
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA porous Co–Ru@C shell as a bifunctional catalyst for lithium–oxygen batteries†
Xiang Chena,
Xiuhui Zhangb,
Chunguang Chenb,
Tao Huang*a and
Aishui Yu *ab
*ab
aLaboratory of Advanced Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Institute of New Energy, Collaborative Innovation Center of Chemistry for Energy Materials, Fudan University, Shanghai 200433, China
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Institute of New Energy, Collaborative Innovation Center of Chemistry for Energy Materials, Fudan University, Shanghai 200433, China. E-mail: asyu@fudan.edu.cn; Fax: +86-21-51630320; Tel: +86-21-51630320
First published on 2nd July 2018
Abstract
We use SiO2 as a template and dopamine as a carbon source to synthesize a hollow C shell, and we load Co and Ru nanoparticles onto it to obtain a Co–Ru@C shell composite. The diameter and thickness of the C shell are 100 nm and 5–10 nm, respectively, and numerous holes of different sizes exist on the C shell. Meanwhile, numerous C shells stack together to form macropores, thereby forming a hierarchical porous structure in the material. Brunauer–Emmett–Teller surface area analysis reveals that the specific surface area and pore volume of the Co–Ru@C shell are 631.57 m2 g−1 and 2.20 cc g−1, respectively, which can result in many three-phase interfaces and provide more space for the deposition of discharge products. Compared with Co@C shell and C shell electrodes, the obtained Co–Ru@C shell-based electrodes exhibit the highest discharge capacity, the lowest oxygen reduction reaction/oxygen evolution reaction overpotential and the best cycle stability, indicating the excellent catalytic ability of the Co–Ru@C shell.
1. Introduction
Recently, with the development of personal mobile devices and electric vehicles, the demand for high-efficiency energy storage devices has grown.1,2 As the most promising candidate for such devices, rechargeable lithium–oxygen batteries have extremely high theoretical energy density (5200 W h kg−1), which is similar to that of gasoline and thus, they have attracted attention of the global research community.3–5 However, some serious challenges such as the lack of clarity regarding the reaction mechanism and sluggish reaction kinetics impede their practical applications.6–8During the discharge process of a lithium–oxygen battery, the reaction material passes through channels in the material, which eventually produces insoluble Li2O2, whereas the holes in the oxygen electrode provide space for deposition of the discharge product. Thus, the pore volume and specific surface area of the materials affect the discharge capacity of the battery, which means that the structural design of the electrode material plays an important role in achieving super-high theoretical energy densities in lithium–oxygen batteries.9,10 Carbon materials are good choices for obtaining a specific structure and morphology and thus, they are widely used in the production of electrodes because of their low costs, large specific surface areas, and various controlled morphologies.11–14 Zhang et al. reported a novel air electrode consisting of an unusual hierarchical arrangement of functionalized graphene sheets (with no catalyst), which delivered exceptionally high capacity of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA h g−1 in Li–O2 batteries.15 Nie et al. reported a micron-sized honeycomb-like carbon material. Compared with batteries based on KB electrodes, batteries based on this material showed a significantly improved capacity (5862 mA h g−1).16
000 mA h g−1 in Li–O2 batteries.15 Nie et al. reported a micron-sized honeycomb-like carbon material. Compared with batteries based on KB electrodes, batteries based on this material showed a significantly improved capacity (5862 mA h g−1).16
However, due to the limited catalytic properties of carbon materials, loading a metal catalyst can make better use of their large specific surface area and special morphological structure, and further enhance the performance of the Li–O2 batteries. Transition metals and their oxides are the most commonly used catalytic materials because of their natural abundances and excellent catalytic performances.17–19 Yong et al. synthesized a carbon-nanotube (CNT)/Co3O4 nanocomposite, and the Li–O2 batteries based on this composite electrode showed superior electrochemical performances such as reduced overpotential and high discharge capacity (approximately 5000 mA h g−1).20 It is noteworthy that during the synthesis process Yong et al. used dopamine to assist the metal-particle growth on the CNT surface, because the hydroxyl groups in dopamine can attract metal ions and disperse the metal particles more uniformly during the growth process, improve the utilization rate, and increase the number of catalytically active sites.21 To further improve the catalytic performance, composite catalysts composed of noble metals and transition metals have been extensively studied. Kim et al. reported a one-dimensional RuO2/Mn2O3 hollow architecture as a catalyst in an air electrode; batteries based on this electrode showed extremely low overpotentials, and they could loop over 100 cycles with a limited specific capacity of 1000 mA h g−1.22 Zhu et al. reported a nano-structured RuO2/NiO composite cathode that enabled the operation of Li–air batteries in ambient air over 200 cycles.23
In this study, we use SiO2 as a template and dopamine as a carbon source to synthesize a hollow C shell, and we load Co and Ru nanoparticles onto it to obtain a Co–Ru@C shell composite. For comparison with Co@C shell and C shell, the obtained Co–Ru@C shell-based electrodes are used in Li–O2 batteries for electrochemical testing, and these batteries exhibit the best performance.
2. Experimental
2.1 Co–Ru@C shell synthesis
The synthesis process is illustrated in Fig. 1. All chemicals were used as received without any further purification.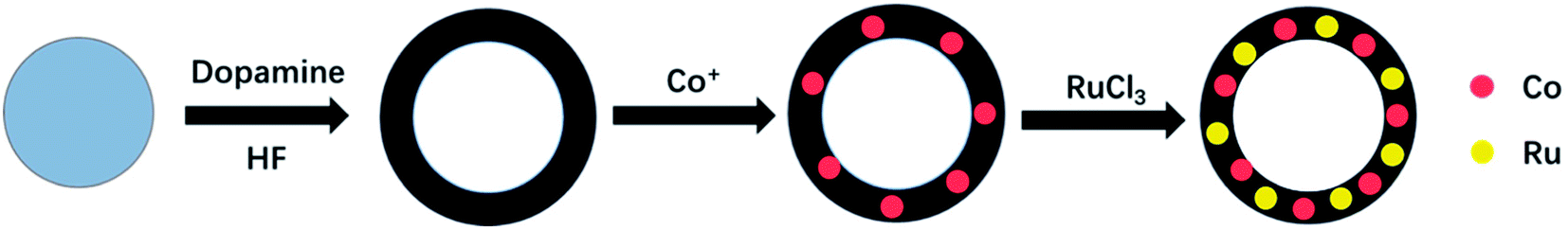 | ||
| Fig. 1 Co–Ru@C shell synthesis process. | ||
2.2 Material characterization
The morphologies of the samples were characterized by field-emission scanning electron microscopy (FESEM, JEOL JSM-6390) and high-resolution transmission electron microscopy (HRTEM, JEM-2100F). The crystal structures of the products were examined by X-ray diffraction (XRD, D8-Advance X-ray diffractometer, Cu-Kα source, λ = 1.5406 Å). N2 adsorption–desorption isotherms were obtained using a Micromeritics Tristar 3000 surface-area and pore-size analyzer. A Kratos Axis Ultra X-ray photoelectron spectrometer (Al-Kα source) was used to record X-ray photoelectron spectroscopy (XPS) spectra.2.3 Electrochemical measurement
Cathodes were prepared by casting a homogeneous mixture of 60 wt% as-synthesized Co–Ru@C shell, 20 wt% Super P (SP), and 20 wt% polytetrafluoroethylene (PTFE) onto a Ni foam (12 mm in diameter). These samples were then dried at 80 °C for 12 h in a dry oven to remove the solvent. The total mass loading of the Co–Ru@C shell, SP, and PTFE was approximately 1.5 mg cm−2. Then, non-aqueous Li–O2 cells were assembled in an Ar-filled glove box (H2O ≤ 0.1 ppm) using Swagelok batteries with an air window of 78.5 mm2, and it contained an O2 electrode, a Li foil anode, a Celgard 3500 membrane, and 1 M lithium bis(trifluoromethanesolphonyl)imide (LiTFSI) in tetraethylene glycol dimethoxyethane (TEGDME) electrolyte.Charge–discharge measurements were performed on a Land cycler (Wuhan Jinnuo Electronic Co., Ltd.) in the voltage range of 2.2–4.3 V versus Li+/Li at different current densities. The specific capacity was based on the amount of the entire composite and SP. Cyclic voltammograms (CVs) were recorded within 2.2–4.3 V at 0.2 mVs−1 on a CHI 660C electrochemical workstation (Shanghai Chenhua, China). Electrochemical impedance spectroscopy (EIS) measurements were performed on the CHI 660C system in the frequency range of 10000–0.1 MHz. All tests were carried out at room temperature.
3. Results and discussion
3.1 Co–Ru@C shell morphology and structure
Fig. 2a–c show XRD patterns of as-synthesized C shell, Co@C shell, and Co–Ru@C shell, respectively. As shown in Fig. 2a, the two broad peaks at 25° and 44° corresponding to the (002) and (101) planes of graphitic carbon demonstrate good crystallinity of the carbon in the as-prepared C shell. After loading Co particles, the three new peaks that appear at 44.3°, 51.6°, and 75.9° are assigned to the pure metallic cobalt phase (Fig. 2b). Meanwhile, Fig. 2c shows the peaks corresponding to Co and Ru. Fig. 2d shows the N2 adsorption–desorption isotherms and pore size distribution image of the Co–Ru@C shell. The Co–Ru@C sample exhibits type-IV isotherms following the International Union of Pure and Applied Chemistry (IUPAC) classification, indicating the existence of a mesoporous structure. The results of pore size distribution of the Co–Ru@C shell show that the pore sizes of the material are mainly distributed between 40 and 120 nm, and the material is mainly composed of mesopores on the C-shell surface and macropores formed by the accumulation of C shells. The specific surface area and cumulative pore volume of the Co–Ru@C shell are calculated to be 631.57 m2 g−1 and 2.20 cm3 g−1, respectively. A large specific surface area provides more active sites for electrochemical reactions, whereas a large pore volume can provide more space for discharge products, which play important roles in improving the battery capacity.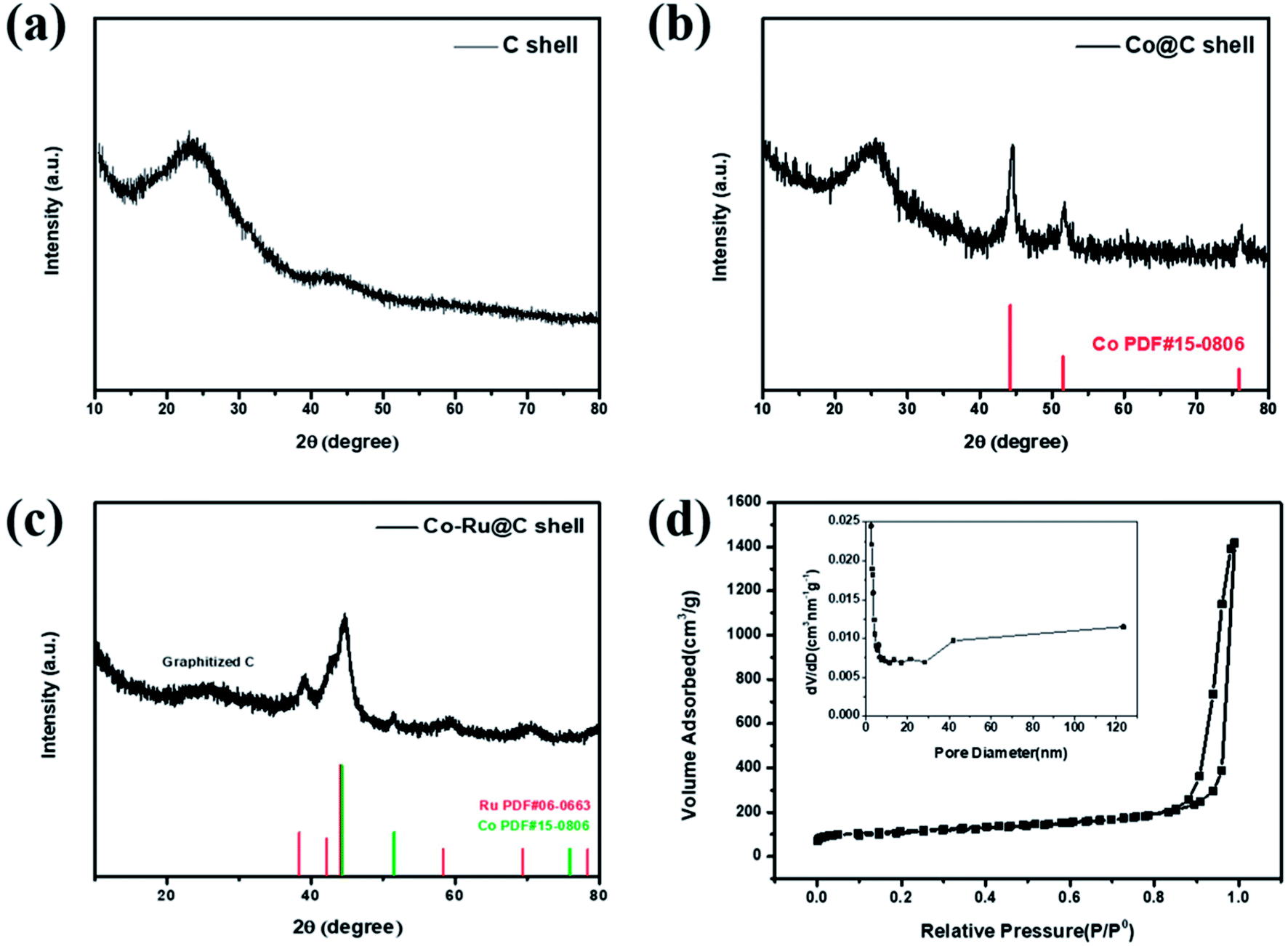 | ||
| Fig. 2 XRD patterns of (a) C shell, (b) Co@C shell, and (c) Co–Ru@C shell. (d) N2 adsorption–desorption isotherms and pore size distribution image of Co–Ru@C shell. | ||
To obtain structural information on the as-prepared materials, SEM and TEM images of the C shells and Co–Ru@C shells are shown in Fig. 3 and 4, respectively. As shown in Fig. 3, the C shell with a clear hollow structure has a diameter of 100 nm and thickness of 5–10 nm. Meanwhile, there are numerous holes of different sizes on the surface. The C shells stack together, resulting in high accumulation of holes; this is consistent with the result of Brunauer–Emmett–Teller (BET) surface area analysis.
 | ||
| Fig. 3 (a and b) SEM and (c and d) TEM images of the C shell. | ||
 | ||
| Fig. 4 (a and b) SEM, (c) TEM, (d–f) HRTEM images, and (g) EDS maps of the Co–Ru@C shell. | ||
Fig. 4 reveals more details regarding the structure of the Co–Ru@C shell. As shown in Fig. 4a and b, after loading Co and Ru particles, the diameter and thickness of the hollow carbon shell did not change, and the structure was well inherited. TEM images show that the metal particles were evenly distributed on the surface of the C shell with sizes of approximately 5–10 nm. The corresponding Co and Ru particles were found by calculating the interplanar spacing. The (002) plane of graphitic C was clearly observed. This result was ascribed to the fact that the metallic Co particles can effectively catalyze the formation of graphitic C, and graphitic C can further enhance the stability of the C shell. The EDS mapping images presented in Fig. 3g further confirmed that Co and Ru particles were uniformly distributed in the C shell, which was consistent with the results of the TEM images. The relative elemental contents of the Co–Ru@C shell obtained from EDS (TEM) are shown in Table S1.† The atomic fractions of C, Co and Ru were 73.32%, 6.74% and 19.93%, respectively.
3.2 Co–Ru@C shell electrochemical performance
The performances of Li–O2 batteries using Co–Ru@C shell electrodes were examined, and C and Co@C shell electrodes were employed for comparison. Fig. 5a shows the initial galvanostatic charge–discharge curves of Co–Ru@C shell, Co@C shell, and C shell electrodes at a current density of 0.1 mA cm−2. The C shell electrode showed a good discharge capacity of 1300 mA h g−1. However, during the charging process, the voltage rapidly increased, reaching the upper limit of the voltage window, which resulted in low coulombic efficiency. After loading Co particles, the charging potential of the batteries reduced significantly, and the charging voltage platform was approximately 3.85 V, indicating good catalytic activity of the Co@C shell for the oxygen evolution reaction (OER). However, the discharge specific capacity and voltage platform of batteries based on Co@C shell electrodes decreased slightly compared with those based on C shell electrodes. There are two reasons for these observations: First, the oxygen reduction reaction (ORR) catalytic activity of Co particles is relatively weak. Second, the quality of Co atoms is far greater than that of C atoms. After loading Co particles onto the C shell, the actual capacity of the battery increased, but the specific capacity decreased. After further loading Ru particles, the batteries based on Co–Ru@C shell electrodes had a great advantage with respect to both discharge capacity and overpotential and thus, they showed a remarkable initial discharge capacity of 2092 mA h g−1. Fig. 5b shows different CV curves at a scan rate of 0.2 mV s−1. The Co–Ru@C shell electrode exhibited the maximum cathode reduction and anodic oxidation peak currents. Compared with the observations for the C shell electrode, the reduction peak current of the Co@C shell electrode showed a slight increase and the oxidation peak current showed a significant advantage, which was consistent with the galvanostatic discharge–charge test results. Furthermore, we found that upon loading Co and Ru particles, the oxidation peak potential of the batteries decreased gradually, corresponding to the gradual decrease in the OER potential in the galvanostatic charge–discharge tests, further demonstrating the effect of the addition of Co and Ru catalysts on the OER catalytic activity.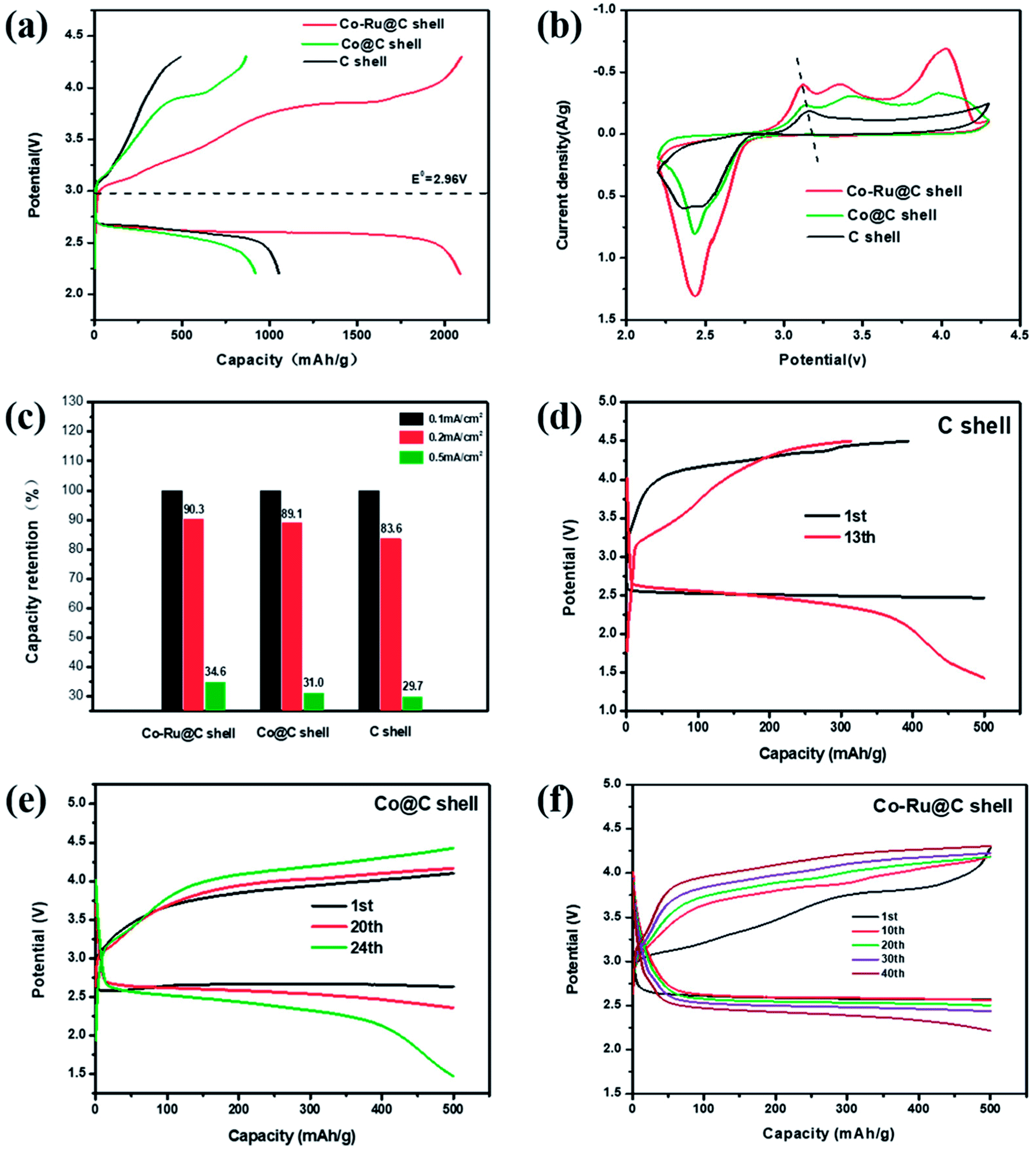 | ||
| Fig. 5 (a) Initial galvanostatic charge–discharge curves at a current density of 0.1 mA cm−2, (b) CV curves at a scan rate of 0.2 mV s−1, (c) capacity retention at different current densities, and (d–f) cycling stabilities under a specific capacity limit of 500 mA h g−1 at 0.2 mA cm−2 of the Li–O2 batteries based on C shell, Co@C shell, and Co–Ru@C shell electrodes, respectively. | ||
Fig. 5c shows capacity retentions at different current densities of batteries based on Co–Ru@C shell, Co@C shell, and C shell electrodes. The specific capacities of the three samples decreased by different degrees with the increase in current density. The Co–Ru@C shell electrode showed the best rate capability, and the Co@C shell electrode showed second highest rate capability; this result may be due to the good catalytic activities of these electrodes. However, in general, their rate capabilities were similar, which can be due to the fact that the C shell having large specific surface area and hierarchical porous structure can increase the number of catalytically actives sites and facilitate reactant transfer during the electrochemical reaction. Fig. 5d–f show cycling stabilities of Li–O2 batteries based on Co–Ru@C shell, Co@C shell, and C shell electrodes at a specific capacity limit for a charge capacity of 500 mA h g−1 at 0.2 mA cm−2. For the C shell electrode, the charging voltage increased rapidly and reached 4.5 V when the charging capacity was less than 500 mA h g−1, indicating that the discharge product could not be decomposed completely. As a result, the circulation was kept only up to 12 cycles when the terminal voltage was maintained above 2 V. The charging voltage of the Co@C shell electrodes was maintained, and the batteries were stable for 24 cycles. However, the Co–Ru@C shell electrodes could function stably for more than 40 cycles, almost twice as long as the Co@C shell electrodes, indicating good cycling stability.
To further explore the reversibilities of the batteries with Co–Ru@C shell electrodes, SEM was used to investigate the discharge products and surface status of the electrodes that went through the first cycle. Fig. 6a–d show SEM images of the pristine Co–Ru@C shell electrode, the electrode after the first discharge process, that after the first charge process, and that after 40 cycles, respectively. The pristine electrodes comprised a Co–Ru@C shell and SP nanoparticles (Fig. 6a) and showed numerous holes of different sizes. After the first full discharge, film-like discharge products grew along the electrode surface and completely covered the holes (Fig. 6b), blocking the transmissions of electrons and various ions; this could be one of the reasons for the termination of discharge. However, after charging, the film-like products disappeared, the porous structure was recovered, and the electrode surface returned to its original state (Fig. 6c), implying decomposition of the products. After 40 cycles, the holes in the electrode surface were no longer as clear as those in the initial state. Some discharge products still remained on the surface, clogging pores and covering the catalytically activity sites, which may be the main reason for the rapid decrease of discharge voltage after 40 cycles of battery circulation. Moreover, the XRD pattern of Co–Ru@C shell electrodes after 1st discharge is shown in Fig. S1;† the result shows no clear peaks corresponding to discharge products such as Li2O2 and Li2CO3, indicating that the film-like discharge product was the typical amorphous Li2O2 compound, which was consistent with previously reported results.24 Additional evidence was also found from the XPS images (Fig. 7b); the main discharge product was Li2O2.
 | ||
| Fig. 6 SEM images of the Co–Ru@C shell electrode: (a) pristine, (b) after first discharge process, (c) after first charge process, and (d) after 40 cycles. | ||
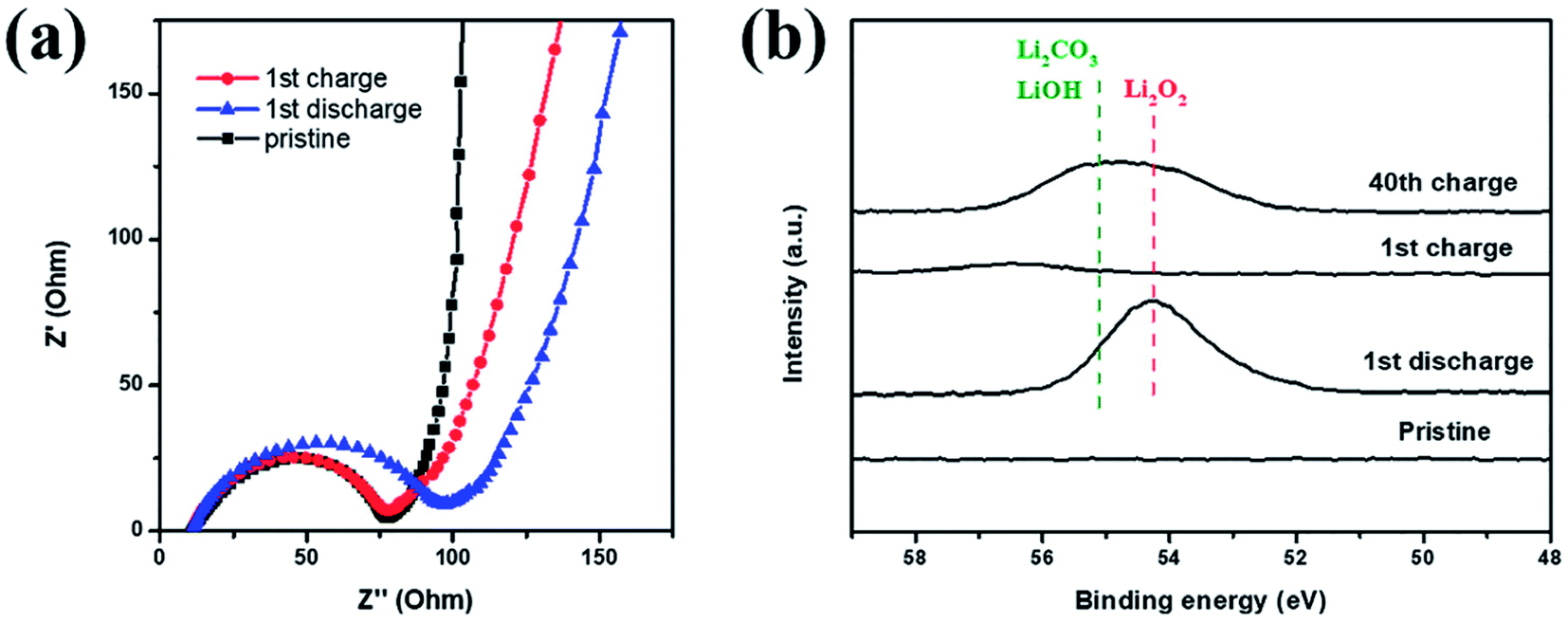 | ||
| Fig. 7 (a) EIS and (b) Li 1s XPS spectrum of Co–Ru@C shell electrodes at different stages. | ||
EIS and XPS testings were also performed to explore the reversibility of the Co–Ru@C shell electrode in different states of charging and discharging. The corresponding impedance variation is shown in Fig. 7a. The charge-transfer resistance increased after discharge, which could be due to the generation of insulating discharge products. After recharge, the charge-transfer resistance decreased, and its value was almost identical to that of the initial state. Similar results could also be observed in the Li 1s XPS spectrum. As shown in Fig. 7b, after charging, the peaks for Li2O2 disappeared, indicating that Li2O2 had completely decomposed.25 However, after 40 cycles, some amount of Li2O2 remained. In addition, LiOH and Li2CO3 peaks were observed; these LiOH and Li2CO3 peaks may have originated from the decomposition of electrolyte. As shown in Fig. 5f, the charging potential of Co–Ru@C shell electrode after 40 cycles was close to 4.3 V, and the electrolyte inevitably decomposed in the presence of ruthenium at this voltage. On the one hand, a catalyst with high catalytic activity can reduce overpotential; on the other hand, it can also make electrolyte decomposition easier. This result was consistent with the conclusions based on SEM analysis. The accumulation of these by products resulted in a series of performance degradations during the cycles such as increased overpotential and decreased capacity.
4. Conclusions
In this study, we used SiO2 as a template and dopamine as a C source to synthesize a hollow C shell, and we loaded Co and Ru nanoparticles onto it to obtain a Co–Ru@C shell composite. The diameter and thickness of the C shell were 100 nm and 5–10 nm, respectively, and there were numerous holes of different sizes on the C shell. Meanwhile, numerous C shells stacked together to form macropores, which resulted in the hierarchical porous structure of the material. The BET results showed that the specific surface area and pore volume of the Co–Ru@C shell were 631.57 m2 g−1 and 2.20 cc g−1, respectively, which provided many three-phase interfaces and more space for the deposition of the discharge products. In comparison with Co@C and C shells, the Co–Ru@C shell electrode showed the highest discharge capacity, the lowest ORR/OER overpotential, and the best cycle stability, indicating the excellent catalytic ability of the Co–Ru@C shell.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding support from the 973 program (2014CB932301), the National Natural Science Foundation (No. 21473040) and Science & Technology Commission of Shanghai Municipality (No. 08DZ2270500), China. In addition, we would like to thank LetPub (http://www.letpub.com) for providing linguistic assistance during preparation of this manuscript.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- K. M. Abraham, J. Phys. Chem. Lett., 2015, 6, 830–844 CrossRef PubMed.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143(1), 143 CrossRef.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef PubMed.
- S. C. Ma, Y. L. Zhang, Q. H. Cui, J. Zhao and Z. Q. Peng, Chin. Phys. B, 2016, 25, 1 Search PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef PubMed.
- L. Grande, E. Paillard, J. Hassoun, J.-B. Park, Y.-J. Lee, Y.-K. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784–800 CrossRef PubMed.
- J. Xiao, D. Wang, W. Xu, D. Wang, R. E. Williford, J. Liu and J.-G. Zhang, J. Electrochem. Soc., 2010, 157, A487–A492 CrossRef.
- C. T. Zhao, C. Yu, S. H. Liu, J. Yang, X. M. Fan, H. W. Huang and J. S. Qiu, Adv. Funct. Mater., 2015, 25, 6913–6920 CrossRef.
- J. Liu, Z. Wang and J. Zhu, J. Power Sources, 2016, 306, 559–566 CrossRef.
- H. Nie, C. Xu, W. Zhou, B. Wu, X. Li, T. Liu and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 1937–1942 CrossRef PubMed.
- X. Xin, K. Ito and Y. Kubo, Carbon, 2016, 99, 167–173 CrossRef.
- R. Yu, W. Fan, X. Guo and S. Dong, J. Power Sources, 2016, 306, 402–407 CrossRef.
- J. Xiao, D. Mei, X. Li, W. Xu, D. Wang, G. L. Graff, W. D. Bennett, Z. Nie, L. V. Saraf, I. A. Aksay, J. Liu and J.-G. Zhang, Nano Lett., 2011, 11, 5071–5078 CrossRef PubMed.
- J. Li, H. Zhang, Y. Zhang, M. Wang, F. Zhang and H. Nie, Nanoscale, 2013, 5, 4647–4651 RSC.
- L. Liu, J. Wang, Y. Hou, J. Chen, H. K. Liu, J. Wang and Y. Wu, Small, 2016, 12, 602–611 CrossRef PubMed.
- Y. Luo, F. Lu, C. Jin, Y. Wang, R. Yang and C. Yang, J. Power Sources, 2016, 319, 19–26 CrossRef.
- F. E. Niu, N. N. Wang, J. Yue, L. Chen, J. Yang and Y. T. Qian, Electrochim. Acta, 2016, 208, 148–155 CrossRef.
- T. H. Yoon and Y. J. Park, J. Power Sources, 2013, 244, 344–353 CrossRef.
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4, 5864–5867 RSC.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef PubMed.
- P. Tan, Z. H. Wei, W. Shyy, T. S. Zhao and X. B. Zhu, Energy Environ. Sci., 2016, 9, 1783–1793 RSC.
- Y. L. Zhang, Q. H. Cui, X. M. Zhang, W. C. McKee, Y. Xu, S. G. Ling, H. Li, G. M. Zhong, Y. Yang and Z. Q. Peng, Angew. Chem., Int. Ed., 2016, 55, 10717–10721 CrossRef PubMed.
- P. L. Lou, C. L. Li, Z. H. Cui and X. X. Guo, J. Mater. Chem. A, 2016, 4, 241–249 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04144j |
| This journal is © The Royal Society of Chemistry 2018 |