
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal free, late-stage, regiospecific, aromatic fluorination on a preparative scale using a KF/crypt-222 complex†
Jimmy Erik Jakobsson *a and
Patrick Johannes Rissabc
*a and
Patrick Johannes Rissabc
aRealomics Strategic Research Initiative, Department of Chemistry, Faculty for Mathematics and Natural Sciences, University of Oslo, Norway. E-mail: j.e.jakobsson@kjemi.uio.no
bDepartment of Surgery and Neuroscience, OUS-Rikshospitalet HF, Oslo, Norway
cNorwegian Medical Cyclotron AS, Nydalen, Oslo, Norway
First published on 11th June 2018
Abstract
We herein report the development of a convenient, regioselective, aromatic fluorination method of hypervalent iodonium ylides for synthesising fluoro-arenes on a preparative scale. This transition metal free, nucleophilic methodology provides good yields for sterically hindered substrates, irrespective of activation. The methodology simplifies reference synthesis for PET imaging.
Fluorinated organic molecules are of importance in crop protection and materials science and as pharmaceuticals due to their unique properties. Aryl fluorides are of particular interest in medicinal chemistry1 due to their exceptional metabolic stability. For instance, fluorine is often used as a tool in medicinal chemistry for fine tuning the physicochemical properties of drug candidates in late stage development.2 The presence of fluorine in drug molecules also allows for further investigation by positron emission tomography (PET) via incorporation of radioactive fluorine-18 (18F, t1/2 = 110 min). The radionuclide 18F is ideal for PET imaging, due to its very advantageous half-life and decay characteristics.3
Current late stage fluorination methodologies of arenes and heteroarenes are often based on expensive, potentially toxic transition-metals (Ag/Cu,4 Cu,5 Ni,6 Pd7) or involve electrophilic fluorination reagents and may necessitate the use of additional, advanced equipment such as gloveboxes to achieve more anhydrous conditions. Therefore a mild, versatile and selective fluorination methodology that utilises cheap nucleophilic fluoride for late stage formation of fluoroarenes is highly sought after.8 In addition, we aim to progress into greener, more environmentally friendly alternatives with easy to handle chemicals by moving away from toxic transition metals. In order to be independent from classical substrates of the SNAr reaction, we chose hypervalent iodine species as fluorination precursors. The hypervalent iodine chemistry utilises the plethora of well-investigated and described iodination protocols and iodonium ylide forming reactions as a basis for constructing aryl fluorides. Such a method would also bridge the gap between preparative organic chemistry and radiochemistry thus applying the same precursor for synthesising both radiotracer and reference compound in the last step (Scheme 1). Radiofluorination of iodonium ylides is well described,9 however in radiochemistry reagent stoichiometry is vastly different with the precursor used in >1000 fold excess over no carrier added fluorine-18, the limiting reagent.‡ For the methodology to be useful on preparative scale the precursor should ideally be the limiting reagent and a cheap fluorination reagent such as KF, CsF or TBAF used in excess. We therefore employed a reverse translational approach developing a preparative organic protocol for the fluorination reaction under stoichiometric conditions. Through this new protocol, PET chemistry is merged with preparative organic chemistry enabling use of iodonium ylides as versatile fluorination precursors.
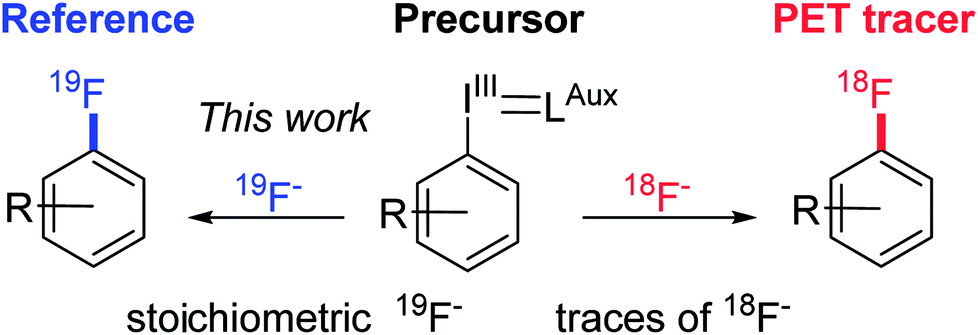 | ||
| Scheme 1 Illustrate a back-translational approach, developing an organic chemistry protocol for synthesising reference compound on preparative scale. | ||
Electron deficient substrates can undergo a classical SNAr reaction10 therefore for this investigation we decided to use 2-fluoroanisole as model compound since it is both sterically hindered and electron rich and therefore challenging to produce. All reactions were performed without access to glove box using standard equipment and precautions to allow for good reproducibility in between labs. We began by screening the most common nucleophilic fluorination reagents in organic, dipolar aprotic solvents (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Reagent | Solvent | Yielda% (2) | Yielda% (3) |
| a 19F NMR yield using 4-fluorobiphenyl as internal standard. Standard deviation is given for reactions performed in duplicate. Product identities were confirmed from spiking with reference compound.b 1 h reaction time. PC = propylene carbonate. | ||||
| 1 | CsF | DMF | Traces | 0 |
| 2 | TBAF·3H2O | DMF | 9% | 0.6% |
| 3 | TBAF(t-BuOH)4 | DMF | 31 ± 4% | 1.4 ± 0.0% |
| 4 | TBAF(t-BuOH)4 | DMFb | 32%a | 1.4% |
| 5 | TBAF(t-BuOH)4 | PC | 11% | 1% |
| 6 | TBAF(t-BuOH)4 | DMSO | 5% | 1% |
| 7 | TBAF(t-BuOH)4 | PhCl | 12% | 25% |
| 8 | TBAF(t-BuOH)4 | MeCN | 5% | 9% |
| 9 | 1.5 eq. TBAF(t-BuOH)4 | DMF | 30 ± 0% | 0.3 ± 0.1% |
| 10 | 4 eq. TBAF(t-BuOH)4 | DMF | 5 ± 0.2% | 6 ± 0.2% |
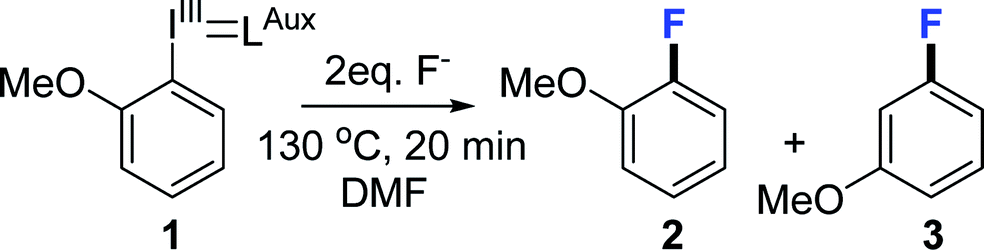
Caesium fluoride produced mere trace amounts of product (2), possibly owing to its high hygroscopicity and difficulty of excluding moisture (Table 1, entry 1). Tetrabutylammonium fluoride trihydrate yielded a higher conversion (2, 9%) but also 3-fluoroanisole (3) as side product (0.6%) (Table 1, entry 2). We have previously observed that the reaction is moderately sensitive to moisture9b on radiochemical scale so we synthesised the anhydrous TBAF(t-BuOH)4 complex,11 which although it increased the yield fourfold (Table 1, entry 2) also produced more of the undesired constitutional isomer that would render purification considerably more difficult (Table 1 entry 3). Heating the reaction mixture for a longer time made no difference (Table 1, entry 4) meaning that the constitutional isomer formation is unrelated to product degradation, varying the temperature did not resolve the selectivity problem either (ESI†). Earlier literature reports suggest that fluorination of aryl bromides goes through an aryne intermediate when using the very basic tetraalkylammonium fluorides12 in DMSO as well as radiofluorination of iodonium ylides using KF/crypt-222 in MeCN.9a We assumed that since reagent basicity is solvent dependent choosing another solvent could circumvent the problem. However, we found that use of either propylene carbonate or DMSO (Table 1, entries 5 and 6) yielded a comparable ratio of 2-fluoroanisole (2) to 3-fluoroanisole (3) (11 + 1% and 5 + 1% respectively) and that the reaction in both chlorobenzene and MeCN (Table 1, entries 7 and 8) proceeded primarily through an aryne intermediate (12% + 25% and 5 + 9% respectively). We concluded that we have two competing reaction pathways and that the aryne intermediate formation is facilitated by TBAF (Scheme 2). By reducing the equivalents of TBAF(t-BuOH)4 from 2 to 1 the yield was marginally decreased to 30% but the formation of 3-fluoroanisole (3) reduced by three quarters to 0.3 ± 0.1% (Table 1, entry 9). This finding suggest that use of tetraalkylammonium fluorides in radiofluorination reactions does not suffer from formation of constitutional isomers since only trace quantities of fluoride is used, which is in agreement with Rotstein et. al.9c By increasing the equivalents of TBAF(t-BuOH)4 complex to 4 equivalents, 5% desired 2-fluoroanisole (2) and 6% undesired 3-fluoroanisole (3) (Table 1 entry 10) were obtained, further supporting our hypothesis.
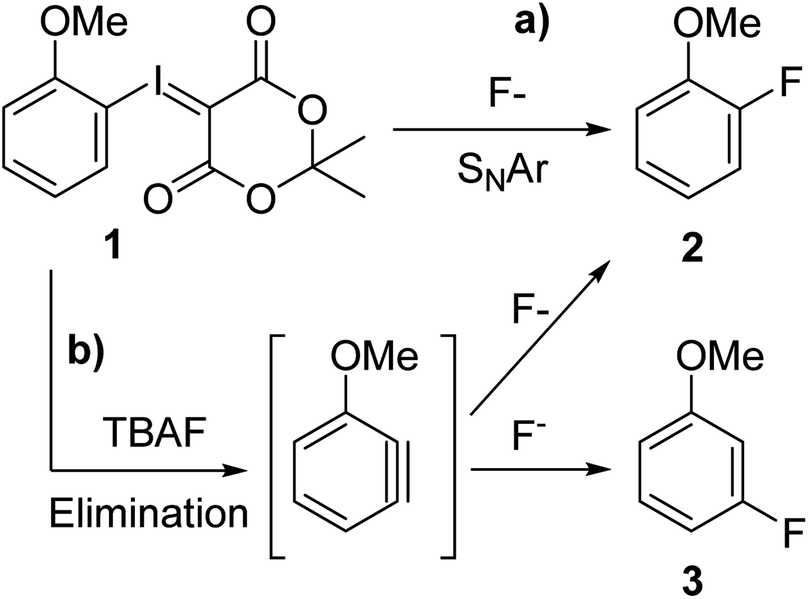 | ||
| Scheme 2 Shows two competing reaction pathways for fluorination of 1 using TBAF(tBuOH)4. (a) Nucleophilic substitution, (b) TBAF mediated aryne formation and subsequent fluorination yielding both 2 and 3. | ||
With this finding, we decided to move away from tetraalkylammonium fluorides and investigate metal fluorides in combination with phase transfer catalysts (Table 2) to enhance both solubility and nucleophilicity.
| Entry | Reagent | Yielda% (2) | Yielda% (3) |
|---|---|---|---|
| a 19F NMR yield using 4-fluorobiphenyl as internal standard. Where standard deviation is given, the reaction was performed in duplicate. Product identities were confirmed from spiking with reference compound.b Preformed KF/crypt-222 complex containing 33 mol% K2CO3 to avoid formation of HF during drying of KF/crypt-222 complex. Crypt-222 = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane. | |||
| 1 | KF/18C6 | 1 ± 0% | 0 |
| 2 | CsF/crypt-222 | 1 ± 1% | 0 |
| 3 | KF/crypt-222 | 8 ± 2% | 0 |
| 4 | KF/crypt-222 | 46 ± 2%b | 0 |
| 5 | 1.5 eq. KF/crypt-222 | 36%b | 0 |
| 6 | 4 eq. KF/crypt-222 | 43%b | 0 |
Addition of conventional phase transfer catalyst potassium complexing crown ether 18-crown-6 to KF produced only traces of product (Table 2, entry 1). Surprisingly neither CsF nor KF in combination with cryptand crypt-222 produced satisfactory results, 1 ± 1% and 8 ± 2% of 2-fluoroanisole (2) respectively (Table 2, entries 2 and 3) but with a 19F NMR spectrum devoid of the 3-fluoroanisole signal (3). We hypothesised that the limited solubility of potassium fluoride13 in DMF made formation of KF/crypt-222 complex too slow to allow for an efficient fluorination reaction since the iodonium ylide substrate slowly degrade under the fluorination conditions.9b 13C-NMR shows only partial formation of crypt-222/K+ complex after 2 hours of heating KF and crypt-222 in DMF at 130 °C (ESI†) strengthening our hypothesis. Adding water to the reaction mixture to facilitate KF/crypt-222 complex formation would destroy the substrate and strongly diminish the nucleophilicity of fluoride. Therefore, we anticipated that the fluorination complex need to be formed in advance. We made the KF/crypt-222 complex from a solution of MeCN/H2O adding 33 mol% K2CO3 to hinder evaporation of HF. The complex was thoroughly dried for several days under high vacuum. In line with our hypothesis, the pre-formed KF/crypt-222 complex produced a significantly higher yield (46 ± 2%) gratefully without formation of any constitutional isomers (Table 2, entry 4). The pre-made KF/crypt-222 complex is not visibly hygroscopic and is an easy to weigh free flowing powder (ESI†) that can be stored for at least several weeks in a closed vial at room temperature without any impairment of the fluorination yield. Increasing or reducing the equivalents of KF/crypt-222 complex did not improve the yield further (Table 2, entries 5 and 6). The complex readily dissolves in DMF and 13C-NMR gives three new signals upfield of the host crypt-222.
With the optimised fluorination conditions in hand, we sat out to screen a range of substrates to examine the reaction scope (Fig. 1). 4-Fluoroanisole (4) was produced in a significantly lower yield (10 ± 1%) than 2-fluoroanisole (2, 46 ± 2%), the large difference is likely due to the substrate being both deactivated and lacking an ortho-substituent thus not benefitting from the ortho-effect.14 Fluorination of non-activated substrates produced fluorobenzene (5) and 4-chloro-1-fluorobenzene (6) in modest yields (33 ± 2% and 26 ± 4%) but good enough to be useful for synthesising sufficient material of reference compounds where milligram quantities is sufficient. Fluorination constructing 5-fluoro-m-xylene (7) proceeded in an acceptable yield (36 ± 1%) whereas sterically hindered constitutional isomer 2-fluoro-m-xylene (8) was produced in a high yield (78 ± 7%) demonstrating the benefit of ortho-substitution. Activated substrates 4-fluorodimethylbenzamide (9) and 4-fluorobenzonitrile (10) were as expected obtained in high to excellent yields (70 ± 3% and 52 ± 18%) albeit under milder conditions then used for SNAr-fluorinations. Activated drug compound tropane (11) was produced in near quantitative yield (88 ± 7%) (90% isolated yield) and the challenging to access morphinane (12) fluorinated well in 30 ± 1% (20% isolated yield), sufficient for testing its potential use as an NMDA–PET radioligand. The radiotracer [18F] 12 was synthesised in 38% isolated yield (150 MBq > 95% RCP after cartridge purification). Activated tropane was readily fluorinated in room temperature yielding 33% of 11. Encouraged by the positive results we decided to investigate how well the pre-dried KF/crypt-222 complex fluorinated activated nitro arenes. We synthesised the NO2-precursor of (11) for a direct comparison and attempted fluorination under standard conditions obtaining 11 in 34% yield compared to 88 ± 7% when using the iodonium ylide precursor. Fluorination at room temperature produced only traces of product after 1 hour (ESI†).
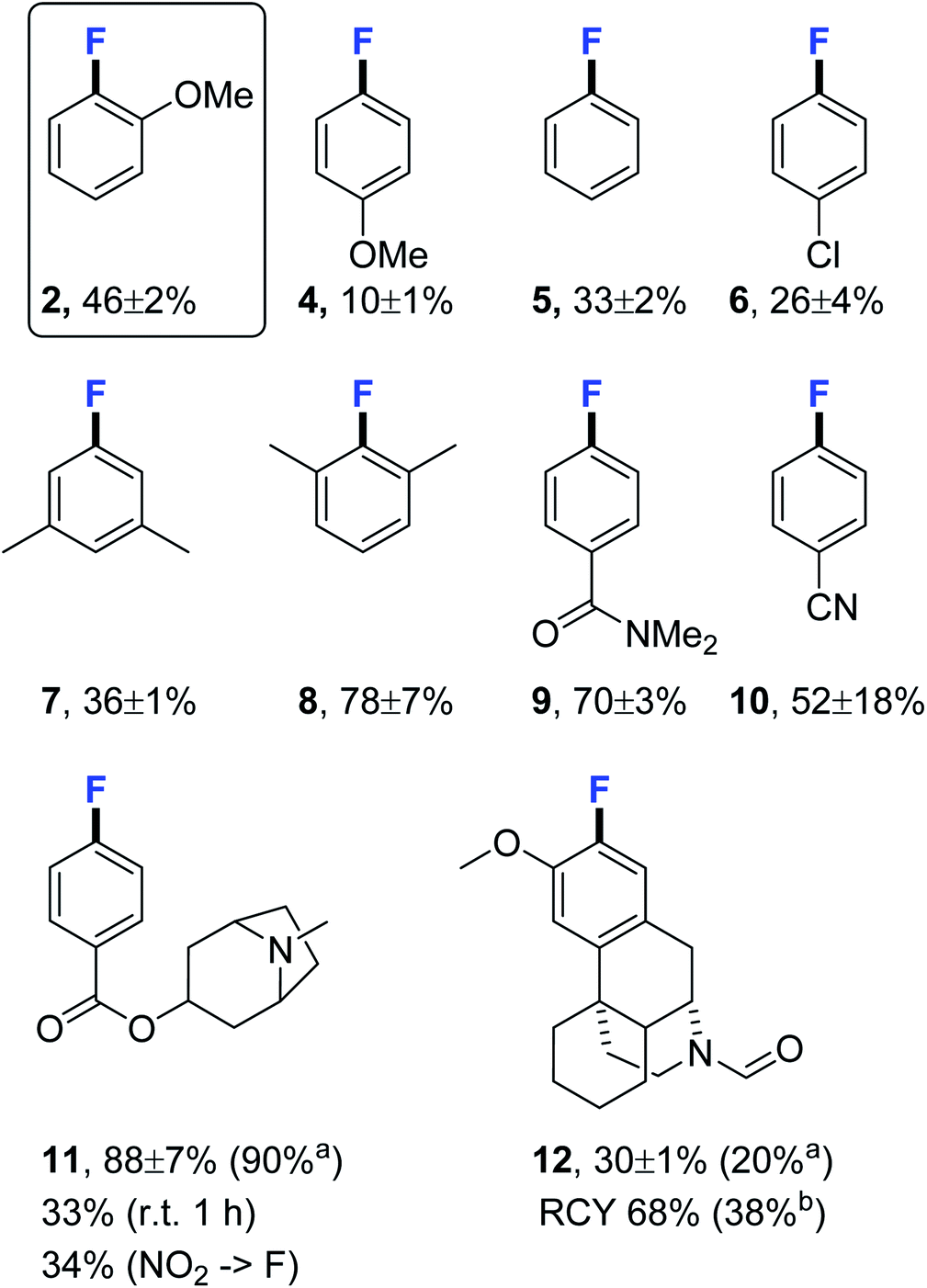 | ||
Fig. 1 Fluorination substrate scope under the developed conditions. 19F NMR yields using 4-fluorobiphenyl as internal standard. Where standard deviation is given, the reaction was performed in duplicate. Product identities were confirmed from spiking with reference compound. Reaction performed on ylide (5 μmol) and crypt-222/KF/K2CO3 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) (6.6 mg, 10 μmol of KF) in DMF (0.5 ml) at 130 °C for 20 min. Radiofluorination performed using ylide (3.7 μmol), crypt-222 (5 mg, 13 μmol) K2CO3 (0.92 mg, 5.5 μmol) and 18F− (400 MBq) in DMF (0.5 ml) at 130 °C for 20 min. aIsolated yield using tracer principle after cartridge purification (C18 and Si). bNon-decay corrected isolated yield after cartridge purification (C18 and Si) in >95% RCP. RCY = radiochemical yield. Crypt-222 = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane. Entry for 2 is identical to Table 2 entry 4. :3:1) (6.6 mg, 10 μmol of KF) in DMF (0.5 ml) at 130 °C for 20 min. Radiofluorination performed using ylide (3.7 μmol), crypt-222 (5 mg, 13 μmol) K2CO3 (0.92 mg, 5.5 μmol) and 18F− (400 MBq) in DMF (0.5 ml) at 130 °C for 20 min. aIsolated yield using tracer principle after cartridge purification (C18 and Si). bNon-decay corrected isolated yield after cartridge purification (C18 and Si) in >95% RCP. RCY = radiochemical yield. Crypt-222 = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane. Entry for 2 is identical to Table 2 entry 4. | ||
A transition metal free fluorination method based on an anhydrous fluorine complex and iodonium ylides useful for preparative aromatic fluorination reactions is successfully demonstrated. Merging radiochemistry and conventional organic chemistry by employing the same precursor eliminates the need of additional synthetic work. An additional advantage of using a transition metal free methodology is to avoid possible interference during biological characterisation and during translation into clinical radiochemistry. We successfully demonstrate the use of the methodology by synthesising complex derivatives via a late stage fluorination reaction and the radioactive counterparts from the same precursor. The method is a good compliment to existing methodologies and with high yield reproducibility expands the accessible chemical space of fluorinated drug molecules.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partly supported by the Research Council of Norway through the Norwegian NMR Platform, NNP (226244/F50) and a personal grant to PJR (NFR ES 231553) JJ thanks the Realomics SRI for a doctoral fellowship.Notes and references
- (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef PubMed; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef PubMed.
- (a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed. Engl., 2008, 47, 8998–9033 CrossRef PubMed; (b) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef PubMed.
- P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 10795–10798 CrossRef PubMed.
- (a) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134–5137 CrossRef PubMed; (b) P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552–2559 CrossRef PubMed.
- H. Lee, J. Borgel and T. Ritter, Angew. Chem., Int. Ed. Engl., 2017, 56, 6966–6969 CrossRef PubMed.
- H. G. Lee, P. J. Milner and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 3792–3795 CrossRef PubMed.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef PubMed.
- (a) J. Cardinale, J. Ermert, S. Humpert and H. H. Coenen, RSC Adv., 2014, 4, 17293–17299 RSC; (b) J. E. Jakobsson, G. Gronnevik and P. J. Riss, Chem. Commun., 2017, 53, 12906–12909 RSC; (c) B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CrossRef PubMed.
- D. J. Adams and J. H. Clark, Chem. Soc. Rev., 1999, 28, 225–231 RSC.
- D. W. Kim, H. J. Jeong, S. T. Lim and M. H. Sohn, Angew. Chem., Int. Ed., 2008, 47, 8404–8406 CrossRef PubMed.
- V. V. Grushin and W. J. Marshall, Organometallics, 2008, 27, 4825–4828 CrossRef.
- D. A. Wynn, M. M. Roth and B. D. Pollard, Talanta, 1984, 31, 1036–1040 CrossRef PubMed.
- B. H. Rotstein, L. Wang, R. Y. Liu, J. Patteson, E. E. Kwan, N. Vasdev and S. H. Liang, Chem. Sci., 2016, 7, 4407–4417 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03757d |
| ‡ We found no benefit of adding triphenylphosphane as previously reported,9b we argue that the reason is that the iodonium ylide is slowly reduced by the phosphane, and consumed reagent is a problem in preparative chemistry that is not encountered in radiochemistry where substrate is used in >1000 fold excess. 31P NMR show PPh3O as minor side product, ESI.† |
| This journal is © The Royal Society of Chemistry 2018 |