
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-glucose 3D INS-1 cell model combined with a microfluidic circular concentration gradient generator for high throughput screening of drugs against type 2 diabetes†
Yong Luo *ab,
Xiuli Zhangc,
Yujiao Lia,
Jiu Denga,
Xiaorui Lia,
Yueyang Qua,
Yao Luc,
Tingjiao Liud,
Zhigang Gaoa and
Bingcheng Linc
*ab,
Xiuli Zhangc,
Yujiao Lia,
Jiu Denga,
Xiaorui Lia,
Yueyang Qua,
Yao Luc,
Tingjiao Liud,
Zhigang Gaoa and
Bingcheng Linc
aState Key Laboratory of Fine Chemicals, Department of Chemical Engineering & School of Pharmaceutical Science and Technology, Dalian University of Technology, Dalian, 116024, China. E-mail: yluo@dlut.edu.cn
bState Key Laboratory of Bioelectronics, Southeast University, Nanjing, 210096, China
cDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
dSection of Oral Pathology, College of Stomatology, Dalian Medical University, Dalian, 116044, China
First published on 16th July 2018
Abstract
In vitro models for screening of drugs against type 2 diabetes are crucial for the pharmaceutical industry. This paper presents a new approach for integration of a three-dimensionally-cultured insulinoma cell line (INS-1 cell) incubated in a high concentration of glucose as a new model. In this model, INS-1 cells tended to aggregate in the 3D gel (basement membrane extractant, BME), in a similar way to 3D in vivo cell culture models. The proliferation of INS-1 cells in BME was initially promoted and then suppressed by the high concentration of glucose, and the function of insulin secretion also was initially enhanced and then inhibited by the high concentration of glucose. These phenomena were similar to hyperglycemia symptoms, proving the validity of the proposed model. This model can help find the drugs that stimulate insulin secretion. Then, we identified the difference between the new model and the traditional two-dimensional model in terms of cell morphology, inhibition rate of cell proliferation, and insulin secretion. Simultaneously, we developed a circular drug concentration gradient generator based on microfluidic technology. We integrated the high-glucose 3D INS-1 cell model and the circular concentration gradient generator in the same microdevice, tested the utility of this microdevice in the field of drug screening with glipizide as a model drug, and found that the microdevice was more sensitive than the traditional device to screen the anti-diabetic drugs.
Introduction
Type 2 diabetes is a long-term metabolic disease that is characterized by high blood sugar, lack of insulin (type I diabetes), insulin resistance (type II diabetes), etc.1,2 Type 2 diabetes occurs frequently in obese people3,4 or middle and upper age groups, who often have been diagnosed with congenital genes, or lack physical activity. As the obesity rate increased,5 by the year 2013, the number of people with type 2 diabetes increased to 368 million.6 Later stages of type 2 diabetes are often accompanied by heart disease, stroke, diabetic retinopathy, kidney failure, poor circulation and the need for amputation, etc.,7 and they are associated with a ten-year-shorter life expectancy.Development of drugs against type 2 diabetes always is a significant area of development in the pharmaceutical industry. In vivo and in vitro models of type 2 diabetes have been widely used in this field. In vivo models mainly are experimental animals, like mouse8,9 and rat.10,11 In vitro models include isolated β-cells, pancreatic islets, insulin-secreting cell lines, hepatocyte, muscle and adipose cell lines, etc. In vitro models are majorly used in large-scale, high throughput, high-efficiency preliminary screening.12 For example, pancreatic islets isolated from adult mice and rats have been used to compare the insulin-releasing activity of novel (nonsulphonylurea) insulin secretagogues to various sulphonylureas.13,14
Insulin-secreting cell lines, such as insulinoma cell line (INS-1 cell),15–17 have been previously used to develop in vitro models for drug development against type 2 diabetes. INS-1 cell is derived from a rat insulinoma induced by X-ray irradiation, which is responsive to glucose within physiological range, and of relatively high insulin content.18 Traditionally, INS-1 cells were two-dimensional cultured in the Petri dish or well plate, in which the cells adhered and spread on the bottom surface.15 However, currently, many studies revealed three-dimensional culture of cells were advantageous over the two-dimensional cultures.19,20 General protocols for 3D culture included spheroid culture21 and forming cell-laden hydrogel either by mixing22 or 3D-printing.23 For example, hepatocytes spheroids have been established as a cutting-edge model for evaluation of drug-induced hepatotoxicity, taking over the two-dimensional culture of hepatocytes.24
In this study, we cultured the INS-1 cells in a hydrogel matrix, as well as performed 2D biofilm cell culture in a Petri dish, and studied the differences between two-dimensional culture and three-dimensional culture of INS-1 cells as models for screening of drugs that stimulates insulin secretion. We validated that our three-dimensionally-cultured cell models indeed produced more insulin production response than comparable two-dimensionally-cultured models. On the basis of this observation, we developed a novel drug screening device, incorporating a 3D INS-1 cell culture model and a microfluidic concentration gradient generation platform.
Microfluidic devices have had extensive use in the field of drug screening over the past few decades.25–27 For example, Kim et al. described the development of a fully automatic and programmable microfluidic cell culture array that integrates on-chip generation of drug concentrations and pairwise combinations with parallel culture of cells for drug candidate screening applications.27 The advantages of microfluidic drug screening include, on-line detection,28 automatic generation of drug concentration gradient,29 high throughput,30 high-level integration,31 minute sample consumption,32 etc. In particular, drug concentration gradient can be automatically generated on a microfluidic device. This function has been widely used in various microfluidic applications.33–35 In this study, we proposed a circular concentration gradient generator, coupled it with the three-dimensional INS-1 cell model in a microdevice, and investigated the potential of the microdevice in screening of drug against type 2 diabetes.
Materials and methods
Materials
INS-1 cells, RPMI 1640 medium and 15% fetal bovine serum were purchased from National Infrastructure of Cell Line Resource, Chinese Academy of Medical Sciences. Penicillin and streptomycin were from Hyclone, USA. Basement membrane extractant (BME) was from R&D Systems, MN, USA. Sodium fluorescein and glipizide were from Sigma, St Louis, MO, USA. Calcein-AM and PI dyes were from Keygen biotech, Nanjing, China. Ultrasensitive Rat Insulin ELISA kit was from Mercodia, Sweden. PDMS prepolymer (Sylgard Silicone elastomer 184) was from Dow Corning, Midland, MI, USA. SU8 3035 negative photoresist was from Microchem, Newton, MA, USA.INS-1 cell culture in BME
We thawed BME at 4 °C in a refrigerator, trypsinized the INS-1 cells (0.25% trypsin in PBS, 2 min), centrifuged them (RMPS: 800, volume: 5 mL, duration: 5 min), and resuspended them at 106 cells per mL in the ice-cold BME (0 °C). Then cell–BME mixture was loaded into the cell culture chambers located in the microdevice shown in Fig. 4 by pipetting.22 After filling the chambers with cell–BME mixture, we moved the chambers to room temperature for 30 min to allow the cell–BME mixture to gel. The gel were then cultured at 37 °C with 5% CO2 and 95% relative humidity in perfused RPMI 1640 medium supplemented with 15% fetal bovine serum and 100 U mL−1 penicillin and 100 U mL−1 streptomycin.Characterization of high-glucose 3D INS-1 cell model
The RPMI 1640 media containing 5.6 mmol L−1 and 25.5 mmol L−1 glucose were perfused on the surface of BME–cell gel, at a flow rate of 1.0 μL min−1 for 24 h, 36 h, 48 h, 72 h, 96 h, respectively. 5.6 mmol L−1 and 25.5 mmol L−1 corresponded to control and high-glucose experimental groups, respectively. Then, we carried out MTT experiment, Calcein-AM/PI staining experiment, and insulin secretion measurement.Microfluidic device fabrication
The microchannels and chambers in the PDMS layers of microfluidic device were fabricated by replicate molding on the master. The master was prepared by spin coating SU8 3035 negative photoresist (Microchem, Newton, MA, USA) onto a plasma-treated glass wafer and patterned by photolithography. Sylgard 184 PDMS base and curing agent (Dow corning, Midland, MI, USA) were mixed thoroughly (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by mass), degassed under vacuum, and poured onto the silane-treated master. The assembly was cured in an oven (80 °C, 1 h). After cooling, the PDMS was gently peeled from the master and trimmed to size. Holes were punched out of the PDMS to form reservoirs for introduction of liquid.
:1 by mass), degassed under vacuum, and poured onto the silane-treated master. The assembly was cured in an oven (80 °C, 1 h). After cooling, the PDMS was gently peeled from the master and trimmed to size. Holes were punched out of the PDMS to form reservoirs for introduction of liquid.
Testing of microfluidic concentration gradient
Driven by the syringe pump, 5 × 10−6 mol L−1 sodium fluorescein solution and sterile water were respectively injected from the two injection ports of the chip at a flow rate of 1.0 μL min−1. Until a stable concentration gradient was formed, we took the solution from each channel outlet to measure the fluorescence intensity with a microplate reader.Testing of the microdevice with glipizide
Perfuse RPMI 1640 medium containing 25.5 mmol L−1 glucose through the microchannel network for 96 h at a flow rate of 1.0 μL min−1, followed by loading of RPMI 1640 medium containing 0.24 μM glipizide and 25.5 mmol L−1 glucose at the inlet and perfusion for another 48 h. At 24 h and 48 h after addition of glipizide, carry out MTT experiments and insulin secretion assays as described previously.Results and discussions
Three-dimensional culture of INS-1 cells
We mixed INS-1 cells with basement membrane extractant (BME), and placed the mixture in a perfusion chamber for long-term cell culture. It can be observed from Fig. 1B that the cells demonstrated spheroid shape in the BME, and these cells tended to aggregate together instead of evenly distributed in the BME. In contrast, the INS-1 cells attached to the bottom surface and spread, when they were cultured in a two-dimensional well plate (Fig. 1A). The aggregation of cells in three-dimensional matrix was also reported with hepatocytes36 and tumor cells.22 In these literatures, the difference in cell morphology between two-dimensional and three-dimensional cultures was reported to affect the physiological behaviours of cells.22,36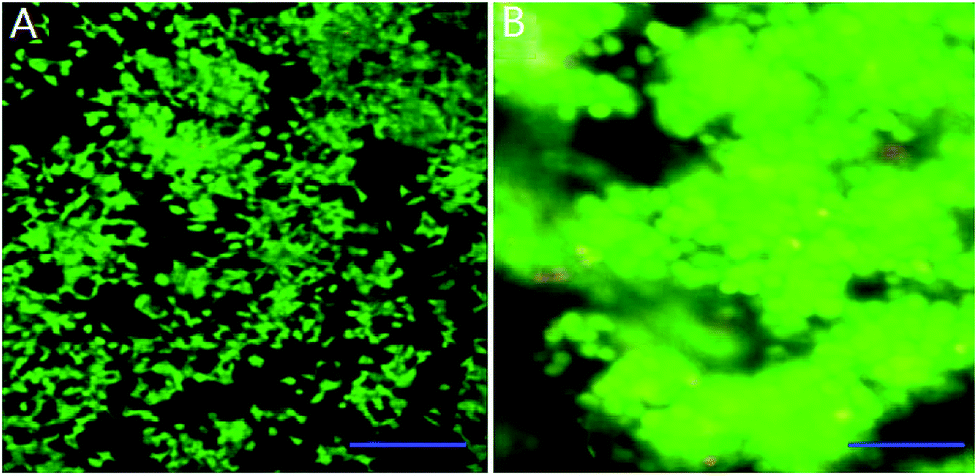 | ||
| Fig. 1 Observation of cell growth of INS-1 cells in three-dimensional and two-dimensional culture conditions. (A) Fluorescence image of INS-1 cells in well plate (6 days). Scale bar = 200 μm; (B) fluorescence image of INS-1 cells in BME (6 days). Scale bar = 50 μm. | ||
Characterization of high-glucose INS-1 cell model
One of the characteristics of type 2 diabetes is insulin insufficiency leading to hyperglycemia. Hyperglycemia in turn acts back on islet cells. The effect is complex. In brief, at the beginning, hyperglycemia promotes the proliferation of islet cells, which in turn increases insulin secretion to lower blood sugar levels. However, after a period of time, hyperglycemia may damage islet cells and make diabetes more serious.We observed the effect of high concentration of glucose on the viability of 3D INS-1 cells in the BME by Calcein-AM/PI staining experiment (Fig. 2) and in the well plate (Fig. S1 in ESI†). As shown in Fig. 2, at 24 h, 36 h and 48 h, there was no observable difference between the control and experimental groups; however, at 72 h and 96 h, obvious apoptosis of INS-1 cells in the experimental group occurred, while the control still had a good viability. This phenomenon was also observed in Fig. S1.† This result implied that the effect of high glucose on the 3D INS-1 cells may not be consistent over time. Thus, we measured the high-glucose-induced inhibition of the cell proliferation in BME. As shown in Fig. 3A (black solid line), at 24, 36, and 48 h, the inhibition rate of experimental group were lower than that of control, which meant that high concentration of glucose promoted the proliferation of 3D INS-1 cells within 48 h; however, at 72 and 96 h, the inhibition rate of experimental group was higher than that of control, which meant high glucose stimulation inhibited the proliferation of 3D INS-1 cells. These data can explain the phenomena in Fig. 2. In addition, it also can be observed that, with the incubation time increased, the inhibition rate increased. All of these phenomena are similar to the in vivo phenomena described in the first paragraph in this section.
 | ||
| Fig. 2 Time-resolved fluorescence images of INS-1 cells cultured in the BME. The green and red colors indicated live and dead cells, respectively. Scale bar = 50 μm. | ||
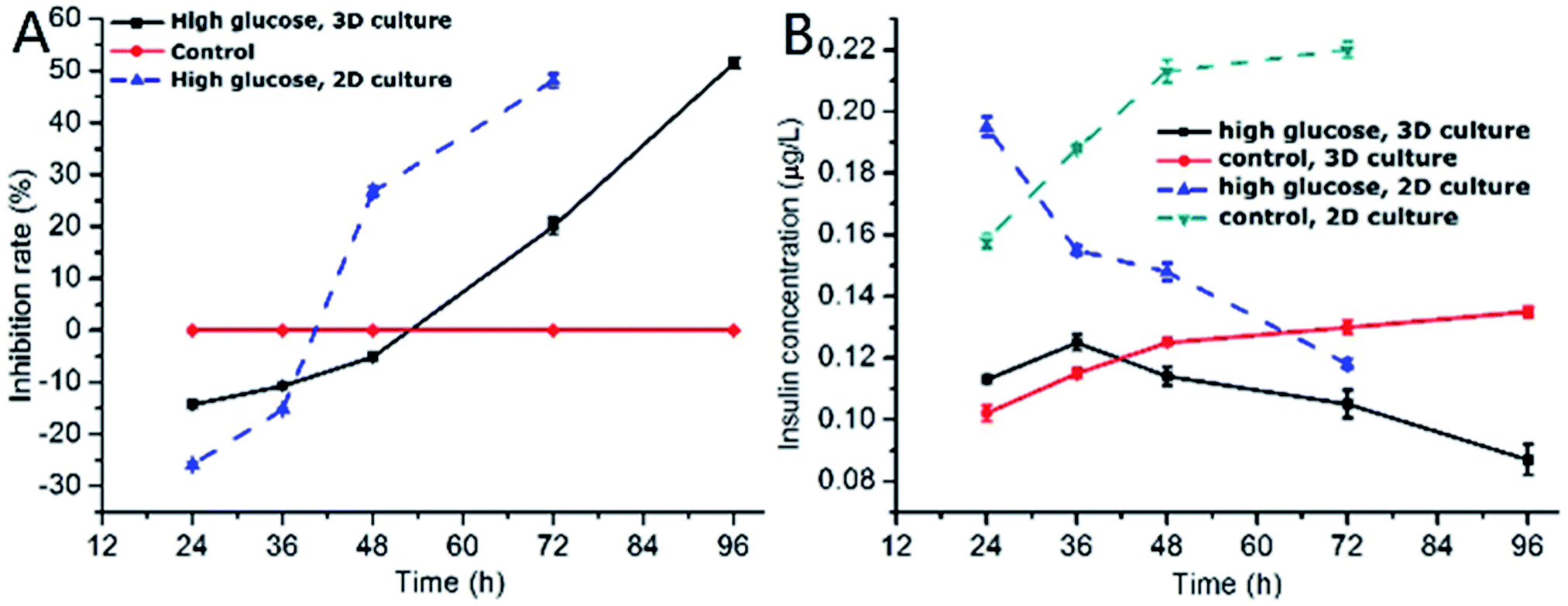 | ||
| Fig. 3 (A) Time-dependence of inhibition rate of INS-1 cell proliferation in 3D BME and 2D well plate, respectively. The high glucose concentration was 25.5 mmol L−1 and 25.0 mmol L−1 in BME and well plate, respectively. The control was 5.6 mmol L−1. The inhibition rates of control in BME and well plate were same. n = 3; (B) time-dependence of insulin secretion of INS-1 cells cultured in 3D BME and 2D well plate, respectively. The high glucose concentration was 25.5 mmol L−1 and 25.0 mmol L−1 in BME and well plate, respectively. The control was 5.6 mmol L−1. n = 3. | ||
As comparison, we also investigated the effect of high concentration of glucose on inhibition rate of INS-1 cell proliferation in two dimensional well plate, as shown in Fig. 3A (dashed line). As compared to the inhibition rate in BME, that in well plate ramped up more drastically with time. In addition, at 48 h, high glucose promoted the proliferation of INS-1 cells in BME, however, inhibited the proliferation in well plate. These differences implied that the phenotypes of INS-1 cells in well plate and BME were different, and undermined the drug screening model based on two-dimensional culture of INS-1 cells.
Subsequently, we evaluated the effect of high glucose on insulin secretion function of INS-1 cells in BME. As shown in Fig. 3B (solid line), at 24 and 36 h, the insulin secretion of high-glucose experimental group was larger than control group, which meant that high concentrations of glucose stimulated 3D INS-1 cells to secrete insulin within 36 h; but with prolonged culture time, we found the insulin secretion of high-glucose experimental group was lower than control group, which meant high concentration of glucose showed inhibitory effects on insulin secretion of 3D INS-1 cells. In addition, with the increase of culture time, inhibition effect increased, and maximized at the end (96 h). All of these phenomena agreed well with the in vivo phenomena described in the first paragraph in this section.
We also investigated the effect of high concentration of glucose on insulin secretion function of INS-1 cells cultured in two dimensional well plate, as shown in Fig. 3B (dashed line). As compared to the insulin secretion of INS-1 cells in BME, that in well plate ramped down more drastically with time. In addition, at 36 h, high glucose stimulated 3D INS-1 cells to secrete insulin; however, inhibited 2D INS-1 cells to secrete insulin. These differences strengthened the viewpoint that the phenotypes of INS-1 cell in well plate and BME are different, and undermined the drug screening model based on two-dimensional culture of INS-1 cells.
Notably, within 48 h, high concentration of glucose promoted the proliferation of 3D INS-1 cells (Fig. 3A, solid line), however, high concentration of glucose stimulated 3D INS-1 cells to secrete insulin only within 36 h (Fig. 3B, solid line). This fact meant proliferation of INS-1 cells in BME did not definitely lead to increase in insulin secretion. A possible explanation is that, long-time stimulation of high glucose causes stress-induced expansion of INS-1 cells, leading to cell hypertrophy and decreased activity.
Circular drug concentration gradient generator on a microfluidic device
Fig. 4A shows the channel design of the on-chip concentration gradient generator. The drug was pumped into the microchannel network via red inlet (point b) in the center, and the cell culture medium via white inlet (point a) in the center. After a radial splitting–mixing–splitting–mixing process in the microchannel network, five evenly-distributed concentrations were produced at 20 outlets. In detail, assuming the drug concentration at the point b was C0, the concentration at outlet 9, 10, 11, 12 were equal to that at the point c which was equal to that at the point b (=C0). Similarly, the concentrations at outlet 1, 2, 19, 20 was equal to that at the point a (=0). The solution at outlet 5, 6, 15, 16 were the 1:1 mixture of drug solution from the point b and the culture medium from point a, therefore their concentration was C0/2. The solution at outlet 7, 8 were the 1:1 mixture of the solution from the point e (=C0/2) and c (=C0), therefore their concentration was 3C0/4. Similarly, the solution at outlet 13, 14 were the 1:1 mixture of the solution from the point i (=C0/2) and c (=C0), therefore their concentration was 3C0/4. The solution at outlet 3, 4 were the 1:1 mixture of the solution from the point e (=C0/2) and g (=0), therefore their concentration was C0/4. Similarly, the solution at outlet 17, 18 were the 1:1 mixture of the solution from the point i (=C0/2) and g (=0), therefore their concentration was C0/4. Experimentally, C0 = 5 × 10−6 mol L−1, and its corresponding fluorescence intensity was 1490. Thus, the theoretical fluorescence intensity at outlet 9, 10, 11, 12 was 1490, outlet 7, 8, 13, 14 was 1490 × 3/4 = 1117.4, outlet 5, 6, 15, 16 was 1490/2 = 745, outlet 3, 4, 17, 18 was 1490/4 = 372.5, and outlet 1, 2, 19, 20 was 0. These theoretical value agreed well with the measured value, as demonstrated in Fig. 4B. In this design, each concentration corresponded to four outlets, which meant 4 parallel experiments can be carried out simultaneously in a single chip, increasing the experimental efficiency. The concentration variation between four parallel outlets were negligible (RSD = 0.5%). The concentration at each outlet were measured by fluorescence microscopy with sodium fluorescein as “drug”. The experimental and theoretical value was plotted in Fig. 4B. A good agreement was observed (R2 was 0.9983). This concentration gradient generator can use the space on a microdevice efficiently, and is especially suitable for applications on a circular chip.
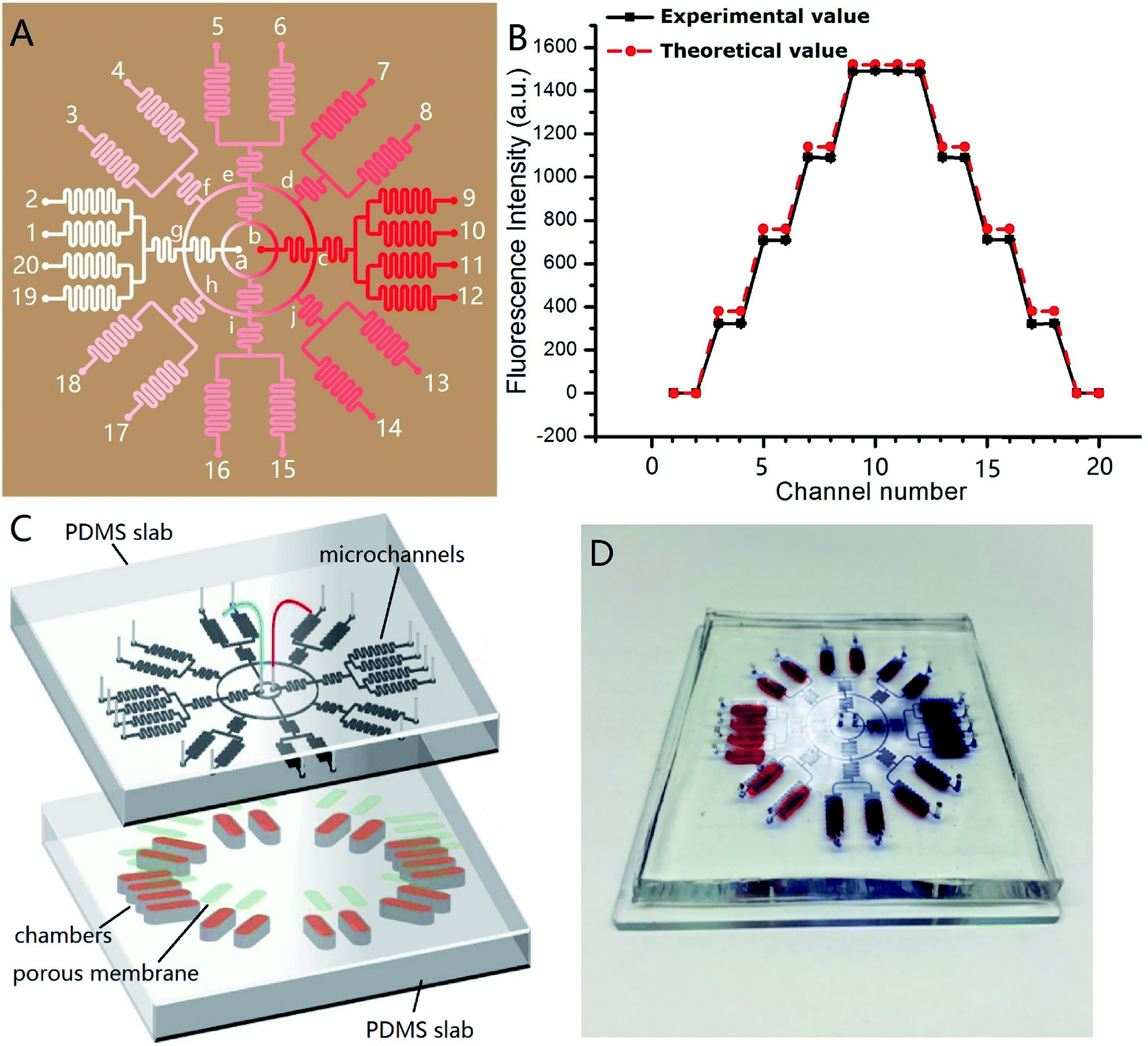 | ||
| Fig. 4 (A) Illustration of channel design of the circular concentration gradient. The red and white spots in the center were solution inlets, and the spots labelled with numbers were solution outlets. The color gradient was simple illustration to show the concentration distribution within the microdevice; (B) plot of the concentration at each outlet, theoretical value and experimental value; n = 3. (C) Exploded view of the integrated microdevice. The top PDMS layer was concentration gradient generator. The bottom PDMS layer embedded 20 identical chambers (indicated by brown color), in which 3D INS-1 cells were cultured. The light green color indicated porous membrane sandwiched in between; (D) photograph of the real microdevice. | ||
Microdevice coupling high-glucose 3D INS-1 cell model and circular concentration gradient generator for screening of anti-diabetic drug
Fig. 4C shows the design of the integrated microdevice. 20 chambers were radially arranged on the substrate. Each chamber was loaded with 100 μL of the mixture of INS-1 cells and BME. The top of the chamber was covered with a porous polycarbonate membrane. The polycarbonate porous membranes (10 μm pore size, Whatman, cyclopore™) were aligned with the exit microchannels of the concentration gradient generator above. The real microdevice was shown in Fig. 4D. The blue color indicated concentration gradient of blue dyes, and the red colors indicated the locations of culture chambers.We tested the efficacy of this microdevice in the field of screening drugs that stimulates insulin secretion with glipizide as model drug. Physiologically, glipizide binds to the sulfonylurea receptor on the cell membrane of pancreatic β-cells, resulting in the K+ channel closure on the cell membrane and depolarization of the cell membrane, which in turn opens Ca2+ channels on the cell membrane and promotes the Ca2+ flux, triggering insulin release.
As shown in Fig. 5A, after addition of glipizide into the INS-1 cell culture medium at 96 h, the increase of inhibition rate of 3D INS-1 cell proliferation became slower than that from 24 h to 96 h, indicating that glipizide had the effect of promoting the proliferation of 3D INS-1 cells. We also tested the traditional high-glucose 2D INS-1 cell model. As shown in Fig. 5B, after addition of glipizide into the INS-1 cell culture medium at 72 h, the increase of inhibition rate of 2D INS-1 cell proliferation also became slower than that from 24 h to 72 h, indicating that glipizide had the effect of promoting the proliferation of 2D INS-1 cells. However, it can be easily observed from Fig. 5A and B that, the variation of inhibition rate with glipizide concentration in 3D model was higher than that in 2D model. We quantitatively plotted the relationship between inhibition rate and glipizide concentration in Fig. 5C and D. At 24 h after addition of glipizide, the decrease rate of inhibition rate with glipizide concentration was 0.5916 and 0.3183 for 3D and 2D models, respectively (Fig. 5C). At 48 h after addition of glipizide, the decrease rate of inhibition rate with glipizide concentration was 0.9133 and 0.4817 for 3D and 2D models, respectively (Fig. 5D). No matter at 24 or 48 h after glipizide addition, the decrease rate of 3D high-glucose INS-1 cell model was larger than that of 2D model, which meant that the 3D high-glucose INS-1 cell model is more sensitive than the traditional 2D high-glucose INS-1 cell model in the field of drug screening against diabetes.
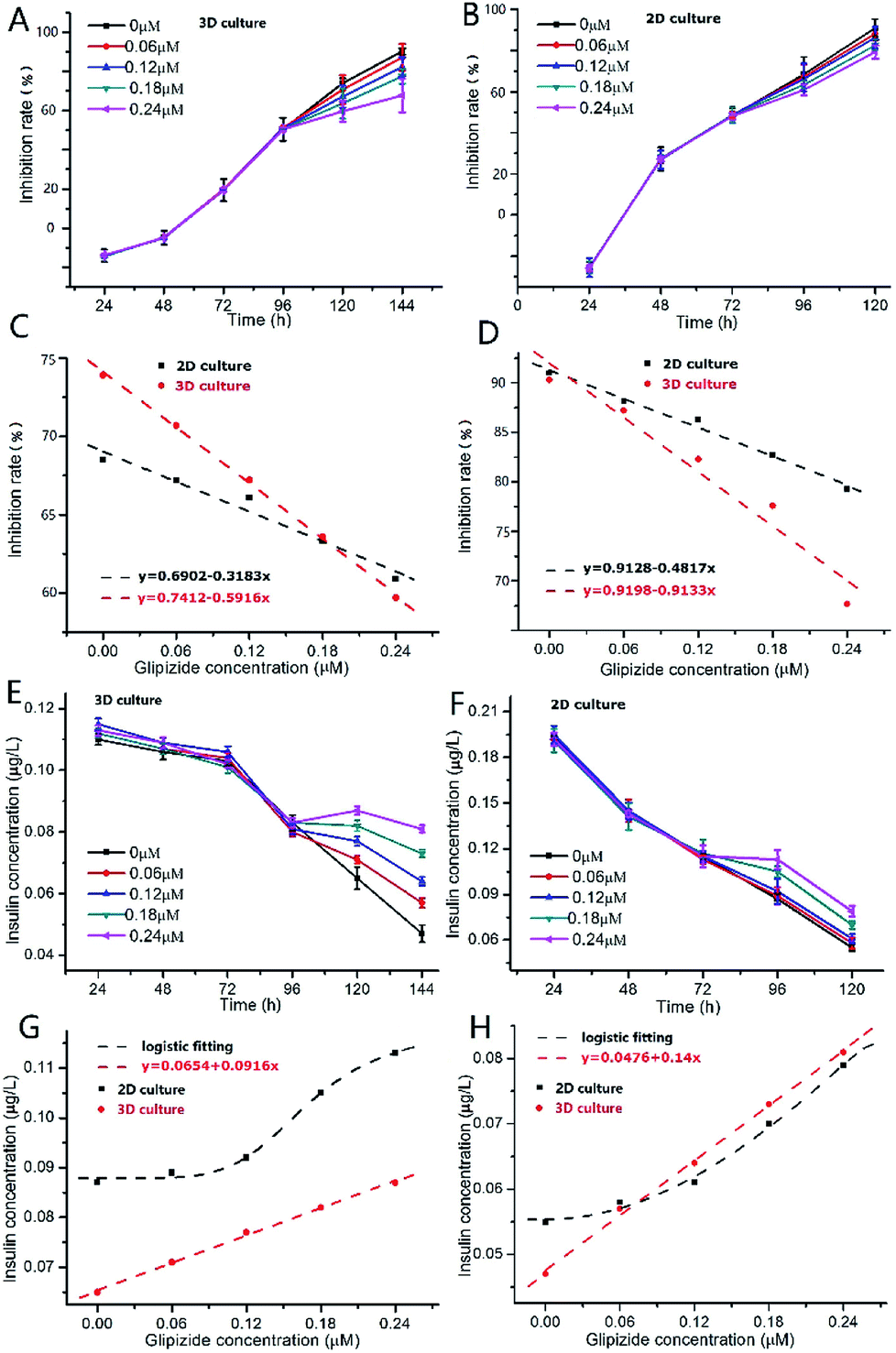 | ||
| Fig. 5 (A) Variation of inhibition rate of 3D INS-1 cell proliferation with culture time in the presence of high glucose (25.5 mmol L−1). At 96 h, glipizide was added into the culture medium; n = 4. (B) Variation of inhibition rate of 2D INS-1 cell proliferation with culture time in the presence of high glucose (25.5 mmol L−1). At 72 h, glipizide was added into the culture medium; n = 3. (C) Variation of inhibition rate with glipizide concentration in 3D and 2D high-glucose models at 24 h after addition of glipizide; (D) variation of inhibition rate with glipizide concentration in 3D and 2D high-glucose models at 48 h after addition of glipizide; (E) variation of insulin secretion function of 3D INS-1 cells with culture time in the presence of high glucose (25.5 mmol L−1). At 96 h, glipizide was added into the culture medium; n = 4. (F) Variation of insulin secretion function of 2D INS-1 cells with culture time in the presence of high glucose (25.5 mmol L−1). At 72 h, glipizide was added into the culture medium; n = 3. (G) Variation of insulin secretion with glipizide concentration in 3D and 2D high-glucose models at 24 h after addition of glipizide; (H) variation of insulin secretion with glipizide concentration in 3D and 2D high-glucose models at 48 h after addition of glipizide. | ||
As shown in Fig. 5E, after addition of glipizide into the INS-1 cell culture medium at 96 h, the decrease of insulin secretion of 3D INS-1 cell incubated in high glucose became slower than that from 24 h to 96 h, indicating that glipizide had the effect of stimulating 3D INS-1 cells to secret insulin. We also tested the traditional high-glucose 2D INS-1 cell model. As shown in Fig. 5F, after addition of glipizide into the INS-1 cell culture medium at 72 h, the decrease of insulin secretion of 2D INS-1 cells incubated in high glucose also became slower than that from 24 h to 72 h, indicating that glipizide had the effect of stimulating 2D INS-1 cells to secret insulin. However, it can be easily observed from Fig. 5E and F that, the variation of insulin secretion with glipizide concentration in 3D model was higher than that in 2D model. We quantitatively plotted the relationship between insulin secretion and glipizide concentration in Fig. 5G and H. At 24 h after addition of glipizide, insulin secretion varied linearly with glipizide concentration in 3D high-glucose model, and the slope was 0.0916 (red line, Fig. 5G). As compared, at 24 h after addition of glipizide, insulin secretion varied with glipizide concentration in an S-shape curve in 2D high-glucose model (black curve, Fig. 5G). At 48 h after addition of glipizide, insulin secretion varied linearly with glipizide concentration in 3D high-glucose model, and the slope was 0.14 (red line, Fig. 5H). As compared, at 48 h after addition of glipizide, insulin secretion varied with glipizide concentration in an S-shape curve in 2D high-glucose model (black curve, Fig. 5H). No matter at 24 or 48 h after glipizide addition, the increase rate of insulin secretion in 3D high-glucose model was larger than that of 2D model, especially at high concentration of glipizide, which strengthened the viewpoint that the 3D high-glucose INS-1 cell model is more sensitive than the traditional 2D high-glucose INS-1 cell model in the field of drug screening against diabetes. Notably, in 3D high-glucose model, both inhibition rate and insulin secretion are linearly related to the glipizide concentration, which facilitate the evaluation of efficacy of an anti-diabetic drug candidate.
Conclusion
We developed a 3D high-glucose insulinoma cell line (INS-1) model to screen the drug that stimulates insulin secretion. Further, we developed an integrated microdevice enabling high-throughput screening of anti-diabetic drugs, featuring the 3D INS-1 cell model. In a single run on a single chip, we can test 5 concentrations and implement 4 parallel experiments. Experimental data showed that the microdevice developed produced a greater insulin production response than a comparable two-dimensionally-cultured well plate.Conflicts of interest
The authors declare neither conflict of interest nor competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21675017), the National Key Research and Development Program of China (No. SQ2017YFC170204-001), the Fundamental Research Funds for the Central Universities, China (No. DUT17LK25), and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University. Yong Luo and Xiuli Zhang contributed equally to this work.Notes and references
- S. Brauer, J. Physiother., 2012, 58, 63 Search PubMed.
- A. J. Garber, Med. Clin. North Am., 2004, 88, XV–XVI CrossRef.
- A. Brown, N. Guess, A. Dornhorst, S. Taheri and G. Frost, Diabetes, Obes. Metab., 2017, 19, 1655–1668 CrossRef PubMed.
- L. Szablewski, Int. J. Diabetes Dev. Countries, 2017, 37, 407–418 CrossRef.
- S. Smyth and A. Heron, Nat. Med., 2006, 12, 75–80 CrossRef PubMed.
- T. Vos, R. M. Barber, B. Bell, A. Bertozzi-Villa, S. Biryukov, I. Bolliger, F. Charlson, A. Davis, L. Degenhardt, D. Dicker, L. Duan, H. Erskine, V. L. Feigin, A. J. Ferrari, C. Fitzmaurice, T. Fleming, N. Graetz, C. Guinovart, J. Haagsma and G. M. Hansen, Lancet, 2015, 386, 743–800 CrossRef.
- “Diabetes Fact sheet N°312”, World Health Organization, August 2011, Archived from the original on 26 August 2013, Retrieved 2012-01-09.
- K. P. Hummel, M. M. Dickie and D. L. Coleman, Science, 1966, 153, 1127–1128 CrossRef PubMed.
- H. Chen, O. Charlat, L. A. Tartaglia, E. A. Woolf, X. Weng, S. J. Ellis, N. D. Lakey, J. Culpepper, K. J. Moore, R. E. Breitbart, G. M. Duyk, R. I. Tepper and J. P. Morgenstern, Cell, 1996, 84, 491–495 CrossRef PubMed.
- B. D. White and R. J. Martin, Proc. Soc. Exp. Biol. Med., 1997, 214, 222–232 CrossRef PubMed.
- N. C. Turner and P. White, J. Cardiovasc. Pharmacol., 1996, 27, 884–890 CrossRef PubMed.
- M. J. Reed and K. A. Scribner, Diabetes, Obes. Metab., 1999, 1, 75–86 CrossRef.
- J. Fuhlendorff, P. Rorsman, H. Kofod, C. L. Brand, B. Rolin, P. MacKay, R. Shymko and R. D. Carr, Diabetes, 1998, 47, 345–351 CrossRef PubMed.
- T. Ikenoue, M. Akiyoshi, S. Fujitani, K. Okazaki, N. Kondo and T. Maki, Br. J. Pharmacol., 1997, 120, 137–145 CrossRef PubMed.
- S. Tao, Y. Ren, H. Zheng, M. Zhao, X. Zhang, Y. Zhu, J. Yang and S. Zheng, Eur. J. Pharmacol., 2017, 814, 56–62 CrossRef PubMed.
- S. Karandrea, H. Q. Yin, X. M. Liang and E. A. Heart, Environ. Toxicol. Pharmacol., 2018, 56, 29–34 CrossRef PubMed.
- X. T. Feng, H. M. Duan and S. L. Li, Int. J. Mol. Med., 2018, 40(3), 922–930 CrossRef PubMed.
- M. Asfari, D. Janjic, P. Meda, G. D. Li, P. A. Halban and C. B. Wollheim, Endocrinology, 1992, 130, 167–178 CrossRef PubMed.
- Z. Li and Z. Cui, Biotechnol. Adv., 2014, 32, 243–254 CrossRef PubMed.
- W. MuellerKlieser, Am. J. Physiol.: Cell Physiol., 1997, 273, C1109–C1123 CrossRef.
- Q. Meng, Expert Opin. Drug Metab. Toxicol., 2010, 6, 733–746 CrossRef PubMed.
- T. J. Liu, C. Y. Li, H. J. Li, S. J. Zeng, J. H. Qin and B. C. Lin, Electrophoresis, 2009, 30, 4285–4291 CrossRef PubMed.
- Y. Zhao, R. Yao, L. Ouyang, H. X. Ding, T. Zhang, K. T. Zhang, S. J. Cheng and W. Sun, Biofabrication, 2014, 6, 3 Search PubMed.
- P. Eribol, A. K. Uguz and K. O. Ulgen, Biomicrofluidics, 2016, 10(1), 011502 CrossRef PubMed.
- N. N. Ye, J. H. Qin, W. W. Shi, X. Liu and B. C. Lin, Lab Chip, 2007, 12, 1696–1704 RSC.
- E. Mancera-Andrade, A. Parsaeimehr, A. Arevalo-Gallegos, G. Ascencio-Favela and R. Parra Saldivar, Front. Biosci., 2018, 10, 74–91 CrossRef.
- J. Kim, D. Taylor, N. Agrawal, H. Wang, H. Kim, A. Han, K. Rege and A. Jayaraman, Lab Chip, 2012, 12, 1813–1822 RSC.
- S. Grist, L. Yu, L. Chrostowski and K. C. Cheung, in Microflu-idics, Biomems, and Medical Microsystems X, ed. H. Becker and B. L. Gray, 2012 Search PubMed.
- N. L. Jeon, H. Baskaran, S. K. W Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CrossRef PubMed.
- C. W. Chi, A. H. R. Ahmed, Z. Dereli-Korkut and S. Wang, Bioanalysis, 2016, 8, 921–937 CrossRef PubMed.
- N. Ye, J. Qin, W. Shi, X. Liu and B. Lin, Lab Chip, 2007, 7, 1696–1704 RSC.
- M. Courtney, X. Chen, S. Chan, T. Mohamed, P. P. N. Rao and C. L. Ren, Anal. Chem., 2017, 89, 910–915 CrossRef PubMed.
- S. K. W. Dertinger, X. Y. Jiang, Z. Y. Li, V. N. Murthy and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12542–12547 CrossRef PubMed.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef.
- X. Y. Jiang, Q. B. Xu, S. K. W. Dertinger, A. D. Stroock, T. M. Fu and G. M. Whitesides, Anal. Chem., 2005, 77, 2338–2347 CrossRef PubMed.
- Y. Miyamoto, M. Ikeuchi, H. oguchi, T. Yagi and S. Hayashi, Cell Med., 2015, 8, 47–56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04040k |
| This journal is © The Royal Society of Chemistry 2018 |