
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-initiated atom transfer radical polymerization grafting from nanoporous cellulose gels to create hydrophobic nanocomposites†
Dan Chenga,
Pingdong Weia,
Lina Zhang a and
Jie Cai*ab
a and
Jie Cai*ab
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: caijie@whu.edu.cn; Tel: +86-27-6878-9321
bResearch Institute of Shenzhen, Wuhan University, Shenzhen 518057, China
First published on 31st July 2018
Abstract
Here, we present the preparation of hydrophobic nanoporous cellulose gel-g-poly(glycidyl methacrylate) (NCG-g-PGMA) nanocomposites by surface-initiated atom transfer radical polymerization (SI-ATRP) of glycidyl methacrylate (GMA) monomers and hydrophobic modification with pentadecafluorooctanoyl chloride (C7F15COCl) on the cellulose nanofibrils of the NCG. The successful grafting of PGMA and hydrophobic modification of C7F15CO– groups on the NCG was evaluated by Fourier transform infrared (FTIR) spectroscopy. X-ray diffraction (XRD) and scanning electron microscopy (SEM) confirmed that the SI-ATRP and hydrophobic modification did not change the microscopic morphology and structure of the NCG-g-PGMA nanocomposites. Dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA) showed remarkable thermomechanical properties and moderate thermal stability. The method has tremendous promise to use NCG as a platform for SI-ATRP and produce new functional NCG-based nanomaterials.
Introduction
Nanocellulose, such as cellulose nanocrystals/nanowhiskers (CNC/CNW), nanofibrillated cellulose (NFC) and bacterial cellulose (BC), has attracted special attention because of its remarkable chemical and physical properties, such as high reactivity, higher elastic modulus than that of carbon nanotubes of 100 to 200 GPa, and lower thermal expansivity than that of silica of 0.1 ppm K−1 as well as its sustainability, high availability, and low cost.1–3 Benefitting from these unique nanostructures and fascinating properties, nanocellulose-based materials are, therefore, of great interest in new technology fields, such as green flexible electronic devices, biological platforms and energy storage materials, especially when the increasing environmental concerns are considered.4–9Because of the natural advantage created by the abundance of hydroxyl groups, the use of nanocellulose in more advanced applications can typically be achieved by performing modifications to alter or tailor the chemical structure and physical properties of nanocellulose, providing access to new application possibilities. Different chemical modification approaches have been attempted, including non-covalent surface modification, sulfonation, 2,2,6,6-tetramethylpiperidine-1-oxyl-mediated oxidation (TEMPO), esterification, etherification, silylation, click chemistry, polymer grafting and so on.3 Among these surface-modification approaches, surface-initiated atom transfer radical polymerization (SI-ATRP) is an effective method and has been widely used to tailor the chemical and physical properties at interfaces of metals, plastics, inorganic materials, biomolecules and so on.10–14 Compared to reversible addition fragmentation transfer (RAFT) or nitroxide mediated polymerization (NRP) reactions, macroinitiators can be easily immobilized on the hydroxyl groups of cellulose so that different polymer structures can be prepared.15 This reaction tolerates a wide variety of functional monomer and has also been successfully applied to the grafting well-defined synthetic polymers on the surface of nanocellulose.16 Many efforts to prepare polymer-grafted cellulose via SI-ATRP have been reported, and numerous new properties have been achieved. For example, enhanced hydrophobicity was accomplished by grafting various monomers (methyl methacrylate, butyl acrylate, styrene, glycidyl methacrylate and perfluorooctylethyl methacrylate);17–20 stimuli-responsive properties were obtained by grafting poly(poly(ethylene glycol)methylacrylate)s, poly(N-isopropylacrylamide), poly(dimethylaminoethy methacrylate), poly(diethylaminoethyl methacrylate), poly(4-vinylpyridine);21–24 improved biofunctionization (low-adherent wound dressings, DNA hybridization detection, protein purification, and so on) were developed by grafting poly(N,N-dimethylaminoethylmethacrylate);25–27 and reinforced mechanical properties were achieved by preparing polymer nanocomposites.28–31
Although the majority of reported studies deal with CNC, CNW, CNF and BC with native crystallinity (cellulose I), nanoporous cellulose gel (NCG) with cellulose II crystallinity developed from aqueous alkali hydroxide/urea solutions is composed of interconnected cellulose nanofibrils.32 Moreover, the NCG showed remarkable mechanical strength, high porosity, light transmittance, and good formability.33,34 Due to its excellent performance and special three-dimensional structure, NCG become an ideal functional platform for the preparation of cellulose-based nanomaterials with specific functions. For example, NCG has been used as a template or support for the synthesis of inorganic nanomaterials35–37 and conductive polymers,38,39 as a precursor for cellulose bioplastics,40 and as a 3D reinforcing phase for polymer nanocomposites.33,41,42 However, NCG-based materials are inherently hydrophilic, and most cellulosic materials can swell significantly in water, limiting their application range to a large extent. Hydrophobic modification is a practical method for increasing the application field of cellulose-based materials. Herein, we demonstrate that the interconnected nanofibrillar network structure of the NCG was grafted with PGMA. Further hydrophobic modification with C7F15COCl did not change the microscopic morphology and structure of the NCG-g-PGMA nanocomposites, leading to a remarkable improvement in the hydrophobicity and tensile storage modulus (E′) above the glass transition temperature (Tg). To the best of our knowledge, this is the first example of NCG as platform for SI-ATRP. This promising synthetic strategy provides potential opportunities for fabricating functional NCG-based nanomaterials.
Experimental section
Materials
Cellulose (cotton linter pulp) with its viscosity-average molecular weight (Mη) of 9.2 × 104 was supplied by Sanyou Chemical Fibre Co., Ltd. (Tangshan, China) and the material was dried under vacuum at 60 °C for 24 h prior to use. α-Bromoisobutyryl bromide (BiB), C7F15COCl, glycidyl methacrylate (GMA) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) were purchased from Aladdin (Shanghai, China). Triethylamine (TEA), 4-dimethylaminopyridine (DMAP), copper(II) bromide (CuBr2), copper(I) chloride (CuCl), tetrahydrofuran (THF), toluene, concentrated hydrochloric acid (HCl), ethanol, methanol, dichloromethane (DCM), lithium hydroxide hydrate, and urea were purchased from Shanghai Chemical Reagent Co., Ltd., China. GMA was passed through a column of neutral aluminum oxide prior to use to remove the inhibitor. All other chemicals were used as received without any further purification.Fabrication of nanoporous cellulose gel (NCG)
Cellulose was dissolved in an aqueous 4.6 wt% LiOH/15 wt% urea solution precooled to −12 °C to form a 6 wt% transparent cellulose solution according to our previous method.43 After removing air bubbles by centrifugation, the cellulose solution was spread on a glass plate in a 1 mm-thick layer following regeneration by ethanol to give cellulose alcogels. The cellulose alcogels were thoroughly washed with deionized water to achieve NCG hydrogels. The water content of the NCG hydrogel was 90%.Immobilization of initiator on the NCG
The procedure for immobilizing the initiator on the NCG surface was performed according to the method of Carlmark and Malmstrom.44,45 Prior to immobilization of the initiator, the NCG hydrogels (0.72 g, 1.33 mmol OH groups) were converted to NCG-THF organogels by solvent exchange with THF. Then, the available hydroxyl groups on the NCG were converted into SI-ATRP initiators by immersing the NCG-THF organogels in a solution containing BiB (305 mg, 1.33 mmol), TEA (148 mg, 1.46 mmol), and a catalytic amount of DMAP in THF (10 ml). The reaction was allowed to proceed overnight at room temperature on a shaking device. Thereafter, the initiator-immobilized NCG (NCG-BiB) was thoroughly washed in THF to remove residual reactants and byproducts and then subjected to solvent exchange with toluene. The byproduct was the white triethylammonium bromide.46Grafting of GMA on the NCG-BiB by SI-ATRP
The NCG-BiB obtained from the previous step was placed in a 25 ml round-bottomed flask containing toluene (4 ml), GMA (4.30 g, 30.2 mmol), CuBr2 (4 mg, 18 μmol), CuCl (14 mg, 141 μmol), and PMDETA (25 mg, 140 μmol). The flask was sealed with a rubber septum and thereafter evacuated and back-filled with argon three times before being placed in a thermostated water bath. Grafting of GMA on the NCG-BiB was conducted between 0 and 30 °C, and the reaction was allowed to proceed for 5 to 48 h. After polymerization, the obtained NCG-g-PGMA nanocomposite was thoroughly washed in THF, DCM, THF/H2O and MeOH, successively, to remove residual monomer and the catalyst complex.To estimate the weight gain, the specimens were weighed before and after polymerization. The weight gain (WPGMA, wt%) was calculated as follows:
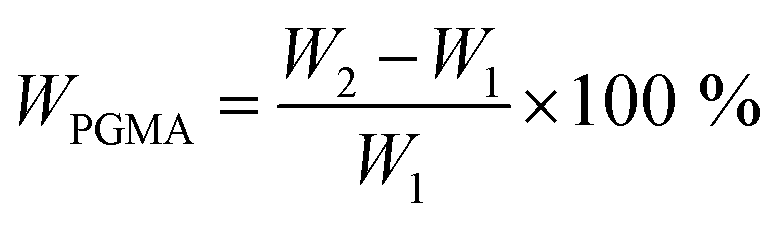 | (1) |
Hydrophobic modification on hydrolyzed NCG-g-PGMA nanocomposites with C7F15COCl
The epoxide groups of PGMA in the NCG-g-PGMA nanocomposites were hydrolyzed with HCl (0.64 mmol l−1) in a THF–H2O mixture. The reaction was allowed to proceed for 12 h; the NCG-g-PGMA nanocomposites were subsequently rinsed with THF and CH2Cl2, successively. The hydrolyzed NCG-g-PGMA nanocomposite was then transferred to a round-bottomed flask containing DCM (10 ml), TEA (74.0 mg, 730 μmol), and DMAP (catalytic amount). C7F15OCl (288 mg, 670 μmol) was added dropwise, and the acylation reaction was allowed to proceed overnight. Thereafter, the NCG-g-PGMA nanocomposites were thoroughly washed in DCM and ethanol, successively, to remove residual reactants and byproducts. The hydrophobic NCG-g-PGMA nanocomposites were finally dried in a vacuum oven at 60 °C. Reference sample of NCG-C7F15 was prepared using C7F15OCl. Prior to reaction, the NCG hydrogel was converted to NCG-DCM gel by solvent exchange with DCM. The acylation reaction was performed in a similar manner using the same molar amounts of C7F15OCl as those in the example above. Reference sample of neat PGMA was prepared using in situ polymerization. The GMA contained 5% (w/v) benzoyl peroxide as initiator was subsequently sealed into glass mold to prevent solution evaporation, and heated at 47 °C for 12 h to polymerize and then 90 °C for 3 h to remove the residual monomer.33 Hydrophobic NCG-g-PGMA nanocomposites were designated as S1–S4 and S3, S5–S8, and the reaction conditions were 0–30 °C for 24 h and 5–48 h at 20 °C, respectively. The reaction condition and properties of nanocomposites is listed in Table 1.| Sample | t, h | T, °C | ρ, g cm−3 | Conv., % | WPGMA, wt% | Mn, g mol−1 | Mw/Mn | CA, ° | Tg, °C |
|---|---|---|---|---|---|---|---|---|---|
| a Monomer conversion (Conv.) calculated from 1H NMR. The WPGMA was calculated by eqn (1). Mn and Mw were number average molecular weight and weight average molecular weight for PGMA side chains calculated from GPC. CA is the water contact angle. The Tg was determined by DSC. | |||||||||
| S1 | 24 | 0 | 0.8 | 17 | 63 | 1500 | 1.03 | 75 | 85 |
| S2 | 24 | 10 | 0.9 | 27 | 67 | 7900 | 1.54 | 112 | 83 |
| S3 | 24 | 20 | 1.1 | 31 | 71 | 8000 | 1.44 | 136 | 82 |
| S4 | 24 | 30 | 1.3 | 54 | 77 | 9000 | 1.53 | 121 | 72 |
| S5 | 5 | 20 | 0.86 | 16 | 63 | 4300 | 1.20 | 79 | 84 |
| S6 | 10 | 20 | 0.96 | 17 | 67 | 6000 | 1.36 | 86 | 83 |
| S7 | 36 | 20 | 1.2 | 34 | 76 | 9400 | 1.83 | 89 | 72 |
| S8 | 48 | 20 | 1.25 | 35 | 80 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
1.95 | 81 | 65 |
Characterization
Fourier transform infrared (FTIR) spectra were recorded at ambient temperature on an FTIR spectrometer (Nicolet 5700 FTIR Spectrometer, MA) in the wavelength range from 4000 to 400 cm−1 with a 2 cm−1 resolution and an accumulation of 32 scans.Wide-angle X-ray diffraction (WAXD) measurements were carried out on a WAXD diffractometer (D8-Advance, Bruker Corporation). The X-rays used were Ni-filtered CuKα radiation with a wavelength of 1.542 Å. The voltage was set at 40 kV, and the current was set at 40 mA. The samples were mounted on a solid circular holder, and the proportional counter detector was set to collect data at a rate of 3° min−1 over the 2θ range from 5° to 40°.
Scanning electron microscopy (SEM) observations of the surface of the NCG, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposites were performed with a Zeiss Sigma microscope. The imbibed water of the NCG was exchanged with tert-butyl alcohol (t-BuOH), and then the hydrogel was freeze dried to give an aerogel. Before observation, the NCG, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposites were coated with gold.
1H NMR spectra were recorded on a Bruker Avance III HD 400 MHz NMR instrument, using CDCl3 as solvent. Dibromomethane peak was used as internal standard.
The molecular weight and molecular weight distributions of the cleaved PGMA obtained by hydrolysis of the NCG-g-PGMA nanocomposites were measured by gel permeation chromatography (GPC, Waters Instruments, American). The hydrolysis procedure was as follows: 0.04 g of the NCG-g-PGMA nanocomposite was placed in a round-bottomed flask containing 15 ml of 1.5 mol l−1 aqueous HCl. The flask was stirred at 90 °C for 72 h. The reaction mixture was filtered to separate the solid cellulose particles, and the aqueous HCl was removed by evaporation. The weight average molecular weight (Mw) and number average molecular weight (Mn) of the cleaved PGMA were determined by GPC on an instrument equipped with a Waters 717 plus autosampler, a Waters 1515 isocratic HPLC pump, a Waters 2414 refractive index detector, and Shodex K-805, K-804, and K-802.5 columns in series. The GPC apparatus was calibrated with polystyrene standards, and THF was used as the eluent (1.0 ml min−1). Measurements were performed at 30 °C (both the columns and the detector).
The water contact angles (CA) of the NCG, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposites were evaluated on a contact angle system (OCA20, Germany). One drop of water (2 μl) was dropped on the sample surface with an automatic piston syringe and photographed.
Differential scanning calorimetry (DSC) was performed using a DSC Q20 differential scanning calorimeter (TA Instruments, USA) to evaluate the glass transition temperature (Tg) of the PGMA and hydrophobic NCG-g-PGMA nanocomposites. Approximately 10 mg of the sample was placed in a DSC cell. Each sample was heated from 30 to 150 °C at 10 °C min−1, kept at 150 °C for 2 min, and then cooled down at same rate.
Dynamic mechanical analysis (DMA) temperature sweeps under oscillatory stress were performed on a DMA Q800 system (TA Instruments, USA) in tensile mode at a heating rate of 3 °C min−1 and with a distance between jaws of 10 mm in the temperature range from 35 to 180 °C with a frequency of 1 Hz. The width of the samples was approximately 10 mm.
Thermogravimetric analysis (TGA) analysis was performed on an STA 449C thermal analyzer (NETZSCH, Germany) under nitrogen at a heating rate of 10 °C min−1 from 30 to 600 °C.
Results and discussion
Fig. 1 shows the synthetic approach toward the hydrophobic NCG-g-PGMA nanocomposites by SI-ATRP of GMA monomer on the surface of the NCG and functionalization of C7F15COCl. The NCG possesses an interconnected cellulose nanofibrillar network structure, resulting in a large pore volume and specific surface area suitable for in situ polymerization. The abundant hydroxyl groups on cellulose reacts with BiB so that the initiator is immobilized on the cellulose surface, becoming a SI-ATRP initiator in the graft polymerization of GMA monomer.47 The NCG hydrogel was subjected to solvent exchange to form a NCG-THF organogel, which was then immersed in a THF solution containing BiB as an initiator and TEA and DMAP as catalysts at ambient temperature to give the initiator-immobilized NCG (NCG-BiB). After removing residual reactants and byproducts, the NCG-BiB was subjected to solvent exchange with toluene. Subsequently, the NCG-BiB toluene organogel was immersed in a solution of GMA monomer in toluene that also contained CuBr2, CuCl and PMDETA as catalysts to obtain an NCG-g-PGMA nanocomposite by SI-ATRP. Then, the NCG-g-PGMA nanocomposite was thoroughly washed with THF, DCM, THF/H2O and MeOH to remove residual monomer and catalysts. The epoxide groups of PGMA in the NCG-g-PGMA nanocomposite were converted to hydroxyl groups by hydrolysis in a solution of THF, H2O and HCl to give the hydrolyzed NCG-g-PGMA nanocomposite. Subsequently, the hydrolyzed NCG-g-PGMA nanocomposite was reacted with C7F15OCl in a DCM solution containing TEA and DMAP as catalysts via acylation. The hydrophobic NCG-g-PGMA nanocomposite was then obtained after thoroughly washing the product with DCM and ethanol and subsequent drying.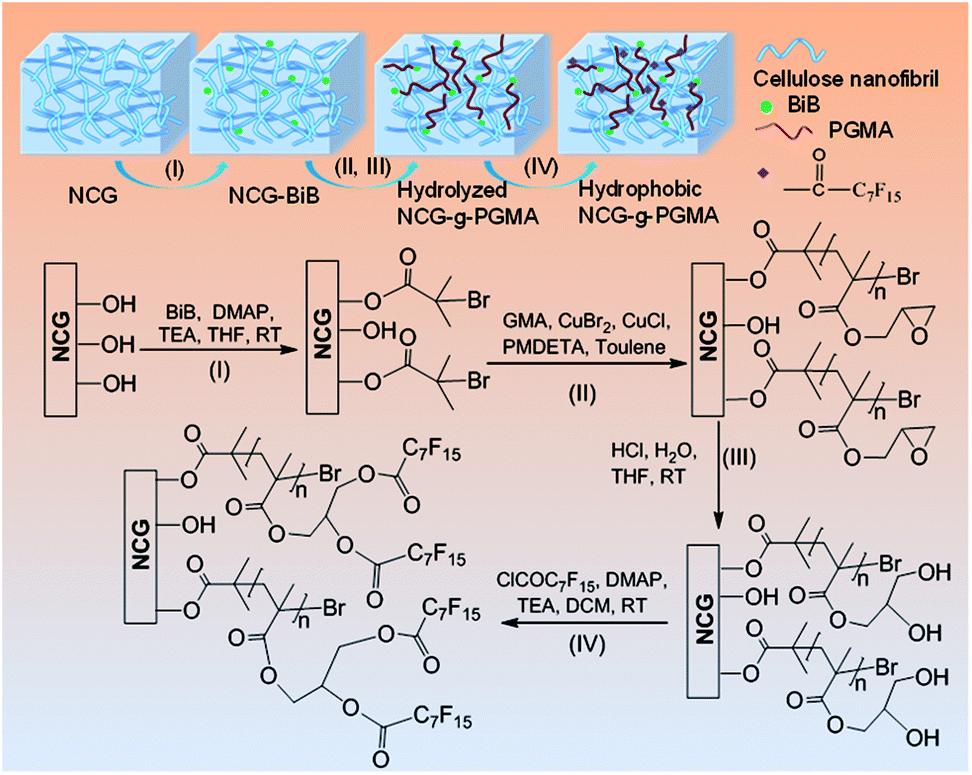 | ||
| Fig. 1 Synthetic approach toward hydrophobic NCG-g-PGMA nanocomposites by SI-ATRP and hydrophobic modification: (I) immobilization of the initiator (BiB) onto the cellulose nanofibrils of the NCG; (II) grafting of GMA on the NCG-BiB by SI-ATRP; (III) hydrolysis of the NCG-g-PGMA nanocomposite; (IV) hydrophobic modification of C7F15COCl on the hydrolyzed NCG-g-PGMA nanocomposite. | ||
Fig. 2 shows the FTIR spectra of the NCG, NCG-BiB, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3). The broad and unstructured band in the OH stretching vibration region (3100 to 3700 cm−1) of the cellulose vibrational spectrum contains information about intra- and intermolecular hydrogen bonding interactions. The typical FTIR bands of the NCG are visible, including the bands attributed to the stretching vibrations of O–H (3413 cm−1), antisymmetric C–O–C glycoside (1161 cm−1), C–O in C–OH groups (1022 cm−1) and C–O–C bridges (897 cm−1) (Fig. 2a).48 The peak observed at 1643 cm−1 was assigned to the H–O–H angle vibration. Compared to the FTIR spectrum of the NCG, the appearance of a new carbonyl ester stretching vibration at 1732 cm−1 in the spectrum of the NCG-BiB indicates the immobilization of BiB on cellulose (Fig. 2b).24,28 Fig. 2c shows the appearance of signals corresponding to the NCG-g-PGMA nanocomposites after SI-ATRP. The strong peak observed at 1732 cm−1 is attributed to the ester carbonyl stretching of the GMA units. The other bands seen at 906 cm−1 and 846 cm−1 were due to asymmetric stretching of the epoxy groups. The peak appearing at 757 cm−1 possibly represents the –CH2 rocking vibration of the oxirane groups. Furthermore, successful polymerization was further confirmed by the appearance of four new strong absorption bands at 3000, 2933, 1483 and 1452 cm−1, which are assigned to the C–H stretching and bending vibrations of the methylene and methyl groups in the PGMA chains.49,50 The band observed at 1265 cm−1 is characteristic of the stretching vibrations of C–O–C in the polymer chains. The shift in this peak from 1643 to 1632 cm−1 and the decrease in its intensity likely results from a decrease in the H–O–H interactions between PGMA and the NCG. Compared with the NCG-g-PGMA nanocomposite, the hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3) exhibited C–O–C stretching vibrations of the polymer chains at 1265 and 1253 cm−1, a C–F symmetrical stretching peak at 1156 cm−1, a C–C skeleton vibration of straight-chain alkanes at 704 cm−1 and a C–Br vibration absorption peak at 617 cm−1 (Fig. 2d).51 Although functionalization with C7F15OCl might also occur at the hydroxyl group of NCG, the primary hydroxyl groups at hydrolyzed NCG-g-PGMA nanocomposite had higher reactivity because the hydroxyl group of NCG were sterically hindered by PGMA chains.16 Moreover, the peak observed at 1732 cm−1 in the spectra of the NCG-BiB, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposite indicates that a large amount of ester groups were introduced into the NCG (Fig. 2b–d), confirming again the successful grafting of BiB, GMA and C7F15COCl.52 The shift in the C–H stretching absorption bands of the methylene and methyl groups in the polymer chains from 3000 and 2933 cm−1, respectively, to 2956 and 2922 cm−1 and the increased intensity of the –CH2 rocking vibration at 749 cm−1 likely result from grafting of the hydrophobic compound (C7F15OCl) on the NCG-g-PGMA nanocomposite. The FTIR spectra of the NCG, NCG-BiB, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposite (sample S3) showed the characteristic stretching vibrations of hydroxyl groups in the range from 3300 to 3500 cm−1. However, this peak and intensity of the hydrogen bonding interactions became narrower and weaker with the grafting of PGMA and C7F15OCl, and the hydroxyl peak shifted from 3421 to 3446 cm−1, indicating that hydrogen bonding interactions occurred between the NCG and PGMA.
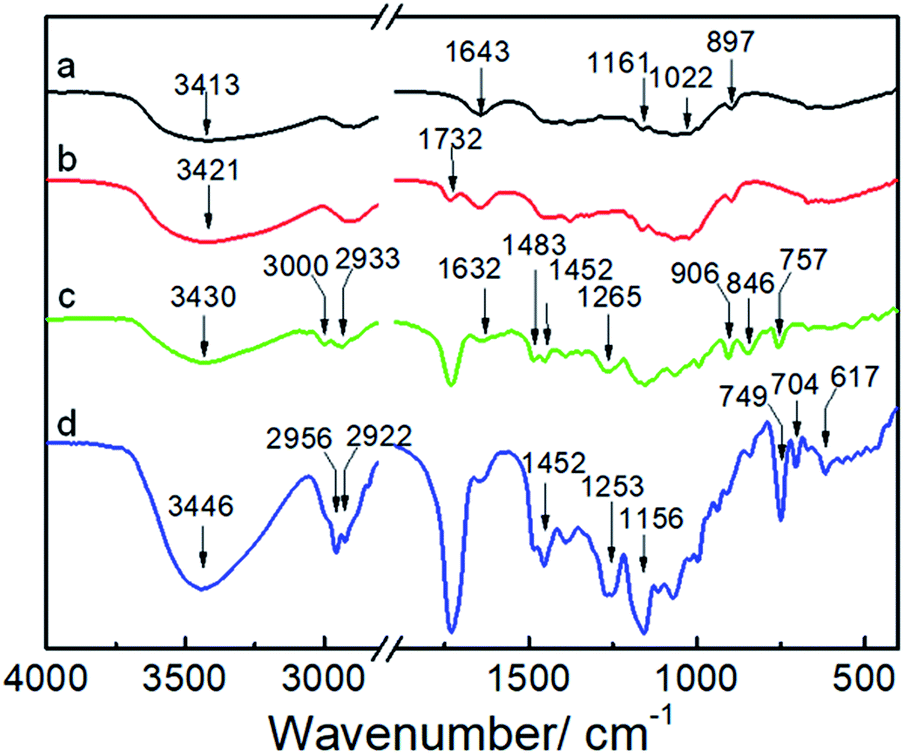 | ||
| Fig. 2 FTIR spectra of the NCG (a), NCG-BiB (b), NCG-g-PGMA nanocomposite (c) and hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3) (d). | ||
X-ray diffraction (XRD) was used to examine the aggregate structure of the NCG, PGMA, and hydrophobic NCG-g-PGMA nanocomposites (Fig. 3). The XRD pattern of the NCG showed three characteristic peaks centered at 12.0°, 20.6° and 21.8°, corresponding to the typical (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (110), and (200) crystalline planes of cellulose II, respectively (Fig. 3a).32 PGMA displayed a diffraction pattern typical of an amorphous polymer with broad bands centered at approximately 18.8° and 30.7° (Fig. 3b). Although the characteristic peak intensity of NCG in hydrophobic NCG-g-PGMA nanocomposites decreases at 12.0°, 20.6° and 21.8°, the (10) and (200) crystal planes of cellulose II still existed obviously. The XRD patterns of the hydrophobic NCG-g-PGMA nanocomposites (Table 1, sample S1–S4) prepared at different temperatures exhibited the characteristics of PGMA and NCG (Fig. 3c–f). These features demonstrate that the XRD patterns of the hydrophobic NCG-g-PGMA nanocomposites (sample S1–S4) nearly become a superposition of those of neat NCG and PGMA, indicating that nothing interfered with the formation of the nanocomposite structure by SI-ATRP and hydrophobic modification.41,42,53
0), (110), and (200) crystalline planes of cellulose II, respectively (Fig. 3a).32 PGMA displayed a diffraction pattern typical of an amorphous polymer with broad bands centered at approximately 18.8° and 30.7° (Fig. 3b). Although the characteristic peak intensity of NCG in hydrophobic NCG-g-PGMA nanocomposites decreases at 12.0°, 20.6° and 21.8°, the (10) and (200) crystal planes of cellulose II still existed obviously. The XRD patterns of the hydrophobic NCG-g-PGMA nanocomposites (Table 1, sample S1–S4) prepared at different temperatures exhibited the characteristics of PGMA and NCG (Fig. 3c–f). These features demonstrate that the XRD patterns of the hydrophobic NCG-g-PGMA nanocomposites (sample S1–S4) nearly become a superposition of those of neat NCG and PGMA, indicating that nothing interfered with the formation of the nanocomposite structure by SI-ATRP and hydrophobic modification.41,42,53
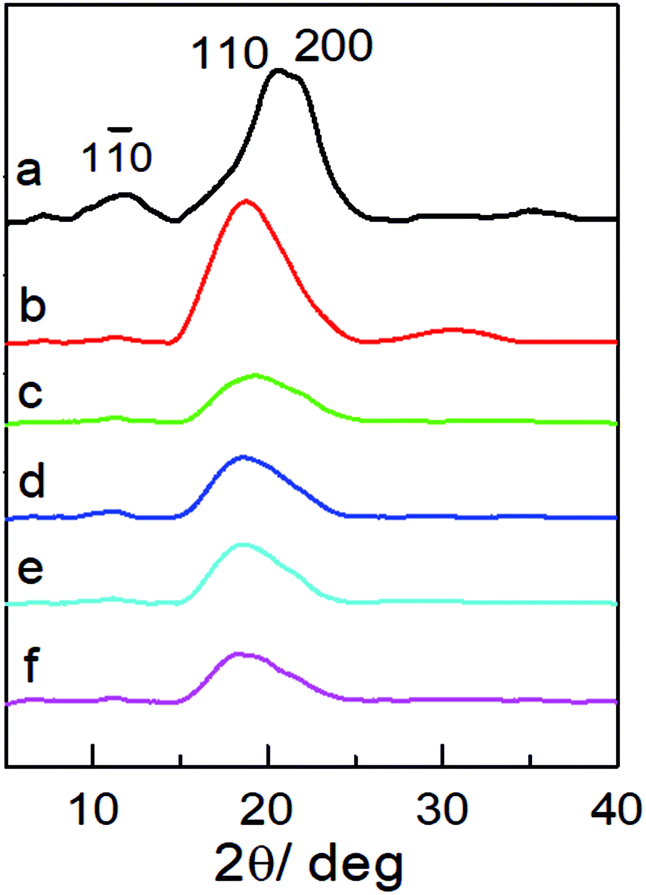 | ||
| Fig. 3 XRD patterns of the NCG (a), PGMA (b) and the hydrophobic NCG-g-PGMA nanocomposites of S1 (c), S2 (d), S3 (e) and S4 (f). | ||
The microscopic morphologies and structures of the NCG, NCG-g-PGMA nanocomposite and the hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S1–S4) were examined by SEM (Fig. 4). The characteristic three-dimensional (3D) network of the cellulose nanofibrils was clearly visible on the surface of the NCG, NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposites. Obviously, the NCG showed a homogeneous nanoporous structure consisting of pores approximately 50 to 150 nm in diameter and interconnected cellulose nanofibrils with a typical diameter of approximately 10 to 20 nm (Fig. 4a and b). After SI-ATRP of GMA monomers, the SEM images of the NCG-g-PGMA nanocomposite showed features consistent with grafting of PGMA (Fig. 4c and d). The NCG-g-PGMA nanocomposite consisted of rough nanofibrils with diameters of approximately 50 to 100 nm and large nanopores with diameters of 100 to 200 nm, which were obviously associated with chemical grafting by PGMA. This result is also consistent with theoretical evidence suggesting that the mean pore size increases with an increase in the fiber diameter.54 Further hydrophobic modification with C7F15COCl did not change the microscopic morphology and structure of the NCG-g-PGMA nanocomposite (Fig. 4i and l, Table 1, sample S4). However, the hydrophobic NCG-g-PGMA nanocomposite (sample S4) prepared at 30 °C exhibited a smoother surface, higher density (1.3 g cm−3), and smaller nanopores with diameters of 20 to 100 nm than the NCG-g-PGMA nanocomposite. This change in the microstructure resulted from the smoothing effect that the grafted polymers had on the surface, making the microstructure less rough.17 A very similar phenomenon was observed in the SEM images of the hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S3, S5–S8) prepared with different grafting times (see the ESI, Fig. S1†). Therefore, the grafting temperature and time played an important role in the grafting of PGMA on the NCG and hence dramatically affected the microscopic morphology and structure of the hydrophobic NCG-g-PGMA nanocomposites.
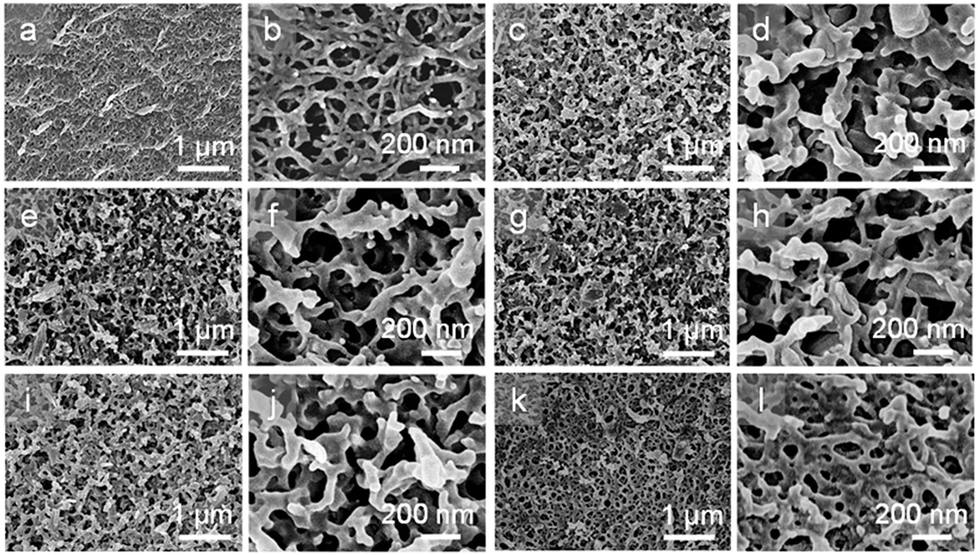 | ||
| Fig. 4 SEM images of the surface of the NCG (a and b), the NCG-g-PGMA nanocomposite (c and d) and the hydrophobic NCG-g-PGMA nanocomposites of S1 (e and f), S2 (g and h), S3 (i and j) and S4 (k and l) at low- and high-magnification. | ||
The hydrophobic behavior of the hydrophobic NCG-g-PGMA nanocomposites was further investigated by measuring the water contact angles (Fig. 5), which was strongly affected by the grafting temperature and time. The hydrophobicity of the NCG-g-PGMA nanocomposite increased considerably following SI-ATRP grafting of PGMA and hydrophobic modification with C7F15OCl. Water droplets stained with toluidine blue could be absorbed on the NCG aerogel freeze dried from water because of the capillary effect of the nanopores and the hydrophilicity of cellulose (Fig. 5a and c), and droplets exist and maintain a droplet shape on the surface of the hydrophobic NCG-g-PGMA nanocomposites (Table 1, sample S3) without being absorbed for a long time, which provides clear evidence of the high hydrophobicity of these materials (Fig. 5b and d). The polymerization of PGMA on the NCG to form the NCG-g-PGMA nanocomposite resulted in an increase in the water contact angle to 67°, indicating a significant improvement in the hydrophobicity. Expectedly, the low surface energy compound (C7F15OCl) improved the hydrophobicity of the NCG-g-PGMA nanocomposite (Fig. 5e; Table 1, sample S3) because the water contact angle of air is considered to be 136°. The water contact angles of the hydrophobic NCG-g-PGMA nanocomposites gradually increased as the grafting temperature increased from 0 to 30 °C (Table 1, samples S1–S4), and a maximum value of 136° was achieved at a grafting temperature of 20 °C. At a grafting temperature of 30 °C, the water contact angle of the hydrophobic NCG-g-PGMA nanocomposite decreased slightly to 121°, which was most likely due to the reduced micro- and nano-pore size of this hydrophobic NCG-g-PGMA nanocomposite, as shown in Fig. 4k and l. Additionally, a very similar trend was observed for the hydrophobic NCG-g-PGMA nanocomposites when the grafting time was increased from 5 to 48 h (Table 1, samples S3, S5–S8). The hydrophobic NCG-g-PGMA nanocomposites exhibited a maximum water contact angle of 136° at a grafting time of 24 h, and then the contact angle decreased dramatically to 81° at 48 h. Compared with the hydrophobic NCG-g-PGMA nanocomposites, the water contact angle of NCG-C7F15 was 84° (not shown). The probable reason was that the amount of reactive hydroxyl groups of NCG was insufficient when using the same amount of hydrolyzed NCG-g-PGMA nanocomposite in C7F15OCl, and the hydrophobic compound did not cover the entire the surface of NCG. Nevertheless, the water contact angles of the hydrophobic NCG-g-PGMA nanocomposites were dramatically increased via the grafting of PGMA but were not directly affected by the PGMA content, confirming again that the water contact angles of the hydrophobic NCG-g-PGMA nanocomposites were affected by the chemical composition of the surface and the micro- and nano-pore size.17
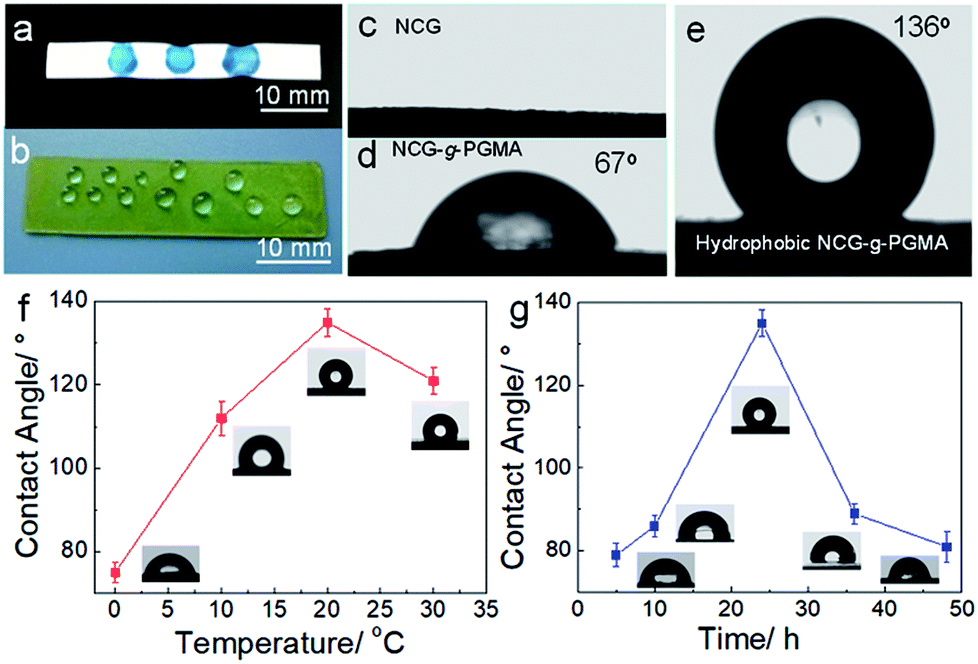 | ||
| Fig. 5 (a and b) Photographs of a NCG aerogel (a) and a hydrophobic NCG-g-PGMA nanocomposite (b) with water droplets. (c–e) Photographs of the water contact angles on the surfaces of the NCG (c), NCG-g-PGMA nanocomposite (d) and hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3) (e). Water contact angles of the hydrophobic NCG-g-PGMA nanocomposites prepared at different temperatures for 24 h (Table 1, samples S1–S4) (f) and with different reaction times at 20 °C (Table 1, samples S3, S5–S8) (g). The insets are the corresponding water contact angle photographs of the hydrophobic NCG-g-PGMA nanocomposites. All error bars represent the s.d. of at least three replicate measurements. | ||
The DSC thermograms of the neat PGMA and the hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S1–S4) are shown in Fig. 6. The Tg of the neat PGMA in the nanocomposites was established by DSC, and the results are also summarized in Table 1. While neat PGMA showed a typical Tg value of approximately 68 °C, the values shifted to higher temperatures in the presence of the NCG in the hydrophobic NCG-g-PGMA nanocomposites. For instance, the Tg of sample S1 was 85 °C, and this value gradually decreased to 72 °C with increasing PGMA content in sample S4 (63 wt% vs. 77 wt%). Additionally, a very similar trend was observed as the PGMA content increased from 63 wt% to 80 wt% in the hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S3, S5–S8), in which the Tg value decreased from 84 to 65 °C (Table 1). Here, the restriction of mobility was weakened as the proportion of PGMA in the nanocomposite increased, thus resulting in a decrease in the Tg.22 This change is also ascribed to the grafting of PGMA chains on cellulose nanofibrils by SI-ATRP and the confinement effect of the interconnected cellulose nanofibril networks of the NCG.41,42
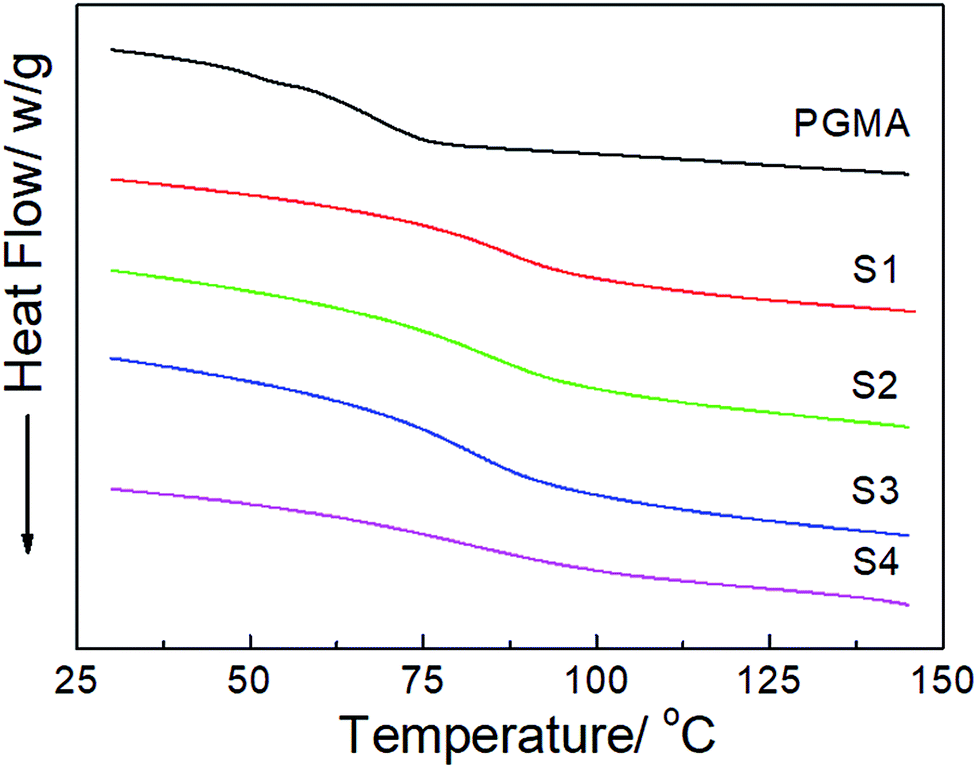 | ||
| Fig. 6 DSC thermograms of neat PGMA and the hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S1–S4). | ||
Fig. 7 shows the dynamic mechanical behavior of the NCG, PGMA and hydrophobic NCG-g-PGMA nanocomposites (Table 1, samples S1–S4) determined by DMA at 1 Hz. Neat PGMA showed behavior typical of an amorphous polymer. Below 60 °C, neat PGMA was in the glassy state and exhibited a slight decrease in the tensile storage modulus (E′) from 35 to 60 °C because of the restriction of molecular motion. A sudden drop in E′ occurred above 60 °C, which resulted from the motion of the PMGA chain segments. In comparison, most of the hydrophobic NCG-g-PGMA nanocomposites exhibited a modest increase in E′ below the Tg of PGMA. Remarkably, the incorporation grafting time of 24 h, and then the contact angle decreased dramatically to 81° at 48 h. Therefore, the contact angles of the hydrophobic NCG-g-PGMA nanocomposites were dramatically increased via the grafting of PGMA but were not directly affected by of the NCG drastically changed the mechanical behavior of the hydrophobic NCG-g-PGMA nanocomposites above their Tg, where they displayed significant mechanical reinforcement behavior as a result of the grafting from the NCG. For example, at 180 °C, the E′ of hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S1) was 337-fold higher than that of PGMA (117.9 MPa vs. 0.35 MPA). In the rubbery state, the E′ of hydrophobic NCG-g-PGMA nanocomposite S4 with 23 wt% NCG was lower than that of sample S1 with 37 wt% NCG. This difference could be attributed to stronger NCG-polymer adhesion in the interfacial zone due to the presence of additional interactions. Similar reinforcement effects of polymers above the Tg have also been reported in bacterial cellulose-g-poly (methyl methacrylate) nanocomposites53 and cotton fabric-g-poly(tert-butyl acrylate).55 Notably, the PGMA grafted in the hydrophobic NCG-g-PGMA nanocomposites had a significantly lower Mn (less than 9.4 × 103 g mol−1) than neat PGMA (1.3 × 104 g mol−1). Moreover, the molecular weight of the grafted PGMA appeared to have no direct effect on the mechanical properties of the hydrophobic NCG-g-PGMA nanocomposites. Thus, the significant reinforcement effect of the hydrophobic NCG-g-PGMA nanocomposites is attributable to the strong mobility was weakened as the proportion of PGMA in the intermolecular hydrogen bonding interactions and the confinement effects of the entangled cellulose chains within the interconnected nanofibrillar network structure of the NCG.41,42 Moreover, the maximum loss tangent (tanδ) exhibited a higher α-relaxation temperature in the hydrophobic NCG-g-PGMA nanocomposites (Tα of approximately 100 °C) than in neat PGMA (approximately 94 °C) (Fig. 7b). Additionally, the intensity of the tanδ peak decreased for the hydrophobic NCG-g-PGMA nanocomposites, confirming the grafting of PGMA on cellulose nanofibrils of the NCG and the confinement effect of the cellulose nanofibril networks of the NCG, which substantially reduced the chain relaxation of PGMA.33,41,42
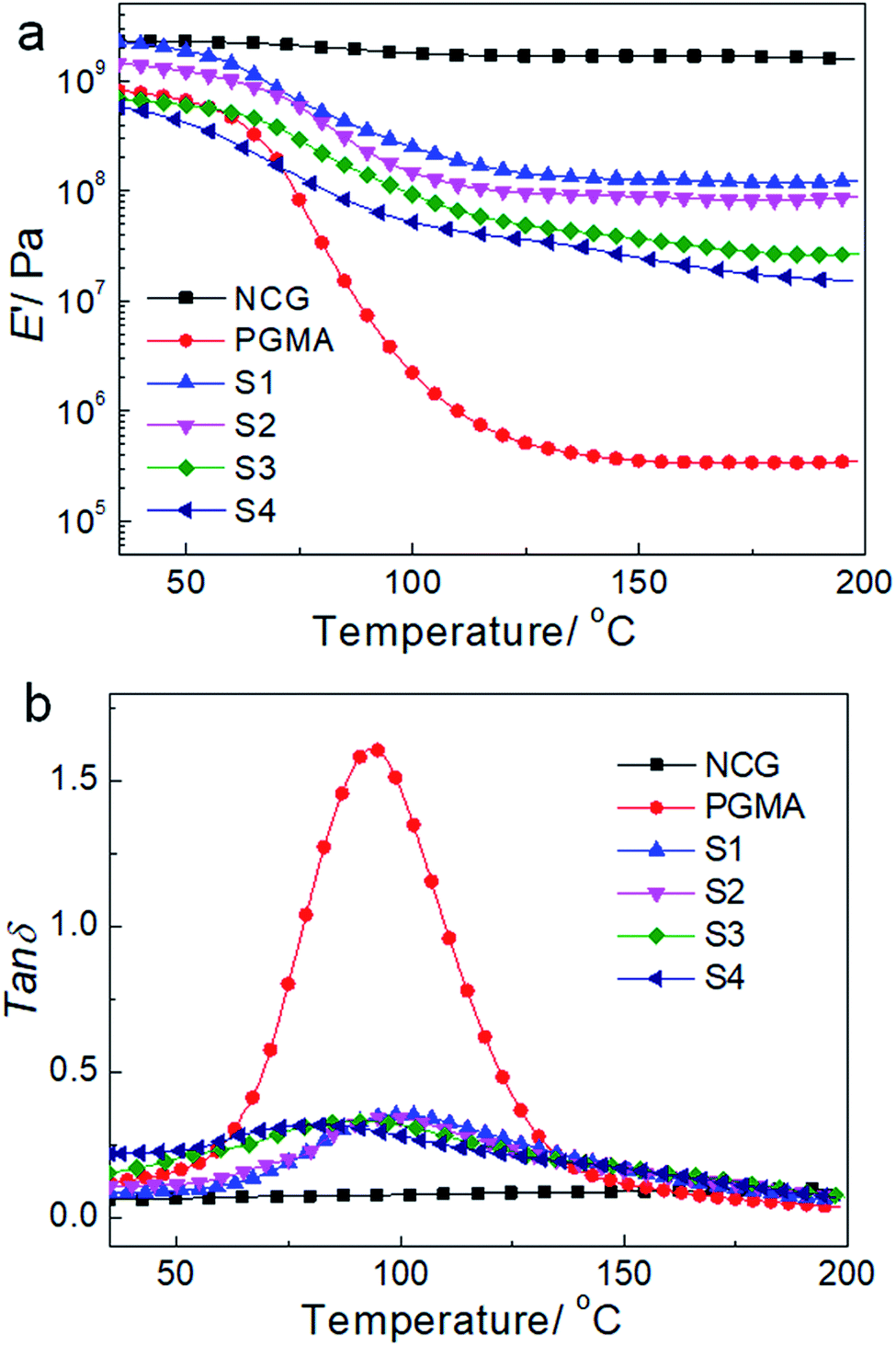 | ||
| Fig. 7 DMA temperature sweeps for the NCG, PGMA and the hydrophobic NCG-g-PGMA nanocomposites. Storage modulus E′ (a) and loss tangents tanδ (b) of the NCG, PGMA and nanocomposites as a function of temperature. | ||
We further examined the thermal decomposition of the NCG, NCG-BiB, NCG-g-PGMA nanocomposite and a hydrophobic NCG-g-PGMA nanocomposite (sample S3) by TGA under nitrogen, as shown in Fig. 8. Although thermal decomposition of the cellulose nanowhiskers occurred below 150 °C because of the presence of sulfate groups, the NCG withstood heating up to 290–370 °C, which is the decomposition temperature of pristine cellulose.41,42 The introduction of BiB groups resulted in a decrease in the thermal stability of the NCG, with the decomposition peak observed at 205 °C. The decrease in thermal stability for NCG-BiB was attributed to the degradation of the bromoalkyl groups to release HBr upon heating, which induces cellulose degradation at lower temperatures.29,53 In contrast, the thermograms of the NCG-g-PGMA nanocomposite and hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3) showed a substantial increase in stability relative to the corresponding NCG-BiB. The NCG-g-PGMA nanocomposite exhibited two decomposition peaks at approximately 305 °C and 415 °C, which can be attributed to the thermal decomposition of the NCG and PGMA components of the NCG-g-PGMA nanocomposite.56 After functionalization with C7F15COCl, the decomposition peaks of NCG and PGMA of hydrophobic NCG-g-PGMA nanocomposite slightly decreased to 276 °C and 411 °C. Since the hydrophobic NCG-g-PGMA nanocomposites did not melt or decompose up to 250 °C, they can be used at much higher temperatures than the neat polymers.
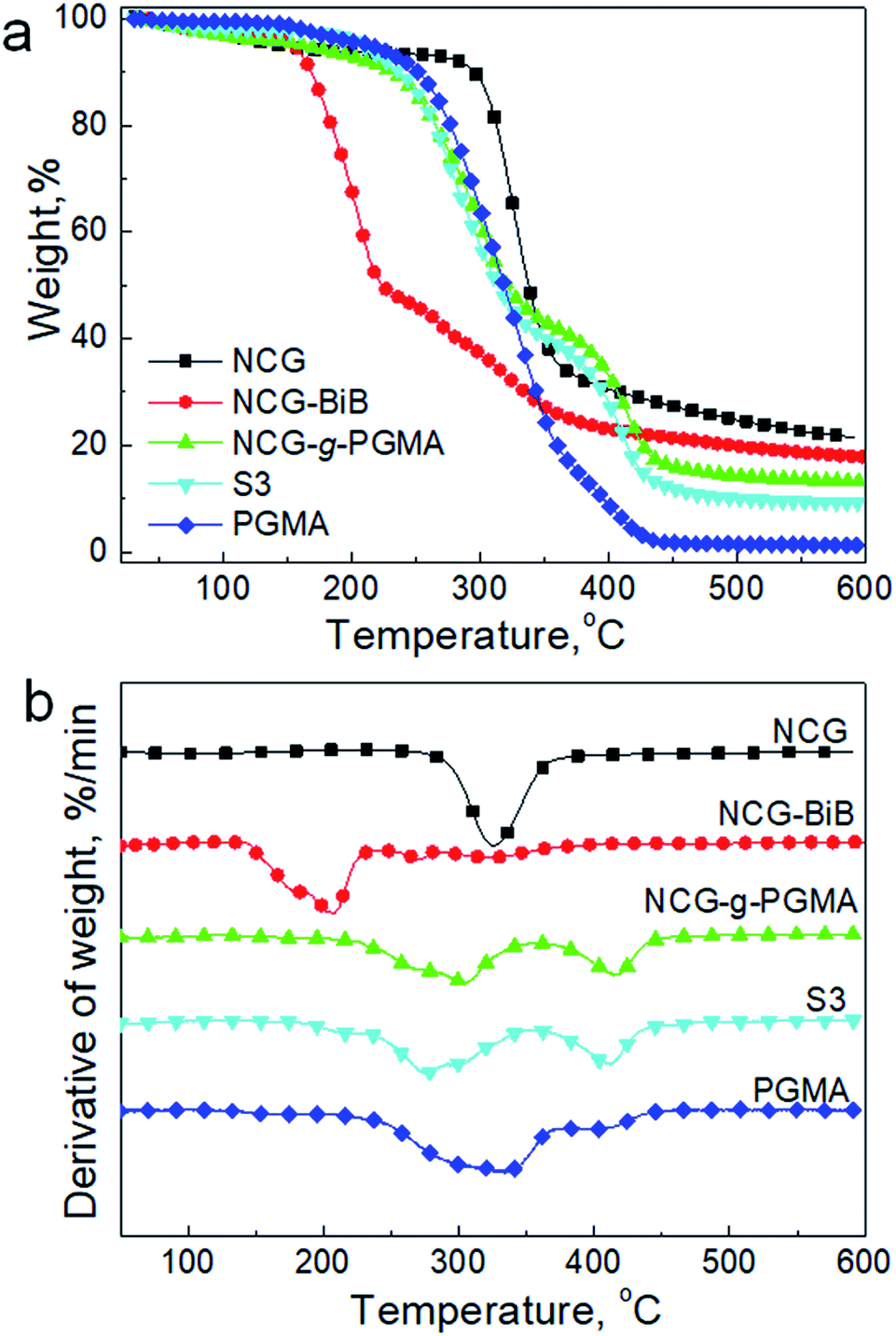 | ||
| Fig. 8 TGA results (a) and derivative thermogravimetry (DTG) curves (b) of the NCG, NCG-BiB, NCG-g-PGMA nanocomposite, hydrophobic NCG-g-PGMA nanocomposite (Table 1, sample S3) and PGMA under a nitrogen atmosphere. | ||
Conclusions
In summary, the hydrophobic NCG-g-PGMA nanocomposites were fabricated via SI-ATRP of GMA monomers and hydrophobic modification with C7F15COCl on the cellulose nanofibrils of the NCG. The grafting temperature and time played an important role in grafting PGMA on NCG and hence dramatically affected the microscopic morphology and structure of the hydrophobic NCG-g-PGMA nanocomposites. The interconnected nanofibrillar network structure of the NCG was deposited with PGMA after SI-ATRP. Further hydrophobic modification with C7F15COCl did not change the microscopic morphology and structure of the NCG-g-PGMA nanocomposites. Moreover, the hydrophobic NCG-g-PGMA nanocomposites demonstrated a remarkable improvement in the tensile storage modulus above the glass transition temperature. The presented results illustrated the usefulness of NCG as platform for SI-ATRP and provided potential opportunities for fabricating functional NCG-based nanomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (21422405, 51373125) and the Major Program of National Natural Science Foundation of China (21334005). The authors thank to the facility support of the Wuhan Morning Light Plan of Youth Science and Technology (2017050304010312), the Special Fund for the Development of Strategic Emerging Industries of Shenzhen City of China (JCYJ20170818112409808), and Fundamental Research Funds for the Central Universities (2042017kf0195, 2042015kf0259, 2042017kf0227).References
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef.
- D. Klemm, F. Kramer, S. Moritz, T. Lindstrom, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef PubMed.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- J. G. Torres-Rendon, T. Femmer, L. De Laporte, T. Tigges, K. Rahimi, F. Gremse, S. Zafarnia, W. Lederle, S. Ifuku, M. Wessling, J. G. Hardy and A. Walther, Adv. Mater., 2015, 27, 2989–2995 CrossRef PubMed.
- H. Zhu, W. Luo, P. N. Ciesielski, Z. Fang, J. Y. Zhu, G. Henriksson, M. E. Himmel and L. Hu, Chem. Rev., 2016, 116, 9305–9374 CrossRef PubMed.
- R. Weishaupt, G. Siqueira, M. Schubert, M. M. Kämpf, T. Zimmermann, K. Maniura-Weber and G. Faccio, Adv. Funct. Mater., 2017, 27, 1604291 CrossRef.
- S. Wang, T. Li, C. Chen, W. Kong, S. Zhu, J. Dai, A. J. Diaz, E. Hitz, S. D. Solares, T. Li and L. Hu, Adv. Funct. Mater., 2018, 28, 1707491 CrossRef.
- M. Zhao, F. Ansari, M. Takeuchi, M. Shimizu, T. Saito, L. A. Berglund and A. Isogai, Nanoscale Horiz., 2018, 3, 28–34 RSC.
- A. Sydney Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413 CrossRef PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef PubMed.
- J. Ran, L. Wu, Z. Zhang and T. Xu, Prog. Polym. Sci., 2014, 39, 124–144 CrossRef.
- O. Garcia Valdez, P. Champagne and M. F. Cunningham, Prog. Polym. Sci., 2018, 76, 151–173 CrossRef.
- V. S. Vo, S. Mahouche Chergui, J. Babinot, V. H. Nguyen, S. Naili and B. Carbonnier, RSC Adv., 2016, 6, 89322–89327 RSC.
- A. Khabibullin, K. Bhangaonkar, C. Mahoney, Z. Lu, M. Schmitt, A. K. Sekizkardes, M. R. Bockstaller and K. Matyjaszewski, ACS Appl. Mater. Interfaces, 2016, 8, 5458–5465 CrossRef PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- S. Hansson, E. Ostmark, A. Carlmark and E. Malmstrom, ACS Appl. Mater. Interfaces, 2009, 1, 2651–2659 CrossRef PubMed.
- D. Nyström, J. Lindqvist, E. Östmark, P. Antoni, A. Carlmark, A. Hult and E. Malmström, ACS Appl. Mater. Interfaces, 2009, 1, 816–823 CrossRef PubMed.
- H. Wu, L. Wu, S. Lu, X. Lin, H. Xiao, X. Ouyang, S. Cao, L. Chen and L. Huang, Carbohydr. Polym., 2018, 181, 419–425 CrossRef PubMed.
- L. Zhao, L. Li, Y. Wang, J. Wu, G. Meng, Z. Liu and X. Guo, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 9 CrossRef.
- J. Lindqvist, D. Nystrom, E. Ostmark, P. Antoni, A. Carlmark, M. Johansson, A. Hult and E. Malmstrom, Biomacromolecules, 2008, 9, 2139–2145 CrossRef PubMed.
- J. O. Zoppe, Y. Habibi, O. J. Rojas, R. A. Venditti, L. Johansson, K. Efimenko, M. Österberg and J. Laine, Biomacromolecules, 2010, 11, 2683–2691 CrossRef PubMed.
- R. Alosmanov, K. Wolski and S. Zapotoczny, Cellulose, 2017, 24, 285–293 CrossRef.
- J. Arredondo, P. G. Jessop, P. Champagne, J. Bouchard and M. F. Cunningham, Green Chem., 2017, 19, 4141–4152 RSC.
- K. Ou, X. Wu, B. Wang, C. Meng, X. Dong and J. He, Cellulose, 2017, 24, 5211–5224 CrossRef.
- A. Aied, Y. Zheng, A. Pandit and W. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 826–831 CrossRef PubMed.
- B. V. Bhut, K. A. Conrad and S. M. Husson, J. Membr. Sci., 2012, 390–391, 43–47 CrossRef.
- A. Boujemaoui, C. C. Sanchez, J. Engstrom, C. Bruce, L. Fogelstrom, A. Carlmark and E. Malmstrom, ACS Appl. Mater. Interfaces, 2017, 9, 35305–35318 CrossRef PubMed.
- Z. Zhang, G. Sèbe, X. Wang and K. C. Tam, ACS Appl. Nano Mater., 2018, 1, 632–641 CrossRef.
- M. Morits, J. R. McKee, J. Majoinen, J. M. Malho, N. Houbenov, J. Seitsonen, J. Laine, A. H. Gröschel and O. Ikkala, ACS Sustainable Chem. Eng., 2017, 5, 7642–7650 CrossRef.
- J. R. McKee, J. Huokuna, L. Martikainen, M. Karesoja, A. Nykänen, E. Kontturi, H. Tenhu, J. Ruokolainen and O. Ikkala, Angew. Chem., Int. Ed., 2014, 53, 5049–5053 CrossRef PubMed.
- J. Cai, S. Kimura, M. Wada, S. Kuga and L. Zhang, Chemsuschem, 2008, 1, 149–154 CrossRef PubMed.
- Z. Shi, J. Huang, C. Liu, B. Ding, S. Kuga, J. Cai and L. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 22990–22998 CrossRef PubMed.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279–6287 CrossRef.
- J. Cai, S. Liu, J. Feng, S. Kimura, M. Wada, S. Kuga and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2076–2079 CrossRef PubMed.
- J. Cai, S. Kimura, M. Wada and S. Kuga, Biomacromolecules, 2009, 10, 87–94 CrossRef PubMed.
- Q. Wang, J. Cai and L. Zhang, Cellulose, 2014, 21, 3371–3382 CrossRef.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- Z. Shi, H. Gao, J. Feng, B. Ding, X. Cao, S. Kuga, Y. Wang, L. Zhang and J. Cai, Angew. Chem., Int. Ed., 2014, 53, 5380–5384 CrossRef PubMed.
- Q. Wang, J. Cai, L. Zhang, M. Xu, H. Cheng, C. C. Han, S. Kuga, J. Xiao and R. Xiao, J. Mater. Chem. A, 2013, 1, 6678–6686 RSC.
- K. Li, J. Huang, H. Gao, Y. Zhong, X. Cao, Y. Chen, L. Zhang and J. Cai, Biomacromolecules, 2016, 17, 1506–1516 CrossRef PubMed.
- K. Li, J. Song, M. Xu, S. Kuga, L. Zhang and J. Cai, ACS Appl. Mater. Interfaces, 2014, 6, 7204–7213 CrossRef PubMed.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef PubMed.
- A. Carlmark and E. Malmstrom, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef PubMed.
- A. Carlmark and E. E. Malmstrom, Biomacromolecules, 2003, 4, 1740–1745 CrossRef PubMed.
- D. Daniel, X. Chunlin, L. Otto, H. Jarl, B. Peter and W. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5100–5110 CrossRef.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef PubMed.
- Y. Maréchal and H. Chanzy, J. Mol. Struct., 2000, 523, 183–196 CrossRef.
- S. Li, M. Xiao, A. Zheng and H. Xiao, Biomacromolecules, 2011, 12, 3305–3312 CrossRef PubMed.
- X. Sui, J. Yuan, M. Zhou, J. Zhang, H. Yang, W. Yuan, Y. Wei and C. Pan, Biomacromolecules, 2008, 9, 2615–2620 CrossRef PubMed.
- Z. Zhang, Q. Wu, K. Song, T. Lei and Y. Wu, Cellulose, 2015, 22, 2431–2441 CrossRef.
- D. Nyström, J. Lindqvist, E. Ostmark, A. Hult and E. Malmström, Chem. Commun., 2006, 34, 3594 RSC.
- P. S. S. Lacerda, A. M. M. V. Barros-Timmons, C. S. R. Freire, A. J. D. Silvestre and C. P. Neto, Biomacromolecules, 2013, 14, 2063–2073 CrossRef PubMed.
- S. J. Eichhorn and W. W. Sampson, J. R. Soc., Interface, 2005, 2, 309–318 CrossRef PubMed.
- X. Dong, H. Bao, K. Ou, J. Yao, W. Zhang and J. He, Fibers Polym., 2015, 16, 1478–1486 CrossRef.
- N. Misra, N. K. Goel, S. A. Shelkar, L. Varshney and V. Kumar, J. Mol. Catal. B: Enzym., 2016, 133, S172–S178 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04163f |
| This journal is © The Royal Society of Chemistry 2018 |