that significantly affects the biodistribution, protein adsorption and pharmacokinetics of i.v. administered nanoparticles. Even though smaller particles generally tend to circulate longer,1,7 the design of multi-compartmental drug delivery devices (e.g. liposome aggregates31) brings certain limitations into the applicable range of particle diameters, especially from below. Silica nanoparticles possess an advantage of tunable size and porosity and thus are intensively studied as carriers for drug delivery.32 Such nanoparticles can be functionalized with targeting moieties and transported to the area of interest.33,34 When modified with an antibody fragment or a whole antibody, silica nanoparticles can accumulate in the tumour tissue, which can be used not only for treatment, but also for diagnostic purposes.35–37 The transported drug or diagnostic probe can be adsorbed to the particle surface or encapsulated in the pores sealed by a gatekeeper system. Moreover, silica is classified as “Generally Recognized as Safe” (GRAS) by the FDA and is used in cosmetics and as a food-additive.32 At present, a phase 2 study is conducted with targeted mesoporous silica nanoparticles for image-guided operative sentinel lymph node mapping.38
Hence, the present work focuses on the preparation, in vitro characterization, in vivo biodistribution and tumour targeting of 170 nm fluorescent silica nanoparticles coated by pHPMA with a covalently attached monoclonal antibody IgG M75 as a new, actively targeted drug delivery system.
Results and discussion
Physico-chemical properties
Fluorescent silica nanoparticles with covalently bound pHPMA (SiO2–pHPMA), pHPMA with antibody IgG M75 (SiO2–pHPMA-M75) or irrelevant isotypic antibody (SiO2–pHPMA-ISO) were prepared and characterized. The nanoparticles exhibited mean diameters ranging from 160 to 180 nm with a polydispersity index under 0.1 (Table 1). The zeta potential of uncoated amino-silica nanoparticles was positive (+27 mV) due to the presence of amino-groups on the nanoparticle surface. Polymers based on pHPMA are generally considered of having zeta potential values close to zero. The covalent coupling of pHPMA chains to the nanoparticle surface is done via the aminolysis of amino-reactive thiazolidine-2-thione (TT) groups. These TT groups might be also hydrolyzed, as a side reaction, with the resulting carboxylic groups in the polymeric side-chain introducing a negative charge to the nanoparticle surface (Table 1). However, when the antibody is attached to the polymer-modified nanoparticles via the TT-groups that were not consumed by the reaction with the nanoparticle surface, the measured zeta potential (Table 1) of the final antibody-functionalised nanoparticles is less negative than in the absence of the antibody (which is also slightly positively charged).| Nanoparticle type | Mean diameter [nm] | Polydispersity index (PDI) [−] | Zeta potential [mV] |
|---|---|---|---|
| SiO2–NH2 | 168 ± 18 | 0.011 | +27 ± 4 |
| SiO2–pHPMA | 169 ± 41 | 0.058 | −20 ± 7 |
| SiO2–pHPMA-M75 | 180 ± 35 | 0.038 | −15 ± 8 |
| SiO2–pHPMA-ISO | 165 ± 36 | 0.004 | −13 ± 7 |
The final amount of polymer and antibody was measured via amino acid analysis and found to be 36.1 μg of pHPMA and 9.5 μg of antibody per 1 mg of nanoparticles (3.6 wt% and 1.0 wt%, respectively), which represents approximately 195 antibody molecules and 2460 pHPMA polymer chains per one nanoparticle, on average. The details of the amino acid analysis and the antibody and polymer quantification are available in ESI 3.†
The results of the morphology and size distribution measurement by Transmission Electron Microscopy (TEM) were in accordance with the data obtained by Dynamic Light Scattering (DLS). The TEM size measurement provided the average nanoparticle diameter of 140 ± 18 nm. Based on the TEM analysis, it can be concluded that nanoparticles were spherical and possessed a narrow size distribution (Fig. 1).
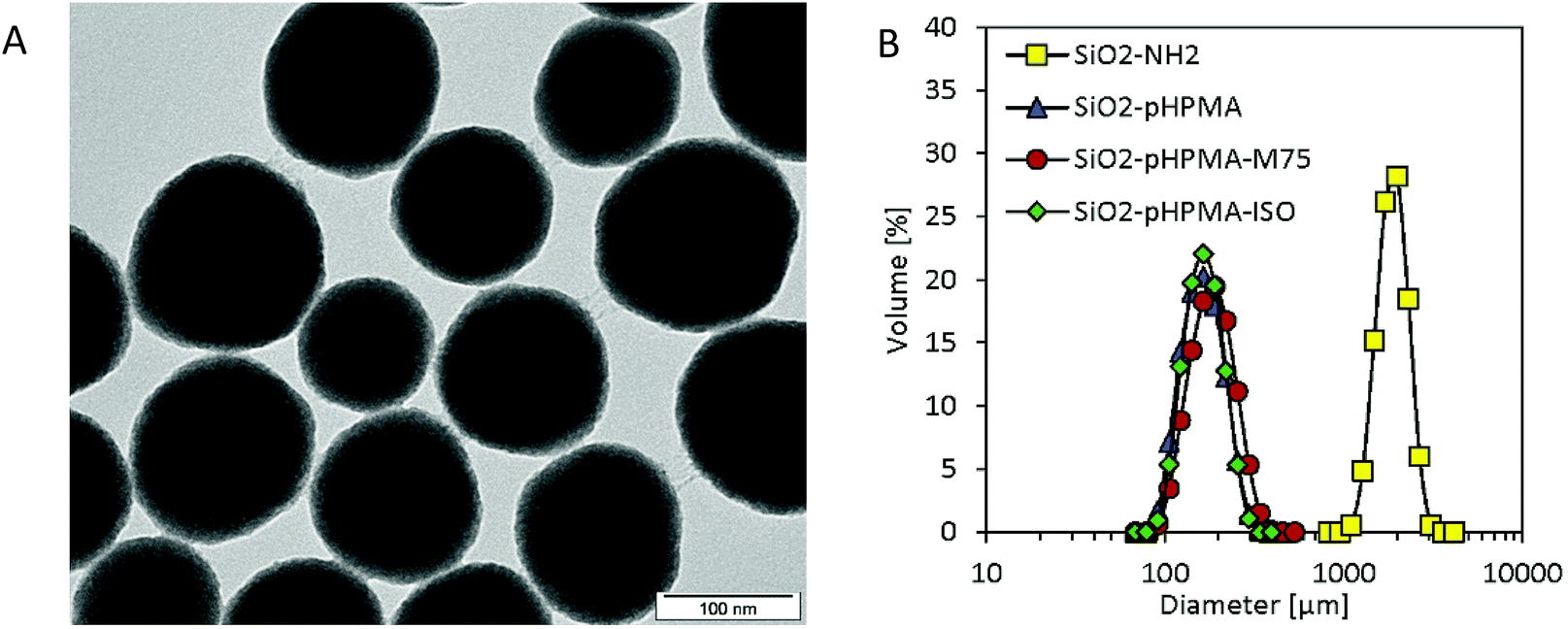 | ||
| Fig. 1 (A) TEM micrograph of SiO2-pHPMA-M75 nanoparticles; (B) DLS measurements in PBS show colloidal stability when pHPMA is present on the nanoparticle surface. | ||
Nanoparticle colloidal stability in the presence of salts was also of interest due to their intended physiological application and was examined by particle size distribution measurements employing DLS (Fig. 1). While uncoated amino-silica nanoparticles aggregated in phosphate buffer saline (PBS, 10 mM; pH 7.4), all three types of pHPMA-modified nanoparticles formed stable colloidal dispersions due to the steric stabilization caused by the hydrophilic pHPMA polymer chains attached to the nanoparticle surface. Even though these particles sediment after two weeks of storage at 4 °C due to their density, they can be quantitatively re-suspended with no change in the particle size distribution.
In vitro behaviour
The ELISA-like test was used to examine the specific interaction of the antibody IgG M75 with the proteoglycan domain of CA IX in fusion with the Maltose Binding Protein (PG-MBP). Fig. 2A shows the data obtained from fluorescence intensity measurement confirming the functionality of the antibody IgG M75 on the SiO2–pHPMA-M75 nanoparticles. At the same time, nanoparticles without the specific antibody (SiO2–pHPMA and SiO2–pHPMA-ISO) did not bind to the PG domain of CA IX. No non-specific interactions of either particle type were observed.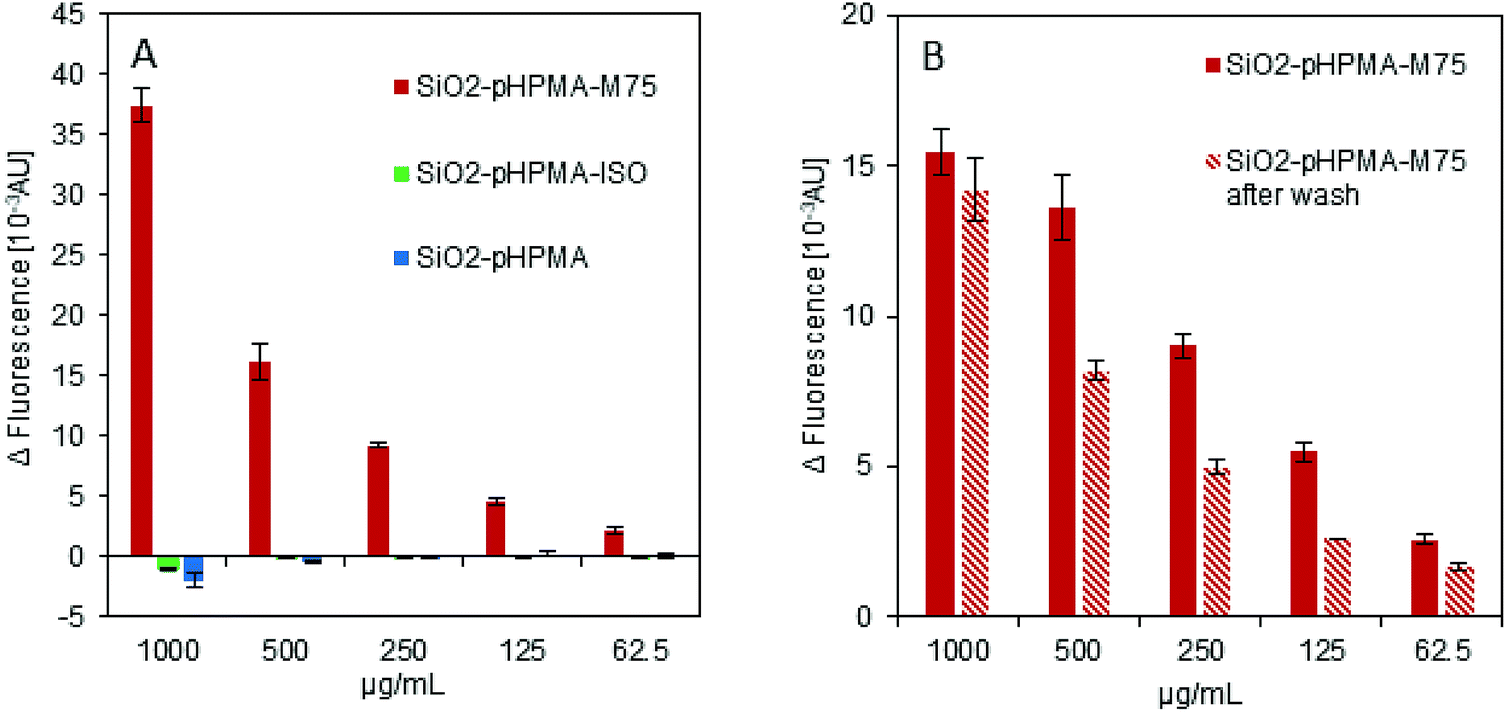 | ||
| Fig. 2 In vitro characterisation of nanoparticle surface functionalisation. (A) ELISA-like test confirms the presence of the active antibody IgG M75 (SiO2–pHPMA-M75). Nanoparticles without the specific antibody (SiO2–pHPMA, SiO2–pHPMA ISO) do not adhere to the antigen-modified surface. (B) ELISA-like test. Filled columns – SiO2–pHPMA-M75 nanoparticles after the synthesis, stripped columns – the same batch of the nanoparticles after their repeated wash with PBS (3×). | ||
Fig. 2B demonstrates the covalent attachment of the antibody to the nanoparticles examined by repeated washing with a buffer (3 times in 1 mL of PBS). The active antibody is present after the washing and therefore it might be concluded that the antibody is covalently attached to the nanoparticle surface rather than simply adsorbed. The fluorescence intensity decrease at some dilutions might be due to the partial nanoparticle loss during the washing steps caused by the fact that pHPMA coating increases the dispersion stability and thus complicates the nanoparticle centrifugation.
The flow cytometry experiments (summarized in Fig. 3) were carried out to prove that only the specific antibody–antigen interactions are responsible for binding of the SiO2–pHPMA-M75 nanoparticles to the target HT-29 cells. In the case of fluorescent nanoparticles adhered to the cell surface, the measured fluorescence signal of the cell is increased. Positive control experiments (free IgG M75 and a secondary antibody GAM-FITC) confirmed the presence of CA IX antigen on the HT-29 cells. At the same time, there were no interactions between the control antibodies and a DLD-1 cell line (which does not express CA IX).
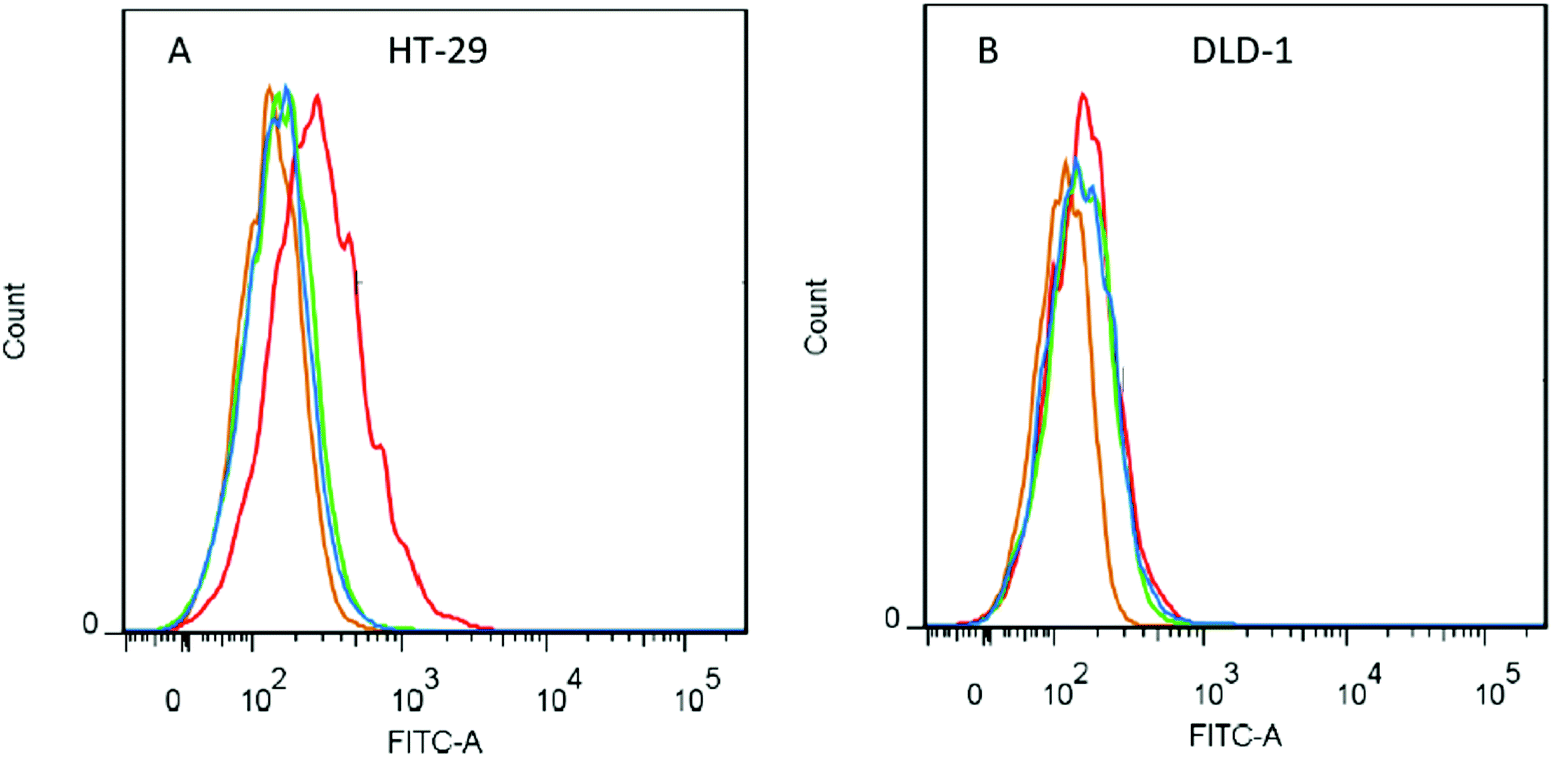 | ||
| Fig. 3 In vitro characterisation of nanoparticle interaction with cells. Flow cytometry experiment with (A) HT-29 cells expressing CA IX, and (B) DLD-1 cells not expressing CA IX. Red – SiO2–pHPMA-M75 nanoparticles; green – SiO2–pHPMA-ISO nanoparticles; blue – SiO2–pHPMA nanoparticles; orange – HT-29(A) or DLD-1(B). | ||
Fig. 3A shows the results of testing with the HT-29 cell line. Since the SiO2–pHPMA-M75 nanoparticles specifically adhere to the cell surface, an increase of fluorescence intensity was detected. There was no signal shift observed in the case of either SiO2–pHPMA or SiO2–pHPMA-ISO nanoparticles. In the case of DLD-1 cells (Fig. 3B), which do not express CA IX, no significant difference in the fluorescence intensity among the tested nanoparticles was observed. The obtained data indicate that the tested nanoparticles do not interact with the cells non-specifically.
The fluorescence microscopy experiments (Fig. 4) showed results consistent with the flow cytometry experiments. Nanoparticles bearing the specific antibody IgG M75 (SiO2–pHPMA-M75) adhered only to the HT-29 cells and not to DLD-1 cells. The uneven distribution of the nanoparticles on the cell surface can be due to the variable level of CA IX expression on the surface of the individual cells. Nanoparticles without the specific antibody did not interact with the HT-29 cells as shown in ESI 1.†
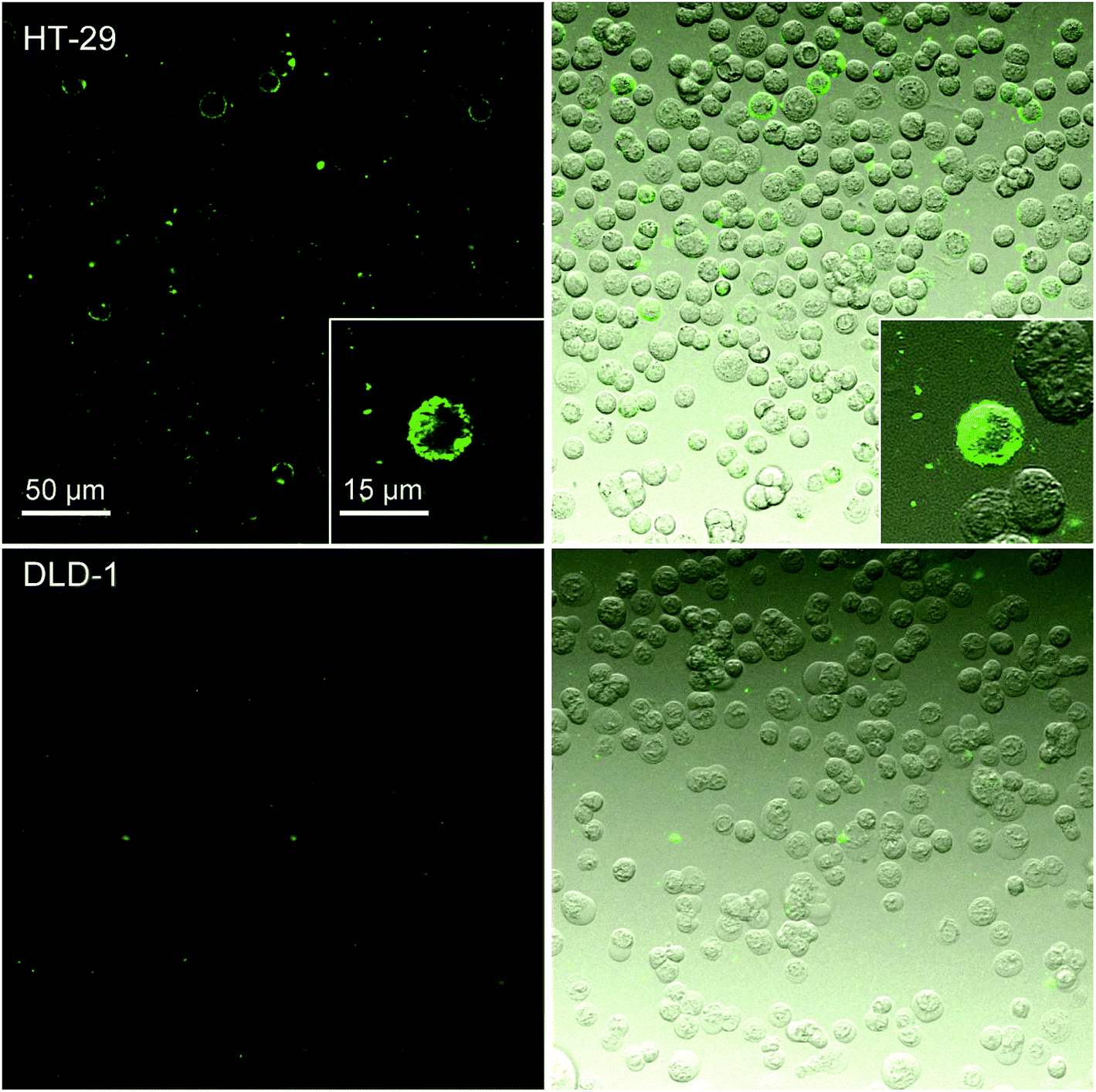 | ||
| Fig. 4 Fluorescence microscopy experiments with SiO2–pHPMA-M75 nanoparticles show specific interaction with HT-29 cells and no interaction with DLD-1 cells (left – fluorescence mode; right – overlay of fluorescence and optical mode). | ||
In vivo behaviour
The nanoparticle biodistribution and specific accumulation in tumour tissue was investigated in athymic NU/NU nude mice bearing HT-29 tumours in two separated in vivo studies. First (in vivo study I), we focused on the circulation times and blood clearance of the stealth SiO2–pHPMA nanoparticles and the tumour targeting of the SiO2–pHPMA-M75 nanoparticles using an initial dose of 75 mg kg−1, which was based on typical values that can be tolerated by the animals.Both types of nanoparticles were detected in the blood of all mice even after 1 hour from the nanoparticle administration (Fig. 5A), which can be considered as significantly prolonged circulation in comparison to other types of particles.3,39 This finding opens up new possibilities for the development of silica–pHPMA drug-delivery particles.
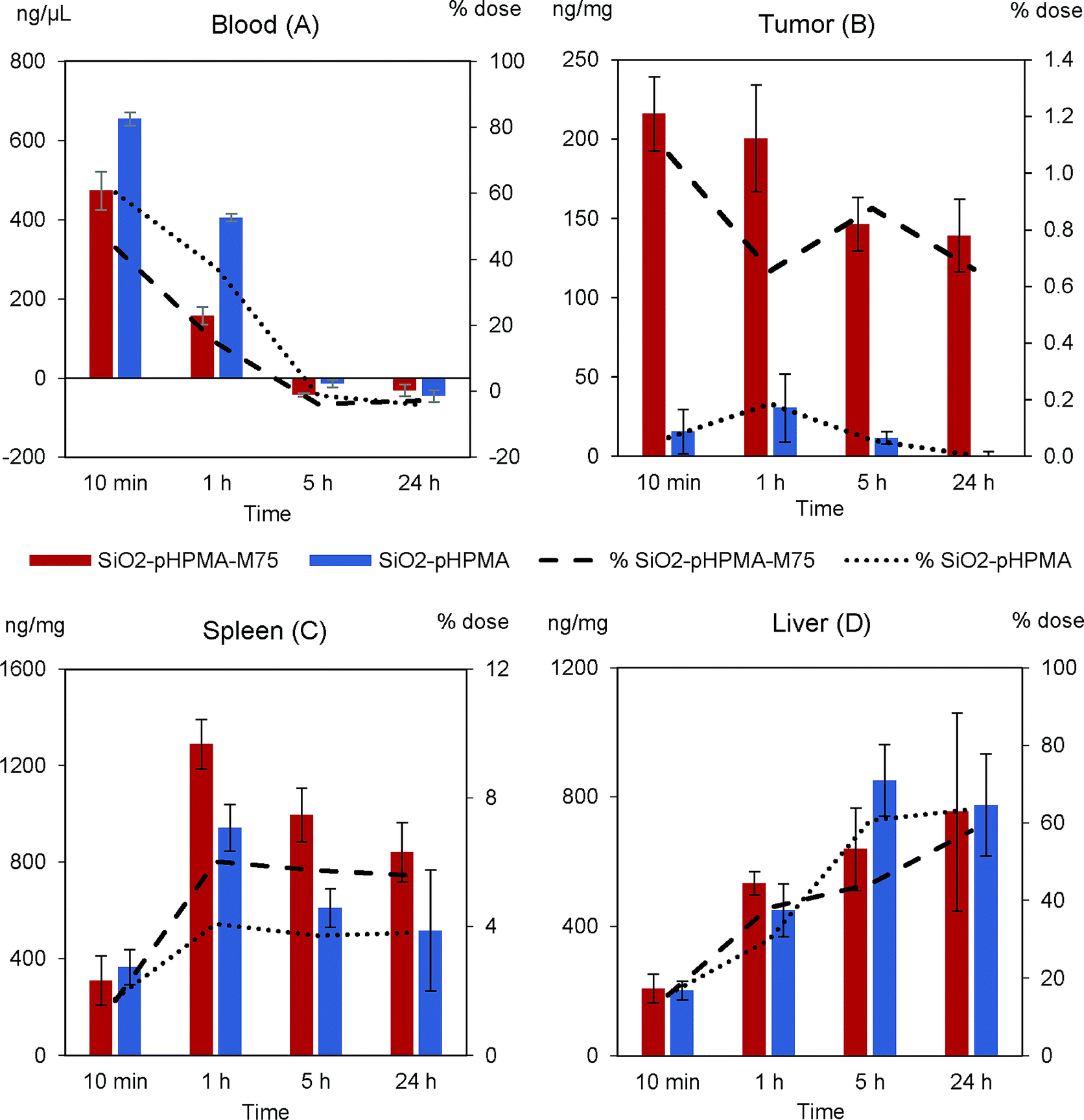 | ||
| Fig. 5 In vivo study I (dose 75 mg kg−1). Primary vertical axis (columns) shows nanoparticle concentration in (A) blood, (B) tumour, (C) spleen and (D) liver in time. Secondary vertical axis (lines) shows the percentage of the total administered dose. Red/dashed line – SiO2–pHPMA-M75; blue/dotted line – SiO2–pHPMA. | ||
Antibody-modified nanoparticles (SiO2–pHPMA-M75) were removed slightly faster than nanoparticles without the antibody (SiO2–pHPMA), which can be attributed to the fact that the antibody may act in a similar way as opsonin, and enhance macrophage phagocytosis6,40 (the random orientation of the antibodies on the nanoparticle surface leads to the exposure of the Fc fragments and their subsequent recognition by the macrophage membrane receptors).
The most important finding concerns the efficiency of the tumour targeting. Actively targeted SiO2–pHPMA-M75 nanoparticles accumulate in the tumour at tenfold higher concentration than the non-targeted ones (Fig. 5B) and reach the maximum measured concentration already within 10 minutes after the administration. Moreover, these nanoparticles are significantly retained in the tumour tissue even after 24 hours, compared to nanoparticles without the antibody (SiO2–pHPMA). In case of drug delivery application, this should provide enough time for the drug release or even in situ drug production, if the particles are loaded with the drug precursors.
High concentrations of nanoparticles were, as expected, detected in the spleen (Fig. 5C) and liver (Fig. 5D), organs of the Reticulo-Endothelial System (RES). The concentration in these organs increases in time, where in the case of the spleen it can be concluded that nanoparticles modified with the antibody are retained more. The overall nanoparticle distribution together with the fact that the maximum concentration in the tumour was reached within 10 minutes after the administration, indicates that the administered nanoparticle dose was probably too high and the binding sites in the tumour were saturated while an excess of nanoparticles remained in circulation and was available to the other organs. Hence, it was hypothesised that by decreasing the dose, a more favourable tumour![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RES ratio should be obtained.
:RES ratio should be obtained.
In order to confirm this hypothesis and to better understand the dose effect on the nanoparticle accumulation, the in vivo study II was carried out. Since the stealth effect of pHPMA nanoparticle coating was already confirmed in in vivo study I, nanoparticles with an irrelevant IgG 2b isotypic antibody (SiO2–pHPMA-ISO) were chosen as a negative control, in order to eliminate possible effects of the particle surface chemistry. The administered nanoparticle dose in study II was 20 mg kg−1, which is approximately 4 times less than in study I but still enables reliable fluorescence detection of the nanoparticles on the background of autofluorescent tissues. The nanoparticle concentration in the blood (Fig. 6A) decreased faster than in the case of the higher nanoparticle dose. This can be attributed to the macrophage phagocytic capacity and kinetics.
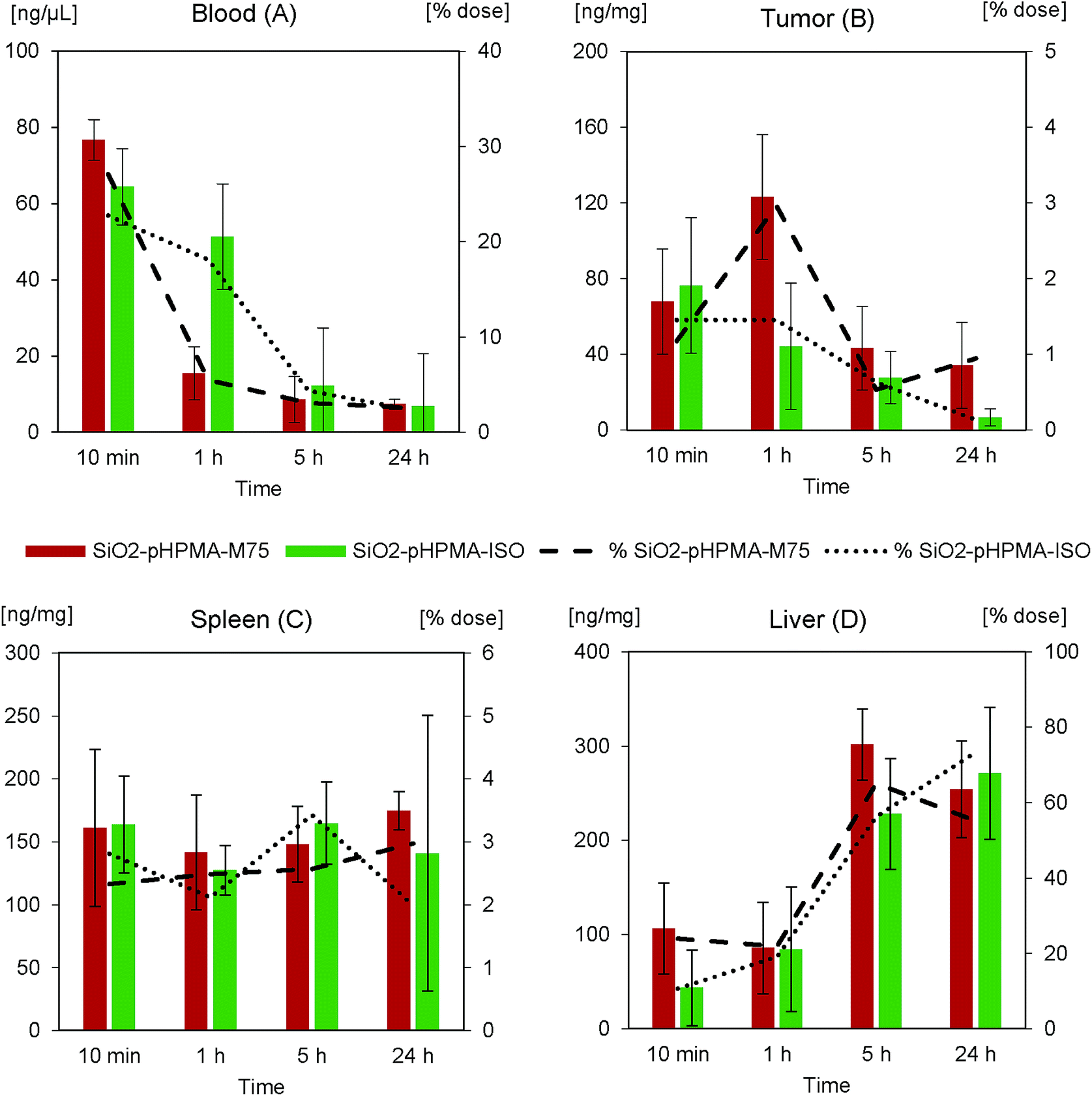 | ||
| Fig. 6 In vivo study II (dose 20 mg kg−1). Primary vertical axis (columns) shows nanoparticle concentration in (A) blood, (B) tumour, (C) spleen and (D) liver in time. Secondary vertical axis (lines) shows the percentage of the total administered dose. Red/dashed line – SiO2–pHPMA-M75; green/dotted line – SiO2–pHPMA-ISO. | ||
The maximum concentration of the specifically targeted SiO2–pHPMA-M75 nanoparticles in the tumour was measured at 1 hour from the administration of the lower dose (Fig. 6B). This indicates a connection between the dose and the tumour accumulation kinetics. The maximum concentration of nanoparticles in the tumour relative to the dose was approximately 3× higher in comparison with in vivo study I. An interesting and a very positive result is that by reducing the dose, the tumour:RES ratio (measured 1 hour after administration) was improved from approx. 1:40 (in vivo study I) to 3:20 (in vivo study II). Hence, exploiting differences in the tumour accumulation and macrophage uptake kinetics offers possibilities for further optimization of the nanoparticle targeting.
Nanoparticles without specific targeting (SiO2–pHPMA-ISO) do not significantly accumulate in the tumour tissue, however their concentration in the 10th minute after the administration was approximately 2× higher than in the case of SiO2–pHPMA nanoparticles in the first in vivo study. This might be attributed to the presence of protein molecules on the nanoparticle surface, which increases their zeta potential and thus can lead to enhanced nanoparticle extravasation or retention in the tumour tissue in comparison to nanoparticles without the antibody (SiO2–pHPMA).41 SiO2–pHPMA-ISO nanoparticles were not significantly retained and their concentration in the tumour decreased proportionally with their concentration in the blood.
The nanoparticle accumulation in the spleen (Fig. 6C) shows lower differences between the used nanoparticle types than in the first study, which is due to the higher surface similarity (both types bear antibody molecules on the surface). The total nanoparticle concentration in the liver (Fig. 6D) after 24 hours reaches approximately 60% of the administered dose, which is the same as in the first study.
Conclusion
For the first time, fluorescent silica nanoparticles (170 nm) coated by a covalently attached polymeric chain based on amino-reactive multivalent poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) and an antibody IgG M75 coupled via the polymer were prepared and characterized in a series of physico-chemical, in vitro and in vivo tests. The amount of the polymer and the antibody, determined by amino acid analysis, was established as 3.6 wt% and 1.0 wt%, respectively. Nanoparticle stability in physiological media was established by DLS measurements.ELISA-like tests revealed specific interaction between nanoparticles bearing the IgG M75 antibody and an antigen comprising of the proteoglycan domain of Carbonic Anhydrase IX (CA IX) in fusion with the Maltose Binding Protein (PG-MBP). Further in vitro characterization employing flow cytometry demonstrated that SiO2–pHPMA-M75 nanoparticles were able to bind selectively to the tumour cells HT-29, which express CA IX.
In vivo study carried out on athymic NU/NU nude mice bearing HT-29 tumours showed that nanoparticles coated with poly(N-(2-hydroxypropyl)methacrylamide) could circulate in the blood longer than 60 minutes, which indicates the stealth effect of pHPMA and increases the possibility of reaching the target tissue. Further, it was shown that actively targeted SiO2–pHPMA-M75 nanoparticles accumulated in the tumour at tenfold higher concentration compared to SiO2–pHPMA nanoparticles and were retained in the tumour tissue even 24 hours after the administration of a 75 mg kg−1 dose. The study of the nanoparticles with irrelevant isotypic antibody (SiO2–pHPMA-ISO) administered at 20 mg kg−1 dose indicates that the presence of the protein molecule on the nanoparticle surface enhanced nanoparticle extravasation in the fenestrated tumour vasculature in comparison to SiO2–pHPMA nanoparticles, but these nanoparticles were not retained in the tissue due to the absence of a specific antibody.
It was shown that the lowering of the nanoparticle dose can lead to a change in the tumour accumulation kinetics. While the high dose caused a fast saturation of the tumour tissue binding sites (the maximum concentration was measured in the 10th minute), the administration of the lower dose lead to the maximum concentration at 1 hour after administration. Consequently, the nanoparticle dose reduction resulted in a 6× improvement of the tumour:RES ratio. In summary, silica nanoparticles coated with pHPMA and IgG M75 were shown to be a promising tumour targeting system with potential for further functionality enhancement (e.g. drug delivery).
Methods
Chemicals
The following chemicals were used for the synthesis, modification and characterization of the nanoparticles: (3-aminopropyl)triethoxysilane (APTES), 3-sulfanylpropanoic acid, β-alanine, albumin from bovine serum (BSA), ammonium hydroxide (28–30%), fluorescein 5(6)-isothiocyanate (FITC), methanol, o-phthalaldehyde, phosphate buffered saline (PBS) tablets, sodium acetate, tetraethyl orthosilicate (TEOS) and triethylamine from Sigma-Aldrich, 1-aminopropan-2-ol from TCI Europe N.V., propidium iodide from Fluka and ethanol for UV, sodium chloride and hydrochloric acid from Penta. The isotypic IgG 2b antibody was purchased from Exbio Praha, a.s. and goat anti mouse IgG-FITC (GAM-FITC) antibody was purchased from Jackson Immunoresearch Laboratories. Water was produced by a demineralized water generator Aqual 25. Polymer p(HPMA-co-Ma-βAla-TT) (pHPMA) (Mw = 44000) with amino-reactive thiazolidine-2-thione (TT) groups (6.8%) was prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization as described earlier.42 The synthesis and characterisation of pHPMA is described in ESI 2.† The monoclonal antibody IgG M75 (M75)43 and the proteoglycan (PG) domain of Carbonic Anhydrase IX (CA IX) in fusion with the Maltose Binding Protein (PG-MBP) were provided by the Laboratory of Structural Biology, Institute of Molecular Genetics of the Czech Academy of Sciences, Prague, Czech Republic.
Cells and media
The cell line HT-29 (human epithelial; tissue: colon; disease: colorectal adenocarcinoma; ATCC) and the cell line DLD-1 (human epithelial; tissue: colon; disease: Dukes' type C, colorectal adenocarcinoma; ATCC) were grown in DMEM (D6429, Sigma-Aldrich) and RPMI-1640 medium (R8758, Sigma-Aldrich), respectively. Both media were supplemented with 10% heat inactivated FBS (10270-106, Invitrogen) and 1% mix of antibiotics and antimycotics anti–anti (15240, Invitrogen). The cells were cultured at 37 °C in a 5% CO2 atmosphere until 70–80% confluency.Nanoparticle synthesis
Synthesis of fluorescently labeled amino-silica nanoparticles was carried out according to the Stöber method.44 Briefly, 7.7 mL of ammonium hydroxide was mixed with 100 mL of ethanol. The mixture was heated to 30 °C under continuous stirring with a magnetic stirrer in a two-neck round flask equipped with a reflux condenser. After reaching 30 °C, the solution was equilibrated for 30 min and subsequently 3 mL of TEOS was added under continuous stirring (500 rpm). Simultaneously, 3 mL of FITC solution (20 mg of FITC and 123 mg of APTES in 10 g of ethanol, stirred for 48 h in a dark at room temperature) was added to the mixture.45 The condensation reaction was allowed to proceed for 22 h at a constant temperature 30 °C. The colloidal dispersion was then cooled down to room temperature and cleaned by repeated centrifugation (10 min at 10000g) and washing (3-times with ethanol) and re-suspended in demineralized water. The prepared nanoparticles were dialyzed against a sodium chloride solution (0.1 M in water) for 2 days in dark to remove unbound FITC and unreacted TEOS, and then against demineralized water for one more day in dark to remove all salts and thus prevent nanoparticle aggregation. The dispersion was then centrifuged and re-suspended in ethanol to a final concentration of 10 mg mL−1. Further nanoparticle modification in order to introduce reactive amino groups was done by a reaction of APTES solution (3 mL of ethanol, 50 μL of demineralized water, 63 μL of APTES adjusted to pH ≈ 2 with HCl and stirred for 15 minutes) with previously prepared nanoparticles in ratio 0.2 mL of APTES solution to 5 mL of nanoparticle suspension. The reaction was carried out in a dark at room temperature for 24 hours. After the reaction, triethylamine was added to the reaction container to obtain pH 8.0, then the nanoparticles were washed 3-times with ethanol (centrifugation at 10000g for 10 min) and re-suspended in ethanol (20 mg mL−1) for further use. The sample was stored in the dark.
Nanoparticle surface modification
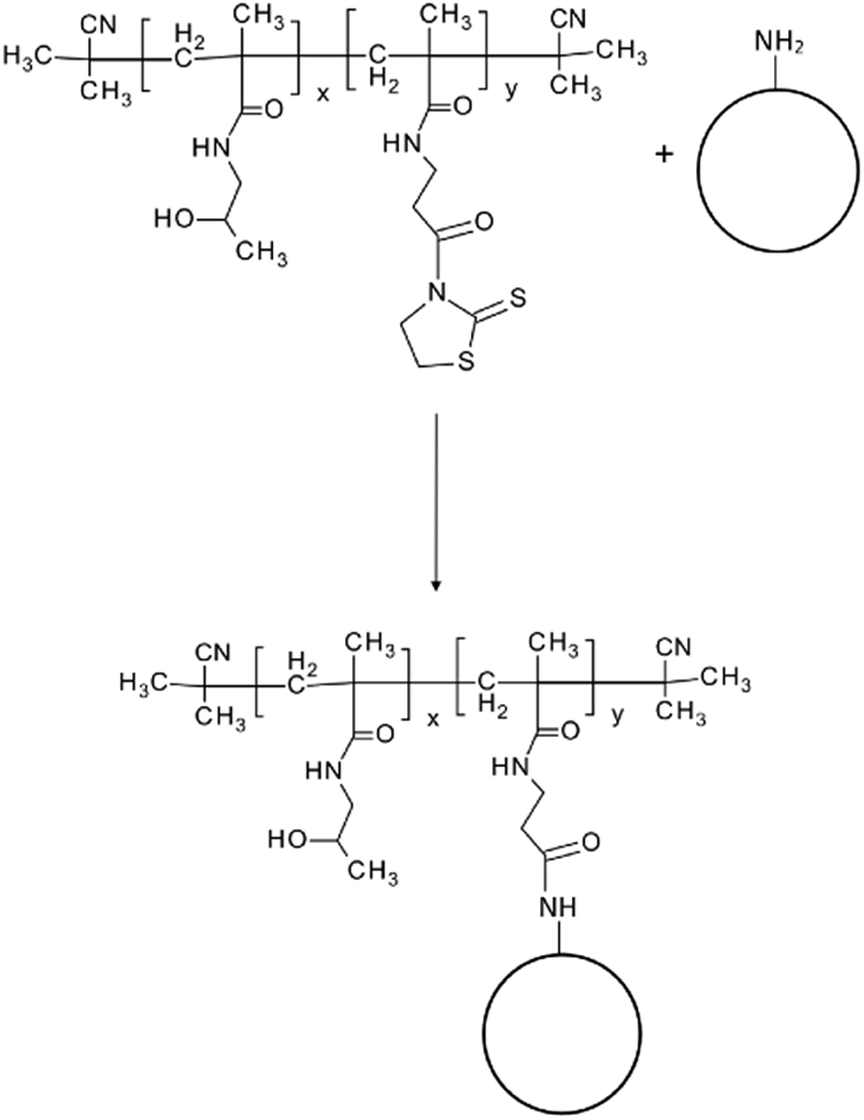 | ||
| Scheme 1 Illustration of reaction between amino-group bearing object and amino-reactive TT group of pHPMA. X = 93.2, Y = 6.8. | ||
000g) and re-suspended in 1 mL of PBS. In the next step, 0.67 mg of the antibody IgG M75 or the isotypic antibody IgG 2b was added in a PBS solution. The amount of the antibody was estimated based on previous experience with antibody coupling to nanoparticles.46 The coupling reaction of IgG M75 with available amino-reactive TT groups from pHPMA that were not consumed by the polymer coupling to the silica nanoparticles was carried out under continuous stirring at room temperature for 20 minutes. Then, 1 μL of 1-amino-2-propanol was added and the suspension was stirred for 10 more minutes to complete the aminolysis of the residual amino-reactive TT groups. In the final step the nanoparticles with covalently bound IgG M75 were washed 3-times with PBS, re-suspended in 1 mL of 1% BSA/PBS (final concentration 20 mg mL−1), and stored in the fridge (4 °C).Physico-chemical characterisation of nanoparticles
The morphology of the prepared silica nanoparticles was examined by Transmission Electron Microscopy (TEM; Jeol-JEM-1010). The particle size distribution and the zeta potential were determined by Dynamic Light Scattering (DLS, Zetasizer Nano-ZS). For the DLS measurement, the nanoparticles were dispersed in demineralized water to a final concentration of 0.5 mg mL−1 and each measurement was carried out in triplicate, at 25 °C and at a detector angle of 173°.In vitro characterisation
100 μL of the PG-domain fused with the Maltose Binding Protein (PG-MBP) solution (2.5 μg mL−1) was added to the desired number of wells in a polystyrene 96-well plate. As a control, the MBP solution of the same concentration and volume was used in the same well-plate. The well-plate was stored in the fridge (4 °C) overnight to achieve sufficient adhesion of the protein to the surface of the well-plate. After a 5-times repeated wash with 200 μL of PBS, a solution of 1% BSA in PBS was added (200 μL) to block the free, unmodified surface of the wells (1 hour incubation, room temperature). Each well was then washed 3-times with 200 μL of PBS. In the next step, the nanoparticle suspensions (100 μL of SiO2–pHPMA, SiO2–pHPMA-M75 and SiO2–pHPMA-ISO; each in a concentration series 1 mg mL−1, 0.5 mg mL−1, 0.25 mg mL−1, 0.125 mg mL−1 and 0.0625 mg mL−1) were added and incubated for one hour at room temperature. Then, all wells were washed 5-times with 200 μL of PBS and the fluorescence was analyzed using a fluorescent well-plate reader. The analysis was performed via gain optimal mode (excitation 488 nm, emission 516 nm), which assigns the maximum numerical value to the well with the highest fluorescence intensity and then calculates the relative values of fluorescence intensity for all other wells.
The samples for flow cytometry were prepared in a 96-well plate. 300000 cells (HT-29 or DLD-1) were added to each well. Then the antibody IgG M75 (15 μL, 14.4 μg mL−1) was added to the wells for the positive control (tests the CA IX presence) – one well per each cell type. After 25 minutes of incubation on ice (to prevent the internalization of the antibody) the cells were washed twice with 200 μL of 1% BSA/PBS by repeated centrifugation (5 min, 280g). Then the fluorescent secondary antibody (goat anti mouse IgG antibody conjugated with FITC (GAM-FITC); 15 μL, 20 μg mL−1) was added to the positive control wells, where IgG M75 was used previously. The secondary antibody was also added to one more well per each cell type as a negative control. In the same step, nanoparticle dispersions were added to the other wells (15 μL; 3.75 μg). After 20 minutes of incubation on ice, the cells were washed twice with 200 μL of 1% BSA/PBS by repeated centrifugation (5 min, 280g). Then, propidium iodide solution in 1% BSA/PBS (0.5 μg mL−1) was added (propidium iodide intercalates to the DNA of dead cells), the samples were transferred to plastic test tubes and subsequently analyzed.
The evaluation of the experiment was performed using the Becton Dickinson FACS Aria II flow cytometer. The fluorescence of 10000 events was measured, using the laser excitation wavelength of 488 nm. Emission was read by a detector with a set of two dichroic mirrors 530/30 Band Pass and 502 Long Pass. The raw data were evaluated by software FlowJo 7.6.1 (Tree Star, Inc.) and presented in the form of histograms. The y-axis shows the number of the cells while the x-axis represents the intensity of fluorescence emitted by the cell (FITC channel).
000 cells (HT-29 or DLD-1) in ice-cold 1% BSA in PBS was added to the each well of a 96-well plate. Then, 20 μL of nanoparticles (SiO2–pHPMA-M75, SiO2–pHPMA-ISO and SiO2–pHPMA, 1 mg mL−1) in ice-cold 1% BSA in PBS was added to the wells and incubated with the cells for 1 hour. After the incubation, samples were centrifuged (250g, 3 min) and re-suspended in 500 μL of ice-cold 1% BSA in PBS. The samples were analyzed using Olympus Fluoview FV1000 confocal system (488 nm laser excitation wavelength).In vivo characterisation
The in vivo study II was performed after the successful study I experiments and explored the effect of lower nanoparticle dose on the tumour accumulation. For the experiments, the nanoparticles with the specific antibody (SiO2–pHPMA-M75) and the nanoparticles with the irrelevant isotypic antibody IgG 2b (SiO2–pHPMA-ISO) were used. Three mice per each time point were injected with 20 mg kg−1 nanoparticle dose (0.65 mg in 300 μL of 1% BSA/PBS) into the tail vein. As a control, three mice were injected with 300 μL of 1% BSA/PBS. No clinical observations were noted during both studies.
After the required time (10 min, 1 h, 5 h, 24 h) the animals were euthanized and the tissues (liver, spleen, brain, and tumour) were collected, weighted and homogenized in Lyzing Matrix A 2 mL Tube (MP Biomedicals) using FastPrep 24 instrument (MP Biomedicals). The blood samples were collected into the heparinized tubes. All tissue and blood samples were stored on ice until the analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Milan Fábry and Irena Sieglová (Institute of Molecular Genetics of the Czech Academy of Sciences, 142 00 Prague 4, Czech Republic) for providing PG-MBP and MBP. Authors also thank Nina Sarvašová and Ondřej Kašpar for the illustrations. In vivo studies were kindly supported by Sotio, a.s. D.L. and M.M. were support by Specific University Research (MSMT No 21-SVV/2018). F.S. was supported by the Agency for Healthcare Research (AZV NV16-34342A). V.K. would like to acknowledge Institutional Support (RVO 68378050). M.P. and R.P. were supported by the Czech Science Foundation (GACR 16-17207S).References
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- S. M. Moghimi and J. Szebeni, Prog. Lipid Res., 2003, 42, 463–478 CrossRef PubMed.
- L. Illum, S. Davis, R. Müller, E. Mak and P. West, Life Sci., 1987, 40, 367–374 CrossRef PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487–495 CrossRef PubMed.
- S. Parveen and S. K. Sahoo, Eur. J. Pharmacol., 2011, 670, 372–383 CrossRef PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef PubMed.
- P. Decuzzi, B. Godin, T. Tanaka, S.-Y. Lee, C. Chiappini, X. Liu and M. Ferrari, J. Controlled Release, 2010, 141, 320–327 CrossRef PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacogn. Rev., 2001, 53, 283–318 Search PubMed.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef PubMed.
- M. Hans and A. Lowman, Curr. Opin. Solid State Mater. Sci., 2002, 6, 319–327 CrossRef.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef PubMed.
- D. Calvo, M. Jesús and D.-H. Lee, Rev. Esp. Cardiol., 2006, 59, 399–400 CrossRef.
- J. Szebeni, Toxicology, 2005, 216, 106–121 CrossRef PubMed.
- A. Chanan-Khan, J. Szebeni, S. Savay, L. Liebes, N. Rafique, C. Alving and F. Muggia, Ann. Oncol., 2003, 14, 1430–1437 CrossRef PubMed.
- J. Szebeni, L. Baranyi, S. Savay, J. Milosevits, R. Bunger, P. Laverman, J. Metselaar, G. Storm, A. Chanan-Khan and L. Liebes, J. Liposome Res., 2002, 12, 165–172 CrossRef PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef PubMed.
- T. Lammers, R. Kühnlein, M. Kissel, V. Subr, T. Etrych, R. Pola, M. Pechar, K. Ulbrich, G. Storm and P. Huber, J. Controlled Release, 2005, 110, 103–118 CrossRef PubMed.
- S. Kunjachan, F. Gremse, B. Theek, P. Koczera, R. Pola, M. Pechar, T. Etrych, K. Ulbrich, G. Storm and F. Kiessling, ACS Nano, 2012, 7, 252–262 CrossRef PubMed.
- M. Pechar, R. Pola, R. Laga, K. Ulbrich, L. Bednárová, P. Maloň, I. Sieglová, V. Král, M. Fábry and O. Vaněk, Biomacromolecules, 2011, 12, 3645–3655 CrossRef PubMed.
- V. Šubr, Č. Koňák, R. Laga and K. Ulbrich, Biomacromolecules, 2006, 7, 122–130 CrossRef PubMed.
- Č. Koňák, V. Šubr, L. Kostka, P. Štěpánek, K. Ulbrich and H. Schlaad, Langmuir, 2008, 24, 7092–7098 CrossRef PubMed.
- D. Klepac, H. Kostkova, S. Petrova, P. Chytil, T. Etrych, S. Kereïche, I. Raska, D. A. Weitz and S. K. Filippov, Nanoscale, 2018, 10, 6194–6204 RSC.
- N. Green, C. W. Herbert, S. Hale, A. Hale, V. Mautner, R. Harkins, T. Hermiston, K. Ulbrich, K. Fisher and L. Seymour, Gene Ther., 2004, 11, 1256 CrossRef PubMed.
- V. Král, P. Mader, R. Collard, M. Fábry, M. Hořejší, P. Řezáčová, M. Kožíšek, J. Závada, J. Sedláček and L. Rulíšek, Proteins: Struct., Funct., Bioinf., 2008, 71, 1275–1287 CrossRef PubMed.
- S. Pastorekova, S. Parkkila, A. K. Parkkila, R. Opavsky, V. Zelnik, J. Saarnio and J. Pastorek, Gastroenterology, 1997, 112, 398–408 CrossRef.
- P. C. McDonald, J.-Y. Winum, C. T. Supuran and S. Dedhar, Oncotarget, 2012, 3, 84 CrossRef PubMed.
- J. Zavada, Z. Zavadova, J. Pastorek, Z. Biesova, J. Jezek and J. Velek, Br. J. Cancer, 2000, 82, 1808 CrossRef PubMed.
- A. Chrastina, J. Zavada, S. Parkkila, Š. Kaluz, M. Kaluzová, J. Rajčáni, J. Pastorek and S. Pastoreková, Int. J. Cancer, 2003, 105, 873–881 CrossRef PubMed.
- V. Tokárová, A. Pittermannová, V. Král, P. Řezáčová and F. Štěpánek, Nanoscale, 2013, 5, 11490–11498 RSC.
- M. Ullrich, J. Haša, J. Hanuš, M. Šoóš and F. Štěpánek, Powder Technol., 2016, 295, 115–121 CrossRef.
- A. Watermann and J. Brieger, Nanomaterials, 2017, 7, 189 CrossRef PubMed.
- Y. Zhang, J. Guo, X.-L. Zhang, D.-P. Li, T.-T. Zhang, F.-F. Gao, N.-F. Liu and X.-G. Sheng, Int. J. Pharm., 2015, 496, 1026–1033 CrossRef PubMed.
- M. Bouchoucha, É. Béliveau, F. Kleitz, F. Calon and M.-A. Fortin, J. Mater. Chem. B, 2017, 5, 7721–7735 RSC.
- T. Mandal, M. Beck, M. Lindén and C. Buske, Blood, 2016, 128, 4713 Search PubMed.
- F. Chen, H. Hong, Y. Zhang, H. F. Valdovinos, S. Shi, G. S. Kwon, C. P. Theuer, T. E. Barnhart and W. Cai, ACS Nano, 2013, 7, 9027–9039 CrossRef PubMed.
- J.-K. Wang, Y.-Y. Zhou, S.-J. Guo, Y.-Y. Wang, C.-J. Nie, H.-l. Wang, J.-l. Wang, Y. Zhao, X.-Y. Li and X.-J. Chen, Mater. Sci. Eng., C, 2017, 76, 944–950 CrossRef PubMed.
- Clinical Trial ID no. NCT02106598, Targeted Silica Nanoparticles for Real-Time Image-Guided Intraoperative Mapping of Nodal Metastases.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- M. Cataldi, C. Vigliotti, T. Mosca, M. Cammarota and D. Capone, Int. J. Mol. Sci., 2017, 18, 1249 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef PubMed.
- T. Etrych, T. Mrkvan, P. Chytil, Č. Koňák, B. Říhová and K. Ulbrich, J. Appl. Polym. Sci., 2008, 109, 3050–3061 CrossRef.
- S. Pastoreková, Z. Závadová, M. Košťál, O. Babušíková and J. Závada, Virology, 1992, 187, 620–626 CrossRef.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. Čejková, J. Hanuš and F. Štěpánek, J. Colloid Interface Sci., 2010, 346, 352–360 CrossRef PubMed.
- Bangs Laboratories, Inc., TechNote 205: Covalent Coupling, 2013 Search PubMed.
- N. Touisni, N. Kanfar, S. Ulrich, P. Dumy, C. T. Supuran, A. Mehdi and J.-Y. Winum, Chem.–Eur. J., 2015, 21, 10306–10309 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: pHPMA synthesis and analysis, additional fluorescent microscopy images, amino acid analysis. See DOI: 10.1039/c8ra03487g |
| This journal is © The Royal Society of Chemistry 2018 |