
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface engineering-modulated porous N-doped rod-like molybdenum phosphide catalysts: towards high activity and stability for hydrogen evolution reaction over a wide pH range†
Liying Chaia,
Wenyu Yuan b,
Xue Cuia,
Haiying Jianga,
Junwang Tangc and
Xiaohui Guo*a
b,
Xue Cuia,
Haiying Jianga,
Junwang Tangc and
Xiaohui Guo*a
aKey Lab of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710069, P. R. China. E-mail: guoxh2009@nwu.edu.cn; Tel: +86-29-81535031
bScience and Technology on Thermostructural Composite Materials Laboratory, Northwestern Polytechnical University, 710072, Xi'an, P. R. China
cDepartment of Chemical Engineering, UCL, Torrington Place, London, WC1E 7JE, UK
First published on 27th July 2018
Abstract
Electrochemical water splitting is an economic, green and sustainable route to produce hydrogen through the hydrogen evolution reaction (HER). Nowadays, noble metal-free phosphides have been widely used as catalysts in the HER, showing potential applications for both renewable energy production and environmental remediation. Nevertheless, developing surface self-doped MoP electrocatalysts with high HER performances in a wide pH range still remains a challenge. In this work, a novel synthesis strategy was developed to fabricate porous one-dimensional (1D) nitrogen-doped molybdenum phosphide (N-MoP) nanorods. The prepared N-MoP-800 catalyst exhibits a low onset potential of 65 mV and low Tafel slope of 58.66 mV dec−1 in 0.5 M H2SO4, which is almost 2 times higher than that of the pristine MoP nanorod anode. Furthermore, the N-MoP materials show long-term durability for 12 h in a wide pH range. The synergistic effects of pyridinic N and N doping in MoP are responsible for the high catalytic activity of N-MoP under acidic conditions, while the N-Mo component plays a key role in enhancing the HER activity of N-MoP. These interesting findings are helpful for the rational design of highly active HER catalysts. More importantly, this study provides a new strategy to synthesize highly active catalysts with low costs for clean energy conversion.
1. Introduction
The global energy crisis and serious environmental contamination trigger the fast development of renewable, secure and environmentally-friendly energy sources.1–4 Hydrogen energy, as an efficient energy carrier, has attracted extensive attention due to its outstanding energy density, environmental benignity and reliability.5,6 The electrocatalytic HER via electrolysis of water is the most economical and effective route for the future hydrogen economy.7 To date, precious Pt-based materials are the most effective catalysts owing to their low overpotential and good stability. However, the excessive cost and low abundance greatly prevent their practical usage.8–11 Therefore, developing novel non-noble metal catalysts is of great significance for hydrogen production with sufficient efficiency, high stability, and low cost.Recently, many new types of non-precious metal materials have been investigated as highly active electrocatalyst towards HER, including Co-, Ni-, Fe-, Mo-based transition metal chalcogenides, nitrides, phosphides, carbides, and some multiple compounds.12–18 Wherein, a kind of ternary pyrite-type CoPS has been fabricated and act as a high-performance Earth-abundant catalyst for electrochemical hydrogen production with lower catalytic overpotential and outstanding long-term operational stability.19 Among different metal-based compounds, Mo-based catalytic materials, such as MoS2,20,21 Mo2C,22,23 MoN,24 MoSe2,25 MoB,26 MoNi4,27 MoP2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and MoP,29–31 which are considered as promising candidates for hydrogen production owing to their low cost and high catalytic activities. Among them, MoP has attracted special attention because of its high electrical conductivity, adjustable morphology, structure and surface architecture. As a result, many progress of MoP-based electrochemical catalysts has been made recently. Li et al.32 demonstrated a MOFs-assisted strategy to develop MoP @ PC with an onset potential of 77 mV and overpotential of 153 mV at 10 mA cm−2. In particular, the electrocatalytic performance of transition metal-based materials can be improved by introducing metallic or non-metallic elements. In particular, the electrocatalytic performance of transition metal-based materials can be improved by introducing metallic or non-metallic elements. Wang et al.33 proposed the N-doped MoP catalyst by a simple temperature-programmed reduction method, which exhibits outstanding HER activity with a small Tafel slop of 54 mV dec−1 and a good durability in acid condition. Zhang et al.34 proposes an N-doped CoP on carbon cloth as the novel and effective electrocatalyst for HER in acid media, which −42 mV is required to drive a current density of −10 mA cm−2. Wang et al.35 successfully synthesized the P/Co–FeS2 on carbon fiber paper (CFP) and Ti foil via solvothermal, sulfurization, and phosphidation processes. It was found that the P/Co–FeS2/Ti foil catalyst exhibited higher HER activity with a low overpotential, a small Tafel slope, and high durability in acidic solution. Above research progress indicated that HER performances can be improved by providing much more catalytic active sites through a facile doping process.
28 and MoP,29–31 which are considered as promising candidates for hydrogen production owing to their low cost and high catalytic activities. Among them, MoP has attracted special attention because of its high electrical conductivity, adjustable morphology, structure and surface architecture. As a result, many progress of MoP-based electrochemical catalysts has been made recently. Li et al.32 demonstrated a MOFs-assisted strategy to develop MoP @ PC with an onset potential of 77 mV and overpotential of 153 mV at 10 mA cm−2. In particular, the electrocatalytic performance of transition metal-based materials can be improved by introducing metallic or non-metallic elements. In particular, the electrocatalytic performance of transition metal-based materials can be improved by introducing metallic or non-metallic elements. Wang et al.33 proposed the N-doped MoP catalyst by a simple temperature-programmed reduction method, which exhibits outstanding HER activity with a small Tafel slop of 54 mV dec−1 and a good durability in acid condition. Zhang et al.34 proposes an N-doped CoP on carbon cloth as the novel and effective electrocatalyst for HER in acid media, which −42 mV is required to drive a current density of −10 mA cm−2. Wang et al.35 successfully synthesized the P/Co–FeS2 on carbon fiber paper (CFP) and Ti foil via solvothermal, sulfurization, and phosphidation processes. It was found that the P/Co–FeS2/Ti foil catalyst exhibited higher HER activity with a low overpotential, a small Tafel slope, and high durability in acidic solution. Above research progress indicated that HER performances can be improved by providing much more catalytic active sites through a facile doping process.
Although some great process of HER from MoP has been realized to a certain extent, some key modulating parameters such as defects, edge and/or surface architectures how to affect the electrocatalytic performances of MoP have not been investigated so far. To our knowledge, the rationale modulation of the materials' surface architectures is increasingly important for optimizing the performance of electrocatalytic materials. The catalytic reactions occur mainly on the surface of catalysts, namely, most of the catalytic active sites should be located on the catalysts' surface. For this reason, the surface conditions, including surface compositions, microstructures, defects and/or vacancies, greatly impact the activity of the catalyst, as these parameters determine the surface adsorption and activation abilities of particular reactants.36,37 Among them, the N-doping in MoP catalyst can obviously influence the surface electronic state and microstructures so as to better modulate HER performances. Recently, although some process of N-doped molybdenum-based electrocatalysts have been obtained for HER applications,38–40 these reported molybdenum-based (including MoP) materials display HER performances only in acid medium, which certainly limit their practical applications. Therefore, investigating their HER performances in a wide pH range becomes very emergent and necessary.
In this work, we designed and synthesized a novel porous one-dimensional (1D) nitrogen-doped MoP (N-MoP) nanorods through a simple two-step synthetic strategy, which shows high HER catalytic activities over a wide pH range. Besides, the rod-like MoP materials are allowed to possess remarkable long-term electrochemical stability in different conditions. The excellent HER performances were mainly resulted from the modulated surface-active sites derived from N-doping and modulated surface architectures. In a word, the high electrochemical active MoP materials would possess great potential in future energy conversion and utilization.
2. Experimental section
2.1 Materials
All chemical reagents are analytical degree without further purification treatment.2.2 Synthesis of Mo3O10/ethylenediamine (EDA) precursor
Herein, the Mo3O10/EDA nanowires were prepared according to previous literature.41 Typically, 1.24 g of (NH4)6Mo7O24·4H2O was dissolved in 15 mL of deionized water, and 0.8 g of EDA was added into the solution. Then, 1 M of HCl was added into above solution dropwise with magnetic stirring at room temperature, until a white precipitate appeared (pH = 4–5). After stirring for 2 h at 50 °C, the Mo3O10/EDA nanowire was obtained after washing with water and ethanol for several times, and dried at 60 °C for 12 h (Fig. S1a†).2.3 Synthesis of rod-like N-MoP
The one-dimensional N-MoP structures were obtained by controlling the thermal reduction temperature of Mo3O10/EDA nanowires. To obtain N-MoP-800, 65 mg Mo3O10/EDA and 100 mg (NH4)2HPO4 (molar ratios of Mo/P = 1:2) were placed on opposite ends with (NH4)2HPO4 at the upstream side of the porcelain boat, and leave it sealed. The boat was heated in a pipe furnace at 800 °C under Ar/H2 (10%) for 120 min at a heating rate of 5 °C min−1. After naturally cooled down to room temperature, the products could be collected for further measurements. Two different temperatures, 750 °C and 850 °C, are chosen to investigate the effect of the thermal reduction temperature. The obtained materials were denoted as N-MoP-750 and N-MoP-850, respectively. The diagram of the preparation process was described in Fig. 1a.
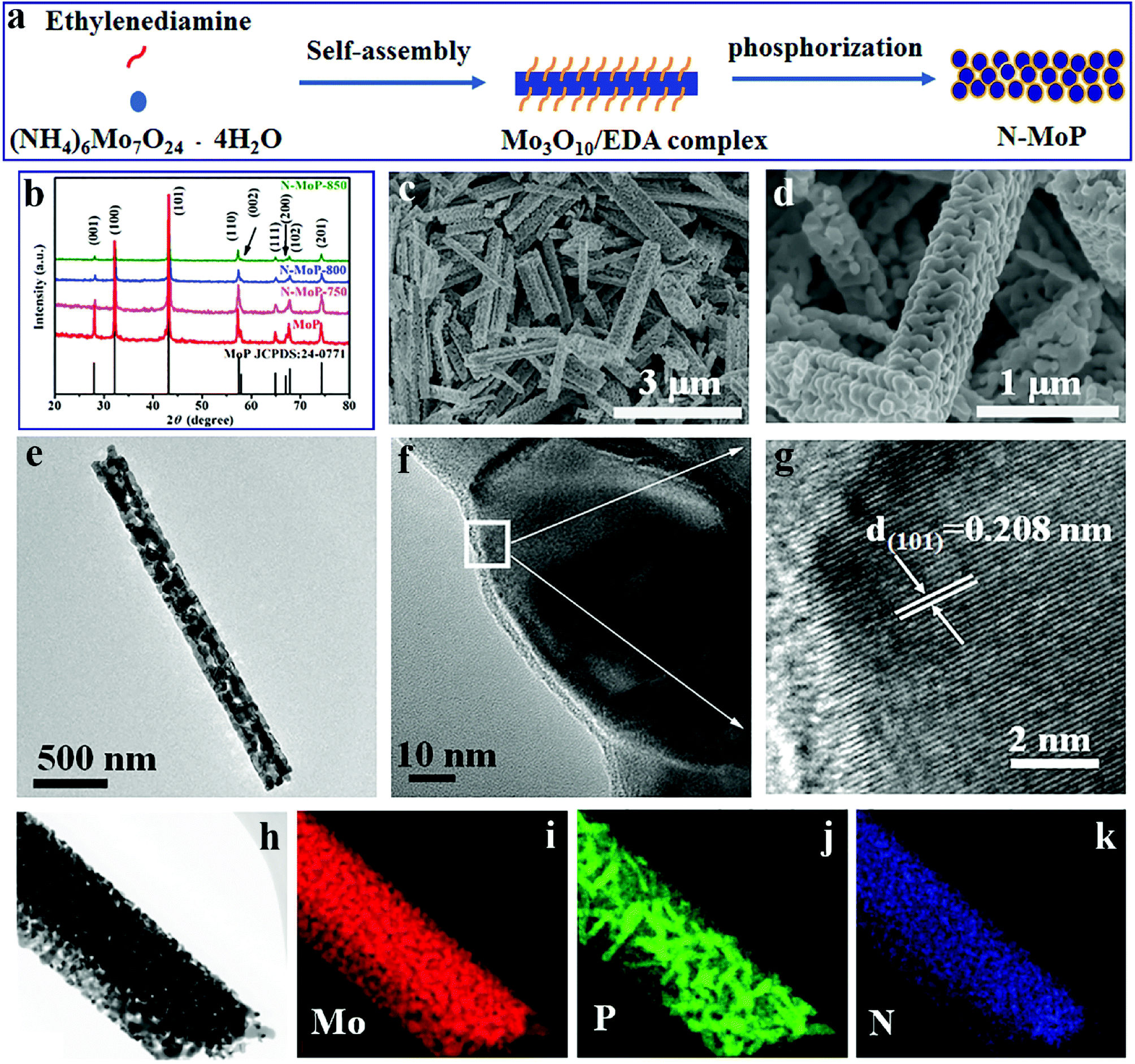 | ||
| Fig. 1 (a) Schematic illustration of the formation process of N-MoP; (b) XRD patterns of MoP, N-MoP-750, N-MoP-800 and N-MoP-850; (c and d) FESEM images of N-MoP-800 with different resolutions; (e and f) TEM images and (g) HRTEM image of a part of N-MoP-800 in (f); (h–k) elemental mapping of Mo, P, N of N-MoP-800. | ||
2.4 Synthesis of MoP
First, the MoO3 product was obtained via calcination of Mo3O10/EDA precursor in air atmosphere at 500 °C for 5 h with a heating rate of 5 °C min−1. And then, the MoP was synthesized with the same molar ratios of the precursors (Mo/P = 1:2) at 800 °C under Ar/H2 (10%) for 120 min with a heating rate of 5 °C min−1.
2.5 Characterization
The phase composition and crystallization of the products were recorded using a diffractometer (XRD, Bruker AXS-D8) with Cu Kα radiation at a scanning step of 0.03° s−1 over the 2θ range from 10° to 80°. The morphologies of the samples were observed by field emission scanning electron microscopy (FE-SEM, SU-8010, Japan), while the information on lattice and fringe were investigated by high-resolution transmission electron microscopy (HRTEM, FEI Tecnai G2 F20). X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe II XPS) was used to analyze the composition and valence states (a Monochromated Al Kα X-ray source, 1486.6 eV).2.6 Electrochemical measurements
The HER activity was studied by a conventional three-electrode method on a Princeton Applied Research Studio P4000 electrochemical workstation (AMETEK Scientific Instruments) at room temperature. A Pt foil, Ag/AgCl (3.0 M KCl) were used as the counter and reference electrodes, respectively. A glass carbon electrode (GCE: diameter = 3 mm) fabricated by drop-casting the catalyst ink was used as the working electrode. 5 mg of catalyst and 100 μL of 5 wt% Nafion solution were dispersed in 900 μL ethanol by at least 30 min sonication, forming a homogeneous ink. Then 4.3 μL of the catalyst ink (containing 21.5 μg of catalyst) was covered on the GCE (loading amount ∼ 0.3 mg cm−2) with a micropipette and dried naturally in air for test. In all measurements, the Ag/AgCl reference electrode was calibrated with respect to a reversible hydrogen electrode (RHE) by adding a value of (0.21 + 0.059 pH) V, and all the potentials reported in our work were vs. RHE. Linear sweep voltammetry (LSV) were conducted in 0.5 M H2SO4 (pH = 0.3), 1 M KOH (pH = 14) and 1 M PBS (pH = 7) at a scan rate of 5 mV s−1. Electrochemical impedance spectroscopy (EIS) analysis was adopted to provide further insight into the electrical conductivity of electrocatalysts in the frequency range of 105 to 0.1 Hz under the amplitude of 10 mV.
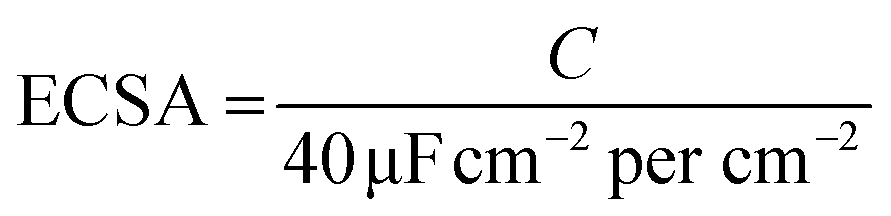 | (1) |
The electrochemical active surface area (ECSA) can be estimated using the capacitance (C). The following formula was used to calculate ECSA.42 To calculate the C values, cyclic voltammetry (CV) curves were measured under different scan rates that from 10 to 100 mV s−1 in the potential range of 0.1–0.3 V (vs. RHE). The durability test was carried out at a static overpotential at a cathodic current density of 10 mA cm−2 for 12 h.
3. Results and discussion
Fig. 1b shows the XRD pattern of the as-prepared sample when the ratio of Mo to P is 1:2. The prepared sample can be assigned to pure phase MoP (JCPDS no. 24-0771), which displays well-resolved diffraction peaks at 2θ = 27.9°, 32.2°, 43.1°, 57.4°, 64.9°, 67.8° and 74.3°, corresponding to the (001), (100), (101), (110), (111), (102) and (201) planes, respectively.43 The XRD patterns display sharp peaks and no impurity peaks can be detected, indicating good crystallization of MoP.
The FESEM images (Fig. 1c and d) show that the N-MoP-800 displays well-defined one-dimensional nanorods structure, meanwhile many bulges and hollows form a porous-like structure on their surfaces. The average diameters and lengths of those nanorods are about 350 nm and 3.2 μm, respectively. A closer observation of N-MoP-800 is provided by TEM and HRTEM testing, as shown in Fig. 1e–g. The nanorods containing numerous nanoparticles can be clearly observed in Fig. 1e. The average diameter of MoP nanoparticles is about 60 nm, as shown in Fig. 1f; it was found that there are obvious humps on the surface of MoP, as shown in Fig. 1d. The HRTEM image in Fig. 1g shows the lattice fringes with a d-spacing of 0.208 nm, which is assigned to (101) plane of MoP and consistent with the XRD result. The elemental mapping images (Fig. 1h–k) indicate that Mo, P and N elements are uniformly distributed in N-MoP-800 sample. It was noted that the reaction temperature is an important parameter that can influence the morphology and/or microstructure of N-MoP catalyst. The results found that N-MoP-750 and N-MoP-850 display similar rod-like morphology with N-MoP-800, but N-MoP-750 exhibits smaller humps, while N-MoP-850 exhibits nearly the same humps on their surfaces with N-MoP-800, details are shown in Fig. S1–S4.† And the HRTEM images in Fig. S2c and 2f† also show the lattice fringes with a d-spacing of 0.213 nm and 0.211 nm for N-MoP-750 and N-MoP-850, respectively. Therefore, all the above characterization results demonstrate that the N-MoP materials were successfully prepared by a simple two-step synthetic strategy.
To evaluate the surface elements and their valence states of N-MoP, XPS was carried out. The XPS survey spectrum of the N-MoP-800 sample is shown in Fig. 2a. The existence of Mo, P and N elements can be clearly observed, which is consistent with the elemental mapping results in Fig. 1h–k. Fig. 2b–d show high resolution XPS profiles of Mo 3d, P 2p and N 1s of the as-synthesized N-MoP-800 samples. As shown in Fig. 2b, the peaks at 231.97 eV/228.30 eV (3d3/2/3d5/2) generally can be assigned to Mo–P bond, which agrees well with the previous reports.44,45 In addition, the peaks at 235.84 eV/233.11 eV (Mo6+ 3d3/2/3d5/2) and 232.53 eV/228.70 eV (Mo4+ 3d3/2/3d5/2) can be assigned to high oxidation state of Mo (MoO3 and MoO2), which is caused by the surface oxidation during the passivation process.46,47 This passivation process is necessary to prevent the pyrophoric oxidation of MoP when it was exposed in air.48,49 Moreover, the XPS spectrum of P 2p displays the peaks at a high binding energy of 134.60 eV/133.83 eV due to the presence of surface PO43− or P2O5.50 Meanwhile, two peaks at 130.62 eV and 129.78 eV, which are attributed to the low valence P in MoP (Fig. 2c).32,46 The N 1s XPS spectrum (Fig. 2d) reveals that the existence of the pyridinic N (397.68 eV), pyrrolic N (399.03 eV) and graphitic N (401.78 eV); in addition, the peak at 394.2 eV can be assigned to N-Mo bond.34 Similar XPS results for N-MoP-750 and N-MoP-850 can be obtained, as shown in Fig. S5 and S6.†
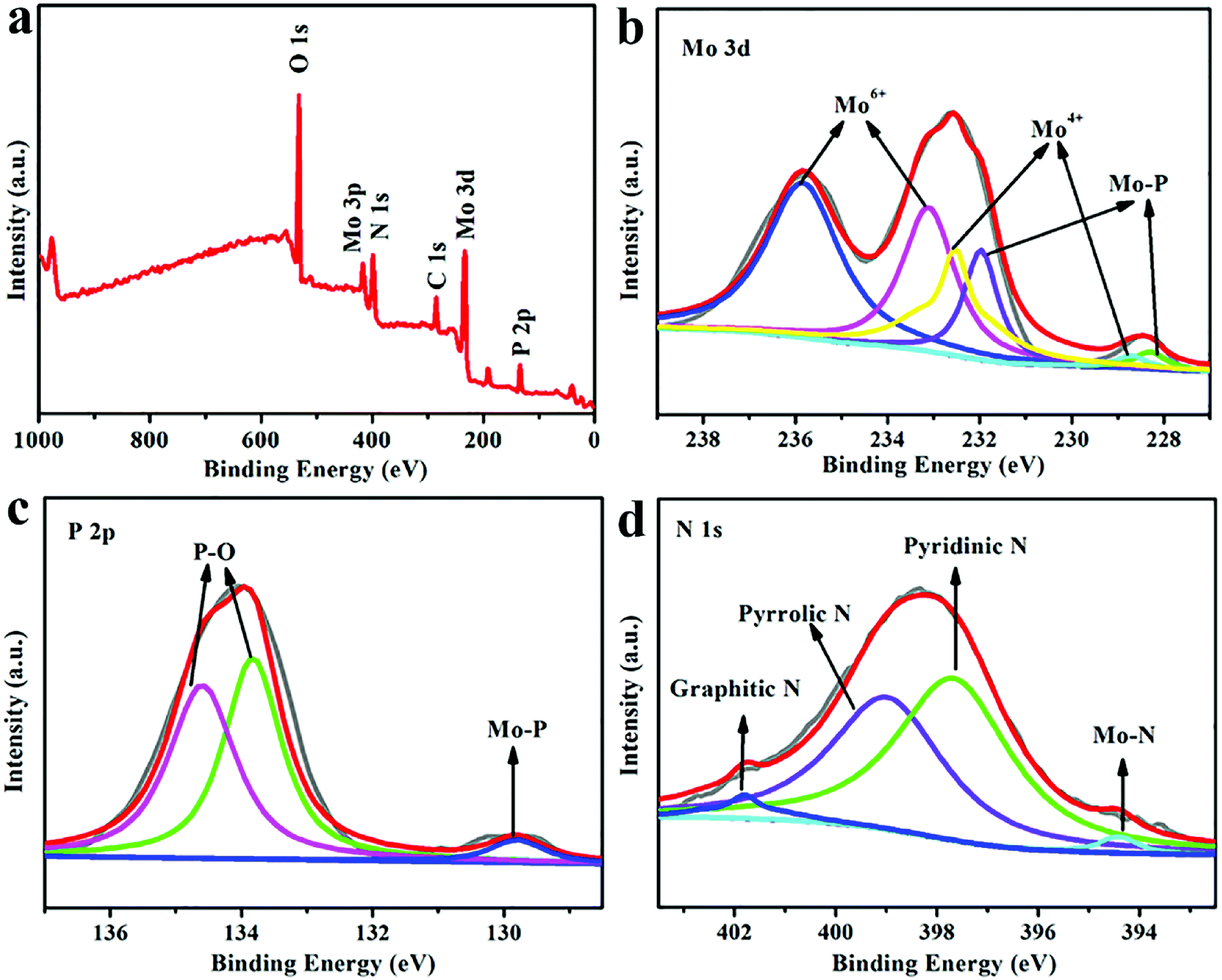 | ||
| Fig. 2 (a) Wide-scan survey XPS spectrum of N-MoP-800. (b–d) High-resolution XPS signals of (b) Mo 3d, (c) P 2p, and (d) N 1s of N-MoP-800. | ||
The electrocatalytic activities of the as-synthesized catalysts for the HER were evaluated in a wide pH range. For comparison, commercial 20 wt% Pt/C was also studied under identical conditions. A pure MoP without N doping which was prepared via the calcination of Mo3O10/EDA precursor in air atmosphere under 500 °C and a following phosphatization process at 800 °C under Ar/H2 atmosphere, was used to show the effects of N doping. No N component was observed from the XPS spectra and EDS results, suggesting the successful synthesis of pure MoP without N doping (Fig. S7†). It was proved that MoP without nitrogen doping was successfully prepared by XPS and EDS (Fig. S7†). All tested samples were deposited on a GCE with the same loading amount of 0.3 mg cm−2. Wherein, the polarization curves of different samples in 0.5 M H2SO4 were shown in Fig. 3a. It was found that N-MoP-800 exhibited optimal activity with the lowest onset overpotential among these samples of MoP (125 mV), N-MoP-750 (68 mV), N-MoP-800 (65 mV) and N-MoP-850 (95 mV). The overpotential (η) at j = 10 mA cm−2 for the N-MoP-800 is 136 mV, which is lower than the other three samples of MoP (207 mV), N-MoP-750 (148 mV) and N-MoP-850 (163 mV). It was believed that the excellent catalytic activity of N-MoP for HER could be attributed to good conductivity, enhanced active sites and modulated surface architectures after N doping.51,52
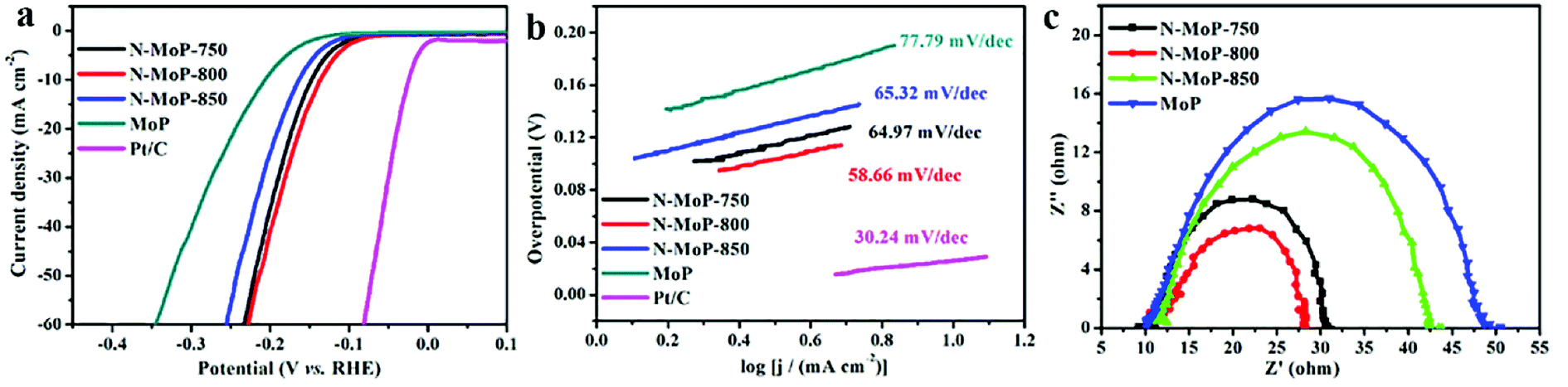 | ||
| Fig. 3 HER performance of the as-prepared materials in 0.5 M H2SO4, (a) polarization curves; (b) Tafel plots for MoP, N-MoP-750, N-MoP-800, N-MoP-850 and Pt/C; (c) Nyquist plots of electrochemical impedance spectra (EIS) of MoP, N-MoP-750, N-MoP-800 and N-MoP-850 recorded at a voltage of −172 mV vs. RHE. | ||
Tafel slope is commonly used to reveal the reaction mechanism and inherent properties of the catalysts for HER. As we know that, for HER in acidic media, the reaction path is mainly composed of proton adsorption and reduction on the catalyst surface to form Hads, followed by H2 formation and desorption.30,53 These include the Volmer mechanism (electrochemical hydrogen adsorption), the Heyrovsky mechanism (electrochemical hydrogen desorption) and the Tafel mechanism (chemical desorption), detailed as follows:
| Volmer reaction: cat + H3O+ + e− → cat|Hads + H2O | (2) |
| Heyrovsky reaction: cat|Hads + H3O+ + e− → cat + H2O + H2↑ | (3) |
| Tafel reaction: 2 cat|Hads → 2 cat + H2↑ | (4) |
According to the classical theory on hydrogen evolution mechanism, the Tafel, Volmer or Heyrovsky slopes can be the rate determining step when the Tafel slopes are of about 30, 40 or 120 mV dec−1, respectively. The Tafel plots for MoP, N-MoP-750, N-MoP-800 and N-MoP-850 are shown in Fig. 3b, their corresponding values are measured to be 77.79, 64.97, 58.66 and 65.32 mV dec−1, respectively. Therefore, the Tafel slopes for all MoP-based catalysts reveal that the HER proceeds a Volmer–Heyrovsky mechanism, and the Heyrovsky (electrochemical desorption) could become the rate determining step.44,45,54 A smaller Tafel slope would lead to a remarkably higher HER rate with a low overpotential.
The enhancement in catalytic performance of N-MoP-800 can be ascribed to the three main factors: (i) nitrogen doping could generate more active sites and accelerate electron transfer; (ii) the humps and edges on the surface of the N-MoP-800 favor the exposure of more active sites for HER;38,40 (iii) The pyridinic N components provide more active sites for HER, further enhanced the activity. According to previous studies, the pyridinic N components can service as the active sites for catalytic reaction. 42,55–57To show the role of pyridinic N components, the ratios of pyridinic N components in these samples (the content of pyridinic N components/the content of all N-contained groups) were calculated. The N-MoP-800 has higher proportion pyridinic N components (52.18%) than N-MoP-750 (23.07%) and N-MoP-850 (43.37%), which can provide more active sites, and therefore enhance catalytic activity of N-MoP for HER. Moreover, N-MoP-800 possess larger amount of humps on the surface compared with N-MoP-750 and N-MoP-850, these obvious humps would contribute high active area that can effectively improve electron transfer and provide desirable active sites for pyridinic N components. As a result, the N-MoP-800 displays optimal HER performances among these samples.
In order to elucidate these reasons, further insight into the electrical conductivity of catalysts by ECSA and EIS analysis, so the double-layer capacitor (Cdl) were investigated. As shown in Fig. 3c, the N-MoP-800 has the smallest value of charge transfer resistance (29 Ω), which indicates that N-MoP-800 has faster electron transfer rate compared to that of MoP (52Ω), N-MoP-750 (32 Ω) and N-MoP-850 (43 Ω). The ECSA of N-MoP-800 was investigated using the Cdl. The results are shown in Fig. 4, the N-MoP-800 has a much higher Cdl (3.72 mF cm−2) and ECSA (93.00 cm2), whereas N-MoP-750 has a relative smaller Cdl (2.63 mF cm−2) and ECSA (65.75 cm2), N-MoP-850 has the smallest Cdl (2.12 mF cm−2) and ECSA (53.00 cm2). Herein, the high ECSA of N-MoP-800 is mainly resulted from the large amounts of humps on the surface. These results fully demonstrate that the surface engineering-modulated microstructure (ca., doping and humps) has dramatic effect on enhancing the HER activity of MoP.
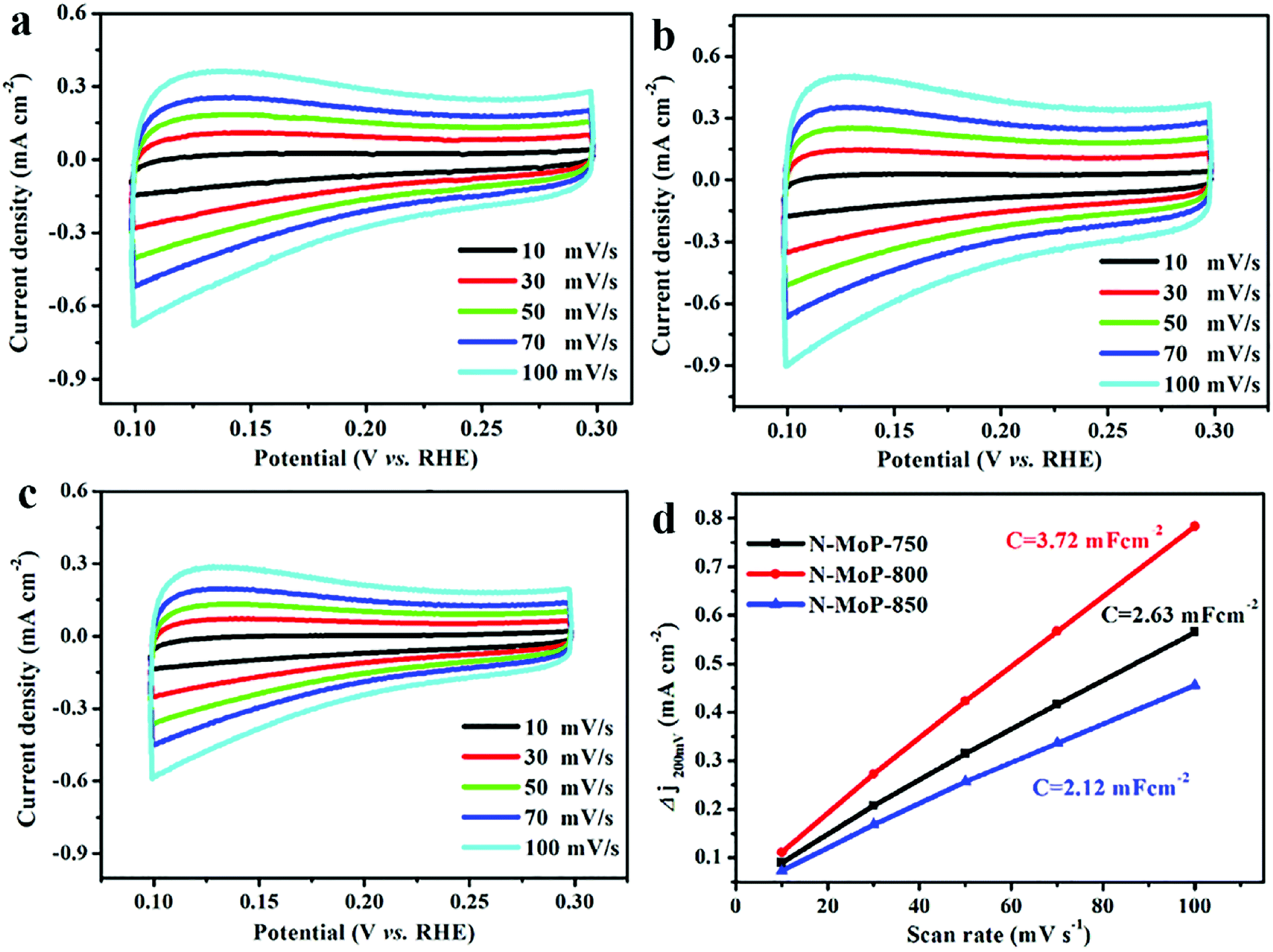 | ||
| Fig. 4 CV curves for (a) N-MoP-750; (b) N-MoP-800 and (c) N-MoP-850 at different rates from 10 to 100 mV s−1 in 0.5 M H2SO4; (d) the relationship curve between capacitive current and scan rate for N-MoP-750, N-MoP-800 and N-MoP-850 at 0.2 V (Δj = ja − jc). | ||
The long-term electrochemical durability is also a critical parameter for HER catalyst. Fig. 5a shows that the polarization curve of N-MoP-800 do not change after 1000 CV cycles at a scan rate of 50 mV s−1. Furthermore, the stability of this catalyst was also evaluated by electrolysis at a constant overpotential of 140 mV, and we also found that the current density only undergoes a slight decrease after 12 h testing (Fig. 5b). This is probably due to the specific 1D aggregate structure and rich N-doping on the N-MoP surface.
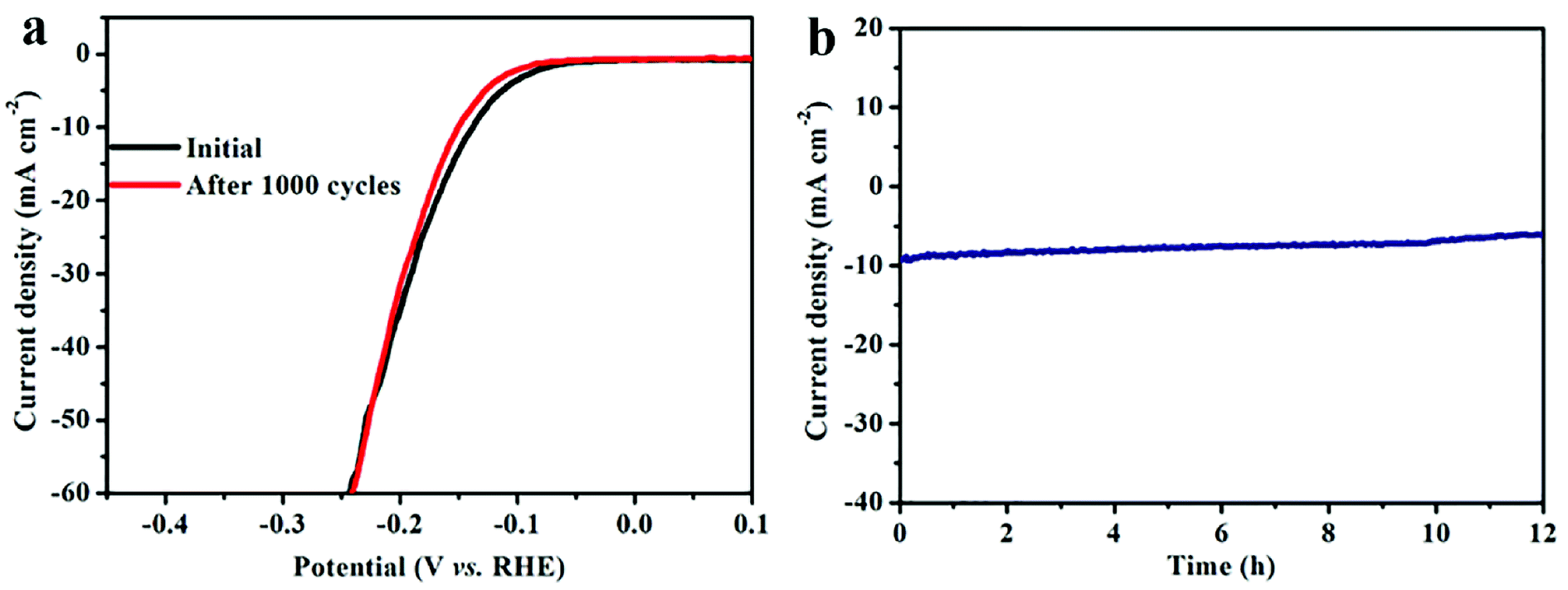 | ||
| Fig. 5 (a) The polarization curves of N-MoP-800 before and after 1000 cycle testing; (b) the relationship plot of time dependence of current density for N-MoP-800 at a static overpotential of −142 mV for 12 h in 0.5 M H2SO4. | ||
The catalytic activity of N-MoP was further conducted in 1 M KOH. Generally, the electrocatalytic HER activity in alkaline media requires more energy to generate H+, which leads to the phenomenon that many catalysts with high activity in acid solution suffer from large activity decay in alkaline solution. It was noted that, the onset potential of MoP, N-MoP-800 and N-MoP-850 are about 157 mV, 70 mV and 84 mV, respectively. And the current density can approach 10 mA cm−2 at an overpotential of 293 mV, 157 mV and 156 mV for MoP, N-MoP-800 and N-MoP-850, respectively. N-MoP-750, exhibits a lower onset potential of 68 mV, and achieves a current density of 10 mA cm−2 at an overpotential of 145 mV (Fig. 6a). The above results show that the N-MoP-750 has relatively high catalytic activity for HER in alkaline media. One suggests that high catalytic activity of N-MoP-750 may be associated with high proportion N-Mo component at sample surface. The formed N-Mo component can optimal adsorption OH− and hence exposure more active sites to boost HER performance in alkaline medium.58 Fig. 6b shows the Tafel slopes for Pt/C, MoP, N-MoP-750, N-MoP-800, and N-MoP-850 are about 58.41, 132.69, 71.15, 78.52 and 71.87 mV dec−1, respectively, which suggests that the Volmer–Heyrovsky mechanism also takes effect on the HER process. To assess the durability of N-MoP-750, accelerated linear potential sweeps were conducted repeatedly on the electrodes. As shown in Fig. 6c, N-MoP-750 exhibits a continuous but small loss of activity before and after 1000 CV cycles, which implies minor corrosion of catalyst in alkaline condition. In addition, it was seen that current density of N-MoP-750 do not take obviously decaying during 12 h cyclic testing, implying high stability of N-MoP-750 (Fig. 6d). Taken together, the enhancement of HER performance of N-MoP-750 in alkaline medium can be ascribed to the two main factors: (i) the formation of high proportion Mo-N (49.87%) component on the surface can provide more active sites in alkaline medium that enhance catalytic dynamics for HER and favor their structural stability; (ii) the humps and edges on the surface of N-MoP-750 can contribute high active areas (ECSA ∼ 62.3 cm2) and further accelerate electron transfer (Fig. S8†), which are responsible for the enhanced HER performance in alkaline solution. For comparison, the catalytic activity in HER of the as-prepared N-MoP nanorod and previously reported MoP-based electrocatalysts are listed in Table S1.† From Table S1,† it was seen that the HER performances of N-MoP are totally higher than most reported results. Based on the above analysis and discussions, it was concluded that desirable design of surface defects (N-doping) and humps on the surface play a key role in enhancing HER performances of N-MoP in both acidic and alkaline medium.
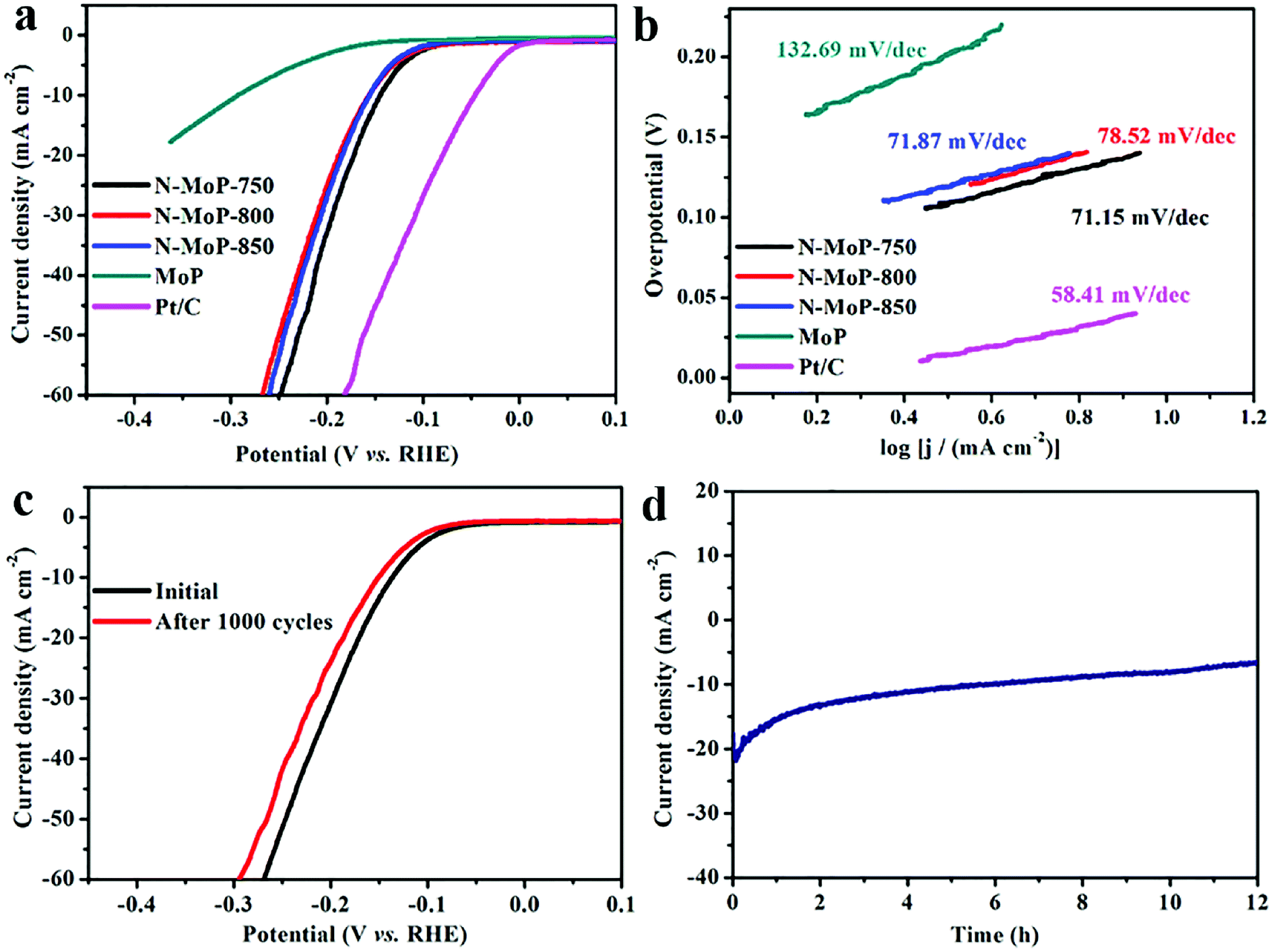 | ||
| Fig. 6 HER performance of the prepared MoP-based materials in 1 M KOH, (a) polarization curves; (b) Tafel plots for MoP, N-MoP-750, N-MoP-800, N-MoP-850 and Pt/C; (c) the polarization curves of N-MoP-750 before and after 1000 cycles. (d) The time dependence of the current density for N-MoP-750 at a static overpotential of −154 mV for 12 h. | ||
4. Conclusions
In summary, we reported a 1D porous N-MoP nanorod structure synthesized through a facile solution reaction, following by a calcination process. The prepared N-MoP exhibited high HER catalytic activities and good stability over a wide pH range. The high HER performance of N-MoP were attributed to the high electrical conductivity, more active sites caused by the N-doping and surface architectures. Our research proves that the N-MoP is a potential candidate for HER and provide a novel, eco-friendly and low-cost method to synthesize other 1D phosphides catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding support from the National Science Foundation of China (NSFC, No. 217031707), the State Key Laboratory of Solidification Processing in NWPU (No. SKLSP201613, and SKLSP201845).References
- V. S. Thoi, Y. J. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- Y. Q. Qu and X. F. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- C. G. Morales-Guio, L. A. Stern and X. L. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed.
- D. Merki and X. L. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 RSC.
- D. Voiry, H. Yamaguchi, J. W. Li, R. Silva, D. C. B. Alves, T. Fujita, M. W. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef PubMed.
- S. Mandegarzad, J. B. Raoof, S. R. Hosseini and R. Ojani, Electrochim. Acta, 2016, 190, 729–736 CrossRef.
- D. Y. Wang, M. Gong, H. L. Chou, C. J. Pan, H. A. Chen, Y. P. Wu, M. C. Lin, M. Y. Guan, J. Yang, C. W. Chen, Y. L. Wang, B. J. Hwang, C. C. Chen and H. J. Dai, J. Am. Chem. Soc., 2015, 137, 1587–1592 CrossRef PubMed.
- T. Wu, M. L. Stone, M. J. Shearer, M. J. Stolt, I. A. Guzei, R. J. Hamers, R. F. Lu, K. M. Deng, S. Jin and J. R. Schmidt, ACS Catal., 2018, 8, 1143–1152 CrossRef.
- S. Tian, X. Li, A. J. Wang, R. Prins, Y. Y. Chen and Y. K. Hu, Angew. Chem., Int. Ed., 2016, 55, 4030–4034 CrossRef PubMed.
- J. Q. Yan, H. Wu, P. Li, H. Chen, R. B. Jiang and S. Z. Liu, J. Mater. Chem. A, 2017, 5, 10173–10181 RSC.
- M. J. Liu, L. M. Yang, T. Liu, Y. H. Tang, S. L. Luo, C. B. Liu and Y. X. Zeng, J. Mater. Chem. A, 2017, 5, 8608–8615 RSC.
- X. Liang, D. Z. Zhang, Z. Z. Wu and D. Z. Wang, Appl. Catal., A, 2016, 524, 134–138 CrossRef.
- J. Q. Chi, W. K. Gao, J. H. Lin, B. Dong, K. L. Yan, J. F. Qin, Z. Z. Liu, Y. M. Chai and C. G. Liu, J. Colloid Interface Sci., 2018, 513, 151–160 CrossRef PubMed.
- M. Cabán-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef PubMed.
- X. Guo, G. L. Cao, F. Ding, X. Y. Li, S. Y. Zhen, Y. F. Xue, Y. M. Yan, T. Liu and K. N. Sun, J. Mater. Chem. A, 2015, 3, 5041–5046 RSC.
- Z. H. Pu, S. Y. Wei, Z. B. Chen and S. C. Mu, RSC Adv., 2016, 6, 11077–11080 RSC.
- F. X. Ma, H. B. Wu, B. Y. Xia, C. Y. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 15395–15399 CrossRef PubMed.
- L. Liao, S. N. Wang, J. J. Xiao, X. J. Bian, Y. H. Zhang, M. D. Scanlon, X. L. Hu, Y. Tang, B. H. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 RSC.
- J. F. Xie, S. Li, X. D. Zhang, J. J. Zhang, R. X. Wang, H. Zhang, B. C. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- B. Qu, X. B. Yu, Y. J. Chen, C. L. Zhu, C. Y. Li, Z. X. Yin and X. T. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 14170–14175 CrossRef PubMed.
- H. Park, A. Encinas, J. P. Scheifers, Y. M. Zhang and B. P. T. Fokwa, Angew. Chem., Int. Ed., 2017, 56, 5575–5578 CrossRef PubMed.
- J. Zhang, T. Wang, P. Liu, Z. Q. Liao, S. H. Liu, X. D Zhuang, M. W. Chen, E. Zschech and X. L. Feng, Nat. Commun., 2017, 8, 15437 CrossRef PubMed.
- W. X. Zhu, C. Tang, D. N. Liu, J. L. Wang, A. M. Asiric and X. P. Sun, J. Mater. Chem. A, 2016, 4, 7169–7173 RSC.
- G. Zhang, G. C. Wang, Y. Liu, H. J. Liu, J. H. Qu and J. H. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef PubMed.
- L. N. Zhang, S. H. Li, H. Q. Tan, S. U. Khan, Y. Y. Ma, H. Y. Zang, Y. H. Wang and Y. G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16270–16279 CrossRef PubMed.
- Z. X. Wu, J. W, J. Zhu, J. P. Guo, W. P. Xiao, C. J. Xuan, W. Lei and D. L. Wang, Electrochim. Acta, 2017, 232, 254–261 CrossRef.
- J. Yang, F. J. Zhang, X. Wang, D. S. He, G. Wu, Q. H. Yang, X. Hong, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2016, 128, 13046–13050 CrossRef.
- A. K. Sun, Y. L. Shen, Z. Z. Wu and D. Z. Wang, Int. J. Hydrogen Energy, 2017, 42, 14566–14571 CrossRef.
- Q. W. Zhou, Z. H. Shen, C. Zhu, J. C. Li, Z. Y. Ding, P. Wang, F. Pan, Z. Y. Zhang, H. X. Ma, S. Y. Wang and H. G. Zhang, Adv. Mater., 2018, 1800140 CrossRef PubMed.
- T. R. Kuo, W. T. Chen, H. J. Liao, Y. H. Yang, H. C. Yen, T. W. Liao, C. Y. Wen, Y. C. Lee, C. C. Chen and D. Y. Wang, Small, 2017, 13, 1603356 CrossRef PubMed.
- T. Ling, D. Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. L. Zheng, J. Mao, X. W. Du, Z. P. Hu, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef PubMed.
- J. Yin, Y. J. Qiu, J. Yu, X. S. Zhou and W. H. Wu, RSC Adv., 2013, 3, 15655–15663 RSC.
- J. Q. Chi, K. L. Yan, W. K. Gao, B. Dong, X. Shang, Y. R. Liu, X. Li, Y. M. Chai and C. G. Liu, J. Alloys Compd., 2017, 714, 26–34 CrossRef.
- R. C. Li, L. J. Yang, T. L. Xiong, Y. S. Wu, L. D. Cao, D. S. Yuan and W. J. Zhou, J. Power Sources, 2017, 356, 133–139 CrossRef.
- Y. Zhao, S. Wang, C. Y. Li, X. B. Yu, C. L. Zhu, X. T. Zhang and Y. J. Chen, RSC Adv., 2016, 6, 7370–7377 RSC.
- Q. S. Gao, S. N. Wang, H. C. Fang, J. W. Weng, Y. H. Zhang, J. J. Mao and Y. Tang, J. Mater. Chem., 2012, 22, 4709–4715 RSC.
- Y. Y. Chen, Y. Zhang, W. J. Jiang, X. Zhang, Z. H. Dai, L. J. Wan and J. S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef PubMed.
- Z. H. Pu, I. S. Amiinu, X. B. Liu, M. Wang and S. C. Mu, Nanoscale, 2016, 8, 17256–17261 RSC.
- A. P. Wu, C. G. Tian, H. J. Yan, Y. Q. Jiao, Q. Yan, G. Y. Yang and H. G. Fu, Nanoscale, 2016, 8, 11052–11059 RSC.
- Z. H. Pu, S. Y. Wei, Z. B. Chen and S. C. Mu, Appl. Catal., B, 2016, 196, 193–198 CrossRef.
- S. M. Yin, J. Y. Han, Y. J. Zou, T. H. Zhou and R. Xu, Nanoscale, 2016, 8, 14438–14447 RSC.
- C. Deng, J. Z. Xie, Y. F. Xue, M. He, X. T. Wei and Y. M. Yan, RSC Adv., 2016, 6, 68568–68573 RSC.
- Q. D. Yue, Y. Y. Wan, Z. J. Sun, X. J. Wu, Y. P. Yuan and P. W. Du, J. Mater. Chem. A, 2015, 3, 16941–16947 RSC.
- P. Xiao, M. A. Sk, L. Thi, X. M. Ge, R. J. Lim, J. Y. Wang, K. H. Lima and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- D. Z. Wang, Y. L. Shen, X. Y. Zhang and Z. Z. Wu, J. Mater. Sci., 2017, 52, 3337–3343 CrossRef.
- R. Jiang, J. H. Fan, L. Y. Hu, Y. P. Dou, X. H. Mao and D. H. Wang, Electrochim. Acta, 2018, 261, 578–587 CrossRef.
- J. Jia, T. L. Xiong, L. L. Zhao, F. L. Wang, H. Liu, R. Z. Hu, J. Zhou, W. J. Zhou and S. W. Chen, ACS Nano, 2017, 11, 12509–12518 CrossRef PubMed.
- J. G. N. Thomas, Trans. Faraday Soc., 1961, 57, 1603–1611 RSC.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef PubMed.
- W. L. Li, B. Herkt, M. Seredych and T. J. Bandosz, Appl. Catal., B, 2017, 207, 195–206 CrossRef.
- M. Sun, X. B. Wu, Z. Y. Xie, X. T. Deng, J. Y. Wen, Q. Z. Huang and B. Y. Huang, Carbon, 2017, 125, 401–408 CrossRef.
- L. J. Li, P. C. Dai, X. Gu, Y. Wang, L. T. Yan and X. B. Zhao, J. Mater. Chem. A, 2017, 5, 789–795 RSC.
- I. S. Amiinu, Z. H. Pu, X. B. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. N. Zhang and S. C. Mu, Adv. Funct. Mater., 2017, 27, 1702300 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03909g |
| This journal is © The Royal Society of Chemistry 2018 |