
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new solvothermal method for the synthesis of size-controlled YAG:Ce single-nanocrystals†
Géraldine Dantelle *a,
Denis Testemalea,
Estelle Homeyerb,
Alexandra Cantaranoa,
Stéphanie Kodjikiana,
Christophe Dujardinb,
Jean-Louis Hazemanna and
Alain Ibaneza
*a,
Denis Testemalea,
Estelle Homeyerb,
Alexandra Cantaranoa,
Stéphanie Kodjikiana,
Christophe Dujardinb,
Jean-Louis Hazemanna and
Alain Ibaneza
aUniv. Grenoble Alpes, CNRS, Grenoble INP, Institut Néel, 38000 Grenoble, France. E-mail: geraldine.dantelle@neel.cnrs.fr
bInstitut Lumière Matière, Univ. Lyon 1 – CNRS – 10, Rue Ada Byron, 69 622 Villeurbanne Cedex, France
First published on 27th July 2018
Abstract
Ce3+-doped Y3Al5O12 (YAG:Ce) nanocrystals were synthesized by a unique solvothermal method, under sub-critical conditions. A home-made autoclave was used, operating in a larger pressure and temperature range than that with conventional commercial equipment and allowing direct in situ photoluminescence (PL) and X-ray absorption characterizations. The study of various synthesis conditions (pressure, temperature, precursor concentration, reaction time) allowed the best reaction conditions to be pinpointed to control YAG:Ce nanocrystal size, as well as crystal quality, and to get efficient optical properties. Without any post thermal treatment, we succeeded in obtaining well-crystallized YAG:Ce nanocrystals (30–200 nm), displaying typical PL properties of YAG:Ce with a maximal emission at 550 nm. The pristine 100 nm-sized YAG:Ce nanoparticles present an internal quantum yield of about 40 ± 5%. In situ X-ray absorption near edge spectroscopy demonstrates the presence of Ce4+ in nanocrystals elaborated at high temperature, resulting from the oxidation of Ce3+ during the crystallization process.
1. Introduction
Y3Al5O12 (YAG) crystal has been widely studied at the macroscopic scale for its high chemical stability, its robustness against radiation (optical and ionizing) and its ability to incorporate luminescent lanthanide ions for the development of optically active materials: bulk single-crystals for solid-state lasers,1 large transparent ceramics for high-power lasers,2,3 single-crystal fibers for optical amplification,4 etc. At the nanoscale, the enthusiasm for this material started nearly 20 years ago5 and remains very strong as rare-earth doped YAG nanoparticles have promising properties for numerous applications: nanoprobes in biology (as luminescent tracers with Ce3+-doped YAG,6 as nanothermometers with Nd3+- or Dy3+-doped YAG7,8) and nanophosphors for LED-based white lighting (Ce3+ or Tb3+-doped YAG9,10). All these applications require a strong control of the YAG crystal quality to ensure efficient optical properties, as well as a good size control with a targeted size in the 50–100 nm range. Indeed, this size range is often considered as a good size compromise for lanthanide-doped luminescent nanoparticles,11–13 allowing minimizing surface effects and optimizing light absorption while preserving a sub-micrometer size, essential for biophotonics and for light-scattering control in solid-state lighting devices. Indeed, in white LEDs, the use of such luminescent nanocrystals, coupled with nanostructured semi-conductors, should allow optimizing phosphor absorption and light extraction, enhancing the external efficiency of the whole device.YAG is a garnet-type crystal, crystallizing in the Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d space group. Typical high-temperature solid-state chemical route (5 h at 1500 °C) leads to the formation of micron-sized YAG powder.14 If the reaction temperature is too low, YAG crystallization remains incomplete and leads to the formation of other phases of the pseudo-binary Y2O3–Al2O3 diagram such as YAlO3 and Y4Al2O9.15 Considering these relatively harsh conditions, the synthesis of pure nanometer-sized YAG particles is not straightforward. Among the different synthesis techniques reported in the literature, most of them (sol–gel, combustion, coprecipitation, etc.) require a post-synthesis heat-treatment to induce YAG crystallization and obtain pure YAG phase.16,17 However, it also induces particle sintering, leading to micrometer-sized particles, which cannot be easily well-dispersed in a medium.
d space group. Typical high-temperature solid-state chemical route (5 h at 1500 °C) leads to the formation of micron-sized YAG powder.14 If the reaction temperature is too low, YAG crystallization remains incomplete and leads to the formation of other phases of the pseudo-binary Y2O3–Al2O3 diagram such as YAlO3 and Y4Al2O9.15 Considering these relatively harsh conditions, the synthesis of pure nanometer-sized YAG particles is not straightforward. Among the different synthesis techniques reported in the literature, most of them (sol–gel, combustion, coprecipitation, etc.) require a post-synthesis heat-treatment to induce YAG crystallization and obtain pure YAG phase.16,17 However, it also induces particle sintering, leading to micrometer-sized particles, which cannot be easily well-dispersed in a medium.
Solvothermal processes, consisting in a wet chemical route under mild temperature and high pressure conditions (sub- or supercritical conditions of the selected solvent), appear to be a very promising technique for the synthesis of nanomaterials.18,19 In the specific case of YAG synthesis, a sub-critical route, first reported by Inoue et al.,5 has been used to large extent and has demonstrated its capability to produce nanometer-sized YAG particles dispersible in an aqueous or alcoholic medium. Obtaining stable colloidal solutions is a strong advantage of this technique with respect to the others, as it produces individual YAG nanocrystals that can be (1) used as nanoprobes for any further development in biology and (2) shaped as nanostructured films20,21 or as scattering-control nanoceramics to obtain enhanced light extraction in solid-state lighting devices.22
In the literature, most reported solvothermal syntheses use commercial autoclaves operating at a maximal temperature of 300 °C with an autogenous pressure, which builds up as the precursor and solvent decomposition happens. Consequently, the reaction occurs under a non-stationary state (pressure increasing up to 60–70 bar upon reaction time) that is detrimental for the nucleation and growth control. Indeed, under these conditions, the size and crystal quality control is highly difficult: the as-grown nanocrystals often present a large polydispersity in size and are described as poorly crystalline, with most of the particles being polycrystalline, rising from the aggregation of primary grains.23–25 As a result, the optical properties of the obtained particles remain much poorer than those of their micron-size equivalent. In the case of cerium doping, the luminescence internal quantum yield (iQY) of the nanoparticles is around 15–20% after a solvothermal synthesis,26,27 whereas it reaches 80% in micron-sized powders.14
Several strategies have been studied to improve particle crystallinity and associated optical efficiency of YAG nanocrystals. Pradal et al. performed post-synthesis annealing treatments on Ce-doped YAG nanoparticles obtained by solvothermal synthesis.27 They show that the crystallinity improvement comes together with sintering, at annealing temperatures higher than 800 °C. It leads to enhanced iQY (from 15% to 40%) but also to the impossibility to redisperse the aggregated particles. Another strategy, used by A. Revaux et al., consists in performing an annealing treatment at 1000 °C of YAG nanoparticles previously dispersed into a silica matrix to avoid their aggregation and sintering.28 In this case, the silica matrix was removed after the thermal treatment by a hydrofluoric acid (HF) washing and the resulting YAG particles, stable in aqueous suspension, exhibited an improved crystallinity associated to a significantly higher iQY (55%). Though efficient, this multi-step process is highly time-consuming and cannot be scaled up due to the use of harmful HF. A more recent method consists in decreasing the precursor concentration and lengthening the reaction time to control more accurately nucleation and growth processes. It successfully produces well-crystallized small nanocrystals (18 ± 5 nm in diam.).29 Although the crystal quality was improved, the iQY did not exceed 25%, probably due to surface traps, numerous at this particle size. Due to temperature and pressure limitations of the commercial autoclave used in that study, larger particles could not be produced. Note that the optical efficiency of Ce-doped YAG nanocrystals could be improved through surface coating of particles, without modifying the crystal quality. For instance, R. Kasuya et al. succeeded in increasing the iQY for 21% to 38% by grafting PEG (polyethylene glycol) groups at the surface of YAG nanoparticles.26 However, in this case, only surface defects are removed, leaving the volume defects unchanged.
In this paper, we propose a new solvothermal method under sub-critical conditions to produce well-crystallized individual Ce3+-doped YAG nanocrystals. We target a crystal size in the 50–100 nm range, which is appropriate to optimize PL while controlling light scattering. The originality of our work, based on the use of a home-made autoclave, lies in two features: (1) the in situ PL and X-ray absorption spectroscopy characterizations, which allow to get a better understanding and control of the nucleation and growth processes, an essential step to master particle size and morphology30 (2) the accessible broad range of thermodynamical factors (pressure and temperature). We explored several synthesis conditions (temperature, pressure, precursor concentrations and reaction time) and discuss the relationship between these reaction conditions and particle size, morphology and optical properties.
2. Experimental work
2.1. Modified-solvothermal syntheses
1.5 mol% Ce3+-doped YAG nanocrystals (YAG:Ce) were synthesized as follow: yttrium acetate hexahydrate (99.99%, Sigma-Aldrich), cerium acetate dihydrate (99.99%, Sigma-Aldrich) and aluminium isopropoxide (98%, Sigma-Aldrich) were mixed in stoichiometric proportions with 1,4-butanediol at room temperature for 48 hours. The Al precursor concentration was varied from 0.02 mol L−1 to 0.27 mol L−1. 0.5 mL of solution was poured into a sapphire tube and placed between two pistons (one in sapphire and one in glass-like carbon). The vessel was placed into a home-made autoclave equipped with optically transparent sapphire windows.31 This autoclave was initially developed for in situ Raman and X-ray spectroscopies for the study of multi-phasic systems under high pressure and high temperature.32 Here, not only in situ photoluminescence (PL) measurements were carried out under high pressure/temperature but the synthesized powder was retrieved for further characterizations. The pressure, controlled by a He gas flux, was first increased up to the set value; then, the temperature was increased. Various synthesis conditions were explored: temperature ranging from 300 °C to 400 °C, fixed external pressure set from 60 to 400 bar and reaction time from 1 to 8 h. The small volume of the autoclave presents the advantage of a rapid heating ramp, with the targeted temperature being reached in less than 10 minutes, which is essential to generate a nucleation burst. In return, only a small volume of solution can be processed (typically 0.5 mL), leading to little quantity of synthesized YAG powder (between 2 and 8 mg according to the precursor concentrations). After the syntheses, the products were retrieved and washed several times with ethanol by successive centrifugations (7000 rpm, 5 min). For solution-based analyses, the nanocrystals were kept in ethanol and sonicated in an ultra-sound bath to ensure good dispersion. Typically, colloidal solutions with a concentration of 10 g L−1 were obtained. For powder-based characterizations, the nanocrystals were dried in air at room temperature.2.2. Structural analysis
X-ray diffraction (XRD) was performed on small quantity of powder deposited onto a silicon wafer using a Siemens D8 Advance diffractometer (λCu = 1.54056 Å, 40 mA, 40 kV) in the 2θ range 15–110°, with a step size of 0.01° and an acquisition time of 5 s per step. Powder X-ray diffraction (PXRD) patterns of YAG:Ce were analyzed by Le Bail method to determine phase purity, unit cell parameters and full-width at half maximum (FWHM) for all diffraction peaks.33 Using the Scherrer's formula,34 the coherence length was calculated. As it takes slightly different values according to the considered diffraction peaks, the one reported hereafter has been calculated using the more intense diffraction peak at 2θ = 33.3°.Samples were also characterized by Transmission Electron Microscopy (TEM, Philips CM300) performed at 300 kV equipped with a TemCam F416 TVIPS camera. The sample preparation consisted in evaporating droplets of nanoparticles strongly dispersed in ethanol onto carbon grids. For sake of clarity, in the following, the word “nanocrystals” will be used to define individual nano-objects. When these nanocrystals do aggregate, we will speak about “aggregates”. Nanocrystal size and their size polydispersity, were extracted from size histograms obtained by analyzing over 100 individual nanocrystals (aggregates were not considered) per sample using ImageJ software.35 Nanocrystal quality was evaluated by observing their atomic planes at high magnification, as well as by correlating the nanocrystal size (determined by TEM) and the coherence length (obtained from PXRD). Energy-dispersive X-ray (EDX, Brüker SDD) analysis was performed on the different samples to determine their chemical composition.
During nano-YAG syntheses, in situ X-ray Absorption Near Edge Spectroscopy (XANES) experiments were performed in high energy resolution fluorescence-detected (HERFD) mode at the Fame-UHD beamline of the European Synchrotron Radiation Facility (ESRF Grenoble) at the L3 edge of cerium. The photon energy was scanned from 5.68 keV to 5.85 keV using a Si(220) double-crystal monochromator. A helium bag was used between the autoclave, the crystal analyzer spectrometer and the detector to avoid partial beam absorption by the air. The signal was recorded with a five-Ge(331) crystal analyzer and a Vortex-Ex detector. The beam size was 300 × 100 μm2 (horizontal × vertical FWHM). The energy calibration was done using the CeO2 spectrum. The same autoclave as the one described above for the synthesis of nano-YAG was used, the only difference being here the use of a glass-like carbon cell and two glass-like carbon pistons (instead of a sapphire cell and one sapphire piston) and the Be windows. The goal of these experiments was to determine the oxidation state of cerium under different synthesis conditions to determine the best solvothermal conditions to preserve Ce3+ ions, responsible for PL. The Ce3+/Ce4+ ratio was determined by linear combination of XANES spectra of standards, i.e. nano-CeO2 as a reference for Ce4+ and Ce3+-doped YAG single-crystal or Ce(acetate)3 in 1,4-butanediol as a reference for Ce3+.36 The experimental data were analyzed using the Demeter software.37
2.3. Optical characterizations
In situ PL was recorded during the solvothermal syntheses through the optical sapphire windows of the autoclave. The excitation was performed at 514.5 nm with an Ar laser (P = 4 mW). The detection was carried out using a liquid nitrogen cooled CCD camera equipped with a T64000 Raman spectrometer. The details of this setup are given in ref. 32.PL emission and PL excitation spectroscopic studies of ex situ YAG:Ce colloidal solutions were investigated using a Safas Xenius spectrofluorometer, at room temperature.
The iQY was measured using a Hamamatsu integrating sphere under a 460 nm excitation and considering the whole emission range (500–700 nm).
PL decays were recorded on YAG:Ce powders using a 444 nm pulsed laser. The emission was selected above 500 nm by a high pass filter and further collected and amplified by a photomultiplier. The signal was then histogrammed using a multichannel counter with a resolution of 800 ps.
3. Results and discussion
3.1. Elaboration of pure YAG:Ce phase
According to XRD, garnet-type YAG phase was obtained, whatever the synthesis conditions. A typical PXRD pattern is shown on Fig. 1, with its Le Bail fit. No crystallized impurity was detected by XRD. The background at low incident angles comes from the scattering of the direct beam and is the result of the low quantity of powder used for the analysis. For all synthesized samples, the coherence length was determined from Le Bail fits of PXRD patterns. It will be systematically compared with the nanocrystal size determined by TEM to evaluate their single-crystal quality. The unit cell parameter a is worth 12.020 ± 0.005 Å for small YAG:Ce, which is consistent with the value obtained in micron-sized powder (12.012 Å![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14). However, for larger YAG:Ce particles (∼100 nm and higher), the unit cell increases (12.080 ± 0.005 Å), which suggests the presence of internal strains. This feature will be discussed hereafter (see paragraph 3.5).
14). However, for larger YAG:Ce particles (∼100 nm and higher), the unit cell increases (12.080 ± 0.005 Å), which suggests the presence of internal strains. This feature will be discussed hereafter (see paragraph 3.5).
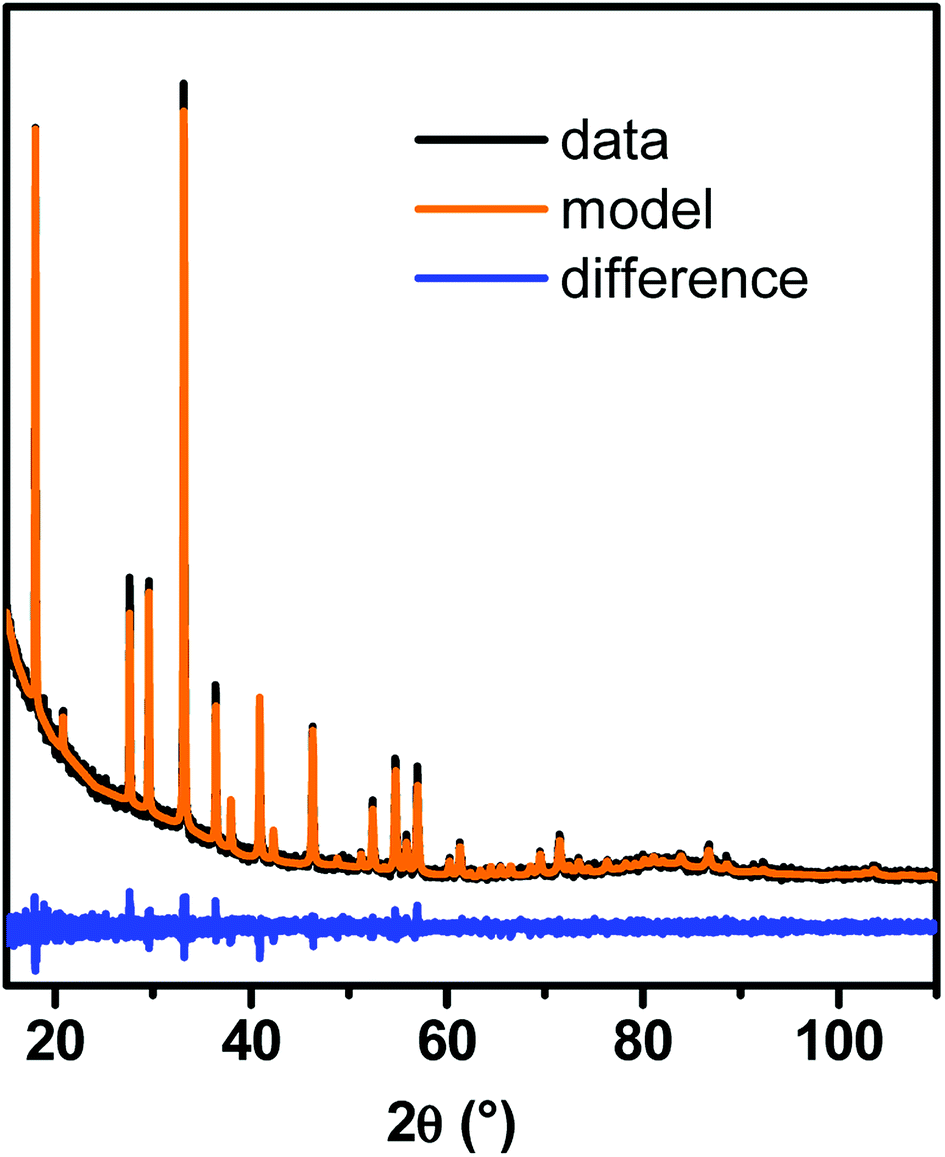 | ||
| Fig. 1 Typical PXRD pattern of YAG:Ce nanocrystals synthesized with our modified solvothermal method and its corresponding Le Bail fit. | ||
3.2. Influence of the reaction pressure on YAG nucleation step
The influence of reaction pressure was investigated during the first steps of YAG crystallization by monitoring in situ PL emission over time during the solvothermal synthesis. The reaction temperature was set at 350 °C, just above the one used in previous studies (300 °C9, 29 and 38). The precursor concentration was fixed at [Al] = 0.09 mol L−1 while the reaction pressure was varied at different values: P = 60, 100, 200 and 400 bar. The maximum PL intensity at 550 nm is reported as a function of time for P = 200 bar (Fig. 2a) and for all the different pressure values (Fig. 2b). We considered, in this experiment, that the typical emission at 550 nm reflects the YAG:Ce formation.
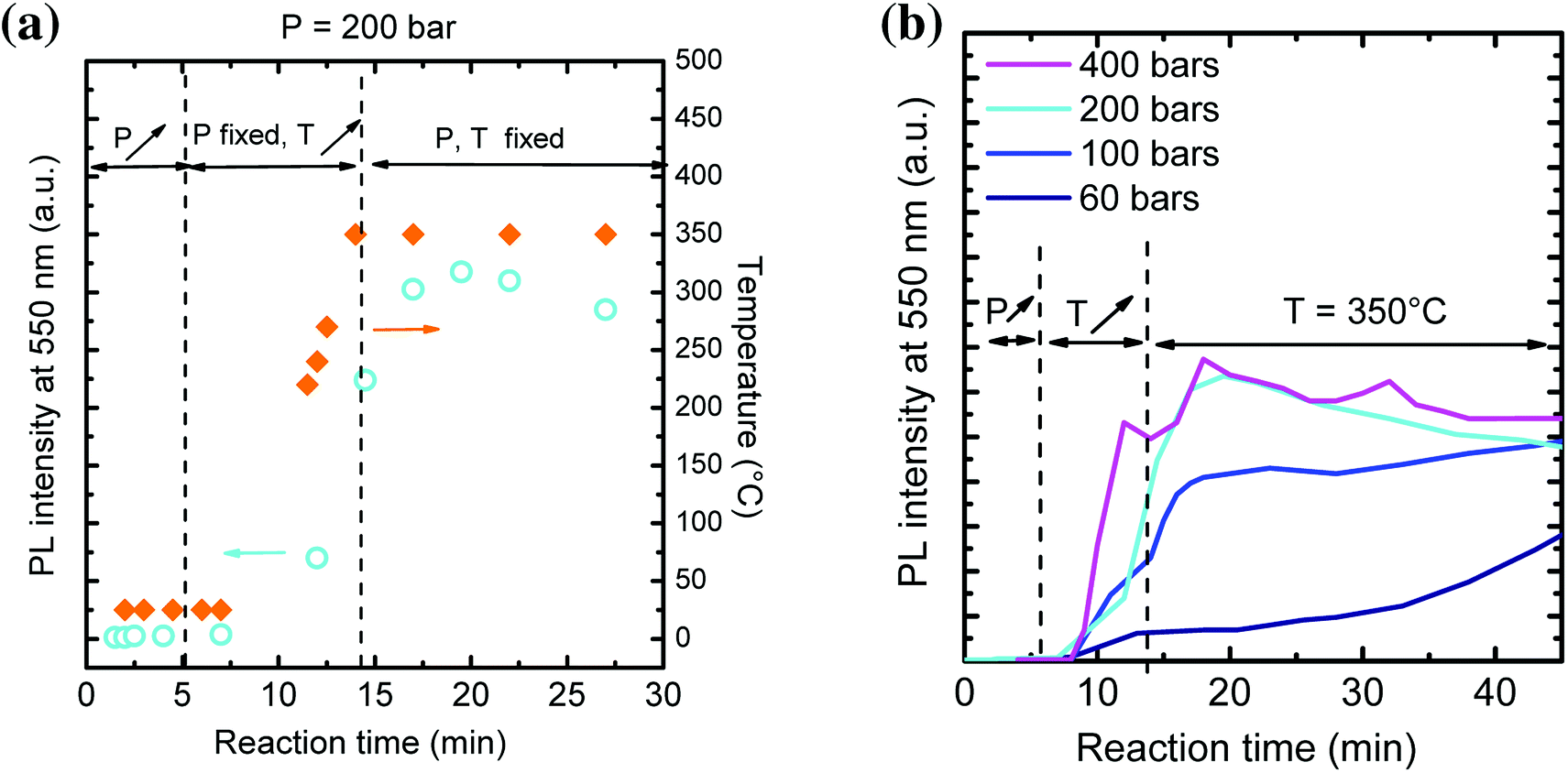 | ||
| Fig. 2 (a) Evolution of PL intensity at 550 nm (cyan circles) and temperature (orange diamonds) over time for a synthesis at P = 200 bar, T = 350 °C and [Al] = 0.09 mol L−1 During the first 5 minutes, the pressure is increased up to 200 bar, then the temperature is increased up to 350 °C. We noted that PL appears when the temperature reaches 250 °C. Finally, pressure and temperature are kept constant, within 0.5 bar and 0.1 °C respectively. (b) Evolution of PL intensity at 550 nm as a function of time for different reaction pressures, at T = 350 °C and [Al] = 0.09 mol L−1. | ||
In Fig. 2a, the PL intensity and the temperature are simultaneously plotted over time. First, pressure is increased up to 200 bar. No PL is observed at 550 nm. Temperature is then increased up to 350 °C (ESI Movie 1†). When it reaches 250 °C, PL appears, indicating the rapid nucleation of YAG:Ce phase. PL intensity keeps on increasing as the temperature stabilizes at 350 °C. However, one can notice a decrease of PL over time. We attribute this decrease to changes in terms of scattering rate of the solution, which makes difficult quantitative PL measurements. Hence, only the evolution of PL at short reaction time, when the solution remains transparent, is further considered.
From Fig. 2b, the higher the external pressure, the faster the PL intensity increases. This can be explained through a faster YAG:Ce nucleation. It correlates well with the density dependence with pressure: the higher the pressure, the higher the density, favouring the interactions between species and thus the crystallization of YAG. Hence, a high external pressure favours a single burst of YAG nucleation and thus allows decoupling nucleation and growth processes: rapid nucleation followed by a second step of the growth of nuclei.
For this set of experiments, the total synthesis duration was set at 150 min. Post-synthesis TEM characterization was performed on the different samples. TEM images (Fig. 3) compare the morphology of YAG nanoparticles for the synthesis at P = 200 bar and at P = 400 bar. When the reaction pressure was fixed at 200 bar, mostly individual nanocrystals can be observed (some of them are circled in yellow in Fig. 3a). For synthesis performed at 400 bar, nanocrystals (also circled in yellow in Fig. 3b) are strongly aggregated, forming large aggregates (circled in white dashed line). The presence of smaller nanocrystals for the synthesis at 400 bar is consistent with the strong nucleation burst (Fig. 2b), creating numerous nuclei leading to smaller particles. Meanwhile, the much stronger aggregation of the nanocrystals is explained by the higher density of the medium where the reaction took place, favouring particle aggregation. Consequently, as a compromise between favouring a nucleation burst and limiting the aggregation state, a reaction pressure of 200 bar is suitable. In the following of the study, all the syntheses will be done at P = 200 bar.
 | ||
| Fig. 3 TEM images of YAG:Ce nanocrystals synthesized at different pressure with T = 350 °C, [Al] = 0.09 mol L−1 and reaction time = 150 min. Some individual nanocrystals are circled with yellow solid lines. An aggregate is circled with a white dashed line. | ||
3.3. Nano-YAG:Ce size and morphology as a function of the synthesis temperature
The influence of the synthesis temperature onto YAG:Ce morphology was studied while the pressure was kept at 200 bar, the aluminium concentration was 0.09 mol L−1 with a reaction time of t = 150 min. As shown on Fig. 4(a–c), particle size drastically changes with the synthesis temperature. For a reaction at 300 °C, very small elongated nanocrystals (6 nm in diameter, pointed by the arrow on Fig. 4a) can be observed, gathering into much larger aggregates, circled in the inset of Fig. 4a. This morphology is very close to that observed for syntheses in commercial autoclaves operating at 300 °C.9,29,38 The nanocrystal size distribution (Fig. S1†), determined without taking into account the aggregates, is very narrow, between 1 and 10 nm. When the synthesis temperature is set at 350 °C, the size of YAG nanocrystals increases (average size ∼23 nm, with higher polydispersity). These nanocrystals are less prone to agglomeration although aggregates are still observed, circled on Fig. 4b. When the synthesis temperature is set at 400 °C, mostly individual YAG:Ce single nanocrystals are observed (Fig. 4c). They present an average size of 47 ± 15 nm. From Fig. S1,† the nanocrystal size distribution broadens as the nanocrystal size increases. This size evolution with temperature is consistent as the growth process, driven either by species diffusion or by surface reactivity, is temperature-dependent and thus activated with temperature.39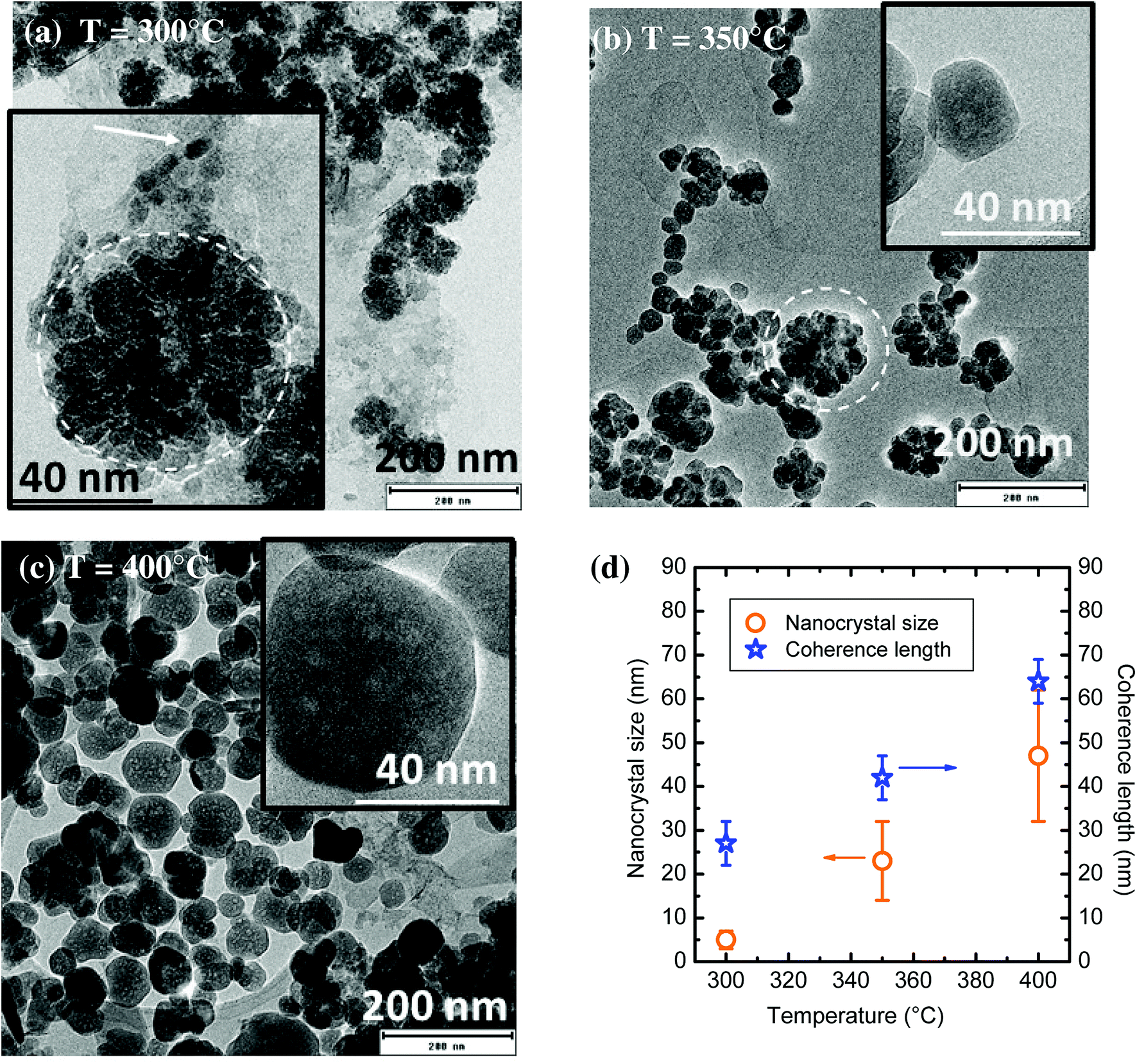 | ||
| Fig. 4 (a–c) TEM images of YAG:Ce nanocrystals synthesized at different reaction temperatures with P = 200 bar, [Al] = 0.09 mol L−1 and t = 150 min. In the inset of (a), the arrow points an individual nanocrystal and the dashed circle emphasizes an aggregate. (d) Evolution of the nanocrystal size (determined by TEM, orange circles, error bars corresponding to size dispersion) and coherence length (determined by PXRD, blue stars) as a function of reaction temperature. | ||
The evolution of nanocrystal size (determined from TEM images) and of the coherence length (calculated from Scherrer's formula using PXRD patterns) is reported in Fig. 4d as a function of the synthesis temperature. In the sample synthesized at 300 °C, the coherence length is much larger than the nanocrystal size. This discrepancy confirms the self-oriented aggregation mechanism of primary particles (small nanocrystals Fig. 4a and b) as previously encountered, which is favoured by the YAG cubic structure.29,40 For the sample synthesized at 350 °C, the difference between nanocrystal size and coherence length significantly decreases but remains. It can be explained by the persistence of some self-oriented aggregates that contribute to increase the coherence length. Finally, the rather good agreement between nanocrystal size and coherence length for the sample synthesized at 400 °C indicates that single nanocrystals are obtained, which is consistent with the TEM observations of well-isolated nanocrystals (Fig. 4c).
One can note that the TEM contrast is not homogeneous within a nanocrystal (inset, Fig. 4c), suggesting the presence of a porosity, resulting in a sponge-like shape. This morphology can be explained by a growth mechanism involving coalescence of small primary particles to form larger nanocrystals, resulting in the encapsulation of voids.41 This coalescence of the initial small nanocrystals is promoted by the self-oriented aggregation mechanism of primary particles. Indeed, the relative crystallographic orientation between aggregated grains is of primary importance for their coalescence as the surface energy associated with grain boundaries depends on grain–grain misorientation.42 Indeed, a low degree of grain–grain misorientation is associated with a low grain boundary energy, thus favouring coalescence.
3.4. Nano-YAG:Ce size and morphology as a function of precursor concentrations
The morphology of YAG:Ce nanoparticles has been studied through the modification of aluminium and yttrium precursor concentrations (Fig. 5 and S2†). As a reference, only the aluminium isopropoxide concentration is indicated, knowing that the reaction stoichiometry is kept. The other parameters, temperature, pressure and reaction time, remain constant (350 °C, 200 bar and 150 min, respectively). When the solvothermal reaction occurs at very low precursor concentrations ([Al] = 0.02 mol L−1) nanocrystals present an average size of around 15 nm in diameter and are strongly aggregated (aggregates circled in white on Fig. 5a). For [Al] = 0.09 mol L−1, YAG:Ce nanocrystals present an average size of 23 ± 9 nm (Fig. 5b, also corresponding to Fig. 4b) while more individual nanocrystals can be observed. As the precursor concentration keeps on increasing, the nanocrystal size increases (Fig. 5c–e) and the formation of aggregates disappears.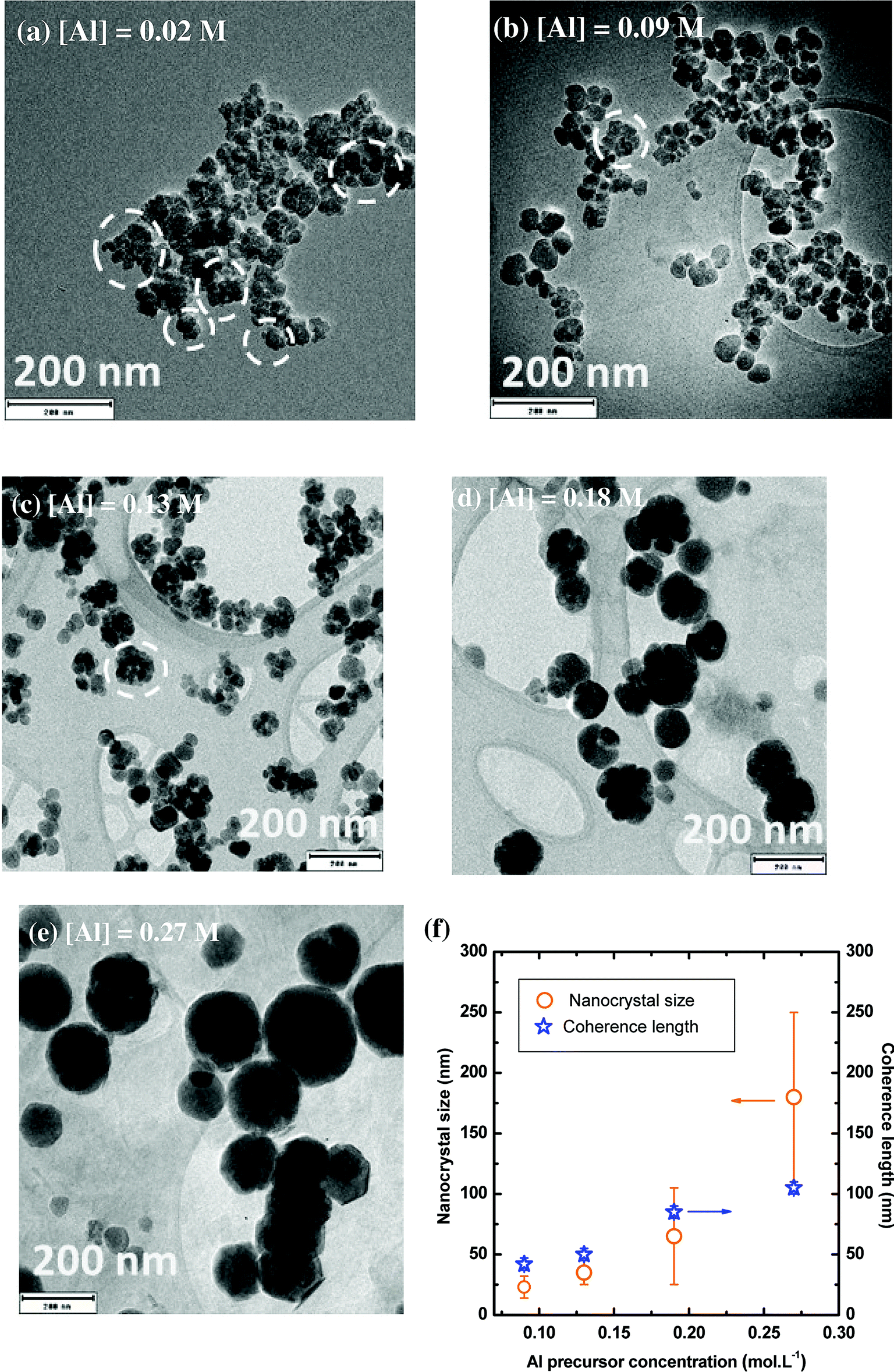 | ||
| Fig. 5 (a–e) TEM images of YAG:Ce nanocrystals synthesized with different precursor concentrations. P = 200 bar, T = 350 °C and reaction time = 150 min. (f) Evolution of the nanocrystal size (orange circle, error bars corresponding to size dispersion) and coherence length (blue star) as a function of the precursor concentration. Coherence length larger than 100 nm cannot be measured to instrumental limitation. | ||
The evolution of nanocrystal size and coherence length as a function of the precursor concentration is reported in Fig. 5f. For [Al] = 0.09 mol L−1, as explained in the previous paragraph, the coherence length is slightly larger than the nanocrystal size, indicating the presence of self-oriented aggregates, which is consistent with TEM observations (Fig. 5b). Rather good agreement between coherence length and nanocrystal size is observed for [Al] = 0.13 and 0.18 mol L−1, evidencing the single-crystal character of the nanocrystals. For the highest concentration, the coherence length appears smaller than the nanocrystal size. This discrepancy can be explained by the intrinsic instrumental broadening of the diffractometer, which does not allow measuring coherence length larger than 100 nm. This lead to underestimated coherence length values. For these nanocrystals ([Al] = 0.27 mol L−1), TEM images at high magnification show atomic planes up to the particle edges, confirming the high crystal quality of these well-isolated nanocrystals (Fig. S3†).
These evolutions with precursor concentrations can be understood by considering the relative prominence of nucleation and growth mechanisms. At low concentration, the nucleation step is predominant inducing a lot of small aggregated nanocrystals: only a single burst of nucleation happens through the precipitation reaction of the formation of YAG phase, followed by a negligible growth mechanism as the precursors were essentially consumed during nucleation. At higher concentrations, the nanocrystal growth is significantly enhanced. This growth by diffusion and self-oriented aggregation and coalescence of primary particles43 leads to the gradual disappearance of aggregates and to larger and individual nanocrystals of high quality, as confirmed by the good agreement between their size and coherence length and TEM characterizations.
3.5. Nano-YAG:Ce size and morphology as a function of the reaction time
The evolution of YAG:Ce size and morphology as a function of the reaction time was studied under two different sets of conditions: T = 350 °C, P = 200 bar, [Al] = 0.09 mol L−1 and T = 400 °C, P = 200 bar, [Al] = 0.18 mol L−1. For the first reaction conditions, some TEM images are presented in Fig. 6a–d. At short reaction time, small nanocrystals can be observed, mainly forming aggregates (circled in white), due to a predominant nucleation step. As the reaction time is lengthened, the number of aggregates diminishes at the benefit of individual nanocrystals exhibiting a larger size, as the nanocrystal growth is significantly enhanced (Fig. 6c and d and S4†). From Fig. 6e, the longer the reaction time, the better the agreement between the nanocrystal size and the coherence length, indicating a reduction of the number of aggregates over time at the benefit of individual well-crystallized nanocrystals. For reaction time of 300 and 480 min, the nanocrystal size and the coherence length are in good agreement and corroborate the TEM images: mainly individual nanocrystals are observed, with faceted edges, indicating their high crystal quality.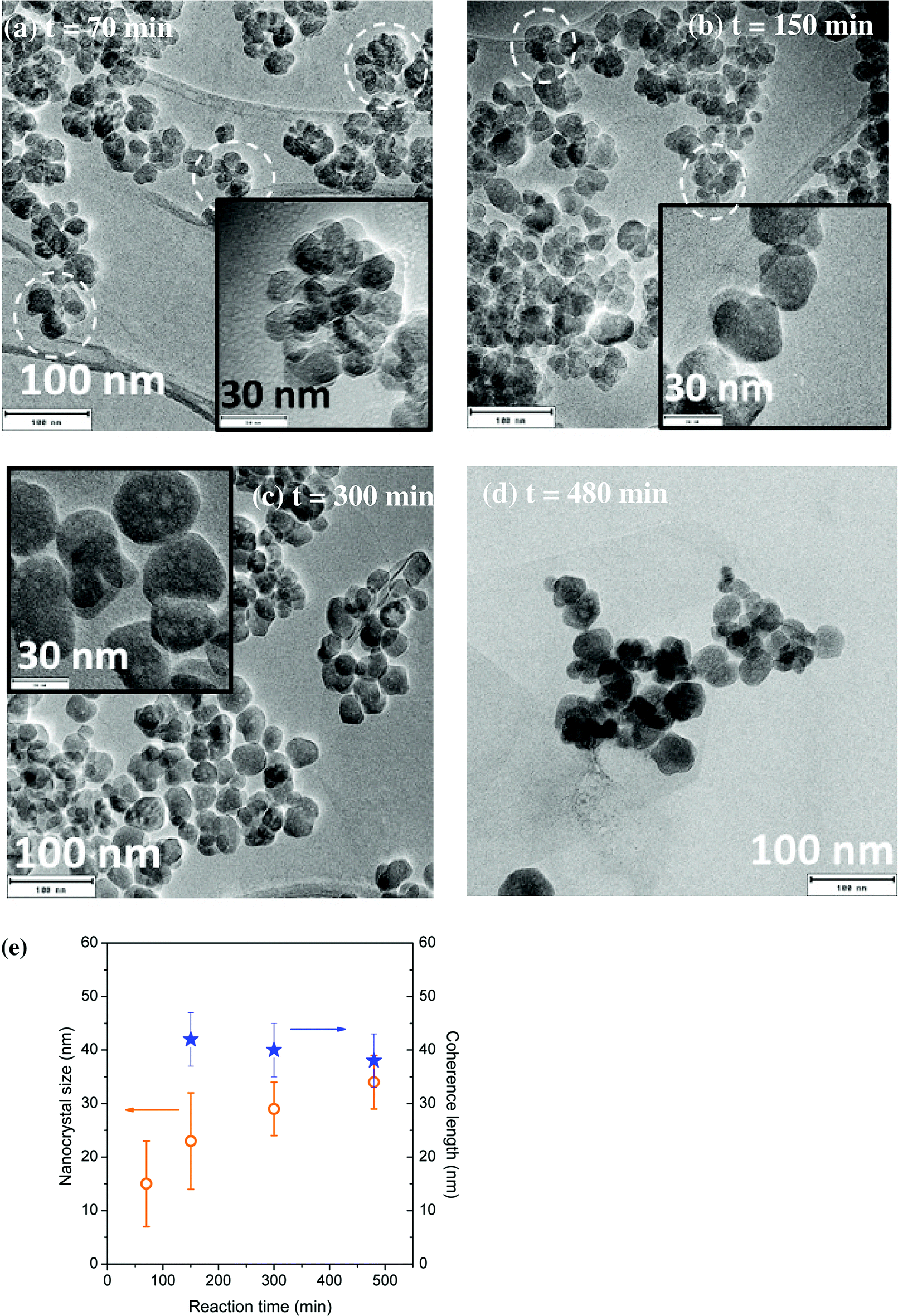 | ||
| Fig. 6 (a–d) TEM images of YAG:Ce nanocrystals synthesized for different reaction times. P = 200 bar, T = 350 °C, [Al] = 0.09 M. (e) Evolution of the coherence length (blue stars) and the nanocrystal size (orange circles, error bars corresponding to size dispersion) as a function of the reaction time under these conditions. | ||
This evolution with time can be explained by the nanocrystal growth through primary particle coalescence, involving also the Ostwald ripening process at long reaction times: the largest nanocrystals continue to grow for longer reaction times at the expense of smaller particles, less stable, exhibiting a higher solubility, which tend thus to disappear.
When the synthesis is performed at higher temperature and concentration (T = 400 °C, [Al] = 0.18 mol L−1), the nanocrystal size is much larger (Fig. 7a and b). For a 300 min reaction time, the average particle size is around 50 nm, with lots of individual nanocrystals. Some of the nanocrystals are faceted, highlighting their single-crystal character. However, some nanocrystals are aggregated in a self-oriented manner (Fig. S5†), which explains why the coherence length is slightly larger than the nanocrystal size (Fig. 7c). From these TEM images (Fig. S5†), nanocrystal sintering can be observed, with the formation of grain boundaries. For a 480 min reaction time, well-crystallized YAG:Ce nanocrystals were obtained, presenting an average diameter of about 100 nm with a relatively narrow size distribution (±20 nm) (Fig. 7b and S6†). The high crystal quality of these crystals is confirmed by high-resolution TEM observations (Fig. 7c) and the very good agreement between the coherence length and the nanocrystal size (Fig. 7d).
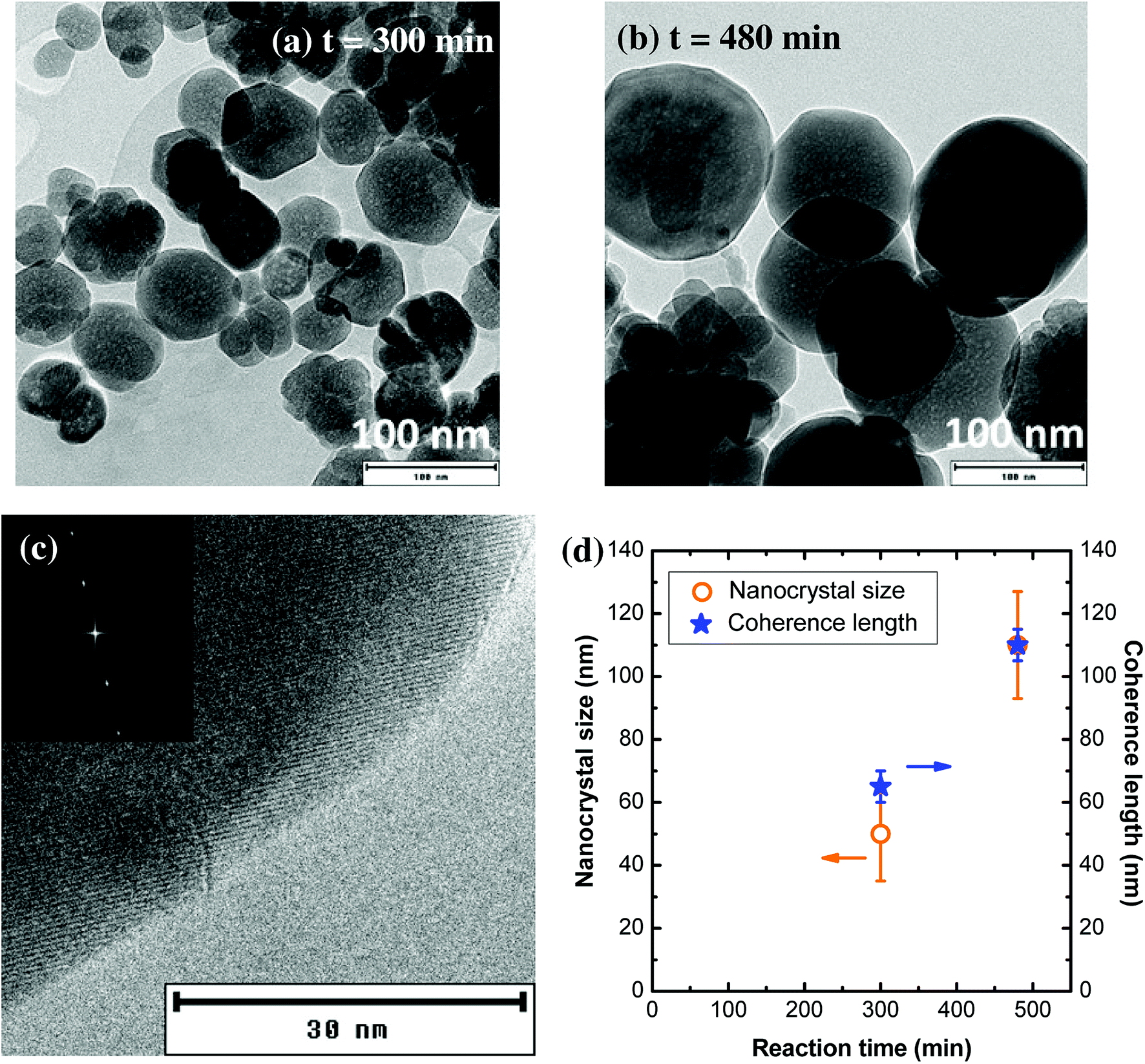 | ||
| Fig. 7 (a and b) TEM images of nanocrystals obtained under the following conditions: P = 200 bar, T = 400 °C, [Al] = 0.18 mol L−1, for two reaction times. (c) High resolution TEM image of a nanocrystal synthesized at 400 °C, 200 bar, [Al] = 0.18 mol L−1, t = 480 nm. The atomic planes are visible, in particular at the edges of the particle. In the inset of (b) is shown the Fourier transform of TEM image evidencing the single-crystal character of the particle. (d) Evolution of the nanocrystal size (orange circles) and coherence length (blue stars) as a function of the reaction time. | ||
A summary of all explored synthesis conditions and of the corresponding nanocrystal morphology has been added in ESI (Table S1†). Through the optimization and the control of various synthesis parameters (temperature, pressure, precursor concentration, reaction time), well-crystallized individual nanocrystals could be obtained using our new solvothermal method:
- 30 nm nanocrystals (Fig. 6d, T = 350 °C, [Al] = 0.09 mol L−1, P = 200 bar, t = 480 min).
- 100 nm nanocrystals (Fig. 7b, T = 400 °C, [Al] = 0.18 mol L−1, P = 200 bar, t = 480 min).
It is worth noting that, although the 30 nm nanocrystals exhibit a unit cell parameter of a = 12.020 ± 0.005 Å, very close to the one of bulk YAG:Ce (12.012 Å14), the 100 nm nanocrystals have a much larger unit cell parameter (a = 12.080 ± 0.005 Å, measured by Le Bail fit of PXRD pattern). This result suggests the presence of internal strains within the 100 nm nanocrystals. The origin of these internal strains can be caused by the coalescence of several grains to form larger nanocrystals, in good agreement with the crystal growth mechanism of self-oriented aggregation and coalescence of primary particles.
3.6. Characterization of the doping concentration
The nominal composition of all studied samples is Y2.955Ce0.045Al5O12, corresponding to a 1.5 mol% Ce doping concentration. At this low concentration and taking into account the internal strains within the particles, it is not possible to the Vegard's law to deduce the exact cerium concentration. EDX analysis, performed in the TEM, allows to get the chemical composition of the samples. In all studied samples, the Ce/(Ce + Y) ratio is worth between 0.012 and 0.015, indicating that a very large majority of the cerium ions initially introduced in the are incorporated into YAG nanoparticles.3.7. Optical properties of nano-YAG:Ce
The study of PL properties of nano-YAG:Ce were performed on powdered samples. Typical PL emission and PL excitation (PLE) spectra are presented on Fig. 8a. All the samples show similar spectra with the typical features of Ce3+ ions in the YAG matrix, with the broad emission band peaking at 550 nm, corresponding to the allowed electric dipole 5d → 4f transition of Ce3+.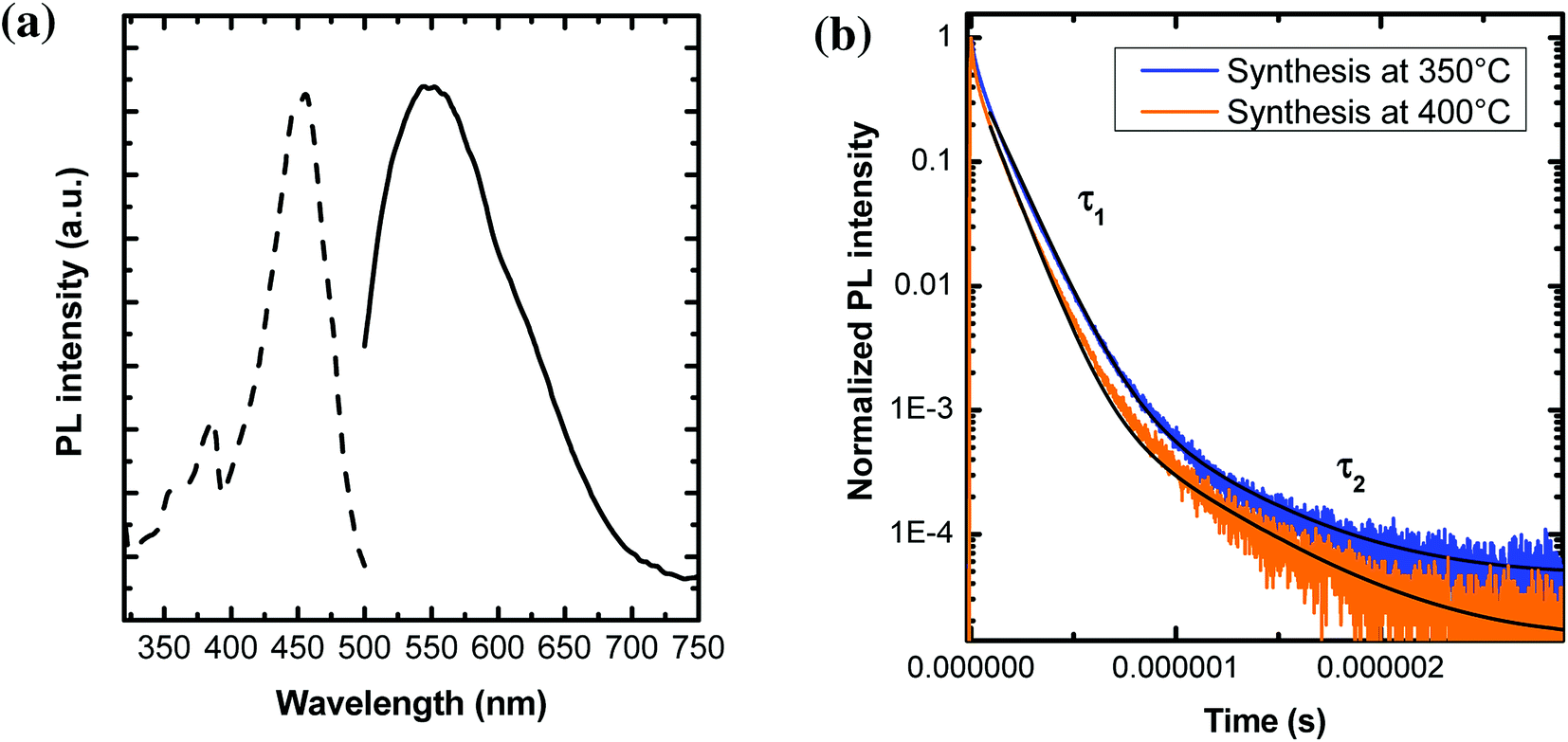 | ||
Fig. 8 (a) PLE (dotted curve) and PL (solid line) spectra of 100 nm YAG:Ce nanocrystals, resulting from a synthesis performed at P = 200 bar, T = 400 °C, [Al] = 0.18 mol L−1 and t = 480 min. The PLE spectrum was recorded when monitoring the 550 nm emission, while the emission spectrum was recorded under a 457 nm excitation. (b) Fluorescence decays of YAG:Ce nanocrystals obtained at 350 °C and 400 °C, under a 444 nm excitation and their associated fit (in black). The fit is performed using a bi-exponential decay: 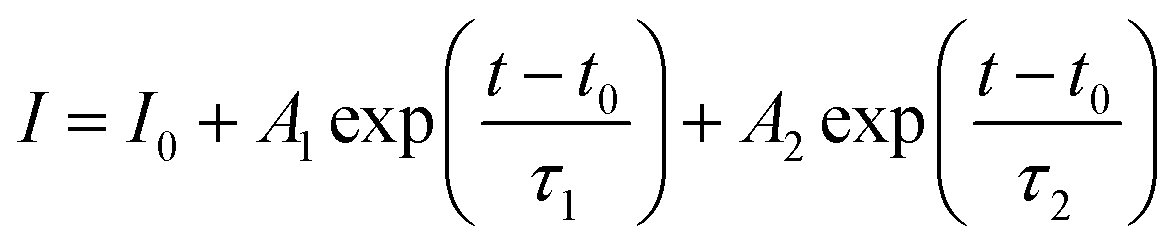 . . | ||
Typical PL decays are represented on Fig. 8b, with their associated bi-exponential fits. The fit gives two characteristic lifetimes: τ1 corresponding to a rapid decay and τ2 at longer time. However, from the fit (and more specifically the A2 pre-factor), it is found that this τ2 value accounts for less than 1% of the total decay. Thus, in the following, we will only report and discuss the values of τ1 (Fig. 9a–c).
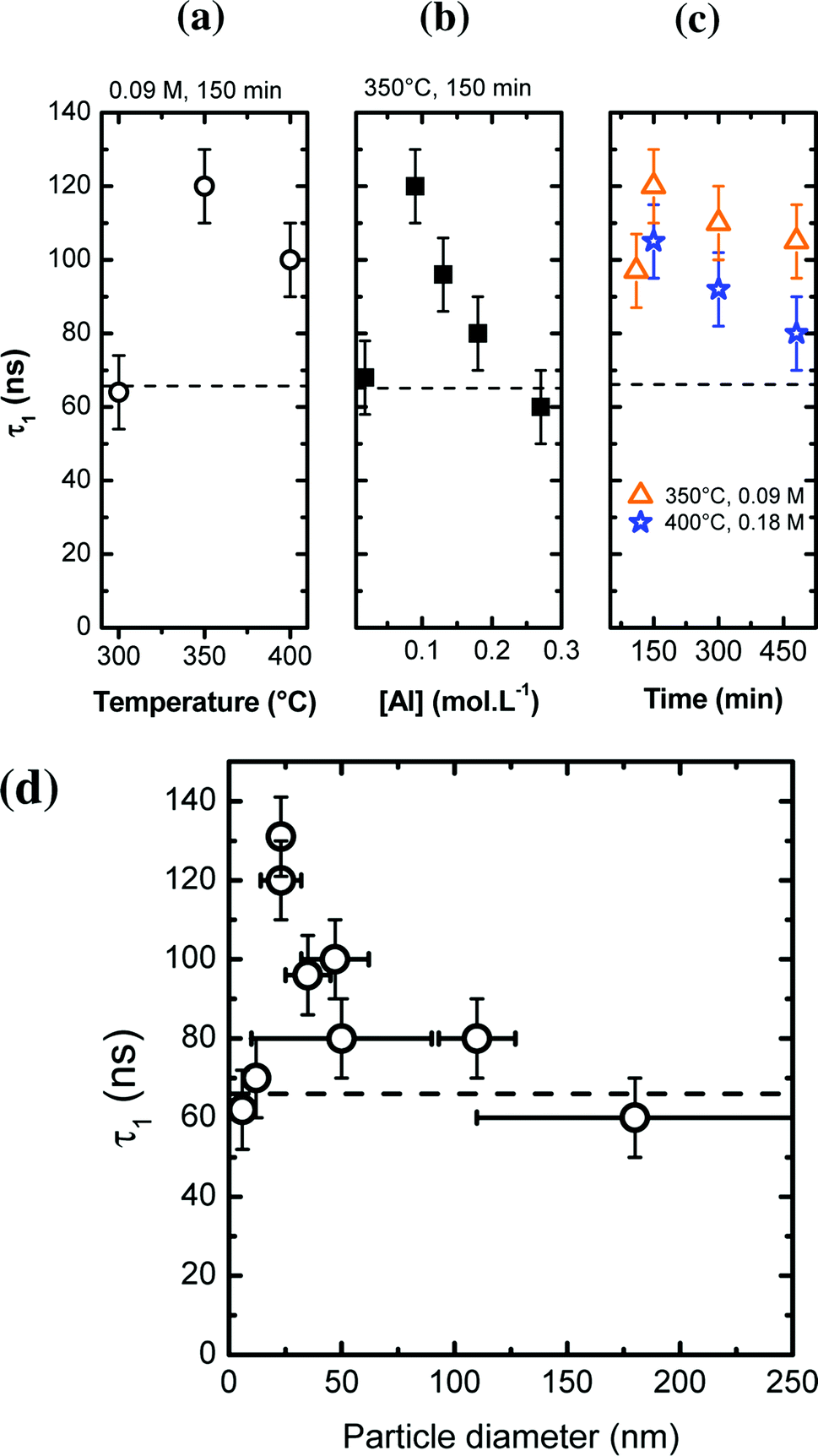 | ||
| Fig. 9 (a–c) Excited state lifetime τ1 of Ce3+ as a function of various synthesis parameters, indicated on the graph (with P = 200 bar). The dashed line represents the excited state lifetime of bulk YAG:Ce. (d) Evolution of τ1 as a function of the particle size. | ||
In most nano-YAG:Ce samples, the excited state lifetime τ1 is longer than in bulk YAG:Ce (τ = 65 ns, represented by a dashed line in Fig. 9a–c). This is commonly observed in nanometer-sized materials as the radiative lifetime is affected by the dielectric environment.44,45 The interpretation of τ1 values as a function of the synthesis conditions is not simple as non-radiative lifetime (τNR) and radiative lifetime (τR) vary in an antagonist manner: when particles become smaller, τR is lengthened due to dielectric effects45 while τNR is shortened due to the increase of surface defect proportion. However, one can notice some trends according to different synthesis parameters (Fig. 9a–c). In nano-YAG:Ce synthesized at 300 °C, τ1 is worth 62 ± 5 ns (Fig. 9a). According to the TEM images (Fig. 4a), YAG:Ce nanocrystals are very small (∼6 nm in diameter). Hence the radiative lifetime should be much longer than in bulk, as obtained by M. Odziomek et al.21 So this expected increase of τ1 is counterbalanced by an enhanced effect of τNR due to strong non-radiative de-excitations associated to surfaces defects and the poor crystal quality of nanoparticles (Fig. 4a). Thus, the value of τ1 (62 ns) evidences strong non-radiative de-excitations. When particles are synthesized at higher temperatures, τ1 increases (Fig. 9a), indicating that the non-radiative de-excitations are less likely, in good agreement with the improvement of crystal quality of nano-YAG:Ce. Between a synthesis at 350 °C and 400 °C, τ1 goes from 120 ns to 100 ns, suggesting a reduction of τR because of size increase.
Similar trend is observed for the syntheses performed at different precursor concentration (Fig. 9b). At very low [Al] concentration ([Al] = 0.02 mol L−1), nanocrystals are very small and strongly aggregated (Fig. 5a). In this case, the non-radiative de-excitations are favoured, leading to a short τ1 (∼65 ns, Fig. 9b). At higher value of precursor concentration ([Al] = 0.09 mol L−1), τ1 is significantly increased, corresponding to an improved crystallinity as well as an increased nanocrystal size. As [Al] is further increased, τ1 decreases. This trend can be explained by the fact that YAG nanocrystals enlarge, leading to a reduction of τR (through the increase of the radiative index). Finally, a similar argument can be given to explain the evolution of τ1 as a function of the reaction time (Fig. 9c).
A better way to discuss the evolution of τ1 is to look at the evolution of τ1 as a function of the nanocrystal size (Fig. 9d). For small nanocrystals (size < 12 nm), τ1 is around 65 ns due to strong non radiative de-excitations. For larger particles (25 nm), longer τ1 is measured (130 ns) due to the lengthening of the radiative index τrad because of the refractive index influence. As the nanocrystal size increases (>25 nm), τ1 decreases down to a value which corresponds to the bulk value. From Fig. 9d, the region of influence of the dielectric medium (the air in our case) is situated around 100–150 nm. In previous studies,46,47 it is comprise between 60 and 80 nm. The difference can be explained by the porosity of our nanocrystals, due to the self-oriented aggregation and coalescence (Fig. 4c, 6c and 7). Such porosity leads to a lower effective refractive index.
The internal quantum yield (iQY) of the 100 nm YAG:Ce nanocrystals was measured to be 40 ± 5%. This value is significantly higher than most of the values reported for as-made YAG:Ce nanophosphors (∼20%).25,27 However, in spite of the high crystal quality of the particles (Fig. 7b and c), it remains about twice lower than in bulk YAG:Ce (80%). Several reasons could explain this difference of optical efficiency between YAG nanocrystals and bulk:
(1) The presence of punctual defects within the nanocrystals. The solvothermal synthesis process, occurring off thermodynamic equilibrium, may induce the formation of these defects.48
(2) The difference of cerium distribution within the materials. Indeed, in bulk or micropowders of YAG doped with a few mol% Ce3+, the annealing process favours the homogeneous distribution of Ce ions within the crystal network.14 In our nanocrystals, due to the burst of nucleation and the nanocrystal sintering, it is likely that the Ce distribution is less uniform, with Ce aggregation being favoured at the grain boundaries.49,50 This would lead to the formation of Ce clusters that could induce luminescence quenching through energy transfers between Ce ions (Ce–Ce cross relaxations), favouring non-radiative de-excitations and inducing iQY decrease. Such segregation would be consistent with the large unit cell parameter measured by Le Bail fit of PXRD in these nanocrystals. Further studies, involving high-resolution scanning TEM analysis, would be required to confirm this suggestion.51
(3) The presence of Ce4+ ions, that could act as luminescence quenchers through energy transfer from Ce3+ to Ce4+.52,53 Bulk YAG:Ce overcome an annealing under reducing atmosphere to force cerium ions into their 3+ state.14 In nanocrystals, Ce3+ ions could get oxidized during the solvothermal synthesis. To confirm this hypothesis, we performed in situ XANES experiments during the modified-solvothermal synthesis.
3.8. X-ray absorption spectroscopy of nano-YAG:Ce
Although it is important to optimize the iQY of YAG:Ce nanocrystals, it is also essential to maximize the concentration of Ce3+ within the material to enhance its absorption and ensure high PL efficiency, while avoiding the problem of Ce quenching. In bulk YAG, the maximal Ce3+ concentration is known to be only 3 mol% due to the unit cell rigidity.14 It is thus important that all cerium ions remain in their 3+ valence state, Ce4+ ions being optically silent. For this, bulk and micropowder of YAG:Ce undergo thermal annealing treatments under reducing atmosphere (typically Ar/H2) to force cerium ions into their 3+ state.14 In nanocrystals, it is known that surface Ce3+ get oxidized into Ce4+ under strong illumination,25,28 or upon post-annealing treatment in air.27 However, thermal annealing cannot be applied to avoid uncontrolled nanocrystal coalescence. The question is whether Ce3+ oxidation occurs during the solvothermal process. To answer it, we performed in situ XANES experiments during the modified-solvothermal syntheses.Fig. 10 shows the experimental in situ XANES spectra as a function of the synthesis temperature, as well as their corresponding fits based on the linear combination of standards for Ce3+ and Ce4+. We used the XANES spectrum of nanopowder of CeO2 as a reference for Ce4+. For Ce3+, we used the XANES spectrum of Ce3+-doped YAG single-crystals, except for the solution of precursors (labelled 30 °C, 20 bar in Fig. 10) for which the XANES spectrum of Ce(acetate)3 was considered.
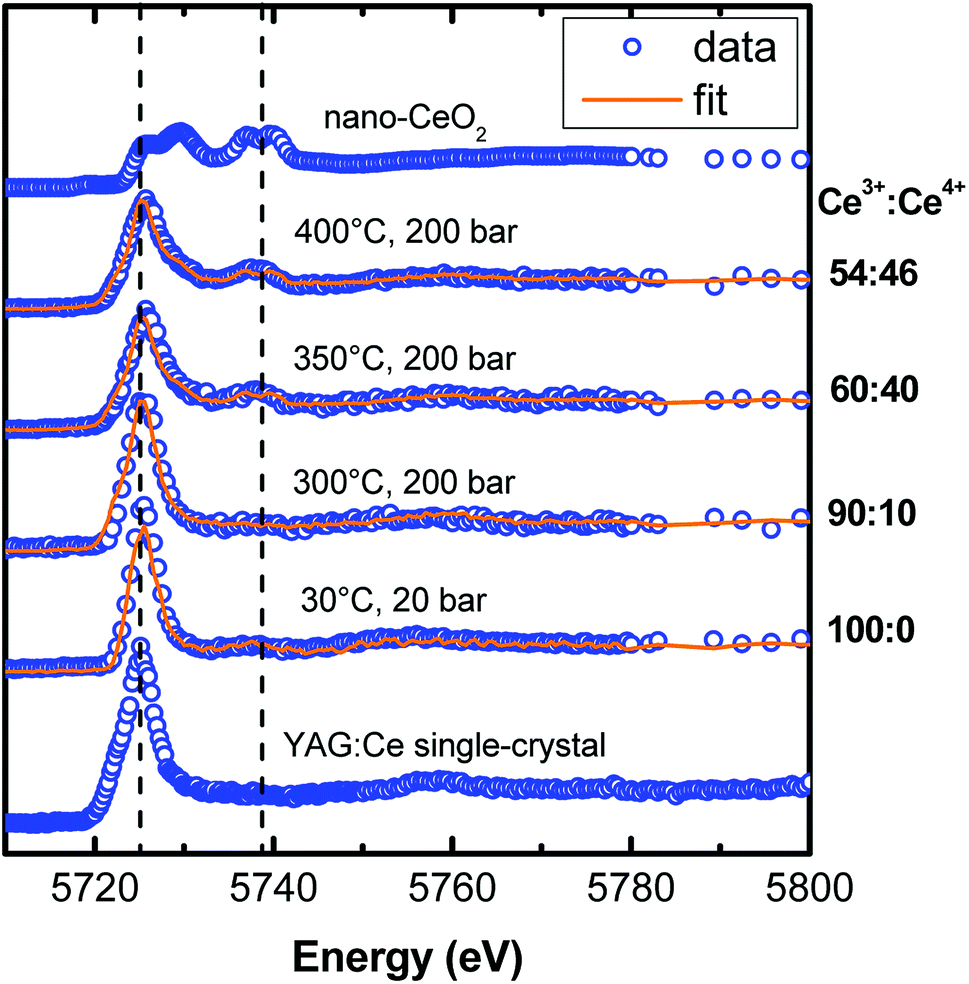 | ||
| Fig. 10 Experimental in situ XANES spectra at the cerium L3 edge (blue open circles), recorded during YAG:Ce nanocrystal solvothermal syntheses under different temperatures and XANES spectra of two powdered references (YAG:Ce single-crystal and nano-CeO2). The fits (orange lines) correspond to a linear combination of XANES spectra of the references, allowing a quantification of the Ce3+/Ce4+ ratio. The references are nano-CeO2 for Ce4+ and YAG:Ce single-crystals for Ce3+, expect for the solution at 30 °C and 20 bar where the Ce3+ reference is Ce(acetate)3 in 1,4-butanediol. For synthesis temperatures higher than 300 °C, strong oxidation of Ce3+ is observed. | ||
For a synthesis of nano-YAG performed at 300 °C, cerium remains in its 3+ state. But, when the synthesis temperature increases, the proportion of Ce4+ raises: the higher the reaction temperature, the more Ce3+ ions get oxidized. In the sample synthesized at 400 °C, only 54 ± 5% cerium ions remain in their 3+ state whereas 46 ± 5% has become Ce4+. The Ce3+ oxidation in the reaction medium can be understood by considering the species in solution: first, it is likely that there is some oxygen gas dissolved in the 1,4-butanediol,54 which thus contributes to the Ce3+ → Ce4+ oxidation; second it has been shown that the 1,4-butanediol solvent dehydrates above 250 °C to form tetrahydrofuran and water.55 Water is likely to favours Ce3+ oxidation, and thus even more than the temperature is high as the oxidation power of water increases with temperature.56
We demonstrated, for the first time to our knowledge, that the cerium oxidation process occurs during the solvothermal synthesis. Under these reaction conditions, there is a trade-off between the high crystal quality, requiring high synthesis temperature (Fig. 4), and the preservation of cerium ions in their 3+ oxidation state, requiring a lower synthesis temperature (Fig. 10).
4. Conclusions
Thanks to this new solvothermal method combining high temperature and high pressure, well-crystallized individual YAG:Ce nanocrystals were prepared. Indeed, the correlations between several synthesis parameters (pressure, temperature, precursor concentration, reaction time) and in situ PL and structural characterizations allowed to obtain a good control of YAG nanocrystallization conditions: a single burst of nucleation followed by the slow growth of nanocrystals through diffusion, involving the Ostwald ripening mechanisms for long reaction times, but also coalescence mechanism of primary particles favoured by self-oriented aggregation. Thus, nanocrystals of the targeted size (∼100 nm), considered as optimal for numerous applications (biophotonics, solid-state lighting), were produced with a narrow size distribution. Thanks to their high crystal quality, these 100 nm YAG:Ce nanocrystals present, without further heat-treatment, a PL iQY of 40%, which is better than most of the already-reported works. Through in situ XANES experiments, we showed that an increase of the synthesis temperature favours Ce3+ oxidation into Ce4+, which thus reduces the absorption and emission capacity of nanocrystals. Further work, involving a reducing atmosphere, should be envisioned to force cerium ions to remain in their 3+ state.This study not only provides a new solvothermal method to obtain size-controlled YAG:Ce nanocrystals with high crystal quality but also paves the way to the development of other garnet-type nanocrystals.57 In addition, the possibility of measuring in situ PL and X-ray absorption during the nanocrystallization opens the door to a better control of the nucleation/growth process of nanomaterials and to the possibility of a direct optimization of their physical properties.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
The French ANR funded this research through the NanophosforLED project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Oliver Leynaud for his assistance in XRD, as well as Sonia De Sousa Nobre for her help in measuring iQY. G. Dantelle acknowledges the ANR for funding through the NanophosforLED project (ANR-17-CE09-0035-01). The FAME-UHD beamline was financially supported by the EQUIPEX project ANR-10-EQPX-27-01 called EcoX, the CEA CNRS CRG consortium and the INSU CNRS institute.References
- J. E. Geusic, H. M. Marcos and L. G. Van Uitert, Laser oscillations in Nd-doped yttrium aluminum, yttrium gallium and gadolinium garnets, Appl. Phys. Lett., 1964, 4(10), 182 CrossRef.
- A. Ikesue, Y. L. Aung and V. Lupei, Ceramic Lasers, Cambridge University Press, Cambridge, 2013 Search PubMed.
- L. Esposito, J. Hostasa, A. Piancastelli, G. Toci, D. Alderighi, M. Vannini, T. Epicier, A. Malchere, G. Alombert-Goget and G. Boulon, Multilayered YAG-Yb:YAG ceramics: manufacture and laser performance, J. Mater. Chem. C, 2014, 2, 10138 RSC.
- C. D. Nie, S. Bera and J. A. Harrington, Growth of single-crystal YAG fiber optics, Opt. Express, 2016, 24(14), 15522–15527 CrossRef PubMed.
- M. Inoue, H. Otsu, H. Kominami and T. Inui, Glycothermal synthesis of rare earth aluminium garnets, J. Alloys Compd., 1995, 226, 146–151 CrossRef.
- R. Asakura, T. Isobe, K. Kurokawa, H. Aizawa and M. Ohkubo, Tagging of avidin immobilized beads with biotinylated YAG:Ce3+ nanocrystal phosphor, Anal. Bioanal. Chem., 2006, 386, 1641–1647 CrossRef PubMed.
- A. Benayas, B. del Rosal, A. Pérez-Delgado, K. Santacruz-Gómez, D. Jaque, G. Adolfo Hirata and F. Vetrone, Nd:YAG Near-Infrared Luminescent Nanothermometers, Adv. Opt. Mater., 2015, 3, 687–694 CrossRef.
- J.-Y. Chong, Y. Zhang, B. K. Wagner and Z. Kang, Co-precipitation synthesis of YAG:Dy nanophosphor and its thermometric properties, J. Alloys Compd., 2013, 581(25), 484–487 CrossRef.
- M. Nyman, L. E. Shea-Rohwer, J. E. Martin and P. Provencio, Nano-YAG:Ce Mechanisms of Growth and Epoxy-Encapsulation, Chem. Mater., 2009, 21, 1536–1542 CrossRef.
- A. Potdevin, G. Chadeyron, D. Boyer, B. Caillier and R. Mahiou, Sol–gel based YAG:Tb3+ or Eu3+ phosphors for application in lighting sources, J. Phys. D: Appl. Phys., 2005, 38, 3251–3260 CrossRef.
- C. Bouzigues, T. Gacoin and A. Alexandrou, Biological Applications of Rare-Earth Based Nanoparticles, ACS Nano, 2011, 5(11), 8488–8505 CrossRef PubMed.
- F. Wang, J. Wang and X. Liu, Direct evidence of a surface quenching effect on size-dependent luminescence of up-conversion nanoaprticles, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef PubMed.
- J.-C. Boyer and F. C. J. M. van Veggel, Absolute quantum yield measurements of colloidal NaYF4: Er3+, Yb3+ upconverting nanoparticles, Nanoscale, 2010, 2, 1417–1419 RSC.
- N. C. George, A. J. Pell, G. Dantelle, K. Page, A. Llobet, M. Balasubramanian, G. Pintacuda, B. F. Chmelka and R. Seshadri, Local Environments of Dilute Activator Ions in the Solid-State Lighting Phosphor Y3−xCexAl5O12, Chem. Mater., 2013, 25, 3979–3995 CrossRef.
- N. J. Hess, G. D. Maupin, L. A. Chick, D. S. Sunberg, D. E. McCreedy and T. R. Armstrong, Synthesis and crystallization of yttrium-aluminium garnet and related compounds, J. Mater. Sci., 1994, 29, 1873–1878 CrossRef.
- A. Potdevin, G. Chadeyron, V. Briois and R. Mahiou, Structural, morphological and scintillation properties of Ce3+-doped Y3Al5O12 powders and films elaborated by the sol–gel process, Mater. Chem. Phys., 2011, 130, 500–506 CrossRef.
- F. A. Selim, A. Khamehchi, D. Winarski and S. Agarwal, Synthesis and characterization of Ce:YAG nanophosphors and ceramics, Opt. Mater. Express, 2016, 2(12), 275266 Search PubMed.
- G. Demazeau, Solvothermal reactions: an original route for the synthesis of novel materials, J. Mater. Sci., 2008, 43(7), 2104–2114 CrossRef.
- F. Cansell and C. Aymonier, Design of functional nanostructured materials using supercritical fluids, J. Supercrit. Fluids, 2009, 47(3), 508–516 CrossRef.
- A. Revaux, G. Dantelle, D. Decanini, A.-M. Haghiri-Gosnet, T. Gacoin and J.-P. Boilot, Synthesis of YAG:Ce/TiO2 nanocomposite films, Opt. Mater., 2011, 33, 1124–1127 CrossRef.
- M. Odziomek, F. Chaput, F. Lerouge, M. Sitarz and S. Parola, Highly luminescent YAG:Ce ultra-small nanocrystals, from stable dispersions to thin films, J. Mater. Chem. C, 2017, 5, 12561 RSC.
- Y. H. Song, E. K. Ji, B. W. Jeong, M. K. Jung, E. Y. Kim and D. HoYoon, High power laser-driven ceramic phosphor plate for outstanding efficient white light conversion in application of automotive lighting, Sci. Rep., 2016, 6, 31206 CrossRef PubMed.
- T. Isobe, Low-temperature wet chemical syntheses of nanocrystal phosphors with surface modification and their characterization, Phys. Status Solidi A, 2006, 203(11), 2686–2693 CrossRef.
- R. Kasuya, T. Isobe and H. Kuma, Glycothermal synthesis and photoluminescence of YAG:Ce3+ nanophosphors, J. Alloys Compd., 2006, 408–412, 820–823 CrossRef.
- Y. Kamiyama, T. Hiroshima, T. Isobe, T. Koizuka and S. Takashima, Photostability of YAG:Ce3+ Nanophosphors Synthesized by Glycothermal Method, J. Electrochem. Soc., 2010, 157(5), J149–J154 CrossRef.
- R. Kasuya, T. Isobe, H. Kuma and J. Katano, Photoluminescence Enhancement of PEG-Modified YAG:Ce3+ Nanocrystal Phosphor Prepared by Glycothermal Method, J. Phys. Chem. B, 2005, 109(47), 22126–22130 CrossRef PubMed.
- N. Pradal, G. Chadeyron, A. Potdevin, J. Deschamps and R. Mahiou, Elaboration and optimization of Ce-doped Y3Al5O12 nanopowder dispersions, J. Eur. Ceram. Soc., 2013, 33, 1935–1945 CrossRef.
- A. Revaux, G. Dantelle, N. George, R. Seshadri, T. Gacoin and J. P. Boilot, Improvement of luminescent properties and photostability of YAG nanoparticle-based films, Nanoscale, 2011, 3, 2015–2022 RSC.
- G. Dantelle, M. Salaün, R. Bruyère, S. Kodjikian and A. Ibanez, Luminescent coatings prepared from optimized YAG:Ce nanoparticles, Thin Solid Films, 2017, 643, 36–42 CrossRef.
- C. Li, S. Bolisetty and R. Mezzenga, Hybrid Nanocomposites of Gold Single-Crystal Platelets and Amyloid Fibrils with Tunable Fluorescence, Conductivity, and Sensing Properties, Adv. Mater., 2013, 25, 3694–3700 CrossRef PubMed.
- D. Testemale, R. Argoud, O. Geaymond and J.-L. Hazemann, High pressure/high temperature cell for X-ray absorption and scattering techniques, Rev. Sci. Instrum., 2005, 76, 043905 CrossRef.
- M. Louvel, A. Bordage, C. Da Silva-Cadoux, D. Testemale, E. Lahera, W. Del Net, O. Geaymond, J. Dubessy, R. Argoud and J.-L. Hazemann, A high-pressure high-temperature setup for in situ Raman spectroscopy of supercritical fluids, J. Mol. Liq., 2015, 205, 54–60 CrossRef.
- A. Le Bail, Whole pattern decomposition methods and applications: a retrospection, Powder Diffr., 2012, 20, 316–326 CrossRef.
- A. L. Patterson, The Scherrer formula for X-ray particle size determination, Phys. Rev., 1939, 56, 978–982 CrossRef.
- https://imagej.nih.gov/ij/.
- M. Tella, M. Auffan, L. Brousset, E. Morel, O. Proux, C. Chanéac, B. Angeletti, C. Pailles, E. Artells, C. Santaella, J. Rose, A. Thiéry and J.-Y. Bottero, Chronic dosing of a simulated pond ecosystem in indoor aquatic mesocosms: fate and transport of CeO2 nanoparticles, Environ. Sci.: Nano, 2015, 2, 653 RSC.
- https://bruceravel.github.io/demeter/.
- M. Vorsthove and U. Kynast, Efficiency issues in Ce3+ doped YAG nanocrystals, Mater. Res. Bull., 2011, 46, 1761–1765 CrossRef.
- T. Sugimoto, Monodispersed Particles, Elsevier, Amsterdam, 2001 Search PubMed.
- P. Ramanujam, B. Vaidhyanathan, J. G. P. Binner, S. Ghanizadeh and C. Spacie, Solvothermal nanoYAG synthesis: mechanism and particle growth kinetics, J. Supercrit. Fluids, 2016, 107, 433–440 CrossRef.
- S. Hosokawa, Y. Tanaka, S. Iwamoto and M. Inoue, Defect structure of rare earth aluminium garnets obtained by the glycothermal method, J. Alloys Compd., 2008, 451, 309–313 CrossRef.
- R. German, Sintering: From Empirical Observations to Scientific Principles Butterworth-Heinemann, Oxford, 2014 Search PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Mechanisms of Nucleation and Growth of Nanoparticles in Solution, Chem. Rev., 2014, 114, 7610–7630 CrossRef PubMed.
- G. Dantelle, R. Calderón-Villajos, C. Zaldo, C. Cascales and T. Gacoin, Nanoparticulate Coatings with Efficient Up-Conversion Properties, ACS Appl. Mater. Interfaces, 2014, 6, 22483–22489 CrossRef PubMed.
- R. S. Meltzer, S. P. Feofilov, B. Tissue and H. B. Yuan, Dependence of fluorescence lifetimes of Y2O3:Eu3+ nanoparticles on the surrounding medium, Phys. Rev. B, 1999, 60(20), R14012 CrossRef.
- A. Pillonnet, P. Fleury, A. I. Chizhik, A. M. Chizhik, D. Amans, G. Ledoux, F. Kulzer, A. J. Meixner and C. Dujardin, Local refractive index probed via the fluorescence decay of semiconductor quantum dots, Opt. Express, 2012, 20(3), 3200 CrossRef PubMed.
- A. Aubret, A. Pillonnet, J. Houel, C. Dujardin and F. Kulzer, CdSe/ZnS quantum dots as sensors for the local refractive index, Nanoscale, 2016, 8, 2317–2325 RSC.
- H. Zheng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Blue Luminescence of ZnO Nanoparticles Based on Non-Equilibrium Processes: Defect Origins and Emission Controls, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef.
- B. Masenelli, O. Mollet, O. Boisron, B. Canut, G. Ledoux, J.-M. Bluet, P. Melinon, Ch. Dujardin and S. Huant, YAG:Ce nanoparticle lightsources, Nanotechnology, 2013, 24, 165703 CrossRef PubMed.
- A. Lyberis, G. Patriarche, D. Vivien and M. Mortier, Origin of light scattering in ytterbium doped calcium fluoride transparent ceramic for high power lasers, J. Eur. Ceram. Soc., 2011, 21(9), 1619–1630 CrossRef.
- G. Boulon, Y. Guyot, M. Guzik, T. Epicier, P. Gluchowski, D. Hreniak and W. Strek, Yb3+ Ions Distribution in YAG Nanoceramics Analyzed by Both Optical and TEM-EDX Techniques, J. Phys. Chem. C, 2014, 118, 15474–15486 CrossRef.
- F. Moretti, K. Hovhannesyan, M. Derdzyan, G. A. Bizarri, E. D. Bourret, A. Petrosyan and C. Dujardin, Consequences of Ca codoping in YAlO3:Ce single crystals, ChemPhysChem, 2017, 18, 493–499 CrossRef PubMed.
- G. Ozen and B. Demirata, Energy transfer characteristics of the hydrogen peroxide induced Ce3+–Ce4+ mixture, Spectrochim. Acta, Part A, 2000, 56, 1795–1800 CrossRef.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Solubility of Oxygen in Organic Solvents and Calculation of the Hansen Solubility Parameters of Oxygen, Ind. Eng. Chem. Res., 2014, 53(49), 19331–19337 CrossRef.
- A. N. Bourns, The vapour phase dehydration of butanediols, PhD Thesis, McGill Univ., 1944.
- T. Naganuma and E. Traversa, Air, aqueous and thermal stabilities of Ce3+ ions in cerium oxide nanoparticle layers with substrates, Nanoscale, 2014, 6, 6637–6645 RSC.
- G. Dantelle, D. Testemale, S. Kodjikian, A. Ibanez, Synthesis of high-quality garnet-type Gd3Sc2Al3O12:Ce3+ nanocrystals, SPIE Proceeding, 2018, vol. 10533, pp. 153322 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM images and nanocrystal size distribution histograms (.pdf file). A movie for the in situ visualization upon YAG nanocrystal synthesis (.mpeg file). See DOI: 10.1039/c8ra05914d |
| This journal is © The Royal Society of Chemistry 2018 |