
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA vanillin-based copper(II) metal complex with a DNA-mediated apoptotic activity†
Wendy M. T. Q. de Medeirosa,
Mayara J. C. de Medeirosa,
Edinilton M. Carvalhob,
Jailma A. de Limac,
Verônica da S. Oliveiraa,
Ana C. F. de B. Pontesa,
Francisco O. N. da Silvaa,
Javier A. Ellena d,
Hugo A. de O. Rochac,
Eduardo H. S. de Sousab and
Daniel de L. Pontes*a
d,
Hugo A. de O. Rochac,
Eduardo H. S. de Sousab and
Daniel de L. Pontes*a
aLaboratório de Química de Coordenação e Polímeros (LQCPol), Instituto de Química, Universidade Federal do Rio Grande do Norte (UFRN), Natal, 59078-970, Brazil. E-mail: pontesdl@yahoo.com
bLaboratório de Bioinorgânica, Departamento de Química Orgânica e Inorgânica, Universidade Federal do Ceará (UFC), Cx. Postal 6021, Fortaleza, 60440-900, Brazil
cLaboratório de Biotecnologia de Polímeros Naturais (BIOPOL), Departamento de Bioquímica, Universidade Federal do Rio Grande do Norte (UFRN), Natal, 59072-970, Brazil
dInstituto de Física de São Carlos, Universidade de São Paulo, 13560-970, São Carlos, SP, Brazil
First published on 10th May 2018
Abstract
Vanillin (vanH) is the major component of vanilla and one of the most widely used flavoring agents. In this work the complex [Cu(phen)(van)2] was prepared and characterized by structural (X-ray), spectroscopic (IR, UV-Vis, EPR) and electrochemical techniques. This compound showed an octahedral geometry with an unusual arrangement of the vanillin ligands, where the methoxy groups of the vanillinate ions are coordinated opposite to each other. The compound promoted DNA cleavage in the presence of glutathione (GSH) and H2O2. At 40 μmol L−1 of complex with GSH (10 mmol L−1), there is a complete cleavage of DNA to nicked form II, while only at 10 μmol L−1 of this complex with H2O2 (1 mmol L−1) an extensive cleavage leading to form III took place. Additionally, we have evidences of superoxide generation upon reaction with GSH. Therefore, DNA fragmentation occurs likely through an oxidative pathway. MTT assays indicated that the complex is highly cytotoxic against three distinct cell lines: B16–F10 (IC50 = 3.39 ± 0.61 μmol L−1), HUH-7 (IC50 = 4.22 ± 0.31 μmol L−1) and 786-0 (IC50 = 10.38 ± 0.91 μmol L−1). Flow cytometry studies conducted with 786-0 cell line indicated cell death might occur by apoptosis. Cell cycle progression evaluated at 5 and 10 μmol L−1 resulted in a clear increase of 786-0 cells at G1 phase and depletion of G2/M, while higher doses showed an expressive increase of sub-G1 phase. Altogether, these results pointed out to a promising biological activity and potential as an anti-cancer agent.
1. Introduction
During the last few decades, the number of cancer cases has increased remarkably pressing for the development of newer and more effective anticancer agents. Although a series of treatments and important drugs are available, many of them face low selectivity,1 unpleasant side effects2,3 and tumor resistance.4,5 These issues represent some of the current challenges still to be overcome.The coordination chemistry has provided relevant contributions to the cancer chemotherapy, where different metal complexes systems with potent antitumor activity have emerged along with many exciting new strategies.6–8 These advances have emerged mainly due to the hallmark discovery of cisplatin anticancer properties.7 In this context, copper(II) compounds have become one promising frontline agent in the development of potential antitumor chemotherapeutics. This can be witnessed by the expressive number of compounds reported and relevant biological properties exhibited by these complexes along with clinical trials.8,9
Biological assays using copper(II) complexes have revealed a close association with redox activity, ROS generation and DNA damage. In this process, Cu(II) is reduced to Cu(I) inside cell and superoxide ion can be generated upon one electron transfer to dioxygen regenerating Cu(II). The production of superoxide is the starting point for other ROS species, which may contribute to increase the oxidative stress in cancer cells. This effect may cause serious damage to the DNA leading to cell death. Additionally, copper complex can also directly hydrolyze DNA further promoting cell death.10
Among other key aspects to be considered on a metal complex, the ligand is also a critical part, which might establish the biological function of the compound. Indeed, some copper complex can exhibit both DNA degradation routes, oxidative and hydrolytic, which can be modulated by the chosen ligand. Cellular uptake, organelles distribution, appropriated structural organization for DNA interaction and overall pharmacological activity can also be adjusted upon selection of suitable ligands.7,11,12 One of the most relevant ligands used in copper biocoordination chemistry is 1,10-phenanthroline (phen) and derivatives. These phenanthroline copper complexes, e.g. [Cu(phen)2]2+, caught larger attention due to the nuclease activity and planar structure appropriated for DNA intercalation.13
Vanillin (vanH), 4-hydroxy-3-methoxybenzaldehyde, is the major component of natural vanilla and one of the most widely used flavoring agents in food, beverage and cosmetics.14 It is also an important raw material for synthesis of several chemical products. Besides its industrial and economic value, vanillin has been recognized also as an important bioactive compound.
Vanillin has been recognized as an effective anti-mutagenic agent. In 1986, Ohta et al. first reported that vanH could reduce 4-nitroquinoline-1-oxide (4-NQO) and furylfuramide (AF-2) that induced mutations in bacteria.15 Subsequently, it was found that vanillin was able to inhibit also spontaneous mutation in bacteria.16,17 Imanishi and collaborators revealed that vanillin reduced significantly mutations caused by ultraviolet light, X-ray and ethylnitrosourea exposure in mammalian cell and hamster lung cell (V79).18 Interestingly, vanillin suppressed in vivo metastasis of mouse breast cancer cells.19 Additionally, it attenuated the expression levels of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) and prevented CCl4-induced hepatic cell alteration and necrosis in rats.20
Durant and Karran reported that the cisplatin cytotoxic effect against A2780 cells was reduced upon co-administration with vanillin, revealing an important synergistic effect. Interestingly, the dose found to yield a survival of 37% of cells (D37) using cisplatin in the absence of vanillin was 9.7 μmol L−1, whereas with vanillin it was reduced to 5.9 and 4.3 μmol L−1 at 100 and 300 μmol L−1 of vanillin, respectively.21 Considering vanillin is widely ingested in the World, any new composition or new compound employing this species would be face very positively by scientific community.
Based on that, we prepared a new copper metal complex, [Cu(phen)(van)2], combining copper and vanillin as a potential anticancer agent, where we investigated its chemical and biological properties.
2. Material and methods
2.1. Materials
The reagents CuCl2·2H2O, 1,10-phenanthroline, vanillin, glutathione, agarose and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. All other common chemicals used were of analytical grade and used without further purification. DMEM and RPMI 1640 culture medium were acquired from Cultilab. DNA plasmid pBR322 was purchased from New England Biolabs Inc. The complex [Cu(phen)Cl2] was prepared according to procedure previously described.222.2. Synthesis of the complex [Cu(phen)(van)2]
The compound [Cu(phen)(van)2] was synthesized dissolving [Cu(phen)Cl2] (200.0 mg, 0.64 mmol) and vanillin (193.2 mg, 1.28 mmol) in distilled water. The pH of the solution was adjusted to 8.0 by the addition of sodium hydroxide (2.0 mol L−1). This reaction mixture was stirred for 2 hours at 60 °C and a dark green solid formed, which was filtered and washed with ethanol. Crystals of [Cu(phen)(van)2]·2H2O were obtained during the recrystallization in water, which were suitable for X-ray structure determination. Yield: 294.0 mg (85%).IR: νmax/cm−1; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O): 1652; ν(CC): 1580, 1545; ν(C–CHO): 1266; ν(O–CH3): 1024; δ(C–H): 851, 722 (KBr disk). UV-Vis: λmax(H2O)/nm (ε/L mol−1 cm−1): 775 (47.8), 650 (55.3), 469 (125.0), 345 (14
O): 1652; ν(CC): 1580, 1545; ν(C–CHO): 1266; ν(O–CH3): 1024; δ(C–H): 851, 722 (KBr disk). UV-Vis: λmax(H2O)/nm (ε/L mol−1 cm−1): 775 (47.8), 650 (55.3), 469 (125.0), 345 (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500), 312 (sh) (16700), 273 (40400), 204 (54300). Elemental analysis (%): calc. for CuC28H22N2O6·3H2O: C, 56.04; H, 4.70; N, 4.67%. Found: C, 56.09; H, 4.74; N, 4.72%. It is a non-electrolyte in water (ΛM = 31.13 S cm2 mol−1 at 25.5 °C).
500), 312 (sh) (16700), 273 (40400), 204 (54300). Elemental analysis (%): calc. for CuC28H22N2O6·3H2O: C, 56.04; H, 4.70; N, 4.67%. Found: C, 56.09; H, 4.74; N, 4.72%. It is a non-electrolyte in water (ΛM = 31.13 S cm2 mol−1 at 25.5 °C).
2.3. X-ray crystal structure determination
Single crystal of the complex was used for data collection on an Enraf-Nonius Kappa-CCD diffractometer, using Mo Kα radiation (λ = 0.71073 Å). Data collection was carried out with the COLLECT program.23 Integration and scaling of the reflections were performed with HKL Denzo-Scalepack24 programs. Absorption corrections were done using the multiscan method. The structure was solved by direct methods with SHELXS25 and refined by fullmatrix least-squares on F2 with SHELXL.262.4. Instrumentation: spectroscopic and electrochemistry characterization
Electronic spectra (UV-Vis) were recorded in solution (water and organic solvents) at 5 × 10−5 to 5 × 10−3 mol L−1 at room temperature on a photodiode-array spectrophotometer, model 8453 (Agilent). Infrared (IR) spectra (400–4000 cm−1) of the compounds dispersed in KBr were recorded on a Shimadzu FTIR-8400S spectrometer. EPR measurements were taken in solid state at room temperature and in DMF frozen solution at 77 K on a Bruker 200D-SRC X-Band spectrometer, equipped with an Oxford ESR 9 Cryostat, operating at 9.412 GHz, 10 db.Molar conductivity measurements were carried out in a Tecnopon MCA-150 conductometer. Electrochemical analysis was performed on an Epsilon potentiostat (BASi – Bioanalytical Systems Inc.). Cyclic voltammetric experiments were done in a three-electrode cell. The working electrode was a glassy carbon, a platinum single-wire electrode was used as the counter electrode and an Ag|AgCl electrode saturated with KCl (3.5 mol L−1) was used as the reference electrode. The cyclic voltammograms were recorded in KCl 0.1 mol L−1 at ν = 100 mV s−1. Oxygen was removed by purging the solutions with argon. All measurements were performed at 25.0 ± 0.2 °C.
2.5. Gel electrophoresis analysis of plasmid DNA cleavage
Agarose gels at 0.8% (w/v) in TAE buffer (tris base 89 mmol L−1, acetate 89 mmol L−1, EDTA 2 mmol L−1) were prepared for these studies. The reactions were performed under aerobic conditions by incubation of different concentrations of [Cu(phen)(van)2] (40 to 0.5 μmol L−1), H2O2 (1 mmol L−1), glutathione (GSH, 10 mmol L−1), 50 μmol L−1 of DTPA and pBR322 DNA plasmid (8.3 ng μL−1). Electrophoreses were conducted with and without H2O2 and GSH after 90 minutes of reaction. All samples were subjected to electrophoresis on agarose gel for 60 minutes under 70 V and variable current 53–64 mA. DNA band was analyzed, after 1 hour of incubation with GelRed™, using a bioimaging system (GelDOC + XR, Bio-Rad). A standard linear DNA marker (1 Kb linear DNA ladder, Life Technologies, Invitrogen) was used as a control. The distinct forms of plasmid DNA were analysed and quantified by densitometry analysis using the Software Quantity One 4.6.9 Basic (Bio-Rad Laboratories, Inc.). In all assays, it was used one or more DNA control lanes, where all conditions were maintained without the presence of the complex.The mechanism of cleavage mediated by the complex (40 μmol L−1) in the presence of GSH (1.5 mmol L−1) was investigated by adding different radical scavengers: histidine (13 mmol L−1), mannitol (13 mmol L−1), superoxide dismutase (4 U μL−1), N-oxyl-2,2,6,6-tetramethylpiperidine (TEMPO, 2.6 mmol L−1) and catalase (3.9 μmol L−1). The reaction was maintained for 30 minutes in the presence and absence of oxygen and applying the method described above.
2.6. Superoxide assay: NBT reduction
The capacity of the complex [Cu(phen)(van)2] to generate superoxide anion was evaluated using the nitro blue tetrazolium (NBT) assay.27 The experiments were conducted in phosphate buffer 0.1 mol L−1, pH 7.4 and 37 °C. The superoxide production was monitored by changes in the absorbance at 560 nm, which is characteristic of formazan originated by the reaction of NBT with O2−. These experiments were done using 100 μmol L−1 of NBT and 10 to 75 μmol L−1 of the metal complex with and without the biological reducing agent glutathione (1.5 mmol L−1).2.7. Cell culture
Human renal adenocarcinoma (786-0) cells (ATCC CRL-1932), murine melanoma (B16–F10) cells (ATCC CRL-6475) and human hepatocarcinoma (HUH-7) cells (JCRB-0403) were kindly donated by Dr Hugo Rocha (Department of Biochemistry, UFRN, Brazil). The cells were grown in RPMI 1640 (786-0 cells) or DMEM (all others) medium supplemented with 10% newborn calf serum (Cutilab) and penicillin–streptomycin (1 mg mL−1), and incubated at 37 °C in 5% CO2.2.8. Cell viability assays (MTT)
The cytotoxicity of the copper(II) complex was evaluated in three different cell lines, B16–F10, HUH-7 and 786-0, using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. The cells were seeded into 96-well plates at a density of 5 × 103 cell per well (100 μL) and allowed to attach overnight in 100 μL medium incubated at 37 °C, 5% CO2. The cells were allowed to adhere for 12 h. After the medium was replaced by 100 mL of fresh medium without fetal calf serum (FCS). Growth-arrested cells were released from the G0 phase by the addition of 10% FCS in the medium in the absence (control) or in the presence of different concentrations of the copper(II) complex (0.001 to 300 μmol L−1) dissolved in the cell culture medium for 24 h at 37 °C and 5% CO2. The medium was removed and MTT (1 mg mL−1) in DMEM or RPMI 1640 were added. After 4 hours, the medium was aspirated and the formazan crystals were dissolved in ethanol 96%. Absorbance was read in a microplate reader (Epoch Biotek Instruments) at 570 nm. Three independent experiments were performed and two replicate cultures were used for each complex concentration in each independent experiment. The number of viable cells was expressed as percentage relative to untreated controls, and the concentrations required for 50% inhibition of cell viability (IC50) were calculated from dose–response curves.2.9. Annexin V-FITC/PI double staining and analysis by flow cytometry
In order to evaluate the effects of the complex on cell death, a FITC/Annexin V Apoptosis Kit was used along with Dead Cell Annexin FITC and propidium iodide (PI) for Flow Cytometry (Invitrogen). For this study only 786-0 cells were evaluated. The cells were transferred to 6-well plates reaching 2 × 105 cells per well. The plates were incubated at 37 °C for 24 hours submitted to humid atmosphere of CO2 at 5%. The cells were then stimulated to exit G0 by adding RPMI 1640 supplemented with 10% FCS, in the presence of [Cu(phen)(van)2] (10 and 50 μmol L−1). A negative control was prepared without the presence of RPMI 1640 and a positive control with cisplatin. After 24 h, 786-0 cells were trypsinized, collected and washed with cold phosphate-buffered saline (PBS). The supernatant was discarded and cells were resuspended in 50 μL of binding buffer. Five microliters of Annexin V-FITC and 1 μL of PI solution (100 μg mL−1) were added in 100 μL of cell suspension. Cells were incubated for 20 min at room temperature and kept under light protection. After the incubation period, 200 μL of binding buffer for annexin V1× was added and cells were analyzed by flow cytometry (FASCANTO II, BD Biosciences), measuring fluorescence emission at 530–575 nm for annexin V and 630/22 nm for PI. A total of 10000 events were acquired. For data analysis, FlowJo Analysis Software version 10.0.7 (Tree Star, Inc.) was used.
2.10. Cell cycle assays
The cell cycle analysis was carried out by flow cytometry. Briefly, 2 × 105 786-0 cells were incubated with the complex [Cu(phen)(van)2] at 5, 10, and 20 μmol L−1 for 24 h at 37 °C in an atmosphere of 5% CO2 air. Cells suspensions were harvested in cold PBS, centrifuged, resuspended and fixed in 70% ethanol and incubated for 1 h at 4 °C. After washing steps with cold PBS, cells were digested by RNase (4.0 mg mL−1) at 37 °C for 30 min, and stained with PI (1.0 mg mL−1) in the dark at room temperature for 20 min. The distribution of the treated cell cycle was measured by FASCANTO II flow cytometer, and data analysis was carried out with FlowJo software (Tree Star, Inc.).3. Results and discussion
3.1. Crystal structure of [Cu(phen)(van)2]
The compound [Cu(phen)(van)2] crystallized in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . An ORTEP view of the complex unit is shown in Fig. 1. Crystallographic data and selected bond lengths and angles are listed in Tables S1 and S2 (ESI†).
. An ORTEP view of the complex unit is shown in Fig. 1. Crystallographic data and selected bond lengths and angles are listed in Tables S1 and S2 (ESI†).
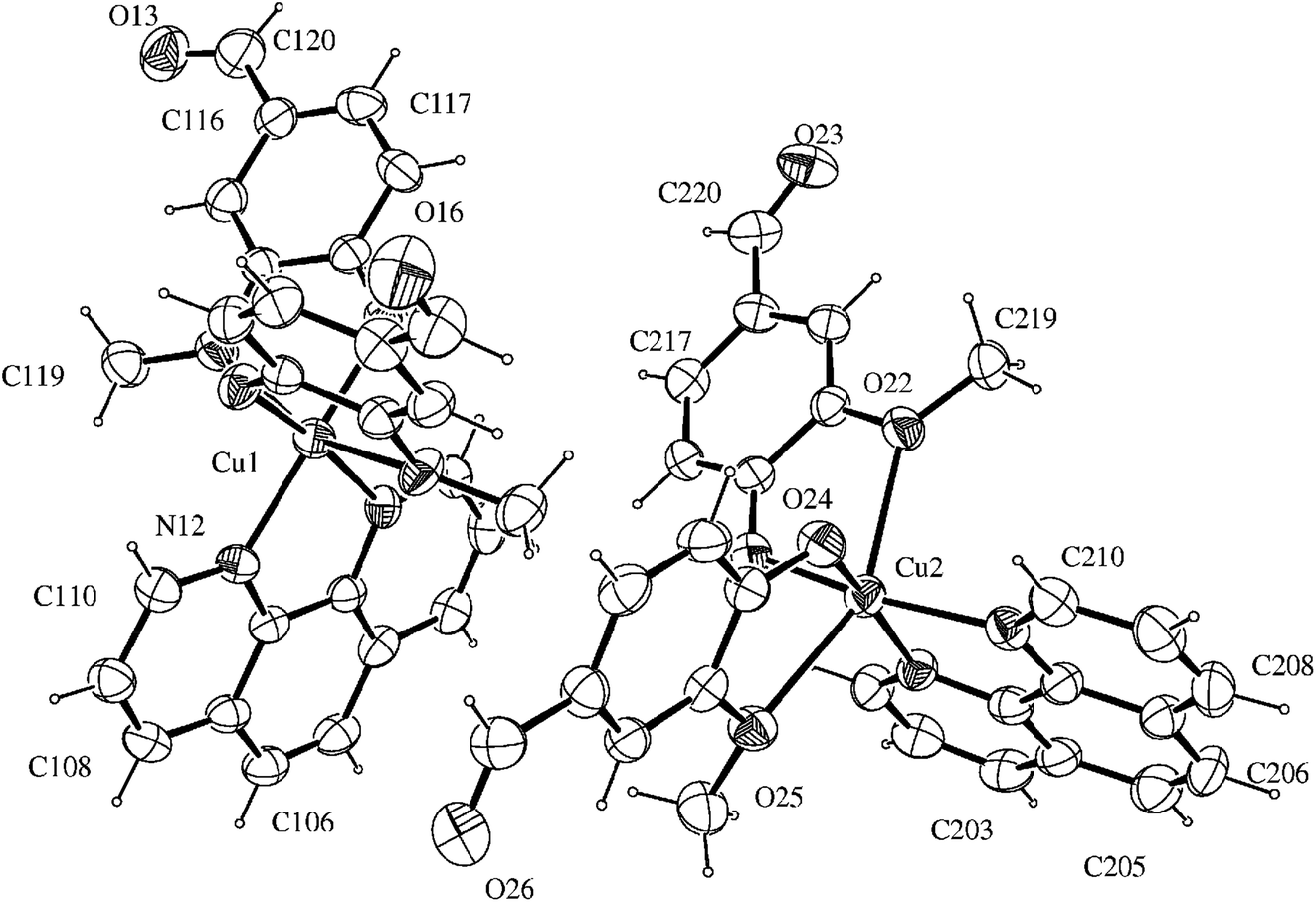 | ||
| Fig. 1 ORTEP view of the complex, showing the atoms labelling and the 50% probability ellipsoids. | ||
There are two independent molecular structures in the asymmetric unit of the complex with N2O4 donor set. Each copper(II) atom is coordinated to 1,10-phenanthroline nitrogen atoms with average distances of 2.020 Å. This value is similar to other Cu(II)–N(phen) distances found for related copper(II) six-coordinated N2O4 complexes in the literature, such as 2.016 Å for [Cu(phen)(2-bromoacetate)2]28 and 1.999 Å for [Cu(phen)(naproxanate)2].29
The distorted octahedral environment of the complex is completed by two deprotonated vanillin molecules that are coordinated to copper(II) in cis positions, due to the bidentate characteristic of the ligands involved. The vanillinate ions are disposed in a way that the methoxy groups are located in opposite sides of the same molecular axis, while the phenolic oxygen atoms are positioned opposite to phen nitrogen atoms. These donor atoms arrangement around the metal gives rise to one symmetrical axis (O(methoxy)–Cu–O(methoxy)) and two asymmetrical ones (N(phen)–Cu–O(phenolate)).
The crystallographic data revealed that Cu–O(methoxy) average distances (2.418 Å) are significantly longer than Cu–O(phenolate) average values (1.932 Å). This expressive difference can be attributed to the higher electronic density over the phenoxide oxygen, leading to a more favorable electronic donation to the metal when compared to the neutral methoxy donor atom. Such behavior is also observed in other metal-vanillinate complexes as for cis-[Fe(van)2(H2O)2] (Fe–O(methoxy) = 2.345 Å, Fe–O(phenolate) = 2.088 Å)30 and cis-[Cu(van)2(H2O)2] (Cu–O(methoxy) = 2.260 Å, Cu–O(phenolate) = 1.909 Å).31 The Cu–O(phenolate) and Cu–N(phen) bonds in the two asymmetrical axes have similar lengths, however, they are considerable different of the symmetrical one. Such arrangement indicates the structure has an elongated tetragonal distortion in z axis that corresponded to the O(methoxy)–Cu–O(methoxy) bonds. For that, the Jahn–Teller effect of Cu(II) may have an important contribution.32
The coordination of the vanillinate ion to Cu(II) led to a slight increase in the CO bond length of the aldehyde group from 1.199(8) Å in vanillin33 to 1.209(5) Å in [Cu(phen)(van)2]. Similar effect can be observed also in other vanillin metal complexes, as for cis-[Cu(van)2(H2O)2], where the aldehyde CO bond increased to 1.217 Å.31
The structural characterization of octahedral complexes having two vanillinate ions and one bidentate or two monodentate additional ligands, in cis or trans structural arrangements, has already been reported for other transition metals.30,34–36 The asymmetrical coordination environment promoted by the vanilloid oxygen atoms makes possible to have up to five different ligand arrangements as described in Fig. 2. Three of them are referred to cis structures, and two to trans isomers, identified in this work as cis-I, II or III and trans-I or II (Fig. 2).
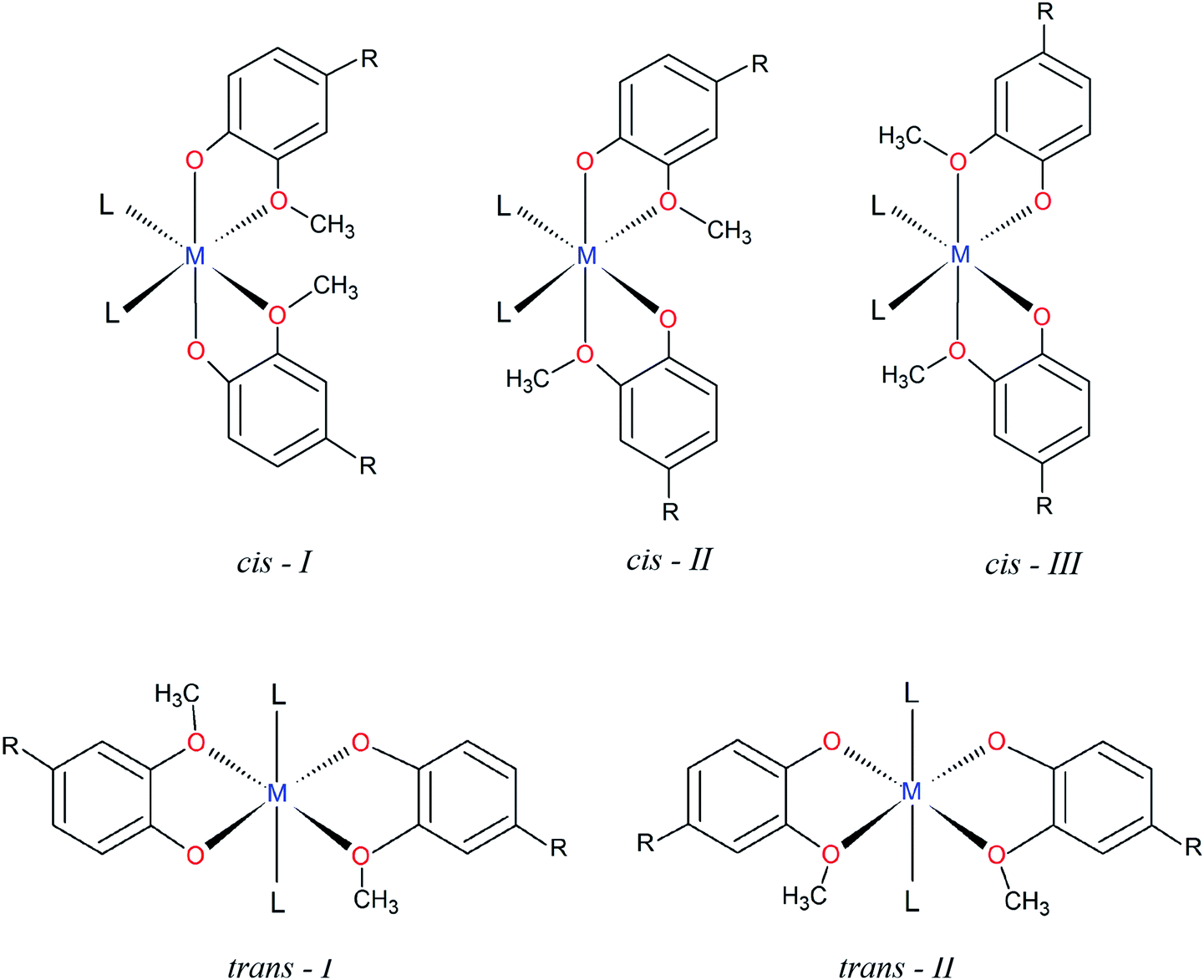 | ||
| Fig. 2 Possible structures of octahedral metal complexes with two vanilloid ligands. | ||
In spite of the number of structural possibilities, a search of this class of compounds carried out on the Cambridge Crystallographic Data Centre, CCDC database,37 revealed that most of the cis divanillin metal complexes are organized preferentially in a cis-I arrangement (7 compounds from a total of 8), with monodentate and bidentate additional ligands completing the coordination sphere. Some cases of this arrangement containing vanillin are as cis-[Cu(van)2(H2O)2] (NEPDAO); cis-[Fe(van)2(H2O)2] (SESGED) and cis-[Cu(van)2(tetramethylene)] (FMPECU).38 However, only one complex with cis-III arrangement was found, [Cu(van)2(tetramethylene)] (FMPECW).39 Therefore, the compound [Cu(phen)(van)2] described in the present work is part of a few cases of vanillin complexes having the cis-III arrangement. No compound was found having cis-II arrangement and vanillin as ligand. Surprisingly, among the trans isomers, there is only trans-I structures, indicating that the arrangement of methoxy groups “trans” to each other is favorable for this type of isomer, but unfavorable for the cis ones.
Once it was extended a search for ligands with a coordination environment similar to vanillin, it was found four compounds with guaicol,40,41 methoxyacetic42 and 4-nitroguaiacol.43 Even those, only the copper(II) complex with 4-nitroguaiacol showed a cis-III configuration, while the others exhibited a cis-I arrangement, supporting the very limited number of cases of this structure.
Interestingly, the different structures solved showed considerable changes in the bond length and tetragonal distortion, exemplified in Fig. S1 (ESI†). For cis-I structures it was observed that the bonds in two of the axes (L–M–OCH3 bonds) had the same length, while the z-axis (O(phenolate)–M–O(phenolate) bonds) was shorter. On the other hand, the cis-III arrangement, having methoxy groups “trans” to each other, exhibited a longer bond length compared to the two L–M–O bonds. Therefore, the different arrangements have a close relationship with the structural tetragonal distortion, where vanilloid metal(II) complexes having cis-I complexes present compressed structures while cis-III present an elongated distortion.
The extension of molecular distortion can be evaluated through the tetragonality parameter T, taken as the ratio of the average M–O and M–L from equatorial bonds (Requatorial) to the average axial M–O bond lengths (Raxial). A plot of T parameter against the average M–Oaxial bond length was obtained from the set of cis divanillin metal complexes available in CCDC database (Fig. 3). The crystallographic data showed a significant data correlation (R2 = 0.977). Additionally, this correlation graph makes possible to group the compounds by their similar arrangement, where these parameters changes quite drastically depending on the arrangement of the complex.
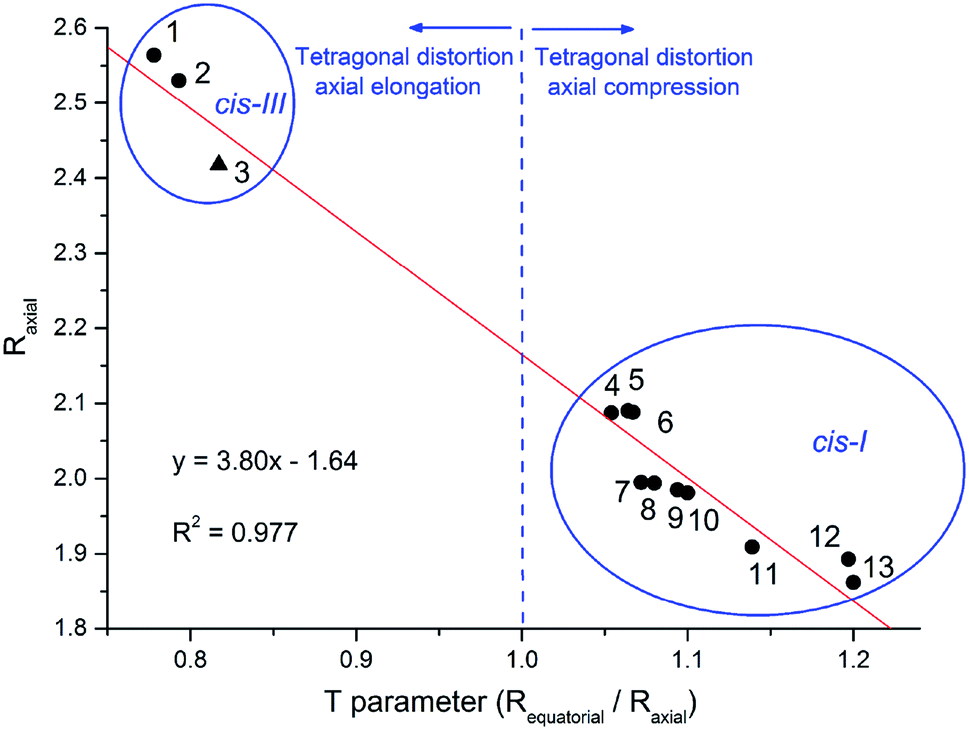 | ||
| Fig. 3 Plot of the T parameter versus the average Cu–Oaxial bond length (Raxial). Compounds: (1) [Cu(4-nitroguaiacol)2(tetramethylene)]; (2) [Cu(van)2(tetramethylene)]; (3) [Cu(phen)(van)2] (▲, this work); (4) [V(guaicol)2(tetramethylene)]; (5) cis-[Mn(van)2(H2O)2]; (6) cis-[Fe(van)2(H2O)2]; (7) cis-[Ni(van)2(H2O)2]; (8) cis-[Co(van)2(H2O)2]; (9) cis-[Zn(methoxyacetic)2(H2O)2]; (10) cis-[Zn(van)2(H2O)2]; (11) cis-[Cu(van)2(H2O)2]; (12) cis-[Ti(guaicol)2(Cl)2]; (13) [Cu(van)2(tetramethylene)]. | ||
The tetragonal distortion was observed for all complexes evaluated, even for those where Jahn–Teller effect is not expected, e.g. nickel(II) and zinc(II) complexes. This indicates that the distortion has a major contribution from the different electronic donation of oxygen atoms on the asymmetrical ligand. Although, this is not so common among the divanillin complexes, the cis-III structure observed for [Cu(phen)(van)2] may have been driven by the π receptor character of the phenanthroline ligand favoring the positioning of electronically rich phenolic oxygen atom trans to each nitrogen atom instead of the methoxide groups. These particular arrangements might have important implications in the chemical and biochemical reactivity as well.
3.2. Spectroscopic characterizations
FTIR spectrum of free vanillin exhibited an intense band at 1666 cm−1, assigned to aldehyde CO stretching, whereas vanillin bound to copper in the [Cu(phen)(van)2] showed 14 cm−1 lower stretching frequency. This result indicated aldehyde group is also affected by the vanillin coordination, which is in agreement with crystallographic data where carbonyl bond length increased. Other vanillin vibrational bands are observed in the FTIR of the complex without any significant changes. Additionally, it was observed typical vibrational modes of phenanthroline at 851 and 721 cm−1, assigned to C–H angular deformation.
The electronic spectrum of [Cu(phen)(van)2] was recorded in water and showed an expressive number of transition bands (ESI, Fig. S2†), spanning almost the entire visible region. These transition bands are due to intraligand (IL) and “d–d” type transitions, whose assignments are presented in Table S3.† Deprotonated vanillin exhibits an electronic transition band at 347 nm, which is shifted to 309 nm upon protonation (pKa = 7.4). Indeed, our copper complex showed a band at 345 nm, which was assigned to the vanillinate ion supporting its binding to the metal as a deprotonated ion as indicated by the crystallographic structure.
Copper(II) complexes show characteristic “d–d” type electronic transitions usually very much dependent on the symmetry and distortions.32,44 The electronic transition of the precursor complex exhibited a broad “d–d” band at 714 nm, which was substantially altered upon vanillinate ion coordination giving origin to three “d–d” bands at 469, 650 and 775 nm (shoulder). Considering an approximation to D4h symmetry group, these bands should correspond respectively to 2B1g → 2Eg (dxz, dyz → dx2−y2), 2B1g → 2B2g (dxy → dx2−y2) and 2B1g → 2A1g (dz2 → dx2−y2) transitions.44,45 Although the band at 775 nm is not well defined in aqueous solution, only observed as a shoulder, it was better resolved in methanol (Fig. S2†). These split bands supported a non-perfect octahedral complex, strongly indicating a distorted octahedral copper(II) coordination environment and the presence of the asymmetrical vanillin ligand.
EPR spectra were taken in frozen DMF solution at 77 K and in the solid state at room temperature. These spectra are shown in Fig. 4 and their parameters summarized in Table 1. The spectra obtained in frozen DMF solution exhibited an anisotropic profile typical for monomeric copper(II) complexes, where four well-defined hyperfine lines arising in the g‖ region from the interaction of the S = 1/2 electron spin with the I = 3/2 copper nucleus. This spectrum showed axially symmetric g-tensor parameters with g‖ (2.300) > g⊥ (2.060) > 2.0023, indicating the copper(II) has a distorted environment with an axial elongation.
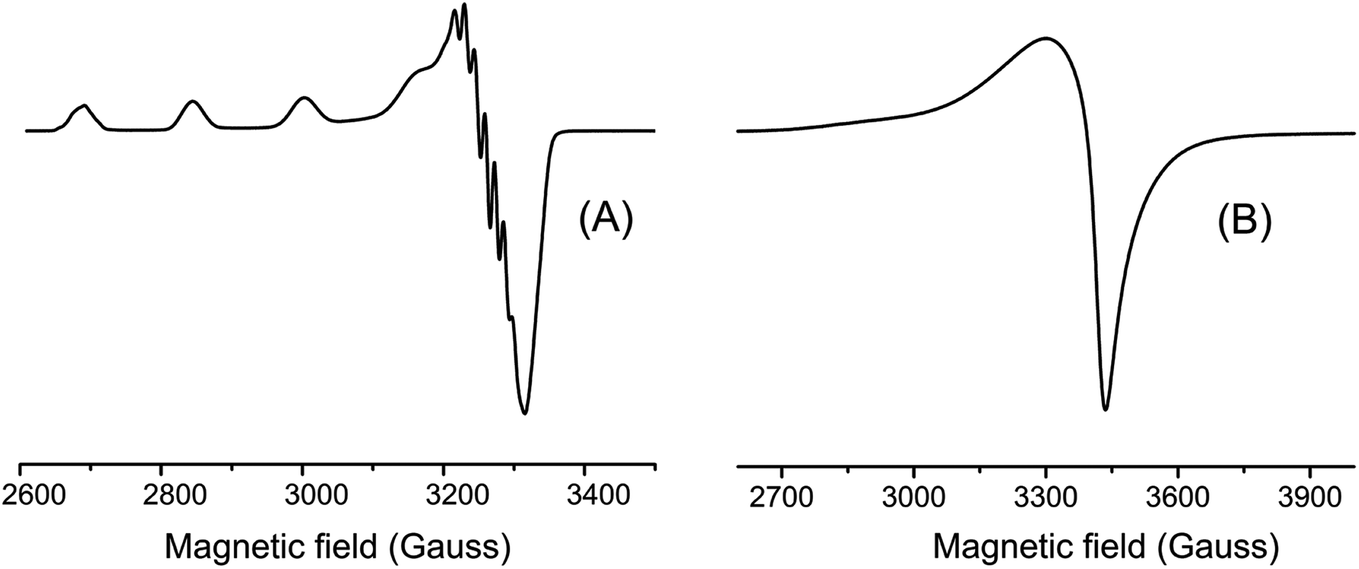 | ||
| Fig. 4 EPR spectra of [Cu(phen)(van)2] in frozen DMF solution (A) and in solid state (B). | ||
| Complex | [Cu(phen)(van)2] | trans-[Cu(bdtbpza)2(pym)(CH3OH)2] | cis-[Cu(bdtbpza)2(tmeda)(H2O)2] |
|---|---|---|---|
| a A‖ (10−4 cm−1) = gβA(G) = 0.46686 gA(G).b G = (g‖ − 2.0023)/(g⊥ − 2.0023). | |||
| Geometry | Octahedral (N2O4) | Octahedral (N2O4) | Octahedral (N2O4) |
| g‖ | 2.300 | 2.316 | 2.260 |
| g⊥ | 2.060 | 2.057 | 2.039 |
| A‖ (10−4 cm−1) | 148 | 169 | 171 |
| A⊥ (10−4 cm−1) | 13 | 19 | 21 |
| g‖/A‖ (cm)a | 155 | 137 | 132 |
| Gb | 5.16 | 5.54 | 6.67 |
The g values suggested the unpaired electron is located in the ground state dx2−y2 (2B1g) of the copper(II) orbital.46 Other EPR parameters are comparable to those reported for complexes containing an octahedral CuN2O4 coordination sphere.46 In contrast, copper(II)–divanillinate complex, [Cu(van)2(H2O)2], organized in a cis-I arrangement, presented an axial symmetry pattern in the solid state at room temperature with g⊥(2.302) > g‖(2.005) ∼2.0023, indicating a compressed geometry.47
Additionally, the five superhyperfine splitting lines (2nIN + 1) in the perpendicular region of the spectrum with coupling value of 13.85 G are due to the interaction with two 14N nuclei (I = 1) from the phenanthroline ligand. On the other hand, the spectrum obtained for the complex in the solid-state did not show any clearly resolved hyperfine splitting neither in parallel nor perpendicular.
The empirical factor f given by the g‖/A‖ ratio has been used to evaluate the tetragonal distortion in copper complex. It has been showed that for planar complexes this ratio is between 110 to 120, while for complexes with slight to moderate distortion between 130 to 150, and for considerable distortion between 180 to 250.46 Our [Cu(phen)(van)2] complex exhibited a f value of 155, indicating a moderately distorted geometry.
3.3. Electrochemical studies
Vanillin, like many other phenolic compounds, has its biological relevance usually related to the antioxidant capacity, which is a consequence of its electrochemical profile.48 The vanillin cyclic voltammetry obtained in aqueous electrolyte (KCl 0.1 mol L−1, pH = 7.0) revealed only one irreversible peak at +720 mV (versus Ag|AgCl), relative to its oxidation (Fig. 5A). This electrochemical process is equivalent to a two-electron oxidation step, which is observed at the same electrochemical potential in aqueous medium as pointed out in literature.49,50 Additionally, it can be observed a decrease in peak current as a consequence of running multiple voltammetric cycles. Li and collaborators described this behavior as due to the polymerization of vanillin oxidation products on the electrode surface.49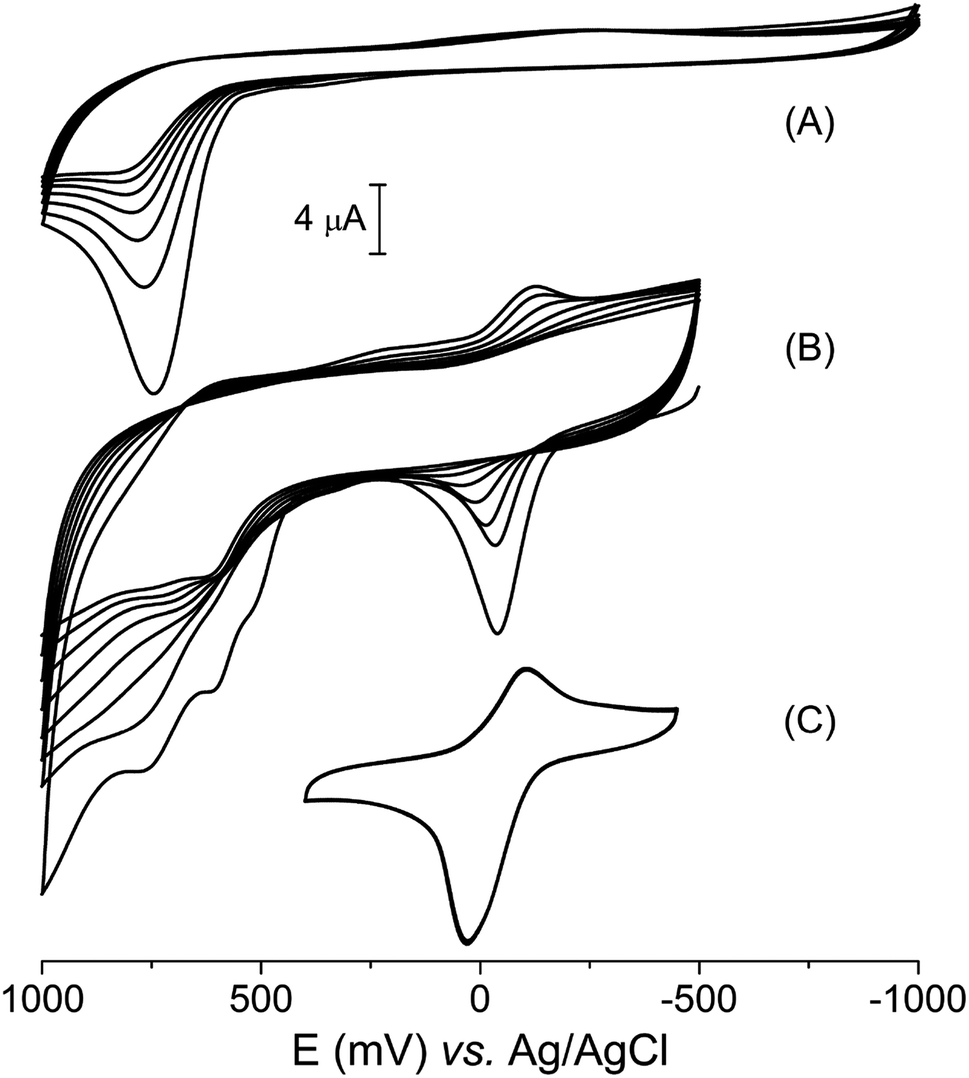 | ||
| Fig. 5 Cyclic voltammogram in KCl 0.1 mol L−1, pH = 7.0 of (A) vanillin, 0.90 mmol L−1 from −1000 to 1000 mV; (B) [Cu(phen)(van)2], 0.90 mmol L−1, from −500 to 1000 mV; (C) [Cu(phen)(van)2] from −400 to 400 mV. For all compounds were conducted eight cycles. | ||
Although the electrochemical behavior of free vanillin is already known, to the best of our knowledge, this is the first work that studied the electrochemical profile of a vanillinate metal complex. The cyclic voltammogram of the [Cu(phen)(van)2] complex showed a quasi-reversible process related to the Cu2+/+ redox couple with E1/2 = −84 mV (vs. Ag|AgCl) along with three oxidation processes at 516, 610 and 760 mV centered on the vanillinate ligands (Fig. 5B).
Multiples voltammetric cycles from −500 to 100 mV were conducted using the copper complex without polishing the electrode between cycles. In this experiment, it was clearly observed a decrease of current to the electrochemical processes centered on both copper and vanillinate ligand, which shared similarity to the electrochemical profile observed for free vanillin. However, the current depletion is strictly dependent on the potential range applied. If the potential range is centered on the metal process from −400 to 400 mV, skipping the ligand redox potentials, then, even multiple cyclic voltammogram showed unchanged profile (Fig. 5C).
In order to investigate the current depletion during the voltammetric experiment K3[Fe(CN)6] was used as a probe. The cyclic voltammogram of K3[Fe(CN)6] (2 mmol L−1, KCl 0.1 mol L−1, pH = 3.5) was obtained using bare glassy carbon and also after multiple voltammetric cycles of [Cu(phen)(van)2] (1 mmol L−1, KCl 0.1 mol L−1, pH = 7.0) without prior polishing. As shown in Fig. S3,† the voltammogram of K3[Fe(CN)6] obtained with the bare electrode exhibited a reversible profile with cathodic potential of 283 mV and difference between peaks (ΔE) of 154 mV. However, after five sweep voltammetric cycles in a [Cu(phen)(van)2] solution, the electrode response to K3[Fe(CN)6] changed. The potentials of the wave peaks were clearly more separated, increasing ΔE to 300 mV in the first cycle, followed by a decrease in the peak currents. This data indicated the electrode surface was modified by the products of [Cu(phen)(van)2] oxidation. Additional multiple voltammetric cycles showed a gradual change with a return of the profile obtained for the probe in bare electrode. These results supported the electrode surface modification and indicated this layer was sensitive to multiples sweeps being likely detached of the electrode surface.
According to Jungwirth et al. the window of accessible redox potential in biological systems can vary from −600 to +610 mV vs. Ag|AgCl, where the strongest reducing agent in cells corresponds to the nicotinamide adenine dinucleotide phosphate (NADP+ + 2e− + H+ → NADPH) with −580 mV. On the other hand, the strongest oxidizing agent is oxygen (O2 + 4H+ + 4e− → 2H2O) at + 610 mV, at pH 7.0.51 Additionally, Betanzos-Lara et al. reported the capacity of Cu+ to generate hydroxyl radical can occurs in biological systems if the metal redox potentials are between −530 to 255 mV vs. Ag|AgCl, which can eventually lead to DNA damage.52 Interestingly, the copper redox potential found for our compound at E1/2 = −84 mV is within that range, indicating it could generate hydroxyl radical (HO˙) in biological systems. The production of this radical can be important if used for therapeutic purposes.
3.4. Nuclease activity
Since DNA is usually reported as an important target for copper(II) compounds exhibiting potential antitumor properties, the activity of the [Cu(phen)(van)2] complex as synthetic nuclease was also evaluated. The ability of the copper(II) complexes to promote DNA cleavage was investigated using supercoiled pBR322 plasmid DNA in agarose gel electrophoresis.DNA cleavage was monitored by following the conversion of the circular supercoiled plasmid DNA form (SC or form I) into the relaxed or nicked circular form due to a single strand breakage (form II), or to the linear form due to a double strand breakage (form III). These features were investigated by evaluating a direct activity of the metal complex or upon stimulation with glutathione (GSH) and hydrogen peroxide. The nuclease efficiency of copper(II) complexes is usually dependent on the type of activator used during DNA cleavage.53 There are some redox activators usually applied for these studies such as thiols (glutathione, 3-mercaptopropionic acid),54 ascorbic acid,55 hydrogen peroxide56 or mixture thereof. The use of these compounds can mimic a chemical environment found in the cytosol of cells.
DNA assay conducted without any activator showed the copper complex did not promote any DNA cleavage even varying the concentration of the complex from 0.5 up to 40 μmol L−1 (Fig. S4†). This result suggests the complex cannot cause DNA cleavage by a hydrolytic pathway under these experimental conditions.
However, there is a remarkable increase in the DNA cleavage in the presence of 10 mmol L−1 of glutathione as noticed by the relaxed form of plasmid (FII), which showed a concentration dependence profile (Fig. 6A). At 20 μmol L−1 of the complex it was observed ca. 50% DNA cleavage, while at the highest concentration, 40 μmol L−1, there was predominantly nicked DNA (FII, 76%) and linear DNA (FIII, 9%). These results clearly indicate the DNA cleavage activity of the copper complex is dependent on the oxidation state of the copper, where generation of copper(I) seems to improve very much the cleavage efficiency. Besides this, it is important to remark GSH is indeed found in millimolar concentration inside cells, which could grant to this complex a nuclease activity supporting the relevance of this investigation.
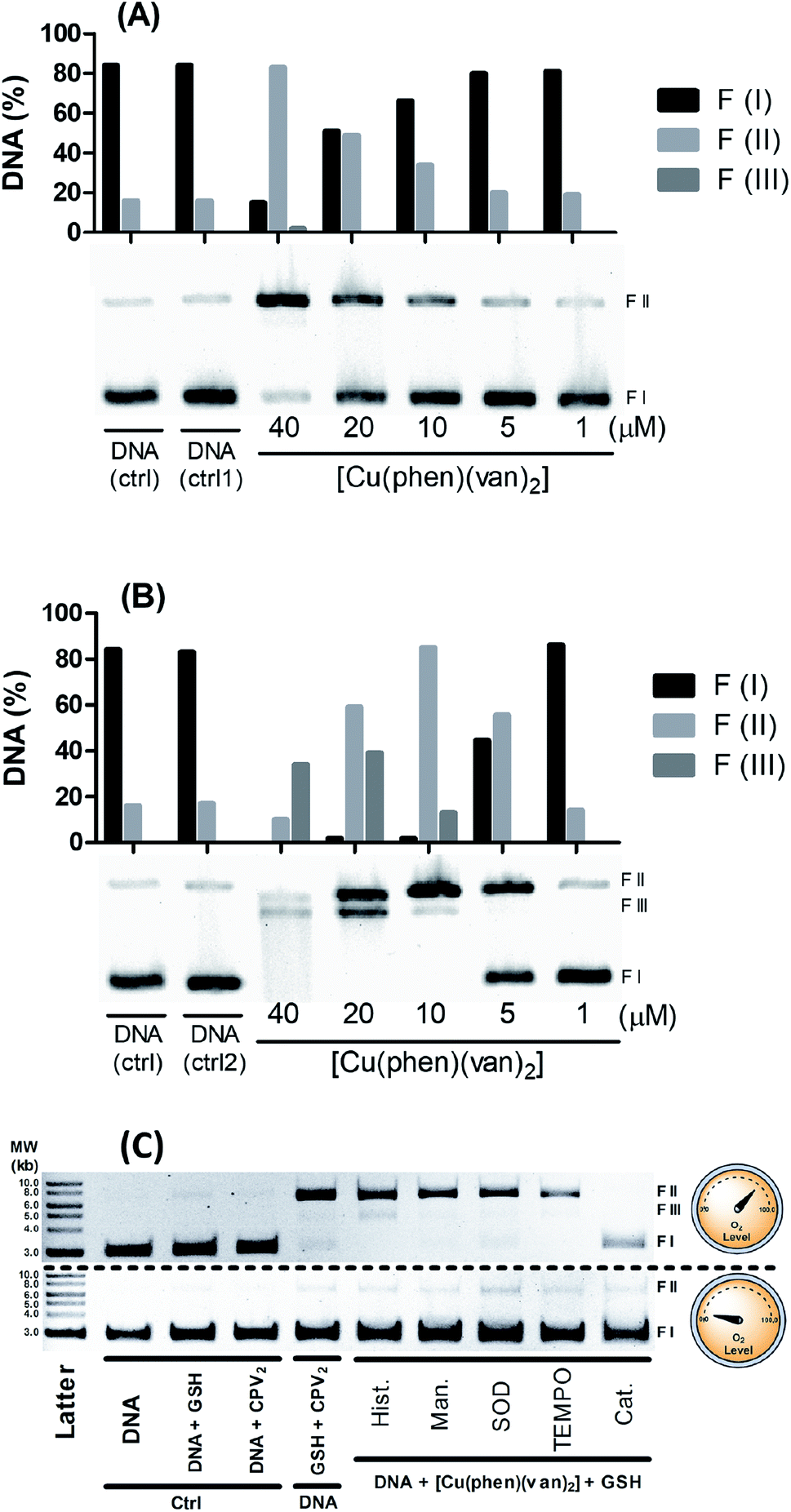 | ||
| Fig. 6 DNA cleavage assay employing plasmid DNA pBR322 (8.3 ng μL−1 per well) with [Cu(phen)(van)2] and 10 mmol L−1 of glutathione (GSH) (A) or 1 mmol L−1 H2O2 (B). Lanes with only DNA (ctrl) and DNA with GSH (ctrl1) or H2O2 (ctrl2) were used as control. Study of oxidizing species involved in the DNA cleavage (C), using GSH (1.5 mmol L−1), [Cu(phen)(van)2] (40 μmol L−1) along with inhibitors of free radicals: histidine (hist.), mannitol (man.), SOD, TEMPO and catalase (cat.). Reactions carried out under aerobic (top gel) and anaerobic (bottom gel) conditions. | ||
Hydrogen peroxide is another potential activator of copper complexes, which could also originate ROS. Here, it was employed 1 mmol L−1 of hydrogen peroxide as activator, where it was observed an expressive cleavage activity originating nicked DNA (FII) at much lower concentration of the complex, e.g. 5 μmol L−1 (Fig. 6B), than that observed with glutathione at 20 μmol L−1. At concentrations above 10 μmol L−1, it was not seen any intact DNA, demonstrating it was entirely cleaved into nicked (FII) and linear DNA (FIII). This latter form was detected more expressively at 20 μmol L−1, which is still at reasonably modest concentration. Additionally, at 40 μmol L−1 of complex it was observed a smeared band consistent with major DNA cleavage producing smaller linear fragments. An attempt to compare these results with precursor complex [Cu(phen)Cl2] was carried out and showed vanillin ligand had improved overall DNA cleavage activity (Fig. S5†).
The results indicate that the DNA cleavage activity of the complex is concentration dependent and also induced by the activator. The most extensive DNA damage was observed when hydrogen peroxide was added. The spectrophotometric monitoring of the complex reactivity with H2O2, in a higher ratio (1:25) than applied in the nuclease activity assay, under the same experimental conditions, did not reveal significant spectral changes (data not showed). This result suggests a structural integrity of the vanillin-based copper compound upon addition of H2O2 as evaluated in the experiment. The DNA cleavage mechanism mediated by [Cu(phen)(van)2] is consistent with DNA breakage induced by polypyridyl copper complexes.57,58
Aiming to shed some light on the mechanism of DNA damage promoted by our complex triggered by glutathione, we carried out a series of measurements with and without oxygen, along with radical scavenger agents. For this study it was mixed 40 μmol L−1 of the metal complexes, 1.5 mmol L−1 of GSH and plasmid DNA with and without oxygen along with radical scavengers (histidine, mannitol, superoxide dismutase – SOD, N-oxyl-2,2,6,6-tetramethylpiperidine – TEMPO, and catalase), which were incubated for 30 minutes at 25 °C. In Fig. 6C, we observed under aerobic conditions there is DNA damage with the production of nicked DNA, whereas no other radical scavenger was able to inhibit cleavage, however catalase had played a significant role in DNA protection.
Interestingly, under anaerobic conditions no DNA cleavage was noticed supporting a full dependence on O2 in the generation of DNA damaging species. A similar mechanistic was also observed for the copper precursor (Fig. S6†), where superoxide dismutase also exhibited a modest inhibitory effect but catalase was the strongest blocker of DNA cleavage under aerobic conditions.
These assays showed there is only oxidative cleavage of DNA, which is promoted by the reaction involving GSH, copper complexes and O2. H2O2 was identified as a key intermediate species involved in the mechanism of cleavage, based on the strong inhibition caused by catalase. Besides that, direct addition of hydrogen peroxide showed cleavage activity only in combination with the copper complex. This agent can also act as a reducing agent during a catalytic cycle of reaction with copper, where it can generate Cu(I) and O2˙−. Copper complexes can still produce HO˙ in a reaction of Cu(I) with H2O2 that may lead to strong DNA damage. Despite the fact neither mannitol nor histidine worked as radical scavengers, this has also been noticed with other copper complex where hydroxyl radical has been clearly identified.12 This lack of protection has been described as due to a likely strong interaction with DNA lowering the effectiveness of radical scavenger action.8,59 The fact that catalase is the only inhibitor that has an efficient action preventing DNA cleavage reinforces the hypothesis the cleavage agents are generated after the reaction of the hydrogen peroxide with the copper(I) complex.
In summary, these data indicated the DNA cleavage mechanism involves the reduction of Cu(II) via GSH reaction, and reduction of O2 to O2˙−. There is also a production of H2O2 through the reaction between copper(I) and O2˙− as observed in other copper complexes.12 In addition, the reaction of copper(I) complex and O2˙− can produce HO˙ as a strong oxidizing agent to damage DNA. A short mechanistic proposal for these species was showed in Fig. S7.†
3.5. Superoxide generation assay
Production of superoxide mediated by the [Cu(phen)(van)2] complex was investigated using nitro blue tetrazolium (NBT) assay following absorbance change at 560 nm.27 The experiment was performed using NBT at 50 μmol L−1 and [Cu(phen)(van)2] complex at 10, 20, 30, 40, 50, 60, and 75 μmol L−1. In order to evaluate the influence of the copper oxidation state in superoxide generation the experiments were conducted with and without the reducing agent GSH (1.5 mmol L−1). The results obtained are shown in Fig. 7, where only a combination of copper complex and GSH led to an intense production of superoxide ion as detected by the gradual increase in absorbance at 560 nm and visual color change. Additionally, there was a concentration dependence during this reaction triggered by GSH addition.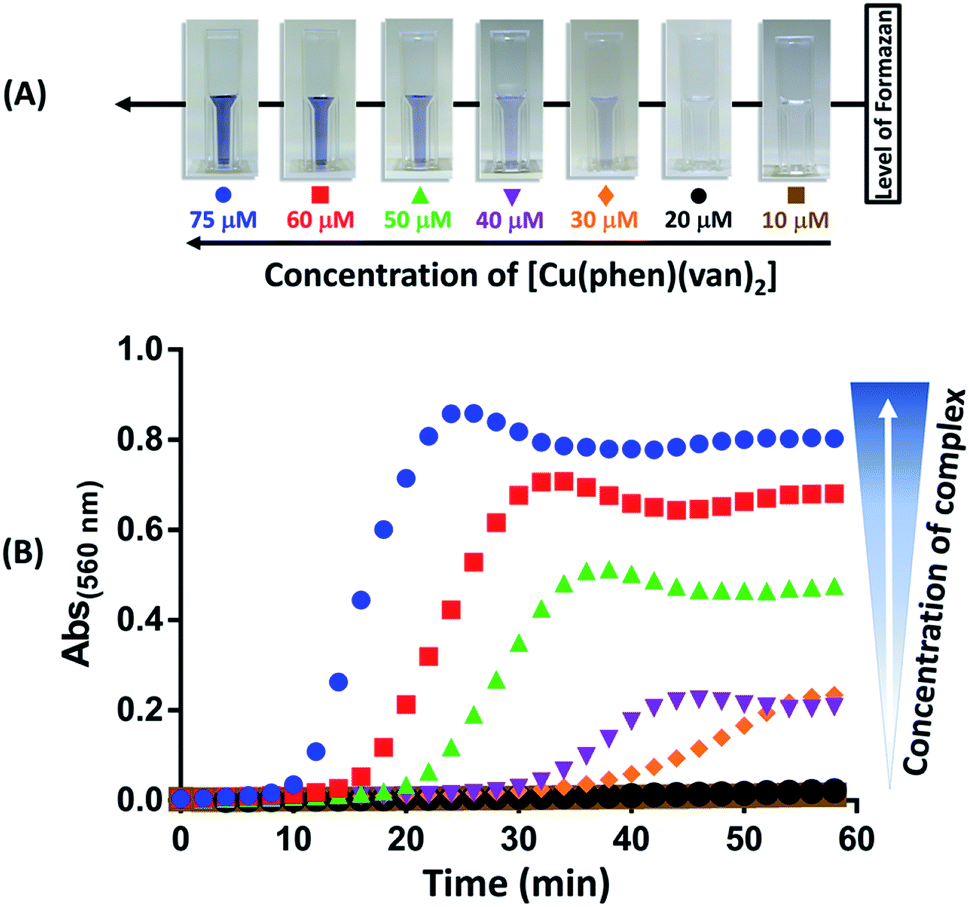 | ||
| Fig. 7 Superoxide detection using NBT (50 μmol L−1) reaction at 37 °C. (A) Color change after 60 min of reaction of GSH (1.5 mmol L−1) with different concentrations of [Cu(phen)(van)2]. (B) Kinetic curves for the reaction of GSH and complex (75 to 10 μmol L−1) using NBT as probe. | ||
This result supported that O2− formation occurs during redox process of copper as proposed in the catalytic cycle depicted in Fig. S7.† Despite many authors have preferred to use ascorbic acid as a co-activator, its physiological concentration is far smaller than glutathione, which reaches up to 15 mmol L−1 while ascorbic acid is only up to 0.2 mmol L−1.60,61 Actually, glutathione is a well-known biological reducing agent found at millimolar concentrations inside cells, it is more likely the key agent to promote ROS generation when our complex reaches cytosol, which could be beneficial for cell death in cancer therapy.
3.6. Cytotoxicity investigations
The cytotoxicity of the complex was evaluated against three different tumor cell lines, mouse melanoma (B16–F10), human hepatocellular carcinoma (HUH-7) and human renal adenocarcinoma (786-0) by MTT assay. The cell viability was measured after 24 h of incubation with [Cu(phen)(van)2] from 0.001 to 300 μmol L−1. In order to assess possible individual contributions of vanillin, 1,10-phenanthroline and CuCl2, they were also evaluated using 786-0 cell line. The IC50 values were calculated from a dose–response curve (Fig. S8 and S9†).Copper complex [Cu(phen)(van)2] showed a dose-dependent behavior and extensive cytotoxicity against all cell lines investigated suggesting its anticancer activity. This complex demonstrated to be effective against melanoma cells with IC50 of 3.39 ± 0.61 μmol L−1, as well as hepatocarcinoma, IC50 of 4.22 ± 0.31 μmol L−1, whereas showed a lower potency for renal carcinoma cells, IC50 of 10.38 ± 0.91 μmol L−1.
The IC50 value for the free ligands and even for CuCl2 in 786-0 cells were higher than 150 μmol L−1, supporting non-cytotoxic effect of these isolated species. Similarly high IC50 values were also reported for these species on other cancer cell lines.13,62,63 This result suggested the cytotoxic activity presented by the complex [Cu(phen)(van)2] is indeed due to changes in copper/ligand reactivity upon formation of the complex and not to any particular individual property.
There are only a few reports of in vitro anticancer activity of copper complexes against B16–F10, HUH-7 and 786-0 cells. Nagababu et al.64 evaluated the cytotoxicity of four copper(II) complexes containing imidazo-phenanthroline (IP) derivatives ligands after 48 h of incubation with B16–F10 cells. The π extended structure of IP ligands have been pointed out as an alternative for increasing the cytotoxicity effect when compared to bpy or phen. However, even using twice the incubation time of our experiment, the IC50 for two of those complexes was over 25 μmol L−1, seven times less efficient than our copper complex. Nevertheless, two of the polypyridyl complexes reported, specifically the ones with the ligands 2-(2-trifluorophenyl)-1H-imidazo[4,5-f][1,10]phenanthroline (TF-PIP) and 2-phenyl-1H-imidazo[4,5-f][1,10]phenanthroline (PIP) had an increased cytotoxicity with IC50 of 2.1 ± 0.2 and 0.5 ± 0.02 μmol L−1, respectively.
Diimine–copper(II) complexes with the phenolate ligand 4-chloro-2-((2-(phenylthio)phenylimino)methyl)phenol were tested against HUH-7 cell line after 24 h of incubation.65 The best result was presented by the 1,10-phenanthroline complex, IC50 of 16.84 μmol L−1, while the complex with 2,2-bipyridine and 4,40-dimethyl-2,2′-bipyridyl were much higher, 23.01 and 31.65 μmol L−1, respectively. Although relevant, these values are also significantly higher than the IC50 of the [Cu(phen)(van)2] for this cell line.
Chen et al.66 evaluated the cytotoxicity of copper(II) complexes with the ligand plumbagin (PLN), a medicinal plant-derived naphthoquinone, against 786-0 cells line after 72 h of incubation. The homoleptic complex, in a square-planar geometry, presented IC50 of 3.4 ± 1.3 μmol L−1, while a binuclear octahedral compound having PLN and bpy in the coordination sphere resulted in a IC50 value of 2.5 ± 0.9 μmol L−1. However, besides the extended exposure to these cells, the ligand itself has a significant cytotoxicity for this cell line (IC50 = 17.9 ± 4.5 μmol L−1). The authors reported also irrelevant cytotoxicity of two other copper–bipyridine complexes, [Cu(bipy)2(H2O)2](NO3)2 and [Cu(acac)(bpy)]NO3, respectively, 5239.3 ± 1411.8 μmol L−1 and 398.7 ± 39.9 μmol L−1.
These results reinforce that the [Cu(phen)(van)2] complex has very good cytotoxicity properties against the cancer cell lines investigated.
3.7. Flow cytometry measurements
Cellular apoptosis studies were conducted using annexin V-FITC/propidium iodide staining in a flow cytometer. In this assay, four different cell populations can be identified after exposure to the copper complex: viable cells (Q4), cells in early apoptosis (Q3), in late apoptosis (Q2) and also in necrosis (Q1). Cells stained only with annexin V-FITC demonstrate the presence of early apoptosis, while cells double labeled with annexin V-FITC and propidium iodide represent late apoptosis and those cells labeled only with propidium iodide are in necrosis.As shown in Fig. 8 treatment of 786-0 cells with 10 μmol L−1 (∼IC50) of copper complex for 24 h resulted in 58.95% of apoptotic cells (Q2 and Q3 quadrants) and a negligible value of necrosis (0.83%). Additionally, it was evaluated the effect of the copper complex at 50 μmol L−1 on the cell death pathway, which corresponded to almost five times its IC50. In this case, apoptotic cells reached 98.77% with an expressive contribution of late apoptosis, 97.40%, and insignificant necrosis rate of 0.64%. Interestingly, the efficiency of this compound is significantly higher than the positive control, cisplatin, for what was found 34.0 and 47.7% of apoptosis at 10 and 50 μmol L−1, respectively. The flow cytometry for untreated cells (negative control) is shown in Fig. S10.† These results suggested that the complex induced cell death mainly by apoptosis.
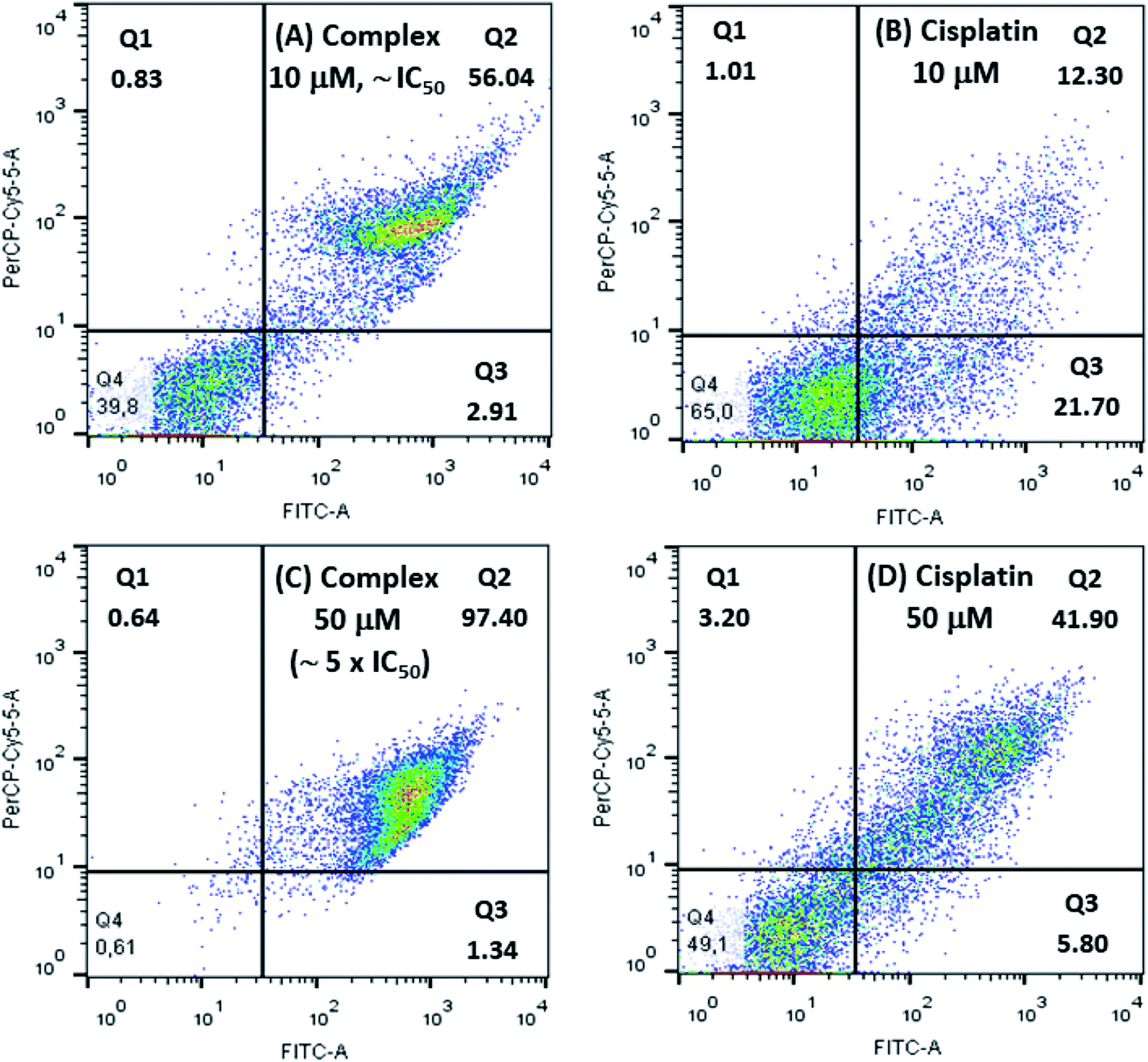 | ||
| Fig. 8 Flow cytometry studies for 786-0 cells treated with 10 μmol L−1 and 50 μmol L−1 of [Cu(phen)(van)2], (A and C), and cisplatin, positive control, (B and D). | ||
González-Álvarez and coworkers reported flow cytometry studies of four copper(II) bipyridine–sulfonamide complexes at 10 μmol L−1 (∼2 × IC50) on Jurkat T lymphocytes cells, after incubation for 48 h. Two of these complexes induced apoptosis in only 25% of cells. The other two complexes caused apoptosis rate between 40 and 50%.67 Comparatively, the [Cu(phen)(van)2] complex showed more efficient apoptosis induction (58.95%) considering also IC50 dose with half of the incubation time.
3.8. Cell cycle analysis
In order to further investigate the effect caused by the [Cu(phen)(van)2] complex on 786-0 cells cycle progression, the DNA content was evaluated by flow cytometry after staining with propidium iodide (PI).68As shown on Fig. 9, the distribution of the different phases (G1, S and G2/M) was significantly altered after incubation of cells with 5, 10, 20 or 50 μmol L−1 of complex during 24 h when compared to untreated control. The concentrations investigated allowed the identification of two different profiles. First of all, at concentrations of 5 and 10 μmol L−1, it resulted in a clear increase of 786-0 cells at G1 phase with concomitant depletion of G2/M. The G1 distribution on untreated cells at 66.25 ± 2.05% gave place to 84.25 ± 1.06% at 10 μmol L−1 of the complex. This G1 arrest may be a consequence of the DNA damage induced by the complex, making it impossible for the cells to be routed to the S and G2/M phases by checkpoint mechanism.69 This control of the cell cycle progression in cancer cells exerted by the complex is considered a potential strategy for the control of tumor growth since these cells are cycling more rapidly than healthy cells.
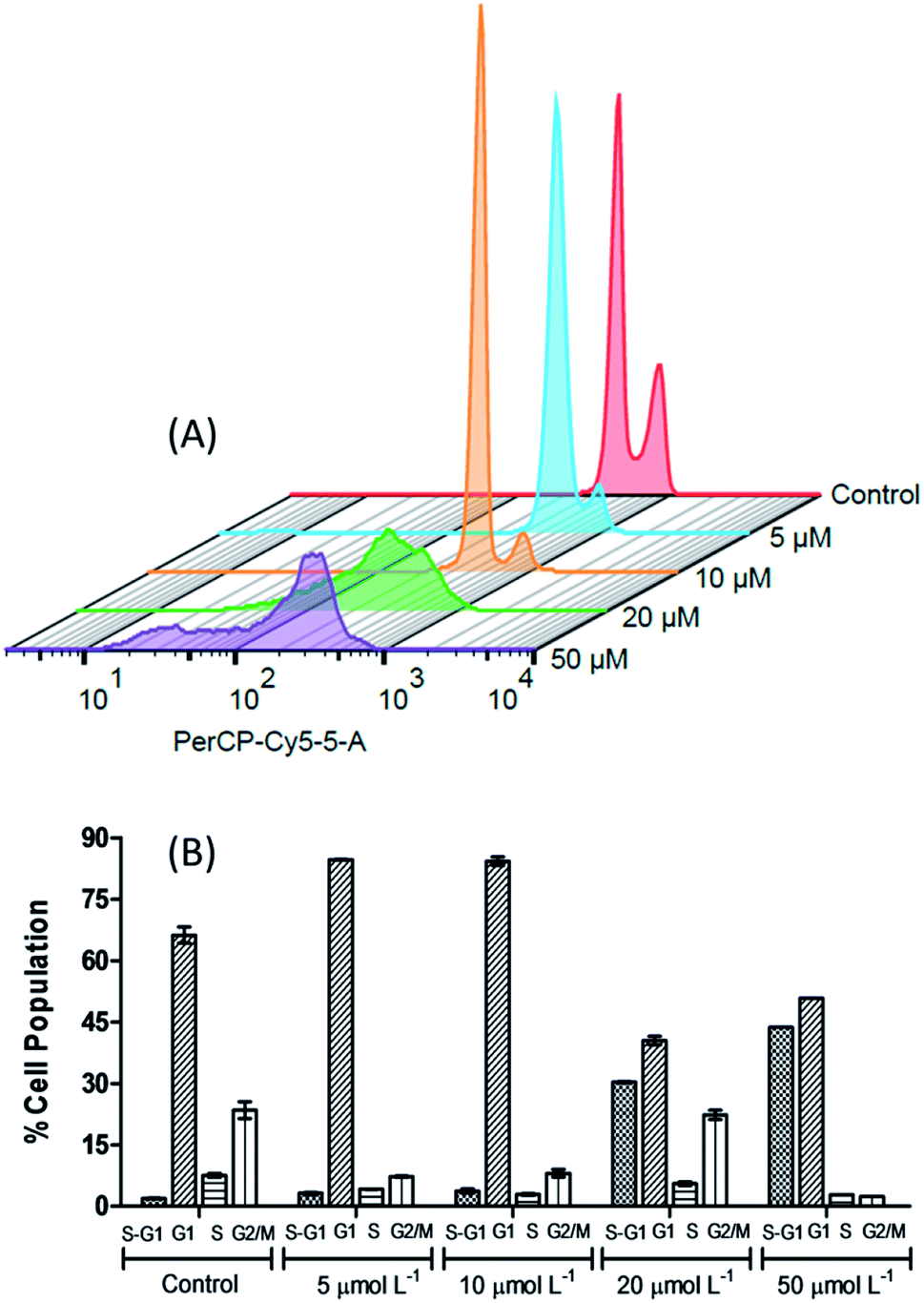 | ||
| Fig. 9 The 3D histogram plot of flow cytometry analysis in 786-0 cells treated without (control) and with [Cu(phen)(van)2] (5, 10, 20 and 50 μmol L−1). | ||
Additionally, the sub-G1 (hypodiploid) content became more significant at 10 μmol L−1 (3.72 ± 0.51%) compared with the control (1.90 ± 0.08%). The presence of sub-G1 cells suggests the activation of cell apoptosis induced by the complex.64,70 On the other hand, if 20 and 50 μmol L−1 of the complex was used then the cell cycle progression showed a clear depletion of G1, S and G2/M content and an expressive increase in the percentage of sub-G1 in comparison with the untreated control cells.
This experiment indicates that the [Cu(phen)(van)2] complex has an antiproliferative activity at 10 μmol L−1, characterized by the cell cycle arrest at G1 phase, as well as, a strong cytotoxic behavior when the cells were submitted to 20 μmol L−1 of the complex. Additionally, there are also evidences of apoptosis induction observed by annexin V- FITC/PI staining for 786-0 cells.
The increment of cells in G1 phase at IC50 dose of [Cu(phen)(van)2] complex, 27.8%, was significantly more expressive than the values found for other copper(II)-phen compounds. These latter compounds also promote G1 cell arrest in MDA-MB-231 breast cancer cells, e.g. [Cu(phen)(aa)(H2O)]NO3 (aa = glycine (12.5%), sarcosine (12.6%) or 2,2-dimethylglycine (14.4%)).69 Additionally, the drugs in clinical use, cisplatin and oxaplatin, are known to induce cell cycle arrest at the S or G2/M phase depending on the cell line and incubation time. Our distinct phase–arrest profile implies in a different mechanistic pathway caused by [Cu(phen)(van)2] when compared to these platinum-based drugs.
Active copper complexes can interact with cellular machinery causing cell arrest in G1,71,72 S,73 or G2/M74 phases depending on the tumor cell and nature of ligands bound to the metal. However, compounds that induce cell cycle arrest in G1 phase may have potential advantages. This phase is determinant to assure the cells have the appropriated genetic material to proceed or interrupt the cell cycle. In the case of defective cell progress from G1 to other stages in the cell cycle there are other subsequent checkpoints in S and G2/M phases before the cell starts the mitosis. In addition to that, since G1 comprises the longest growth period of interphase any interruption in this phase may enable elimination of the defective cells.
4. Conclusion
A new octahedral vanillinate–Cu(II) complex with an unusual cis-III arrangement, where the methoxy groups of the vanillinate ions are coordinated opposite to each other in the structure, was prepared and fully characterized. This compound showed a strong ability to cleave DNA only under co-activation with redox active agents, hydrogen peroxide and glutathione, supporting an oxidative nuclease pathway.The DNA damage promoted by the complex along with GSH was fully dependent on O2, which clearly indicated a key role of electron transfer from Cu(I) to O2 and consequent formation of superoxide radical as an important route of action of this compound. Considering that O2, H2O2, and glutathione are present in widely variable concentrations in some cell types and organelles, they might cause different degrees of reactivity, depending on the cellular localization (e.g. nucleus, mitochondria and cytoplasm) or even the redox state of the cell (e.g. under oxidative stress and normal conditions).
The increase of ROS promotes a major redox unbalance leading to severe damages and cell death. Our results showed low IC50 for three different cancer cells, significantly lower than those found for CuCl2 or the ligands 1,10-phenanthroline and vanillin. This might be due to our complex behave as a much better ROS generator. Indeed, [Cu(phen)(van)2] performed even better then cisplatin as showed by flow cytometry assays against 786-0 cell line, where 10 μmol L−1 (∼IC50) resulted in 58.95% of apoptotic cells (Q2 and Q3) and 98.77% for 50 μmol L−1 (∼IC50), while cisplatin reached only 47.7% at even higher dosage.
In addition to that, increasing cell cycle depletion of S and G2/M, observed at 20 μmol L−1 of complex, and consequent G1 phase arrest, is a great indication that the complex induces DNA damage, making it difficult for the cells to be driven to S and G2/M phases. At concentrations higher than 20 μmol L−1, an expressive increase of sub-G1 cells was observed, indicating that the damage caused by the complex was sufficiently high leading a large number of cells to apoptosis. Altogether, these results grant this compound a particular interest as a promising antitumor chemotherapeutic agent that deserves further biological studies.
Conflicts of interest
There are no conflicts to declareAcknowledgements
The authors thank CNPq and CAPES for their financial support. The Department of Biochemistry at the Universidade Federal do Rio Grande do Norte for allowing us to use its facilities of cell culture. Dra Shirley Nakagaki and Laboratório de Bioinorgânica e Catálise, Universidade Federal do Paraná, for providing EPR analysis. We also acknowledge CNPq for granting award fellowship to E. H. S. S. (process 312030/2015-0) and Universal (process 403866/2016-2).References
- L. Dai, J. Liu, Z. Luo, M. Li and K. Cai, J. Mater. Chem. B, 2016, 4, 6758–6772 RSC.
- N. J. Wheat, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- M. Vitali, C. I. Ripamonti, F. Roila, C. Proto, D. Signorelli, M. Imbimbo, G. Corrao, A. Brissa, G. Rosaria, F. de Braud, M. C. Garassino and G. Lo Russo, Crit. Rev. Oncol. Hematol., 2017, 118, 7–14 CrossRef PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- J. Chien, R. Kuang, C. Landen and V. Shridhar, Front Oncol., 2013, 3, 1–6 Search PubMed.
- E. H. S. Sousa, L. A. Ridnour, F. S. Gouveia Junior, C. D. S. Silva, D. A. Wink, L. G. de F. Lopes and P. J. Sadler, ACS Chem. Biol., 2016, 11, 2057–2065 CrossRef PubMed.
- I. Romero-Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- I. Iakovidis, I. Delimaris and S. M. Piperakis, Mol. Biol. Int., 2011, 2011, 1–13 CrossRef PubMed.
- R. F. Brissos, A. Caubet and P. Gamez, Eur. J. Inorg. Chem., 2015, 2015, 2633–2645 CrossRef CAS.
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS.
- A. I. B. Romo, D. S. Abreu, T. de F. Paulo, M. S. P. Carepo, E. H. S. Sousa, L. Lemus, C. Aliaga, A. A. Batista, O. R. Nascimento, H. D. Abruça and I. C. N. Diógenes, Chem. - Eur. J., 2016, 22, 10081–10089 CrossRef CAS PubMed.
- A. Kellett, O. Howe, M. O'Connor, M. McCann, B. S. Creaven, S. McClean, A. Foltyn-Arfa Kia, A. Casey and M. Devereux, Free Radicals Biol. Med., 2012, 53, 564–576 CrossRef CAS PubMed.
- S.-L. Wu, J.-C. Chen, C.-C. Li, H.-Y. Lo, T.-Y. Ho and C.-Y. Hsiang, J. Pharmacol. Exp. Ther., 2009, 330, 370–376 CrossRef CAS PubMed.
- T. Ohta, M. Watanabe, K. Watanabe and Y. Shirasu, Food Chem. Toxicol., 1986, 24, 51–54 CrossRef CAS PubMed.
- S. De Flora, C. Bennicelli, A. Rovida, L. Scatolini and A. Camoirano, Mutat. Res. Fund. Mol. M., 1994, 307, 157–167 CrossRef CAS.
- D. T. Shaughnessy, R. W. Setzer and D. M. DeMarini, Mutat. Res., 2001, 480–481, 55–69 CrossRef CAS PubMed.
- H. Imanishi, Y. F. Sasaki, K. Matsumoto, M. Watanabe, T. Ohta, Y. Shirasu and K. Tutikawa, Mutat. Res. Lett., 1990, 243, 151–158 CrossRef CAS.
- K. Lirdprapamongkol, H. Sakurai, N. Kawasaki, M. K. Choo, Y. Saitoh, Y. Aozuka, P. Singhirunnusorn, S. Ruchirawat, J. Svasti and I. Saiki, Eur. J. Pharm. Sci., 2005, 25, 57–65 CrossRef CAS PubMed.
- M. Makni, Y. Chtourou, H. Fetoui, E. M. Garoui, T. Boudawara and N. Zeghal, Eur. J. Pharmacol., 2011, 668, 133–139 CrossRef CAS PubMed.
- S. Durant and P. Karran, Nucleic Acids Res., 2003, 31, 5501–5512 CrossRef CAS PubMed.
- D. J. Awad, F. Conrad, A. Koch, U. Schilde, A. Poppl and P. Strauch, Inorg. Chim. Acta, 2010, 363, 1488–1494 CrossRef CAS.
- Enraf-Nonius, COLLECT, Nonius BV, Delft, The Netherlands, 1997–2000 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Academic Press, New York, 1997, vol. 276 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- F. J. Schmitt, G. Renger, T. Friedrich, V. D. Kreslavski, S. K. Zharmukhamedov, D. A. Los, V. V. Kuznetsov and S. I. Allakhverdiev, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 835–848 CrossRef CAS PubMed.
- G. He, J. Sun, R. Zhao and J. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m417 CAS.
- F. Dimiza, F. Perdih, V. Tangoulis, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 476–489 CrossRef CAS PubMed.
- H.-H. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m3325–m3327 CAS.
- B. Kozlevcar, M. Humar, P. Strauch and I. Leban, Z. Naturforsch. B Chem. Sci., 2005, 60, 1273–1277 CrossRef CAS.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
- T. Lee, H. R. Chen, H. Y. Lin and H. L. Lee, Cryst. Growth Des., 2012, 12, 5897–5907 CAS.
- D.-D. Lin and D.-J. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1821–m1822 CAS.
- J. Gao and Y. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1692–m1693 CAS.
- B. Kozlevcar, N. Kitanovski, Z. Jaglicic, N. A. G. Bandeira, V. Robert, B. Le Guennic and P. Gamez, Inorg. Chem., 2012, 51, 3094–3102 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- T. J. Greenhough and M. F. C. Ladd, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2744–2752 CrossRef.
- T. J. Greenhough and M. F. C. Ladd, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2619–2621 CrossRef.
- C. Kuo, T. Huang, M. Shao and H. Gau, Inorg. Chim. Acta, 1999, 293, 12–19 CrossRef CAS.
- R. K. Minhas, J. J. H. Edema, S. Gambarotta and A. Meetsmat, J. Am. Chem. Soc., 1993, 115, 6710–6717 CrossRef CAS.
- K. Prout, A. Edwards, V. Mtetwa, J. Murray, J. F. Saunders and F. J. C. Rossotti, Inorg. Chem., 1997, 36, 2820–2825 CrossRef CAS PubMed.
- M. F. C. Ladd and D. C. Povey, J. Mol. Struct., 1976, 6, 205–215 CrossRef CAS.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- A. B. Lever, Inorganic Electronic Spectroscopy, Elsevier Science, 1986 Search PubMed.
- J. M. Gajewska, W. Ching, Y.-S. Wen and C.-H. Hung, Dalton Trans., 2014, 43, 14726–14736 RSC.
- B. Kozlevcar, A. Golobic and P. Strauch, Polyhedron, 2006, 25, 2824–2828 CrossRef CAS.
- F. B. Daniel Chavarria, T. Silva, D. Martins, J. Bravo, T. Summavielle and J. Garrido, MedChemComm, 2015, 6, 1043–1053 RSC.
- Q. Li, C. Batchelor-Mcauley and R. G. Compton, J. Phys. Chem. B, 2010, 114, 9713–9719 CrossRef CAS PubMed.
- G. Ziyatdinova, E. Ziganshina and H. Budnikov, Anal. Methods, 2013, 5, 4750–4756 RSC.
- U. Jungwirth, C. R. Kowol, B. K. Keppler and G. Christian, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- S. Betanzos-Lara, N. P. Chmel, M. T. Zimmerman, L. R. Barrón-Sosa, C. Garino, L. Salassa, A. Rodger, J. L. Brumaghim, I. Gracia-Mora and N. Barba-Behrens, Dalton Trans., 2015, 44, 3673–3685 RSC.
- D. D. Li, J. L. Tian, W. Gu, X. Liu, H. H. Zeng and S. P. Yan, J. Inorg. Biochem., 2011, 105, 894–901 CrossRef CAS PubMed.
- B. de Souza, A. J. Bortoluzzi, T. Bortolotto, F. L. Fischer, H. Terenzi, D. E. Ferreira, W. R. Rocha and A. Neves, Dalton Trans., 2010, 39, 2027–2035 RSC.
- R. F. Brissos, E. Torrents, F. Mariana dos Santos Mello, W. Carvalho Pires, E. de Paula Silveira-Lacerda, A. B. Caballero, A. Caubet, C. Massera, O. Roubeau, S. J. Teat and P. Gamez, Metallomics, 2014, 6, 1853–1868 RSC.
- S.-P. Y. Jun-Ling Li, L. Jiang, Bi-W. Wang, J.-L. Tian, W. Gu and X. Liu, New J. Chem., 2015, 39, 529–538 RSC.
- X.-F. Zhao, Y. Ouyang, Y.-Z. Liu, Q.-J. Su, H. Tian, C.-Z. Xie and J.-Y. Xu, New J. Chem., 2014, 38, 955 RSC.
- M. Salimi, K. Abdi, H. M. Kandelous, H. Hadadzadeh, K. Azadmanesh, A. Amanzadeh and H. Sanati, BioMetals, 2015, 28, 267–278 CrossRef CAS PubMed.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC.
- S. J. Padayatty, H. Sun, Y. Wang, H. D. Riordan, S. M. Hewitt, A. Katz, R. A. Wesley and M. Levine, Ann. Intern. Med., 2004, 140, 533–538 CrossRef CAS PubMed.
- M. Deponte, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3217–3266 CrossRef CAS PubMed.
- P. Khan, S. Rahman, A. Queen, S. Manzoor, F. Naz, G. M. Hasan, S. Luqman, J. Kim, A. Islam, F. Ahmad and M. I. Hassan, Sci. Rep., 2017, 1–15 Search PubMed.
- C. H. Ng, C. Kong, S. T. Von, P. Balraj, P. Jensen, E. Thirthagiri, H. Hamada and M. Chikira, Dalton Trans., 2008, 447–454 RSC.
- P. Nagababu, A. K. Barui, B. Thulasiram, C. S. Devi, S. Satyanarayana, C. R. Patra and B. Sreedhar, J. Med. Chem., 2015, 58, 5226–5241 CrossRef CAS PubMed.
- S. Kathiresan, S. Mugesh, M. Murugan, F. Ahamed and J. Annaraj, RSC Adv., 2016, 6, 1810–1825 RSC.
- Z.-F. Chen, M.-X. Tan, L.-M. Liu, Y.-C. Liu, H.-S. Wang, B. Yang, Y. Peng, H.-G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 10824–10833 RSC.
- M. González-Álvarez, A. Pascual-Álvarez, L. del C. Agudo, A. Castiñeiras, M. Liu-González and G. Alzuet-Piña, Dalton Trans., 2013, 42, 10244–10259 RSC.
- M. G. Ormerod, Flow Cytometry: A practical approach, Oxford University Press, New York, 3rd edn, 2000 Search PubMed.
- C. H. Ng, S. M. Kong, Y. L. Tiong, M. J. Maah, N. Sukram, M. Ahmad and A. S. B. Khoo, Metallomics, 2014, 6, 892–906 RSC.
- V. Gandin, M. Porchia, F. Tisato, A. Zanella, E. Severin, A. Dolmella and C. Marzano, J. Med. Chem., 2013, 56, 7416–7430 CrossRef CAS PubMed.
- K. Hu, G. Zhou, Z. Zhang, F. Li, J. Li and F. Liang, RSC Adv., 2016, 36077–36084 RSC.
- V. Punj, S. Bhattacharyya, D. Saint-Dic, C. Vasu, E. A. Cunningham, J. Graves, T. Yamada, A. I. Constantinou, K. Christov, B. White, G. Li, D. Majumdar, A. M. Chakrabarty and T. K. Das Gupta, Oncogene, 2004, 23, 2367–2378 CrossRef CAS PubMed.
- K.-B. Huang, Z.-F. Chen, Y.-C. Liu, X.-L. Xie and H. Liang, RSC Adv., 2015, 5, 81313–81323 RSC.
- Q.-P. Qin, Y.-C. Liu, H.-L. Wang, J.-L. Qin, F.-J. Cheng, S.-F. Tang and H. Liang, Metallomics, 2015, 7, 1124–1136 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1486391. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03626h |
| This journal is © The Royal Society of Chemistry 2018 |