
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArsenic removal from water/wastewater using layered double hydroxide derived adsorbents, a critical review
Junya Wang
 a,
Taiping Zhang
a,
Min Li
a,
Ying Yang
a,
Peng Lu
b,
Ping Ning
*a and
Qiang Wang
c
a,
Taiping Zhang
a,
Min Li
a,
Ying Yang
a,
Peng Lu
b,
Ping Ning
*a and
Qiang Wang
c
aFaculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming, 650500, Yunnan, P. R. China. E-mail: 1456149879@qq.com; Tel: +86 13708409187
bSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, 315211, Zhejing, P. R. China
cCollege of Environmental Science and Engineering, Beijing Forestry University, 35 Qinghua East Road, Haidian District, Beijing 100083, P. R. China
First published on 20th June 2018
Abstract
Arsenic pollution has become a worldwide environmental concern. Dangerous arsenic concentrations in natural waters threaten the health of millions of people, and this has received significant attention. Among the various technologies that have been developed for arsenic removal from water, the use of adsorption technology is considered to be a prevailing method, because the adsorption approach usually has high removal efficiency and the advantage of convenience of handling. In recent years, layered double hydroxides (LDHs) have become prime candidates for arsenic removal, due to their hydrophilic nature and cationic layered structures. Research on arsenic removal using LDHs is mainly focused on (1) the influence of the synthesis method and composition of the LDH, (2) the influence of the particle size of the LDH, (3) the influence of the Mg/Al ratio in LDHs, (4) LDH-based hybrids and (5) the competition with other anions. This paper provides a review of the currently available literature focusing on arsenic removal using LDHs for the five parts mentioned above. In addition, based on this overview, a closing section will suggest research efforts for future work. It is expected that this review will provide a summary of the main research in this area, and will also shed light on the direction of future development.
 Junya Wang | Junya Wang received her B.Sc from Heilongjiang University in 2007 and her M.Sc from the Guilin University of Technology in 2010. After that, she worked at the Guangxi Eco-engineering Vocational and Technical College. Following this, she attained her Ph.D from Beijing Forestry University in 2016. Since 2016, she has worked as an Associate Professor in the Faculty of Environmental Science and Engineering at Kunming University of Science and Technology in China. Her research interests are in environmental functional materials. |
 Taiping Zhang | Taiping Zhang is a master’s candidate at the College of Environmental Science and Engineering at Kunming University of Science and Technology in China. She received her bachelor’s degree at Yuxi Normal University in June 2017. Her research interests are in adsorption materials. |
 Min Li | Min Li is currently a master’s candidate in the Faulty of Environmental Science and Engineering at Kunming University of Science and Technology. She received her bachelor’s degree at Hengyang Normal University in June 2017. Her research interest is the study of adsorbents. |
 Ying Yang | Ying Yang is currently a master’s candidate in the Faulty of Environmental Science and Engineering at Kunming University of Science and Technology. She received her bachelor’s degree at China West Normal University in 2016. Her research interests are in layered double hydroxide derived CO2 capture adsorbents. |
 Peng Lu | Peng Lu received his B.Sc. degree (2012) in Environmental Engineering from the North University of China and M.Sc. degree (2014) in hydraulic engineering from the University of Jinan, and his Ph.D. (2017) in Environmental Science and Engineering from Beijing Forestry University in China. Now he holds an Assistant Professor position in the School of Materials Science and Chemical Engineering at Ningbo University in China. His research is in the general area of membrane-based processes for gas/liquid separation, and especially focuses on the preparation of layered double hydroxides (LDHs) as nanofiller modified thin-film composite (TFC) membranes. |
 Ping Ning | Ping Ning received his B.Sc. (1982) and M.Sc. (1985) from Kunming University of Science and Technology in China, and his Ph.D. (1995) from the University of Kaiserslautern in Germany. In 1995, he worked as a Postdoctoral Associate in the Department of Chemistry, University of Kaiserslautern. Since 1996, he has worked in the Faulty of Environmental Science and Engineering at Kunming University of Science and Technology. His research interest is environmental pollution control. He was given the award for nationally outstanding teachers and is a distinguished professor in the Yunnan province, and enjoys the State Council Special Allowance in China. |
 Qiang Wang | Qiang Wang received his B.Sc. (2003) and M.Sc. (2005) from the Harbin Institute of Technology in China, and his Ph.D. (2009) from POSTECH in South Korea. From 2009–2011, he worked as a Research Fellow in the Institute of Chemical and Engineering Sciences under A*STAR in Singapore. From 2011–2012, he worked as a Postdoctoral Associate in the Department of Chemistry, University of Oxford. Since 2012, he has held a full professor position in the College of Environmental Science and Engineering at Beijing Forestry University researching environmental functional materials. He was awarded the China 1000 Talents Plan for Young Scholars in 2013. |
1. Introduction
Arsenic (As), a metalloid which occurs naturally, is the 20th most abundant element in the Earth’s crust, the 14th in seawater and the 12th in the human body, and is a component of more than 245 minerals.1 These are mostly ores containing sulfide, along with copper, nickel, lead, cobalt, or other metals. As and its compounds are mobile in the environment.1 Most environmental As problems are the result of mobilization under natural conditions. However, mining activities, the combustion of fossil fuels, petroleum refining, sewage sludge, the use of As pesticides, herbicides, and crop desiccants, the ceramic manufacturing industries, and the use of As additives in livestock feed increase As concentrations in the environment.2–5 As occurs in the environment mainly as inorganic arsenic oxides, such as arsenite (As(III)), arsenate (As(V)) and methylated forms (e.g. (CH3)3As and (CH3)2AsOOH).6,7 As is a carcinogen for both humans and animals.8,9 Of the various sources of As in the environment, drinking water probably poses the greatest threat to human health.10 As is considered to be one of the most toxic pollutants, and is introduced into natural water in different ways.11 The World Health Organization (WHO) guideline value for As in drinking water was provisionally reduced in 1993 from 50 to 10 μg L−1. The new recommended value was chosen based on increasing awareness of the toxicity of As, particularly its carcinogenicity, and on the ability to measure it quantitatively.12Therefore, dangerous As concentrations in natural water have become one of the major worldwide environmental concerns. Millions of people have been exposed to high As concentrations (through contaminated drinking water), raising severe health problems, which have received significant attention.2,13,14 Up to now, different techniques have been developed and tried out for the removal of As from water (e.g. coagulation and flocculation, precipitation, membrane filtration, ion exchange and adsorption).15–21 Among these techniques, the removal of As from aqueous solutions using adsorption technology is the prevailing remediation method, because the adsorption approach usually has high removal efficiency and the advantage of convenience of handling.22–25 Several materials have already been proposed for the adsorption of As from water, which include biological materials, mineral oxides, activated carbons, polymer resins and LDHs etc.16,26–30 Of these materials, LDHs have become the prime candidates for As removal, with As removal capacities in the range of 5–615 mg g−1, due to their hydrophilic nature and cationic layered structures.31–33 LDHs are a class of ionic lamellar compound, and are made up of stacked positively charged brucite-like layers, with the interlayer spaces containing charge compensating anions and solvation molecules.34–37 Metal cations occupy the centers of octahedra whose vertexes contain hydroxide ions. The octahedra are connected through edge sharing to form an infinite sheet.38 The composition of LDHs is [M1−x2+Mx3+(OH)2][An−]x/n·mH2O, as shown in Fig. 1, where M2+ could be Mg2+, Zn2+, Co2+, Ca2+ or Ni2+ and M3+ could be Al3+, Fe3+, Ga3+ or Mn3+. An− is the anion, which is intercalated in the interlayer for charge compensation, and it can an inorganic or an organic anion, e.g. CO32−, NO3−, Cl− or CH3COO− etc.39–44 In addition, LDHs may also contain M+ and M4+ cations but these are limited to specific examples such as Li+ and Ti4+.34 Due to their well-defined layer structure with its interlayer distance of nanometers, large surface areas, high anion exchange capacities and pH buffering capacities, LDH materials have been intensively studied in a wide range of important areas45–50 e.g. catalysis, photochemistry, gas adsorption, flame retardants, electrochemistry, polymerization, magnetization, biomedical science and metal corrosion inhibition and for environmental applications.34,51–62 Since LDHs often possess large surface areas, high anion exchange capacities, small crystallite sizes and good thermal stabilities and can also be easily recycled, they are excellent candidates for removing anions from aqueous solutions.63–65 LDHs remove As from water mainly via three mechanisms: (1) adsorption, (2) ion exchange, and (3) structure reconstruction.66,67 As can be removed by adsorption onto the external surface of LDHs. Also, the interlayer ions in LDHs can be replaced by As for the positively-charged binding sites in the hydroxide sheets. The latter mechanism is that As can be used as the interlayer ions to reconstruct the calcined LDHs and the LDHs can recover their original structure. The reaction equations for As(III) and As(V) with calcined LDHs are shown in eqn (1) and (2). A schematic illustration of the removal mechanism is shown in Fig. 2.
| M2+ + M3+ + AsO43− → Mx2+M1−x3+(OH)2x+3y−3z(As3−)z·mH2O | (1) |
| M2+ + M3+ + AsO2− → Mx2+M1−x3+(OH)2x+3y−z(As−)z·mH2O | (2) |
 | ||
| Fig. 1 Structure models for LDHs [M1−x2+Mx3+(OH)2][An−]x/n·mH2O (a) 3D structure and (b) 2D structure (reproduced with permission from ref. 68). | ||
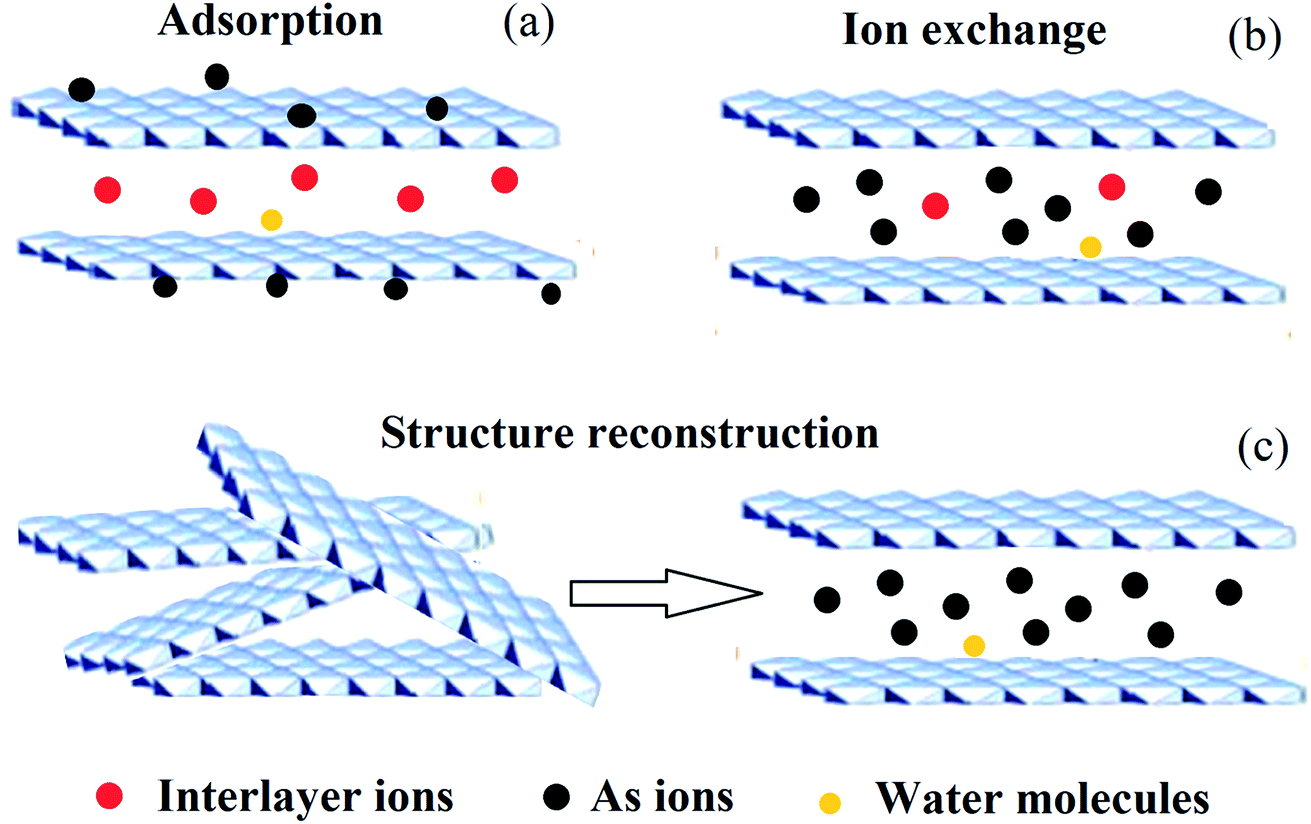 | ||
| Fig. 2 A schematic illustration of the As removal mechanism using a LDH. | ||
Until now, research on LDHs for As removal has mainly focused on (1) the influence of the synthesis method and composition of the LDH, (2) the influence of the particle size of the LDH, (3) the influence of the M2+/M3+ ratio in LDHs, (4) LDH-based hybrids and (5) the competition with other anions, as well as the adsorption mechanism. Overall, the objective of this work is to review the currently available literature focusing on As removal using LDHs for the five parts mentioned above. Finally, based on this overview, a closing section will suggest research efforts for future work.
2. Influence of the synthesis method and composition of the LDH
Many methods have been employed to synthesize LDHs. Co-precipitation is a well-known method which is often used.68 Another well-known synthesis method is the hydrothermal method.69 A third method is urea hydrolysis, from which one can obtain LDHs after slow and homogenous precipitation of metal cations as a result of slow hydrolysis of urea at certain temperatures.70 Moreover, the methods of ion exchange, structure reconstruction, sol–gel and microwave irradiation etc. are also used to synthesize LDHs.71–74 The use of different synthesis methods may influence the morphology of the LDH, thus influencing its adsorption capacity.70 Therefore, starting with conventional Mg–Al–CO3 LDHs, plenty of work has been done to study the influence of the synthesis method and composition of the LDH on As removal. You et al.75 used Mg–Al LDHs as adsorbents to remove As(III) from aqueous solutions for the first time. Uncalcined LDHs and calcined LDHs were compared in their study. It was found that As(III) could be adsorbed on both uncalcined and calcined chloride intercalation LDHs (Mg–Al–Cl LDH), but no adsorption occurred on carbonate intercalated LDHs (Mg–Al–CO3 LDH). The adsorption isotherms of As(III) on a Mg–Al–Cl LDH showed typical L-type curves, which indicated that As(III) was strongly attracted to the LDH through ion-exchange interactions. While on a calcined Mg–Al–Cl LDH, the As(III) adsorption isotherms were typical H-type curves, which suggested the high adsorption affinity of As(III), and As(III) was completely adsorbed at low concentrations. Nevertheless, no adsorption happened on the Mg–Al–Cl LDH, which indicated that CO32− was preferentially adsorbed, preventing As(III) exchange. However, after calcination at 450 °C for 2 h, it was able to reach the highest As(III) adsorption capacity. However, what is the difference between a calcined LDH and an uncalcined one? The structure of the Mg–Al–CO3 LDH will be changed during the calcination process. Upon thermal treatment, LDHs gradually lose their interlayer water, and then dehydroxylate and decarbonate to a large extent, leading to the formation of a mixed metal oxide with a poorly defined 3D network (see the left image in Fig. 2(c)).52 Moreover, calcined LDHs have relatively high surface areas, and exhibit memory effect. Because of their instability and memory effects, calcined LDHs can reconstruct their original layered structures upon adsorption of various anions (see Fig. 2(c)).34 Therefore, the highest As(III) adsorption capacity on the calcined Mg–Al–CO3 LDH may be explained by: (1) initial As(III) adsorption at the beginning of the calcined LDH reconstruction, (2) loss of H2O and CO32− (as CO2) resulting in higher calcined LDH anion exchange capacities, (3) fewer competing anions (e.g., CO32−), as CO2 is lost during the As(III) adsorption process, and (4) higher surface areas as a result of calcination. Then, the influence of shaking time on As(III) adsorption using a calcined Mg–Al–CO3 LDH was also investigated, and it was shown that As(III) adsorption on the calcined Mg–Al–CO3 LDH was a slow process and a quasiequilibrium was established after a 20 h reaction time. The results showed that over 80% of As(III) adsorption happened in the first 8 h, and then the adsorption rate became slow from 8 to 28 h. It was suggested that this two-step kinetic process might be related to the reconstruction mechanism of the calcined LDH. At the beginning of the adsorption process, reconstruction of calcined LDH, release of OH− and adsorption of CO32− coincide. Therefore, the adsorption of As(III) during the reconstruction of the calcined LDH was a fast process and competition with OH− and CO32− was relatively low. However, after LDH reconstruction, As(III) adsorption became a competitive process with OH− and CO32−, due to the increasing concentration of OH− and CO32− in the solution, which led to a slow adsorption process. The results from investigating the influence of pH on As(III) adsorption showed that the LDHs had high pH buffering capacities, while for calcined LDHs, As(III) adsorption was a function of pH.Following this, Yang et al.76 carried out a systematic investigation of removal of As(V) using calcined and uncalcined Mg–Al–CO3 LDHs from dilute aqueous solutions representing “too clean to clean” model power-plant effluent streams, whose contaminant levels are expected to be at trace ppb levels. The results indicated that both calcined and uncalcined Mg–Al–CO3 LDHs exhibited better adsorption performance on As(V), and the adsorption isotherms obeyed the Freundlich model. Similarly, the calcined Mg–Al–CO3 LDH displayed a higher As(V) adsorption capacity than the uncalcined one. In addition, the influence of pH on As(V) adsorption on uncalcined and calcined Mg–Al–CO3 LDHs was also tested. Results showed that the starting solution pH did not obviously affect the adsorption of As(V) on the calcined LDH, as long as it was higher than 4; however, for the uncalcined LDH, the initial pH had a significant influence on the adsorption capacity for As(V). This is because the point of zero charge (pHpzc) for the uncalcined LDH was normally in the range 6.8–8.9, and the surface of the LDH is negatively charged when pH > pHpzc. Therefore, the As(V) anionic species will be repelled by the LDH surface in a higher pH range. However, for pH < pHpzc, the LDH surface is positively charged, which usually favors the uptake of negatively charged anionic species. On the other hand, as reported, a very low pH can negatively influence the stability of the LDH structure and therefore leads to a decrease in the adsorption capacity for As anions. At a higher pH range, the negative effect of the pH may be further compounded by the increasing competitive effect of OH− adsorption on the LDH. Moreover, the impact of the As oxidation state on the adsorption performance on LDHs was also investigated. The results revealed that As(V) is more easily adsorbed on both the calcined and uncalcined LDHs than As(III). In addition, Chetia et al.77 studied the removal of As from contaminated water using calcined Mg–Al–CO3 LDHs as the adsorbents. Results showed that 100 mg of the Mg–Al–CO3 LDH can remove As to the extent of 99.99% from 0.1 ppm As solution under the conditions of adsorption for 90 min at 200 rpm and a pH of 7.5. In addition, the experiment showed that the adsorption process could be described by the Freundlich model.
Moreover, Liu et al.78 studied As(V) adsorption on lithium/aluminum LDHs intercalated by chloride (Li–Al–Cl LDH). In order to investigate the reaction of As(V) on the Li–Al–Cl LDH and gibbsite, adsorption isotherms, envelopes (see Fig. 3) and the extended X-ray absorption fine structure (EXAFS) technique were employed in this paper. The results showed that the Li–Al–Cl LDH displayed a superior adsorption capacity on As(V) over gibbsite. Although the surface area of gibbsite was larger than that of the Li–Al–Cl LDH, the maximum adsorption value of As(V) on the Li–Al–Cl LDH was 24.19 mg g−1, which was approximately six times higher than that of gibbsite (3.86 mg g−1) as shown in Fig. 3(a). This is because the surfaces of gibbsite do not take part in ligand exchange reactions, since the OH− groups on the planar surfaces of gibbsite are completely charge-satisfied and relatively inert. Therefore, the only active sites for adsorption reactions are the pH-dependent edges that have an abundance of under coordinated O atoms which can never be fully charge-satisfied by the addition or removal of a proton. Meanwhile the adsorption envelopes of As(V) on the Li–Al–Cl LDH were very sensitive to the pH when the pH was between 4.0 and 7.0 (see Fig. 3(b)). However, it displayed insensitivity to the pH when the pH was above 7.0, which is approaching the pHpzc of the Li–Al–Cl LDH (7.22). Therefore, from comparing with gibbsite, it was suggested that the superior adsorption capacity of the Li–Al–Cl LDH was mainly because of the two different adsorption sites in the structure. It was found that treatment with LiCl can lead to the intercalation of Li cations into the host structure of Al(OH)3 to form the Li–Al–Cl LDH. The Li cations occupied the vacant octahedral holes within Al(OH)3 and transformed the surface of the Al(OH)3 layers into active adsorption sites with a high affinity for As(V), as a result of the additional positive charge brought by the Li cations and the raising of the pHpzc of the Li–Al–Cl LDH. Then, Liu et al.79 also incorporated both molecular spectroscopic and macroscopic studies to determine the adsorption behavior of As(V) on the Li–Al–Cl LDH, which not only contained the beginning transfer process but also contained the subsequent adsorption mechanisms. The results proved that inner-sphere complexes were the main As(V) adsorption configurations on the planar surfaces and edges of the Li–Al–Cl LDH. From the results of the kinetics study, As(V) adsorption on the Li–Al–Cl LDH can also be separated into two processes. One is the fast reaction process; the other is the slow reaction process. This biphasic As(V) adsorption behavior is partially attributable to: (i) there being two different adsorption sites associated with Li, exposed planar surfaces, and Al, which exists on the edges of the double hydroxyl layers, and (ii) micropore adsorption sites within the Li–Al–Cl LDH surfaces. From the results on the activation energies derived by the Arrhenius equation, it was indicated that the diffusion process was a step involving As(V) adsorption restriction on the Li–Al–Cl LDH.
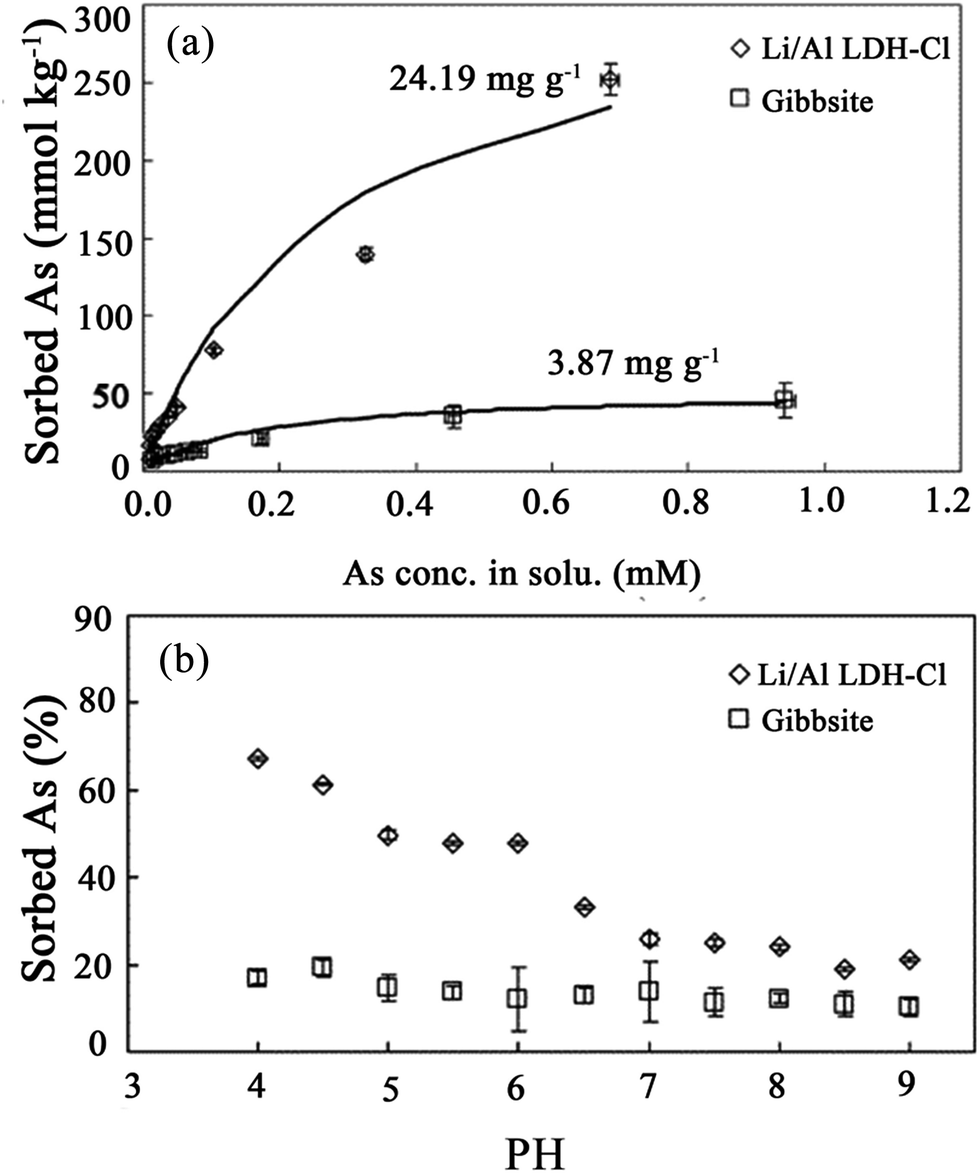 | ||
| Fig. 3 (a) Adsorption isotherms for arsenate reacted with the Li–Al–Cl LDH and with gibbsite at a pH = 5.0, (b) adsorption envelopes for arsenate reacted with the Li–Al–Cl LDH and with gibbsite (adapted with permission from ref. 78). | ||
Furthermore, Grover et al.80,81 did a systematic study on the influence of the synthesis method and composition of the LDH on the As adsorption performance. Two types of LDH, hydrotalcite and hydrocalumite, with different compositions of layers and interlayers, were used in their study to remove As(III) from the solutions. Based on the kinetics study, the results showed that the anion exchange process was very fast and apparently attained a steady state between 8 and 16 h. The synthesis method had an impact on the adsorption capacity of As on the LDHs. The LDH synthesized using the coprecipitation method displayed a higher adsorption capacity than that synthesized using the hydrothermal method. One reason is that the LDHs synthesized using the hydrothermal method had large crystal sizes leading to small surface areas. The other is that LDHs synthesized using the hydrothermal method were more crystalline in comparison with those synthesized using the coprecipitation method and it is harder to break the bonds between the layers in the former than in the latter. The results from XRD showed that the d003 spacing is unchanged after exchange in the hydrothermally synthesized LDH because the exchange was mainly on the surfaces and edges. What’s more, the type of interlayer anion in the LDH had a significant influence on the As uptake capacity of the LDH. Therefore, hydrotalcite and hydrocalumite with the same charge density and different interlayer anions displayed different As uptake capacities. The As(III) uptake capacities for the hydrotalcites were in the following order: Mg–Al–NO3 > Mg–Al–Cl > Mg–Al–CO3. For the hydrocalumites, the As(III) uptake capacity order was Ca–Al–NO3 > Ca–Al–Cl. This was because the d003 spacing reflected that nitrate has a larger size than chloride. Therefore, the nitrate resulted in larger interlayer spacing, which facilitated As(III) exchange. Finally, it was thought that the As(III) adsorption mechanism in the hydrocalumite-type LDH seemed to be topotactic exchange, and then dissolution and reprecipitation. Guo et al.82 also used hydrocalumite as an adsorbent to treat As(V) solutions with a wide range of initial concentrations. The results showed that the maximum uptake capacity of hydrocalumite for As(V) was 361.7 mg g−1. To study the adsorption mechanism, it was found that the process of dearsenication in the solution using hydrocalumite was principally driven by its dissolution and the subsequent precipitation of johnbaumite and other arsenic-bearing minerals (probably sainfeldite or bulachite) in the solid–solution series.
In addition, Guo et al.83 developed a novel LDH, which contains lanthanum (La3+) in it (Cu–Mg–Fe–La LDH) for As(V) removal in aqueous solution. The purpose of the introduction of La3+ into the LDH was to try to improve the adsorption capacity of As(V) and broaden the application field of LDH functional materials. The results showed that the Cu–Mg–Fe–La LDHs displayed highly efficient As(V) adsorption capacities from aqueous solutions. When the initial As(V) concentration was 5 mg L−1 with an adsorbent dosage of 1.5 g L−1, the final As(V) concentration was lower than 10 μg L−1 after adsorption. Moreover, with increasing La3+ content in the LDH, the As(V) adsorption capacity of the LDH increased. The adsorption efficiency of this La3+ containing LDH was dependent on the initial pH of the solution, and the co-existing ions had different negative effects on the As(V) adsorption performance. The main mechanism controlling the adsorption of As(V) onto the LDHs might also be ion exchange with carbonate and ligand exchange with the layer OH group. It was proposed that their high As(V) removal efficiencies would make Cu–Mg–Fe–La LDHs potential adsorbents for the treatment of water contaminated with As.
In addition to changing the divalent Mg, the trivalent Al has also been substituted by other cations in LDHs. Otgonjargal et al.66 prepared a Mn–Fe–CO3 LDH using the co-precipitation method to remove As(III) and As(V) from aqueous solutions. The adsorption capacities of the Mn–Fe–CO3 LDH for As(III) and As(V) were 0.02 and 0.009 mg g−1, respectively, which were higher than those of the iron- and manganese-containing compounds such as the Fe- and Mn-enriched samples (an adsorption capacity of 14 mg g−1 at an effluent concentration of 50 μg L−1).84 Subsequently, Bagherifam et al.85 carried out a study on As(III) and As(V) removal from a simulated soil solution using Zn–Al–SO4 LDHs as adsorbents. The results revealed that the maximum uptake values of the Zn–Al–SO4 LDHs on As(III) and As(V) were 34.24 and 47.39 mg g−1, respectively. The adsorption kinetics of As(V) followed pseudo-second order while those of As(III) uptake showed better correlation with the intraparticle diffusion model. Moreover, the Zn–Al–SO4 LDH displayed higher selectivity for As(V) compared to that with As(III).
Furthermore, Lu et al.86 used Zn–Fe LDHs in both simulated and practical polluted water to achieve removal of As(V). In the single pollution system, the results showed that the maximum adsorption capacity for As(V) was 151.37 mg g−1. In the batch experiments, the residual concentration of As(V) could be decreased from 2 mg L−1 to lower than 0.005 mg L−1, when the adsorbent dosage was 0.2 g L−1. However, in the practical polluted water, after adsorption, the results showed that both the As residual concentrations could be made to meet the limit value for drinking water standards recommended by WHO with a dosage of Zn–Fe LDH of 0.2 g L−1. Subsequently, Huang et al.87 synthesized Mg–Al LDHs with Cl− and CO32− as the interlayer anions via a solvothermal method to remove As(V) and F− in water. The maximum uptake capacities of the Mg–Al LDHs for As(V) and F− were 125.8 and 28.6 mg g−1, respectively, under neutral conditions. In order to investigate the detailed adsorption mechanisms for As(V) and F−, As(V) and F− saturated Mg–Al LDHs were prepared. XPS and XRD were used to characterize the LDHs before and after the adsorption of As(V) and F− (see Fig. 4). As shown in Fig. 4(a), after adsorption, an As signal appeared and the peak for Cl 2p at 198 eV disappeared, which all indicated that the As(V) ions had been successfully adsorbed on the Mg–Al LDHs with an exchange with Cl−. Besides, the high resolution Cls spectra of the LDHs before and after As(V) adsorption (see inset in the Fig. 4(a)) revealed that the intensity of the carbonate located at 288.4 eV, which could be assigned to the intercalated CO32− of the Mg–Al LDHs, decreases significantly after As(V) adsorption, indicating that the partial replacement of the interlayer CO32− had also exchanged with As(V). For F− adsorption, in Fig. 4(b), the peaks at 198 eV (Cl 2p) and 267 eV (Cl 2s), which show that the fluoride had disappeared after F− adsorption, indicate that there had been ion exchange between Cl− and F−. The high resolution F 1s spectrum is shown in the inset of Fig. 4(b). The small peak at 685 eV and the main peak at 689.3 eV can be assigned to the physically adsorbed F− on the surface and the F− in the lattice of the Mg–Al LDHs, respectively. These results suggested that F− had mainly intercalated into the layers rather than adsorbing on the surface. In addition, there was no obvious change in the Cls spectra observed for the Mg–Al LDHs before and after F− adsorption, which indicated that CO32− may not have taken part in ion exchange during the F− adsorption process. Fig. 4(c) shows the XRD patterns of Mg–Al LDHs after the adsorption of As(V). It can be see that there was no obvious change in the interlayer spacing of the Mg–Al LDHs detected at low concentrations. However, at high concentrations, there was an obvious shift observed in the interlayer spacing to a higher diffraction angle, which indicated that As(V) had intercalated into the interlayer and expanded the interlayer. Meanwhile for F− adsorption (see Fig. 4(d)), nearly no change could be seen with a full range of concentrations. The reason was that the only Cl− exchanged with F− and they are nearly the same size. Based on the above results, it can be concluded that the main adsorption mechanism between the interlayer anions in the Mg–Al LDHs and As(V) and F− in the solution was also that of ion exchange.
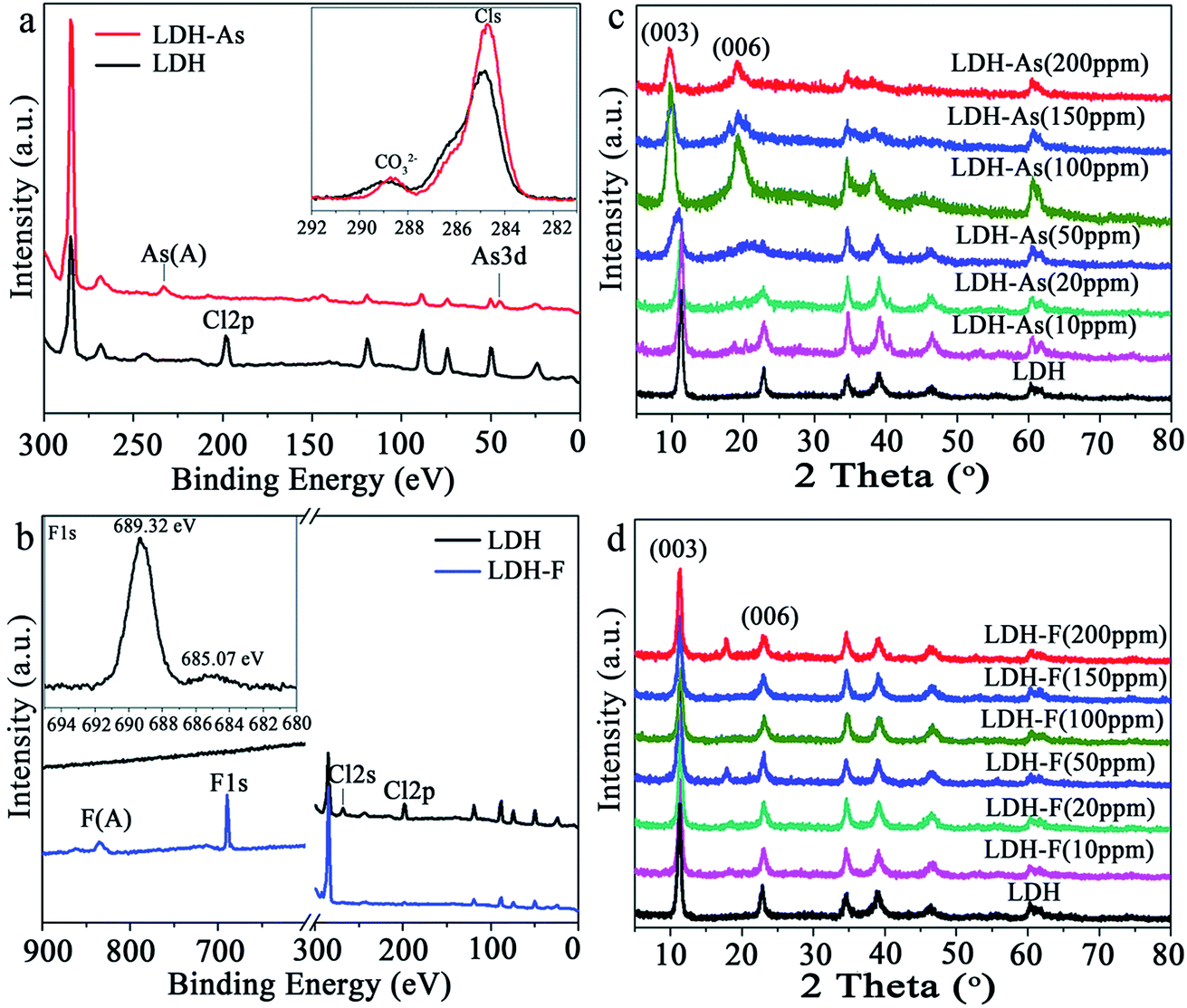 | ||
| Fig. 4 XPS spectra of the Mg–Al LDHs before and after the adsorption of (a) As(V) and (b) F−; XRD patterns of the Mg–Al-LDHs after the adsorption of (c) As(V) and (d) F− with different initial concentrations (adapted with permission from ref. 87). | ||
Recently, Lu et al.88 combined ferric-based LDHs (Mg–Fe–S2O8 LDH) with oxidant anion (persulfate) intercalation using a calcination–reconstruction method. Since the exchangeable persulfate ions in the LDH had strong oxidation abilities, the experimental results showed that the As(III) species in water were almost completely oxidized to the As(V) state and had adsorbed onto the Mg–Fe–S2O8 LDH simultaneously. For As(III) and As(V) in a single-pollutant system, each maximum uptake capacity was able to reach 75.00 and 75.63 mg g−1, respectively. Moreover, in the batch experiment, for the removal of both As(III) and As(V) together in the pollutant system, when the adsorbent dosage was 0.5 g L−1, the residual arsenic concentration in the solution could be reduced from 1 mg L−1 to lower than the limit value for drinking water standards recommended by WHO. The main removal mechanism of As(III) and As(V) using the Mg–Fe–S2O8 LDH was speculated on, and includes two approaches: one is the in situ oxidation of As(III) to As(V) by persulfate and the other is the exchange of S2O82− with As(V). Fig. 5 displays a schematic diagram of the proposed removal mechanism. The overall reaction with respect to the persulfate anion is as follows:
| As(III) + S2O82− → As(V) + 2SO42− |
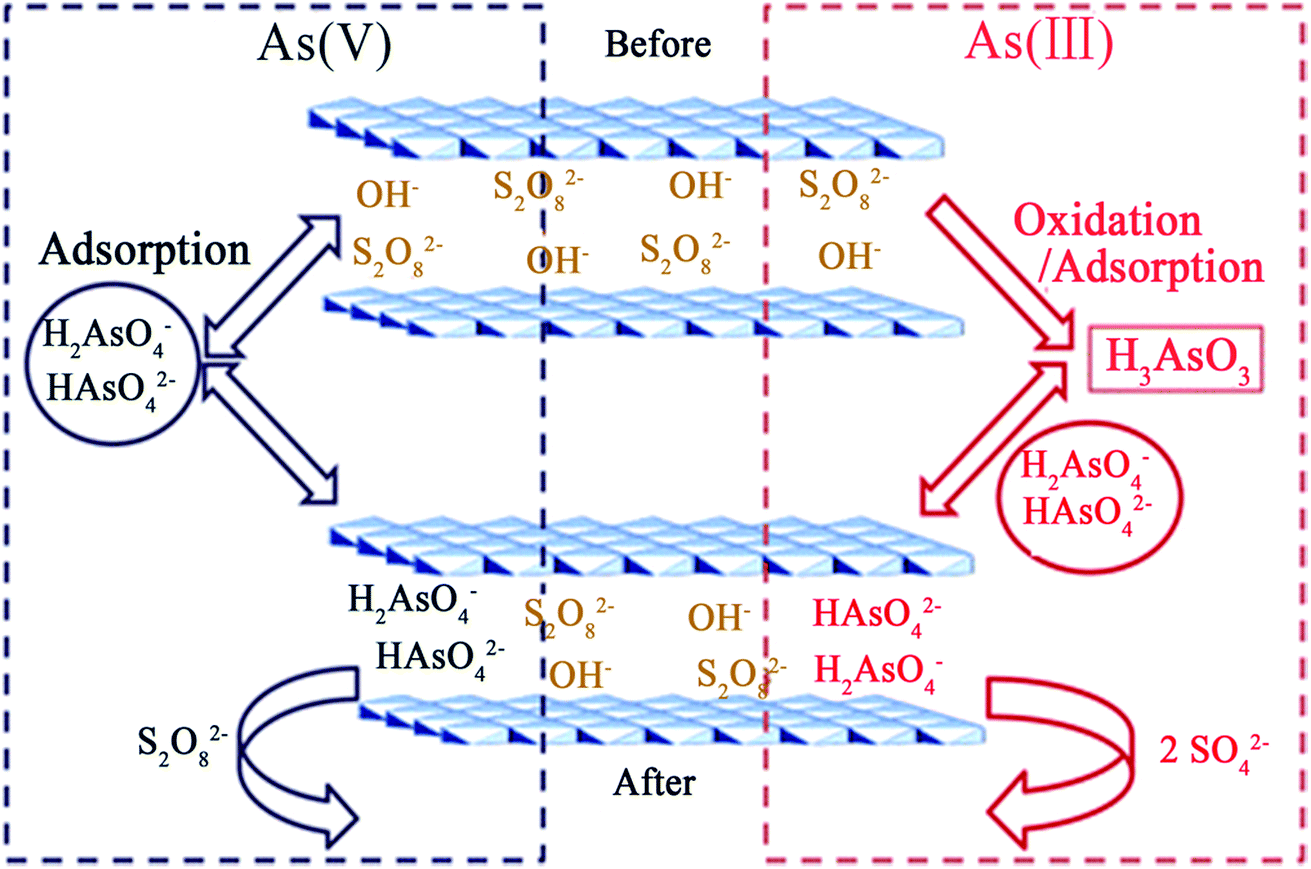 | ||
| Fig. 5 A schematic diagram of the proposed mechanism for As(III) and As(V) adsorption on the Mg–Fe–S2O8 LDH (reproduced with permission from ref. 88). | ||
Very recently, Ma et al.89 synthesized an Mg/Al LDH intercalated with MoS42− for capturing the oxoanions of As(III)/As(V) (HAsO32−/HAsO42−). This MoS4-LDH exhibited very high removal rates for As(III) and As(V) of up to 99% and the maximum uptake capacities for As(III) and As(V) were 99 and 56 mg g−1, respectively. From the SEM images (see Fig. 6), it can be seen that the crystallites of the LDHs fully maintained their hexagonal prismatic shape after the adsorption of As(III) and As(V), which proved that topotactic insertion of the adsorbed anionic complexes during the uptake process had taken place. Therefore, the dominant phases and possible binding modes of MoS42− with HAsO42− in the LDH gallery were speculated on, as shown in Fig. 6(d). When the anion was at low concentrations, the adsorbed amount was not large enough to produce a new discernable phase; thus, the 1.07 nm dbasal was mainly MoS4-LDH (see Fig. 6(d), option A). Meanwhile when the anion was at high concentrations, a 0.87 nm dbasal appeared, which suggests a new phase related to the intercalation of an anionic complex with a [(HAsO4)·(MoS4)·(HAsO4)]6− mode (see Fig. 6(d), option B).
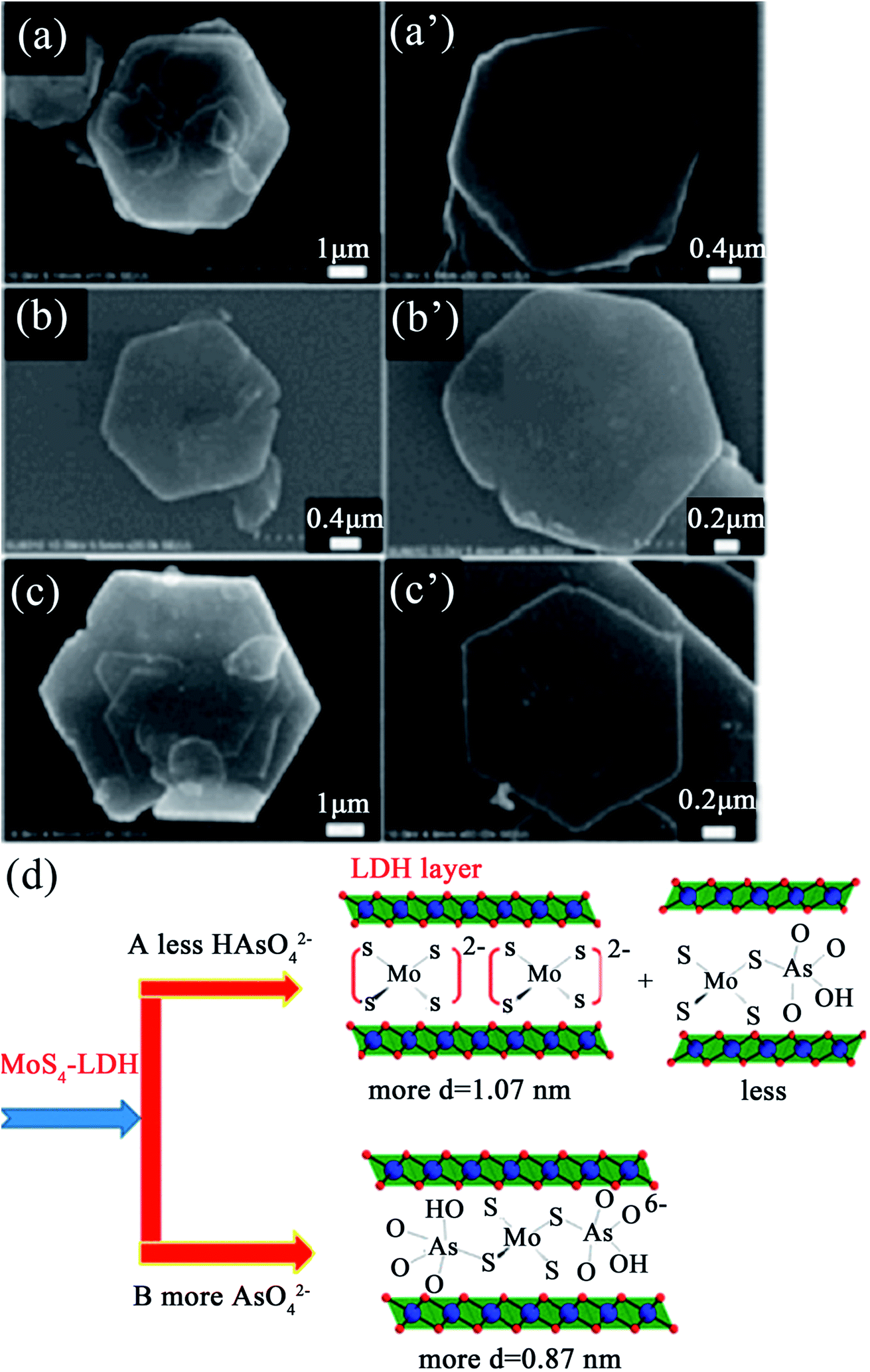 | ||
| Fig. 6 SEM images of the samples after MoS4-LDH adsorption with (a and a′) 10 ppm As(III), (b and b′) 300 ppm As(III) and (c and c′) 10 ppm As(V); (d) a scheme showing the dominant phases and possible binding modes of MoS42− with HAsO42− in the LDH gallery (adapted with permission from ref. 89). | ||
3. Influence of the particle size of the LDH
For the LDH adsorbent, it is believed that reducing its particle size can increase its adsorption capacity. Yang et al.90 investigated the influence of the particle sizes of LDHs on the As(V) adsorption performance in aqueous solution. The results for As(V) adsorption showed that the As(V) adsorption rate on a conditioned LDH was associated with its particle size. The As(V) adsorption rate increased with decreasing adsorbent particle size. However, the adsorption capacity of As(V) on a conditioned LDH had no relationship with the particle size. Moreover, the experimental data was fitted to two models. One was the homogeneous surface diffusion model (HSDM), the other was the bidisperse pore model (BPM). HSDM considers the adsorbent particle to be a single particle, the advantage of which is its simplicity and ease for simulations. When the HSDM was used to describe the experimental data, the estimated diffusivity values were shown to increase with increasing particle size. Nevertheless, BPM assumes that the particle is an agglomerate of a number of equal-size, single-crystal microparticles, and provided a better representation of the true pore structure of the LDH. When the BPM was utilized to fit the experimental data, it predicted that diffusivity is particle-size-independent.4. Influence of the M2+/M3+ ratio
It has been reported that the M2+/M3+ ratio in LDHs can influence their adsorption capacities. Therefore, Wang et al.91 explored the influence on As(V) adsorption using Mg–Al–NO3 LDHs of varying the Mg/Al ratio. Firstly, the Mg–Al–NO3 LDHs were synthesized using a constant-pH co-precipitation method with different Mg/Al ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:1 and 4:1. It was found that the maximum uptake capacities for As(V) on the 2:1, 3:1 and 4:1 LDHs were 117, 81 and 27 mg g−1, respectively, which follows the order of their layer charge densities. Thus, in order to clarify the structural implications of the differences in As(V) adsorption of the various LDHs, the XRD patterns of the LDH samples before and after As(V) adsorption were investigated. It can be observed that the basal spacings of the original LDH with Mg2+/Al3+ molar ratios of 2, 3, and 4, were 8.78, 8.13, and 7.95 Å, respectively. After interacting with As(V), the basal spacings of the 2:1 and 3:1 LDHs became smaller than those of the original, and had shifted to 8.48 and 7.83 Å, respectively. However, there was no significant change for the 4:1 LDH after As(V) adsorption. Thus, the LDH with a Mg2+/Al3+ molar ratio of 4 has a low As(V) adsorption capacity, in combination with no change in the basal spacing after As(V) adsorption, which indicated that As(V) had relatively little access to the small interlayer space in the 4:1 LDH due to the horizontal orientation of the interlayer nitrate with respect to the hydroxide sheets. Therefore, it was speculated that As(V) had adsorbed only on the external surface of the 4:1 LDH particles for the range of initial As(V) concentration used in this study (see Fig. 7(a)). Consequently, the 4:1 LDH had maximum As(V) adsorption at a low initial concentration of As(V), which was much less than those of the 2:1 and 3:1 LDHs. Conversely, for the 2:1 and 3:1 LDHs, there were changes in the basal spacings after As(V) adsorption, which indicated partial replacement of the interlayer nitrate ions by As(V) ions. The results from XRD showed that the 2:1 LDH has the largest basal spacing due to the perpendicular orientation of the interlayer nitrate with respect to the hydroxide layers in the material. The large basal spacing of the 2:1 LDH facilitates the diffusion of nitrate and As(V) molecules into its interlayer space. Thus, in conjunction with adsorption on the external surface, the interlayer nitrate ions could be replaced by As(V) for the positively-charged binding sites in the hydroxide sheets (as shown in Fig. 7(a)), which consequently led to better As(V) uptake performance. It was suggested that the 3:1 LDH is an intermediate phase, containing nitrate ions with both parallel and perpendicular orientations. Therefore, they hypothesized that the interlayer nitrate ions with a perpendicular orientation contribute to As(V) adsorption of the 3:1 LDH, while those with a parallel orientation are not replaced and remain in the structure (see Fig. 7(a)). Therefore, As(V) adsorption on the 2:1 and 3:1 LDHs occurred by the same reaction mechanism, with different surface availabilities for As(V). The local structure of adsorbed As(V) on various LDHs was further determined using results from the XANES and EXAFS as shown in Fig. 7(b). It can be seen that the absorption edges of As adsorbed by various LDHs were all located at 11875 eV, which was the same as the value determined for Na2HAsO4, but different from that for NaAsO2. This indicated that there was no reduction of As(V) to As(III) after As(V) adsorption on the LDHs, and that the corresponding local structures of adsorbed As(V) on various LDHs were not different, according to their XANES spectra. The k3-weighting oscillation function and RDF profile with peak positions (without phase shift correction), derived from the EXAFS of As(V) adsorbed on the 2:1 LDH are shown in Fig. 7(c). The peak positions presented in Fig. 7(c) correspond to the radial distances from As to atoms at different coordination shells. The structural parameters were fitted, including the coordination number (CN), inter-atomic distance (R), and Debye–Waller factor (σ2). The first coordination shell was attributed to the As(V)–O interatomic distance of 1.70 ± 0.02 Å, with a CN of 4. This is consistent with the molecular structure of an As(V) ion. Furthermore, the interatomic distance between As and the second-shell atom was determined to be 2.92 ± 0.07 Å, with a CN of 2.0. These data cannot be fitted to Al or Mg as the second shell atoms from As, but a good fit could be obtained if this interatomic distance of 2.92 Å was attributed to that between As and O. This result reveals that no direct chemical bond is formed between the adsorbed As(V) and the Al or Mg sites in the hydroxide sheets of the LDH. Thus, As(V) ions were adsorbed on the external and internal surfaces of the LDHs by predominantly forming outer-sphere complexes, and the O atom of the second shell might be assigned to the surface OH groups of the hydroxide layers. The results of this study have demonstrated that selective retention of As(V) from water can be maximized by controlling the orientation of interlayer nitrate in Mg–Al–NO3 LDHs.
:1, 3:1 and 4:1. It was found that the maximum uptake capacities for As(V) on the 2:1, 3:1 and 4:1 LDHs were 117, 81 and 27 mg g−1, respectively, which follows the order of their layer charge densities. Thus, in order to clarify the structural implications of the differences in As(V) adsorption of the various LDHs, the XRD patterns of the LDH samples before and after As(V) adsorption were investigated. It can be observed that the basal spacings of the original LDH with Mg2+/Al3+ molar ratios of 2, 3, and 4, were 8.78, 8.13, and 7.95 Å, respectively. After interacting with As(V), the basal spacings of the 2:1 and 3:1 LDHs became smaller than those of the original, and had shifted to 8.48 and 7.83 Å, respectively. However, there was no significant change for the 4:1 LDH after As(V) adsorption. Thus, the LDH with a Mg2+/Al3+ molar ratio of 4 has a low As(V) adsorption capacity, in combination with no change in the basal spacing after As(V) adsorption, which indicated that As(V) had relatively little access to the small interlayer space in the 4:1 LDH due to the horizontal orientation of the interlayer nitrate with respect to the hydroxide sheets. Therefore, it was speculated that As(V) had adsorbed only on the external surface of the 4:1 LDH particles for the range of initial As(V) concentration used in this study (see Fig. 7(a)). Consequently, the 4:1 LDH had maximum As(V) adsorption at a low initial concentration of As(V), which was much less than those of the 2:1 and 3:1 LDHs. Conversely, for the 2:1 and 3:1 LDHs, there were changes in the basal spacings after As(V) adsorption, which indicated partial replacement of the interlayer nitrate ions by As(V) ions. The results from XRD showed that the 2:1 LDH has the largest basal spacing due to the perpendicular orientation of the interlayer nitrate with respect to the hydroxide layers in the material. The large basal spacing of the 2:1 LDH facilitates the diffusion of nitrate and As(V) molecules into its interlayer space. Thus, in conjunction with adsorption on the external surface, the interlayer nitrate ions could be replaced by As(V) for the positively-charged binding sites in the hydroxide sheets (as shown in Fig. 7(a)), which consequently led to better As(V) uptake performance. It was suggested that the 3:1 LDH is an intermediate phase, containing nitrate ions with both parallel and perpendicular orientations. Therefore, they hypothesized that the interlayer nitrate ions with a perpendicular orientation contribute to As(V) adsorption of the 3:1 LDH, while those with a parallel orientation are not replaced and remain in the structure (see Fig. 7(a)). Therefore, As(V) adsorption on the 2:1 and 3:1 LDHs occurred by the same reaction mechanism, with different surface availabilities for As(V). The local structure of adsorbed As(V) on various LDHs was further determined using results from the XANES and EXAFS as shown in Fig. 7(b). It can be seen that the absorption edges of As adsorbed by various LDHs were all located at 11875 eV, which was the same as the value determined for Na2HAsO4, but different from that for NaAsO2. This indicated that there was no reduction of As(V) to As(III) after As(V) adsorption on the LDHs, and that the corresponding local structures of adsorbed As(V) on various LDHs were not different, according to their XANES spectra. The k3-weighting oscillation function and RDF profile with peak positions (without phase shift correction), derived from the EXAFS of As(V) adsorbed on the 2:1 LDH are shown in Fig. 7(c). The peak positions presented in Fig. 7(c) correspond to the radial distances from As to atoms at different coordination shells. The structural parameters were fitted, including the coordination number (CN), inter-atomic distance (R), and Debye–Waller factor (σ2). The first coordination shell was attributed to the As(V)–O interatomic distance of 1.70 ± 0.02 Å, with a CN of 4. This is consistent with the molecular structure of an As(V) ion. Furthermore, the interatomic distance between As and the second-shell atom was determined to be 2.92 ± 0.07 Å, with a CN of 2.0. These data cannot be fitted to Al or Mg as the second shell atoms from As, but a good fit could be obtained if this interatomic distance of 2.92 Å was attributed to that between As and O. This result reveals that no direct chemical bond is formed between the adsorbed As(V) and the Al or Mg sites in the hydroxide sheets of the LDH. Thus, As(V) ions were adsorbed on the external and internal surfaces of the LDHs by predominantly forming outer-sphere complexes, and the O atom of the second shell might be assigned to the surface OH groups of the hydroxide layers. The results of this study have demonstrated that selective retention of As(V) from water can be maximized by controlling the orientation of interlayer nitrate in Mg–Al–NO3 LDHs.
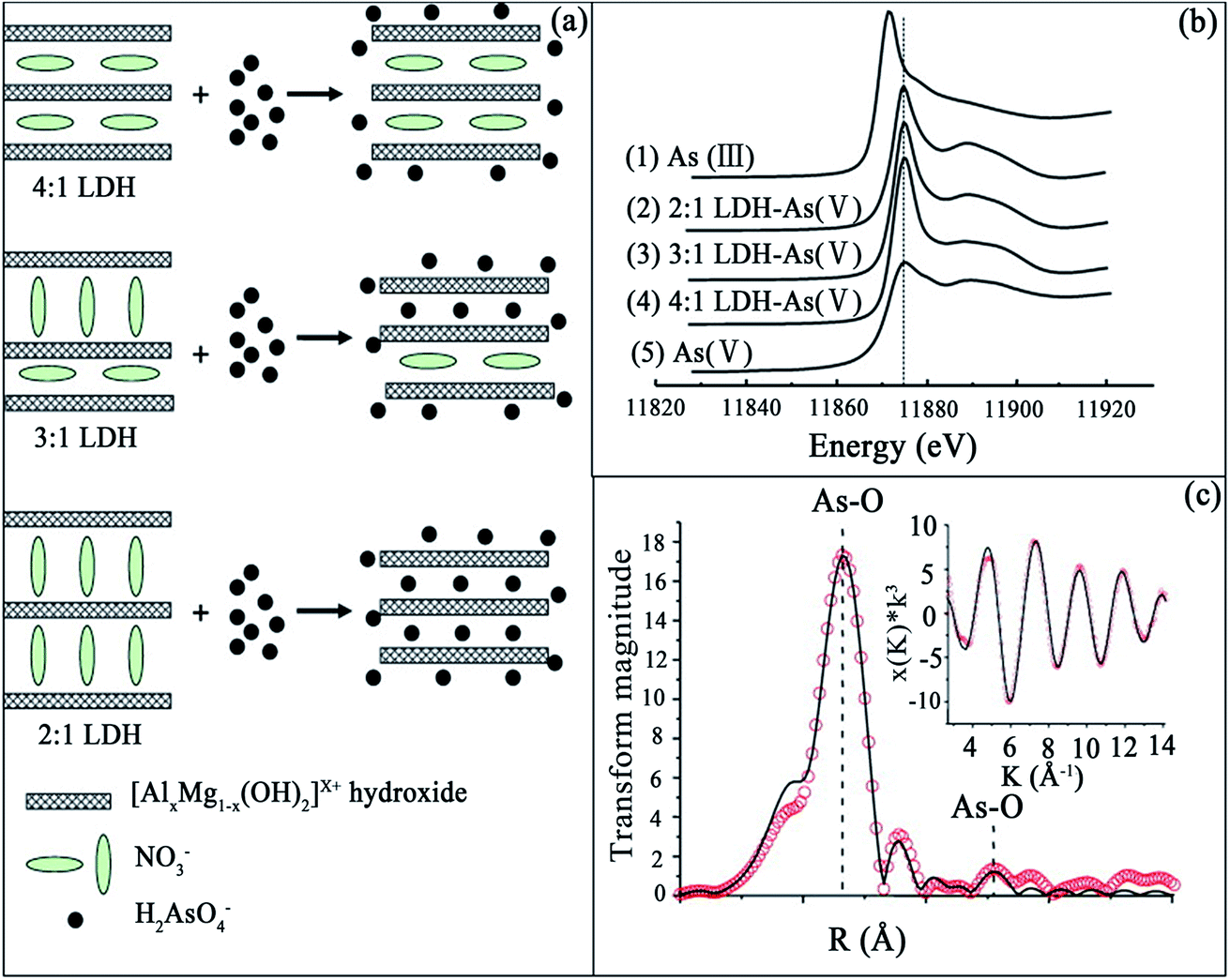 | ||
| Fig. 7 (a) A schematic representation of the As(V) adsorption mechanisms for the 2:1, 3:1 and 4:1 LDHs; (b) the As K-edge XANES spectra of (1) the As(III) standard, (2) the 2:1 LDH-As(V), (3) the 3:1 LDH-As(V), (4) the 4:1 LDH-As(V), and (5) the As(V) standard; (c) k3x(k) spectrum and RDF profile derived from the As K-edge EXAFS of As(V) adsorbed on the 2:1 LDH. The solid lines and open circles represent fitted and experimental data, respectively, without phase shift correction (adapted with permission from ref. 91). | ||
Recently, Guo et al.92,93 used iowaite (a Mg–Fe LDH) for both As(III) and As(V) removal for the first time, and investigated the influence on the uptake capacity of the Mg/Fe ratio. It was found that the Mg/Fe ratio had little influence on As(V) removal with the uncalcined iowaites, but played a crucial role in adsorption of As(III) from the solutions (see Fig. 8). The As(III) removal and reaction rate in solution using iowaite decreased with the increase of its Mg/Fe molar ratio. However, for the calcined iowaites, their initial Mg/Fe ratios and adsorption capacities for both As(V) and As(III) were independent (see Fig. 8). Therefore, it was suggested that the removal of both As(V) and As(III) from solution using uncalcined iowaite may be primarily due to Fe–As complexation occurring on its surface. However, the calcined iowaites generally had higher As adsorption capacities than the uncalcined iowaites, which was because of the high availability of the interlayer regions for the arsenic oxyanions during the process of structural reconstruction.
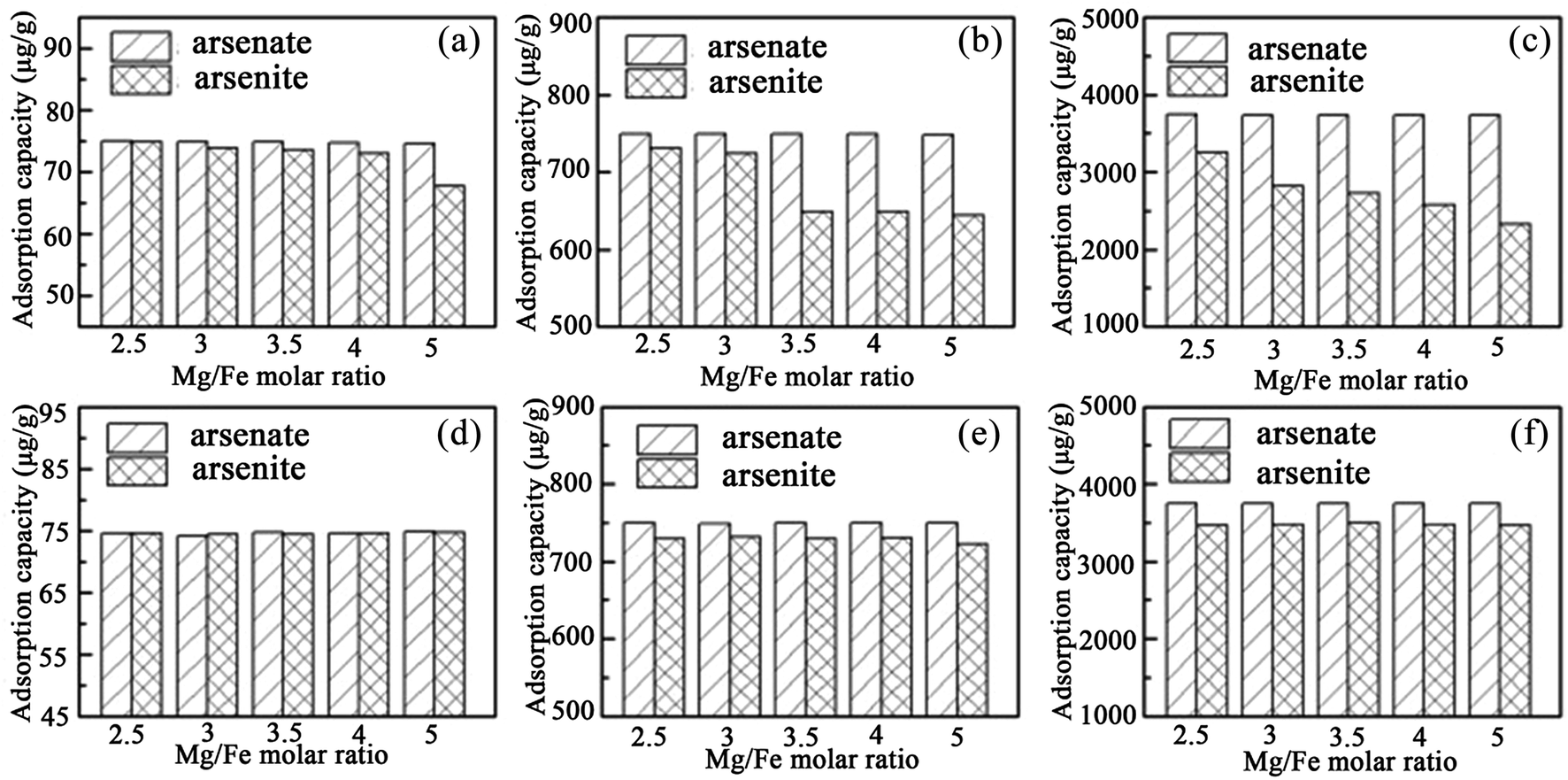 | ||
| Fig. 8 Comparison of the adsorption capacity between the uncalcined (a–c) and calcined (d–f) iowaites. (a–c) or (d–f) correspond to the initial arsenic concentrations in solution of 150, 1500, and 7500 μg L−1, respectively (reproduced with permission from ref. 92). | ||
5. LDH-based hybrids
In recent years, LDH-based hybrids have become promising as adsorbents.53,94,95 Nhat Ha et al.96 immobilized LDHs in a hybrid polymer system [alginate/polyvinyl alcohol (PVA) in the presence of the cross-linker glutaraldehyde] to synthesize spherical poly(LDH) beads with less distortion for As(V) adsorption. Two kinds of LDH polymer hybrids (poly(Mg–Al) and poly(Mg–Fe)) were synthesized for As(V) removal. The As(V) adsorption rate of the poly(Mg–Al) and poly(Mg–Fe) beads could reach 91.2% and 79.1% from an As(V) solution (with an initial concentration of 50 mg L−1), respectively. As calculated from the Langmuir adsorption isotherm, at pH 8, the adsorption capacities of the poly(Mg–Al) and poly(Mg–Fe) beads were from 1.73 to 1.64 mg of As per g. For the regeneration test, the results showed that the poly(LDH) beads had good cycling stabilities, and their adsorption capacities only decreased by approximately 5–6% after 5 adsorption–desorption cycles. The As(V) removal rates of the poly(Mg–Al) and poly(Mg–Fe) beads remained at 85.4 and 72.7%, respectively, after the fifth regeneration cycle. Moreover, in order to investigate the dynamic behavior of the adsorption column, a study of the fixed-bed column was carried out with real-life arsenic-containing water using a column with a 2 cm diameter. Then, the poly(Mg–Al) or poly(Mg–Fe) beads were packed in the column with a 40 cm bed depth. The column was in up-flow mode with a volumetric flow rate of 57.32 cm3 (cm2 h) −1 (3.0 mL min−1), and was charged with real-life arsenic-containing water. Although both the fixed-bed poly(Mg–Al) and poly(Mg–Fe) beads showed decent removal of the problematic As(V), the results showed that the fixed-bed column of poly(Mg–Al) displayed better column performance than that of poly(Mg–Fe), and had longer operation times, treated larger volumes and removed higher quantities of As(V). Following this, Suvokhiaw et al.32 prepared magnetic Mg–Fe–Zr LDH/Fe3O4 hybrids as adsorbents for As(V) removal. The As(V) adsorption ability of the pure Mg–Fe–Zr LDH (LDH), bare Fe3O4, and the Mg–Fe–Zr LDH/Fe3O4 hybrid (m-LDH) and their calcined counterparts were compared. The results showed that the calcined LDH and calcined m-LDH possessed higher adsorption amounts than their noncalcined counterparts due to the increase in surface area after calcination. In the case of the iron oxides, their surface areas decreased dramatically after calcination due to phase transformation and particle aggregation, resulting in lower adsorption capacities. In addition, the calcined m-LDH (188 mg g−1) showed lower adsorption than the calcined LDH (203 mg g−1), because the hybrid contained the Fe3O4 phase. However, the as-synthesized m-LDH hybrid and calcined m-LDH could be separated from the aqueous solution using an external magnetic field. The two-step mechanism of the adsorption behavior was proposed as: (i) the structural reconstruction of the mixed metal oxides to LDH layers, and (ii) the anion exchange of hydroxides inside the reformed LDH galleries.All the LDH derived adsorbents and their performance in As removal available in the literature are summarized in Table 1. Since the As adsorbents’ removal capacities were evaluated at different As concentration ranges, temperatures, pH values, adsorbent doses and As(III)/As(V) ratios etc., it is very difficult to directly compare these adsorption capacities with those of other As adsorbents. In Mohan’s paper, they gave a summary showing that some low-cost adsorbents developed from agricultural waste or industrial waste have outstanding capacities. For instance, the As adsorption capacity of immobilized biomass can reach more than 700 mg g−1, while basic yttrium carbonate and goethite also have high As removal abilities of as much as 400 mg g−1. Some commercial adsorbents, which include resins, gels, silica and titanium dioxide, tested for As removal have an As removal capacity of 100–200 mg g−1.2 However, LDHs also performed well in As removal, and values can reach as high as 361.7 mg g−1, which are comparable to those of the other adsorbents. Besides their removal ability, the cost of the adsorbent is also very significant.97 The cost of the adsorbent is mainly dependent on the availability of each type, and their preparation and applicability in actual field conditions.98 Although waste material derived adsorbents seem inexpensive, their pretreatment and chemical modification costs are seldom mentioned in research papers. In addition, most research has focused on laboratory evaluations. Jessica et al.99 have summarized the cost effectiveness of selected As avoidance methods. Reverse osmosis ($411 annually) was the most cost effective, followed by activated alumina ($518) and 1-gal jugs of water ($321–1285) for households with more than one person.
| Adsorbent | Initial As concentration (mg L−1) | pH | Adsorption capacity/(mg g−1) | Ref. | |
|---|---|---|---|---|---|
| As(III) | As(V) | ||||
| Calcined Mg–Al–CO3 | 997.5 | 10 | 158.25 | 75 | |
| Calcined Mg–Al–CO3 | 0.2 | 10 | 0.17 | 76 | |
| Calcined Mg–Al–CO3 | 0.1 | 7.5 | 0.02 | 77 | |
| Li–Al–Cl | 52.5 | 5 | 24.19 | 78 | |
| Ca–Al–Cl | 1125 | 10.5 | 361.7 | 82 | |
| Cu–Mg–Fe–La–CO3 | 5 | 3 | 75 | 83 | |
| Mn–Fe–CO3 | 100 | 7.8 | 112.5 | 52.5 | 66 |
| Zn–Fe–SO4 | 50 | 4 | 143 | 86 | |
| Mg–Al–Cl | 150 | 7 | 36 | 100 | |
| Mg–Fe–Cl | 187.5 | 7 | 81 | 100 | |
| Cu–Al–Cl | 120 | 7 | 52.5 | 100 | |
| Zn–Al–Cl | 150 | 7 | 35.25 | 100 | |
| Mg–Fe–S2O8 LDH | 5 | 4 | 12.5 | 17.5 | 88 |
| Mg–Al–MoS4 LDH | 306 | ≈11 | 99 | 56 | 89 |
| Mg–Al–NO3 | 87.75 | 5 | 117 | 91 | |
| Calcined Mg–Fe–Cl | 7.5 | 8 | 3.75 | 92 | |
| Calcined Mg–Fe–Cl | 7.5 | 7 | 3.35 | 92 | |
| Mg–Fe–Cl | 750 | 7 | 286.9 | 331.1 | 93 |
| Calcined poly(Mg–Al) LDH | 50 | 8 | 1.73 | 96 | |
| Calcined poly(Mg–Fe) LDH | 50 | 8 | 1.64 | 96 | |
| Calcined Mg–Fe–Zr LDH/Fe3O4 | 200 | 3 | 188.2 | 32 | |
6. Competition with other ions
It is known that environmental As is always accompanied by other ions in contaminated water. When competitive ions exist, the solid–liquid adsorption equilibrium will be changed, and a new set of isotherms will emerge.2 In particular, for LDHs, the effect of competing anions on the adsorption of As may be related to the anion affinity or anion intercalation capability of the LDH,101,102 while the desorption rates correspond to the different anion affinities for the LDH.103 Therefore, the adsorption behavior of As with LDHs in the presence of multicomponent impurities has been studied by researchers. You et al.75 investigated the influence of the presence of additional anions on the adsorption of As(III) using LDHs in water. It was found that other anions present had strong interactions with the calcined LDH, affecting As(III) adsorption. The As(III) adsorption capacity was in the following order: HPO42− < SO42− < CO32− < F− < Cl− < Br− < NO3−. Nevertheless, the desorption results showed that As(III) could be desorbed by different anions after adsorption on calcined LDHs, but there was no systematic relationship between As(III) desorption and the anion affinities for the calcined LDHs. For bivalent anions (CO32− and SO42−), As(III) release can reach a plateau after 8 iterations, however, the desorbed As(III) (78 mg g−1) was less than that of the adsorbed As(III) (165.6 mg g−1) on the calcined LDH. Meanwhile for monovalent anions (NO3− and Cl−), with the desorption times increasing, the desorption of As(III) increased continually. However, in HPO3− solution, the desorption rate was very slow. Following this, Yang et al.76 studied the influence of competitive anions on the removal of As(V) using a calcined LDH. The results indicated that NO3− had almost no influence on the uptake of As(V) on the calcined LDH, while SO42−, CO32− and HCO3− had a modest effect, and HPO42− had the most negative impact on the uptake of As(V) on the calcined LDH. In addition, it was proven that competing ions had a stronger effect on the uptake by uncalcined LDH than on that by calcined LDH.Moreover, Caporale et al.104 studied the competition between AsO43− and organic and inorganic ligands for the adsorption sites on two kinds of LDH, which were Mg–Al LDH and Mg–Fe LDH. Specifically, they examined AsO43− adsorption on the Mg–Al LDH and the Mg–Fe LDH, as affected by the pH. Various concentrations of inorganic [nitrate (NO3−), nitrite (NO2−), phosphate (PO43−) and selenite (SeO32−), sulphate (SO42−)] and organic [oxalate (OX2−) and tartrate (TAR2−)] ligands were used. The adsorption isotherm results showed that the Mg–Fe LDH had better AsO43− adsorption performance than the Mg–Al LDH. Moreover, with different competing anions in the solution, the AsO43− adsorption efficiency of the two LDHs was also different. The efficiency of the anions in competing with AsO43− for the adsorption sites on the Mg–Al LDH was in the order NO3− < NO2− < SO42− < SeO32− < TAR2− < OX2− ≤ PO43− at all molar ratios examined (see Fig. 9A), while on the Mg–Fe LDH the order was NO3− < SO42− ≈ NO2− < TAR2− < OX2− < SeO32− ≤ PO43− (see Fig. 9B). The inhibition of AsO43− adsorption increased with increasing initial ligand concentration and was greater on the Mg–Al LDH than on the Mg–Fe LDH, evidently because the AsO43− anions had a stronger affinity for Fe than Al and due to the presence in the Mg–Fe LDH of short-range-ordered materials on which AsO43− forms very strong inner-sphere complexes which are not easily desorbed by competing ligands.105–107 The effect of residence time on the desorption capacity of AsO43− LDHs due to these ligands was also investigated. At all the examined residence times, the results showed that the ability of the anions to desorb AsO43− from the adsorption sites on the Mg–Al LDH was in the order NO3− < NO2− < SeO32− ≈ SO42− < TAR2− < OX2− ≤ PO43−, while on the Mg–Fe LDH, the order was different: NO3− < SO42− ≈ NO2− < TAR2− ≤ OX2− < SeO32− ≤ PO43−. Nevertheless, the influence of increasing residence time on the desorbing of AsO4 was similar for all the ligands, for both LDHs, varying from 2 to 10%. However, for the Mg–Al LDH systems, with PO43−, TAR2− and OX2−, 10–20% more AsO43− was desorbed compared to the Mg–Fe LDH systems for the same residence time. It was suggested that AsO43− was bonding more strongly to the Mg–Fe LDH than the Mg–Al LDH. Therefore the AsO43− was more easily desorbed on the Mg–Al LDH in comparison with that on the Mg–Fe LDH. Recently, Nhat Ha et al.96 tested the effect of co-existing anions on As(V) adsorption using poly(Mg–Al LDH, Mg–Fe LDH) hybrids. It was found that phosphate and carbonate markedly decreased the removal of As(V). The effects of coexisting anions on the adsorption capacity declined in the following order: HPO42− ≫ HCO3− > SO42− > Cl−. Furthermore, Ardau et al.108 tested synthetic Zn–Al–sulphate LDHs for As removal from aqueous systems through adsorption experiments. In order to assess the influence of competition on As removal, a series of aqueous solutions containing dissolved HAsO42− together with other anions (Cl−, SO42−, MoO42−, HCO3− and CO32−) were prepared. The competitors were added into the solution simultaneously and afterwards with respect to HAsO42− in order to verify the effectiveness and possible reversibility of the As sorption process. The experimental results also confirmed that As adsorption from the solution was mostly by ion exchange processes, due to the strong capability of As in replacing S in the interlayers of the LDH structure, where anionic groups are hosted, giving rise to heteroanionic compounds. With the competitors, the results showed that CO32− had a significant impact on the As adsorption efficacy on LDHs. In fact, up to ∼90% of HAsO42− could be removed from the solution, decreasing to ∼60% in the presence of CO32−, whilst up to ∼30% of HAsO42− could be desorbed when CO32− was added afterwards into the solution. Since a very restricted pH range was used (c. pH 10–11.5), where HAsO42− and CO32− were simultaneously the predominant species in the solution, they thought that Zn–Al–sulphate LDHs could be successfully used for the treatment of As contaminated water with pH values ranging from circum-neutral to moderately alkaline. In addition, Bagherifam et al.85 carried out an investigation of the effects of two kinds of competing divalent anion (SO42− and CO32−) on the adsorption of As(III) and As(V). From Fig. 10, it can be seen that the As(III) removal rate decreased from 69 to 61 and 54%, however, the As(V) uptake rate decreased only from 88 to 86 and 82%, in the presence of 1 mM Na2SO4 or Na2CO3, respectively. This clearly proved that the adsorption capacity for As(III) was more affected by the presence of competing anions than that for As(V). Moreover, CO32− had a greater effect on As(III) and As(V) adsorption than SO42−.
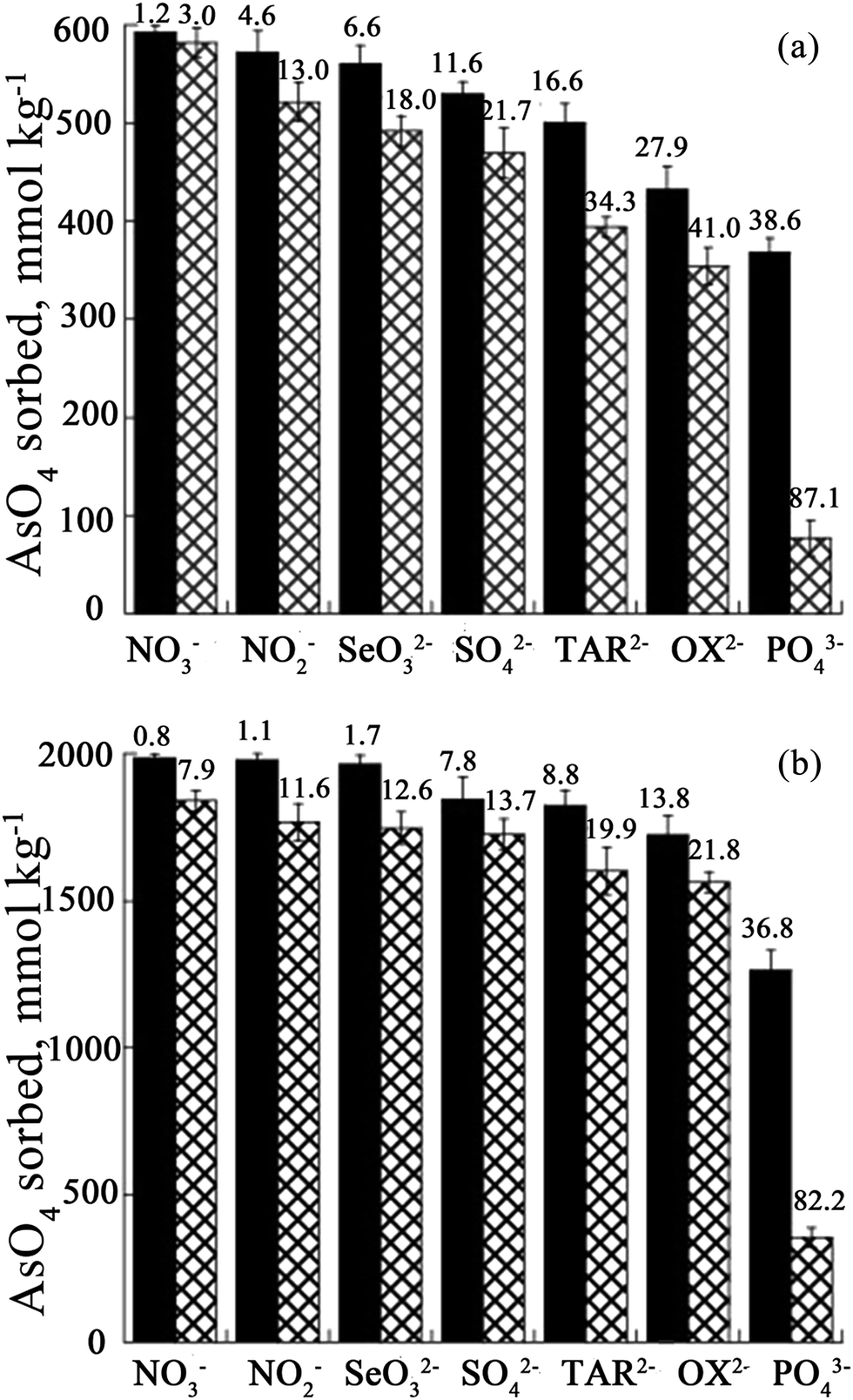 | ||
| Fig. 9 The amount of AsO4 sorbed onto the Mg–Al LDH (A) and Mg–Fe LDH (B) in the presence of increasing concentrations of inorganic and organic ligands after a reaction time of 24 h; initial ligand/AsO4 molar ratios = 1 (black) and 5 (checkered white). The numbers indicate the efficiency (%) of the ligands in preventing AsO4 sorption. The initial AsO4 concentrations for the Mg–Al LDH and the Mg–Fe LDH were 600 and 2000 mmol kg−1, respectively (reproduced with permission from ref. 104). | ||
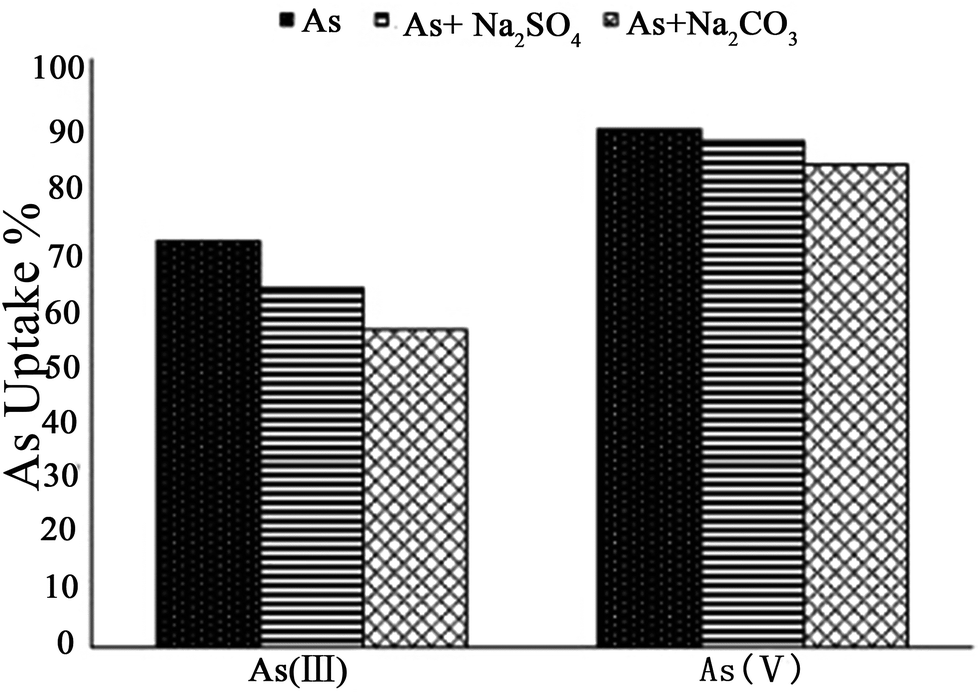 | ||
| Fig. 10 The effect of competitive anions on the As(III) and As(V) uptakes using the Zn–Al–SO4 LDH (reproduced with permission from ref. 85). | ||
Very recently, Pigna et al.100 prepared Cu–Al, Mg–Al, Mg–Fe, and Zn–Al LDHs as As adsorbents to investigate the As(III) removal efficiency in the presence of competing inorganic anions (i.e., CO32−, SO42−, PO43−, Cl− and F−), which are commonly present in bodies of water. The results confirmed that the nature and reactivity of the LDH adsorbents on As(III) removal depended on the composition of the LDH with divalent (Mg vs. Zn or Cu) and trivalent (Fe vs. Al) cations. Moreover, without other competing anions, the efficiency of the competing anions for inhibiting the As(III) adsorption of the LDHs was Cl− ≤ F− < SO42− ≪ CO32− ≪ PO43−, regardless of the initial ligand/As(III) molar ratio or the LDH. The results showed that the Mg–Fe-LDH had the highest As(III) adsorption capacity, nevertheless, the Cu–Al LDH showed a higher affinity for As(III) in the presence of competing anions. Hence, in order to achieve in situ oxidation of As(III) and to remove both As(V) and As(III) simultaneously, the Cu–Al LDH may be a better As adsorbent than the Mg–Fe LDH in contaminated water where competing anions occur in high molar ratios to As(III).
7. Conclusions
In this review, the progress in research on LDHs as As adsorbents has been thoroughly reviewed in five parts. It has indicated that As adsorption can more easily occur on calcined LDHs than on uncalcined LDHs by the anion exchange and reconstruction mechanisms. Moreover, the LDH synthesis method may impact its As adsorption performance. LDHs synthesized using the coprecipitation method displayed higher As uptake capacities than those synthesized using the hydrothermal method. The research results verified that the composition of the LDH influences the As adsorption capacity and corresponds to the cations and anions in the LDH. In addition, the Mg/Al ratio has a strong influence on the adsorption capacity of LDHs, because of the different layer charge densities in LDHs. For LDH-based hybrids, polymer LDH hybrids showed excellent As adsorption and regeneration abilities. Meanwhile, the Mg–Fe–Zr LDH/Fe3O4 hybrid showed a little lower As adsorption capacity because the hybrid contained the Fe3O4 phase. However, the hybrid could be separated from the aqueous solution using an external magnetic field. In addition, the influence of competing anions on the adsorption of As on LDHs relates to the anion affinity or anion intercalation capability of the LDH. Meanwhile, the desorption rate is associated with the different anion affinities for LDHs. Except for the influencing factors, the As adsorption capacity of LDHs also depends on the As concentration and adsorbent dose, as well as the other elements and their concentrations in water. The pH of water can influence the removal ability of As of LDHs. As removal using LDHs is very sensitive to the pH when the pH is relatively low, however, it is insensitive to the pH when the pH is above 7.0.Clearly, LDHs as As adsorbents have been studied across a broad range, however, to further improve the As adsorption capacity is also of first importance for future research. In addition, future research must focus on developing molecular models based on actual pollution water. For the practical use of an adsorbent, the regeneration of the adsorbent is very important, which is associated with the cost. Therefore, the regeneration ability and techno-economic assessment of LDHs as As adsorbents will be focused on in the future, which may provide solutions for understanding the adsorption mechanism and carrying out economic evaluations of LDH derived adsorbents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the Scientific Research Fund Projects of the Yunnan Provincial Department of Education (2017ZZX137) and the National Natural Science Foundation of China (51568067, 51622801 and 51572029). We are very grateful to all member units of the project team for their help.References
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201 CrossRef PubMed.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1 CrossRef PubMed.
- K. Wu, H. Wang, R. Liu, X. Zhao, H. Liu and J. Qu, J. Hazard. Mater., 2011, 18, 990 CrossRef PubMed.
- C. M. Babu, B. Palanisamy, B. Sundaravel, K. Shanthi and V. Murugesan, Sci. Adv. Mater., 2015, 7, 794 CrossRef.
- M. A. Rosa, J. A. Egido and M. C. Márquez, J. Taiwan Inst. Chem. Eng., 2017, 78, 409 CrossRef.
- M. Leist, R. J. Casey and D. Caridi, J. Hazard. Mater., 2000, 76, 125 CrossRef PubMed.
- P. Nakseedee, V. Tanboonchuy, N. Pimpha, P. Khemthong, C. H. Liao and N. Grisdanurak, J. Taiwan Inst. Chem. Eng., 2015, 47, 182 CrossRef.
- J. C. Ng, J. Wang and A. Shraim, Chemosphere, 2003, 52, 1353 CrossRef PubMed.
- K. Wu, R. P. Liu, H. J. Liu, H. C. Lan and J. H. Qu, J. Hazard. Mater., 2012, 239, 308 CrossRef PubMed.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517 CrossRef.
- D. B. Singh, G. Prasad, D. C. Rupainwar and V. N. Singh, Water, Air, Soil Pollut., 1988, 42, 373 Search PubMed.
- WHO, Environmental Health Criteria 224: Arsenic compounds, 2nd edn, 2011 Search PubMed.
- C. K. Jain and I. Ali, Water Res., 2000, 34, 4304 CrossRef.
- M. Vaclavikova, G. P. Gallios, S. Hredzak and S. Jakabsky, Clean Technol. Environ. Policy, 2008, 10, 89 CrossRef.
- U. Shafique, A. Ijaz, M. Salman, W. uz Zaman, N. Jamil, R. Rehman and A. Javaid, J. Taiwan Inst. Chem. Eng., 2012, 43, 256 CrossRef.
- B. Daus, R. Wennrich and H. Weiss, Water Res., 2004, 38, 2948 CrossRef PubMed.
- Y. Sun, G. Zhou, X. Xiong, X. Guan, L. Li and H. Bao, Water Res., 2013, 47, 4340 CrossRef PubMed.
- D. Haraguchi, C. Tokoro, Y. Oda and S. Owada, J. Chem. Eng. Jpn., 2013, 46, 173 CrossRef.
- S. Chakrabortty, M. Sen and P. Pal, Environ. Sci. Pollut. Res., 2014, 21, 3840 CrossRef PubMed.
- A. Dominguez-Ramos, K. Chavan, V. García, G. Jimeno, J. Albo, K. V. Marathe, G. D. Yadav and A. Irabien, J. Ind. Eng. Chem., 2014, 53, 18920 CrossRef.
- B. J. Lafferty and R. H. Loeppert, Environ. Sci. Technol., 2005, 39, 2120 CrossRef PubMed.
- S. Wang, B. Gao, A. R. Zimmerman, Y. Li, L. Ma, W. G. Harris and K. W. Migliaccio, Bioresour. Technol., 2015, 175, 391 CrossRef PubMed.
- S. Wang, B. Gao, Y. Li, A. R. Zimmerman and X. Cao, RSC Adv., 2016, 6, 17792 RSC.
- S. Zhang, H. Niu, Y. Cai, X. Zhao and Y. Shi, Chem. Eng. J., 2010, 158, 599 CrossRef.
- X. Guo, X. Jin, X. Zhao, Z. Yang, X. Lv, J. Zhang and N. Qiu, Sci. Adv. Mater., 2014, 6, 793 CrossRef.
- L. Dambies, T. Vincent and E. Guibal, Water Res., 2002, 36, 3699 CrossRef PubMed.
- M. M. Benjamin, R. S. Sletten, R. P. Bailey and T. Bennett, Water Res., 1996, 30, 2609 CrossRef.
- M. N. Amin, S. Kaneco, T. Kitagawa, A. Begum, H. Katsumata, T. Suzuki and K. Ohta, Ind. Eng. Chem. Res., 2006, 45, 8105 CrossRef.
- D. Kang, X. Yu, S. Tong, M. Ge, J. Zuo, C. Cao and W. Song, Chem. Eng. J., 2013, 228, 731 CrossRef.
- S. Paikaray, M. J. Hendry and J. Essilfie-Dughan, Chem. Geol., 2013, 345, 130 CrossRef.
- K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343 CrossRef PubMed.
- S. Suvokhiaw, A. Imyim and N. Sukpirom, Sep. Purif. Technol., 2016, 51, 2948 Search PubMed.
- S. Duan, W. Ma, Z. Cheng, P. Zong, X. Sha and F. Meng, Colloids Surf., A, 2016, 490, 250 CrossRef.
- Q. Wang and D. O’Hare, Chem. Rev., 2012, 112, 4124 CrossRef PubMed.
- B. Li, Z. Xu, F. Jing, S. Luo, N. Wang and W. Chu, J. Energy Chem., 2016, 25, 1078 CrossRef.
- S. M. N. Mohsin, M. Z. Hussein and S. H. Sarijo, Sci. Adv. Mater., 2014, 6, 648 CrossRef.
- T. Wang, J. Li, Y. Su, C. Wang, Y. Gao, L. Chou and W. Yao, J. Energy Chem., 2015, 24, 432 CrossRef.
- N. D. Hutson, Chem. Mater., 2004, 16, 4135 CrossRef.
- J.-I. Yang and J. N. Kim, Korean J. Chem. Eng., 2006, 23, 77 CrossRef.
- P.-H. Chang, T.-J. Lee, Y.-P. Chang and S.-Y. Chen, ChemSusChem, 2013, 6, 1076 CrossRef PubMed.
- S. Radha and A. Navrotsky, J. Phys. Chem. C, 2014, 118, 29836 CrossRef.
- O. Aschenbrenner, P. McGuire, S. Alsamaq, J. Wang, S. Supasitmongkol, B. Al-Duri, P. Styring and J. Wood, Chem. Eng. Res. Des., 2011, 89, 1711 CrossRef.
- X. Tong, P. Choi, S. Li, Y. Shi and H. Zhang, RSC Adv., 2016, 6, 98804 RSC.
- Q. Wang, Z. H. Wu, H. H. Tay, L. W. Chen, Y. Liu, J. Chang, Z. Y. Zhong, J. Z. Luo and A. Borgna, Catal. Today, 2011, 164, 198 CrossRef.
- R. Rojas, Appl. Clay Sci., 2014, 87, 254 CrossRef.
- F. R. Peligro, I. Pavlovic, R. Rojas and C. Barriga, Chem. Eng. J., 2016, 306, 1035 CrossRef.
- M. A. González, I. Pavlovic, R. Rojas-Delgado and C. Barriga, Chem. Eng. J., 2014, 254, 605 CrossRef.
- R. Rojas, Chem. Eng. J., 2016, 303, 331 CrossRef.
- D. Zhao, G. Sheng, J. Hu, C. Chen and X. Wang, Chem. Eng. J., 2011, 171, 167 CrossRef.
- M. R. Othman, Z. Helwani and W. J. N. Fernando, Appl. Organomet. Chem., 2009, 23, 335 CrossRef.
- W. Bao, J. Wang, Q. Wang, D. O’Hare and Y. Wan, Sci. Rep., 2016, 6, 1 CrossRef PubMed.
- J. Wang, X. Mei, L. Huang, Q. Zheng, Y. Qiao, K. Zang, S. Mao, R. Yang, Z. Zhang, Y. Gao, Z. Guo, Z. Huang and Q. Wang, J. Energy Chem., 2015, 24, 127 CrossRef.
- J. Wang, L. Huang, Q. Zheng, Y. Qiao and Q. Wang, J. Ind. Eng. Chem., 2016, 36, 355 CrossRef.
- L. Wang, Y. Wang and X. Wang, Materials, 2017, 10, 1140 CrossRef PubMed.
- F. Barahuie, M. Z. Hussein and P. Arulselvan, Sci. Adv. Mater., 2016, 8, 501 CrossRef.
- Q. Qin, J. Wang, T. Zhou, Q. Zheng, L. Huang, Y. Zhang, P. Lu, A. Umar, B. Louis and Q. Wang, J. Energy Chem., 2017, 26, 346 CrossRef.
- H. M. Kim, K. M. Kim and B. C. Jung, Sci. Adv. Mater., 2014, 6, 1582 CrossRef.
- X. Shan, Y. Hu and H. Chen, Sci. Adv. Mater., 2015, 7, 1848 CrossRef.
- P. Lu, X. Zhang and Z. Wang, Sci. Adv. Mater., 2016, 8, 1656 CrossRef.
- J. Tedim, M. L. Zheludkevich, A. N. Salak, A. Lisenkov and M. G. S. Ferreira, J. Mater. Chem., 2011, 21, 15464 RSC.
- M. L. Zheludkevich, J. Tedim and M. G. S. Ferreira, Electrochim. Acta, 2012, 82, 314 CrossRef.
- T. L. Galvão, C. S. Neves, M. L. Zheludkevich, J. R. Gomes, J. Tedim and M. G. Ferreira, J. Phys. Chem. C, 2017, 12, 2211 CrossRef.
- Z. Yang, Z. Mo and X. Niu, J. Hazard. Mater., 2015, 180, 401 Search PubMed.
- Z. Yang, Z. Mo and X. Niu, Sep. Sci. Technol., 2015, 50, 99 CrossRef.
- S. Li, Y. Shen and D. Liu, Sci. Adv. Mater., 2015, 7, 756 CrossRef.
- E. Otgonjargal, Y. S. Kim, S. M. Park, K. Baek and J. S. Yang, Sep. Sci. Technol., 2012, 47, 2192 Search PubMed.
- B. Hudcová, V. Veselská, J. Filip, S. Číhalová and M. Komárek, Chemosphere, 2017, 168, 539 CrossRef PubMed.
- L. Huang, J. Wang, Y. Gao, Y. Qiao, Q. Zheng, Z. Guo, Y. Zhao, D. O’Hare and Q. Wang, J. Mater. Chem. A, 2014, 2, 18454 RSC.
- Q. Wang, H. H. Tay, Z. Zhong, J. Luo and A. Borgna, Energy Environ. Sci., 2012, 5, 7526 RSC.
- Z. Zhang, J. Wang, L. Huang, Y. Gao, A. Umar, Z. Huang and Q. Wang, Sci. Adv. Mater., 2014, 6, 1154 CrossRef.
- J. Wang, W. Bao, A. Umar, Q. Wang, D. O’Hare and Y. Wan, J. Biomed. Nanotechnol., 2016, 12, 922 CrossRef PubMed.
- P. Benito, I. Guinea, F. M. Labajos and V. Rives, J. Solid State Chem., 2008, 181, 987 CrossRef.
- W. F. Lee and Y. C. Chen, Eur. Polym. J., 2006, 42, 1634 CrossRef.
- B. Zapata, P. Bosch, G. Fetter, M. A. Valenzuela, J. Navarrete and V. H. Lara, Int. J. Inorg. Mater., 2001, 3, 23 CrossRef.
- Y. W. You, H. T. Zhao and G. F. Vange, Environ. Technol., 2000, 22, 6804 Search PubMed.
- L. Yang, Z. Shahrivari, P. K. Liu, M. Sahimi and T. T. Tsotsis, Ind. Eng. Chem. Res., 2005, 44, 6804 CrossRef.
- M. Chetia, R. L. Goswamee, S. Banerjee, S. Chatterjee, L. Singh, R. B. Srivastava and H. P. Sarma, Clean Technol. Environ., 2012, 14, 21 CrossRef.
- Y. T. Liu, M. K. Wang, T. Y. Chen, P. N. Chiang, P. M. Huang and J. F. Lee, Environ. Sci. Technol., 2006, 40, 7784 CrossRef PubMed.
- Y. T. Liu, T. Y. Chen, M. K. Wang, P. M. Huang, P. N. Chiang and J. F. Lee, Appl. Clay Sci., 2010, 48, 485 CrossRef.
- K. Grover, S. Komarneni and H. Katsuki, Water Res., 2009, 43, 3884 CrossRef PubMed.
- K. Grover, S. Komarneni and H. Katsuki, Appl. Clay Sci., 2010, 48, 631 CrossRef.
- Q. Guo and J. Tian, Chem. Eng. J., 2013, 231, 121 CrossRef.
- Y. Guo, Z. Zhu, Y. Qiu and J. Zhao, J. Hazard. Mater., 2012, 239–240, 279 CrossRef PubMed.
- E. Deschamps, V. S. T. Ciminelli and W. H. Holl, Water Res., 2005, 39, 5212 CrossRef PubMed.
- S. Bagherifam, S. Komarneni, A. Lakzian, A. Fotovat, R. Khorasani, W. Huang, J. Ma and Y. Wang, Appl. Clay Sci., 2014, 95, 119 CrossRef.
- H. Lu, Z. Zhu, H. Zhang, J. Zhu and Y. Qiu, Chem. Eng. J., 2015, 276, 365 CrossRef.
- P. P. Huang, C. Y. Cao, F. Wei, Y. B. Sun and W. G. Song, RSC Adv., 2015, 5, 10412 RSC.
- H. Lu, Z. Zhu, H. Zhang and Y. Qiu, Water, Air, Soil Pollut., 2016, 227, 1 CrossRef.
- L. Ma, S. M. Islam, H. Liu, J. Zhao, G. Sun, H. Li and M. G. Kanatzidis, Chem. Mater., 2017, 29, 3274 CrossRef.
- L. Yang, M. Dadwhal, Z. Shahrivari, M. Ostwal, P. K. Liu, M. Sahimi and T. T. Tsotsis, Ind. Eng. Chem. Res., 2006, 45, 4742 CrossRef.
- S. L. Wang, C. H. Liu, M. K. Wang, Y. H. Chuang and P. N. Chiang, Appl. Clay Sci., 2009, 43, 79 CrossRef.
- Y. Cao, Q. Guo, Z. Shu, Y. Zhuang, Z. Yu, W. Guo, C. Zhang, M. Zhu and T. Ren, Appl. Clay Sci., 2016, 126, 313 CrossRef.
- Q. Guo, Y. Cao, Y. Zhuang, Y. Yang, M. Wang and Y. Wang, Appl. Geochem., 2017, 77, 206 CrossRef.
- L. Zhang, L. Li, X. Sun, P. Liu, D. Yang and X. Zhao, Materials, 2016, 9, 927 CrossRef PubMed.
- M. Wang, J. Lian and X. Zhou, Sci. Adv. Mater., 2017, 9, 602 CrossRef.
- H. N. Nhat Ha, N. T. Kim Phuong, T. Boi An, N. T. Mai Tho, T. Ngoc Thang, B. Quang Minh and C. Van Du, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2016, 51, 403 CrossRef PubMed.
- P. Z. Ray and H. J. Shipley, RSC Adv., 2015, 5, 29885 RSC.
- S. Lata and S. R. Samadder, J. Environ. Manage., 2016, 166, 387 CrossRef PubMed.
- S.-M. Jessica, J. B. Kevin and A. E. Smith, J. Am. Water Resour. Assoc., 2006, 42, 1237 CrossRef.
- M. Pigna, J. J. Dynes, A. Violante, A. Sommella and A. G. Caporale, Environ. Eng. Sci., 2016, 33, 98 CrossRef.
- L. M. Parker, N. B. Milestone and R. H. Newman, Ind. Eng. Chem. Res., 1995, 34, 1196 CrossRef.
- Y. W. You, G. F. Vance and H. T. Zhao, Appl. Clay Sci., 2001, 20, 13 CrossRef.
- R. L. Goswamee, P. Sengupta, K. G. Bhattacharyya and D. K. Dutta, Appl. Clay Sci., 1998, 13, 21 CrossRef.
- A. G. Caporale, M. Pigna, J. J. Dynes, V. Cozzolino, J. Zhu and A. Violante, J. Hazard. Mater., 2011, 198, 291 CrossRef PubMed.
- A. Violante and M. Pigna, Soil Sci. Soc. Am. J., 2002, 66, 1788 CrossRef.
- A. Violante, M. Pigna, S. Del Gaudio, M. Ricciardella and D. Banerjee, Environ. Sci. Technol., 2007, 41, 8275 CrossRef PubMed.
- S. Goldberg and C. T. Johnston, J. Colloid Interface Sci., 2001, 234, 204 CrossRef PubMed.
- C. Ardau, F. Frau and P. Lattanzi, Appl. Clay Sci., 2013, 80, 1 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |