
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpeditious isomerization of glucose to fructose in aqueous media over sodium titanate nanotubes†
Sandeep Kumar‡
ac,
Devadutta Nepak‡b,
Sushil Kumar Kansalc and
Sasikumar Elumalai *a
*a
aChemical Engineering Division, Center of Innovative and Applied Bioprocessing (CIAB), Mohali, Punjab 140306, India. E-mail: sasikumar@ciab.res.in
bDepartment of Chemistry, Indian Institute of Technology Hyderabad (IITH), Kandi, Telangana 502285, India
cDr S. S. Bhatnagar University Institute of Chemical Engineering & Technology, Panjab University, Chandigarh 160014, India
First published on 24th August 2018
Abstract
Isomerization reaction of glucose to fructose over sodium titanate nanotubes (Na-TNTs) as a Lewis base catalyst was studied. Analytical instruments recorded the specific structural, textural and basic properties of the as-synthesized Na-TNTs. Furthermore, studying the catalytic isomerization performance of the Na-TNTs confirmed their high catalytic efficiency and suitability in aqueous media. The catalyst prompted rapid glucose isomerization within 2 min by achieving nearly half of the maximum yield, whereas with a prolonged reaction up to 15 min the maximum glucose conversion could be reached with 31.26% fructose yield and 65.26% selectivity under relatively lower operating conditions (100 °C and 10% wt catalyst dose). However, the recyclability performance of the catalyst was not impressive due to the accelerated leaching of cations and surface retention of carbonaceous content, resulting in ∼16% reduced yield after 4 runs. A simple regeneration technique using NaOH led to the initial catalytic activity being totally regained. Overall, a titania-based catalyst (preferably nanotube structured sodium titanate) was shown as a potential catalyst for large-scale demonstration of glucose isomerization to achieve high fructose productivity.
Introduction
The steadily increasing energy demand, depleting fossil reserves, and the negative environmental impacts of petroleum-derived chemicals all stimulate the search for alternative energy sources. In recent years, lignocellulosic biomass-derived chemicals and energy fuel precursors have been of great interest owing to their long-term sustainability and zero carbon emissions. In this regard, 5-hydroxymethylfurfural (HMF) has been identified as one of the top most building blocks, and it finds potential application in the preparation of fuel grade and other industrial chemicals (e.g., levulinic acid, 2,5-furandicarboxylic acid, 2,5-dimethylfuran).1 In the reconstruction of lignocellulose to HMF, the derived glucose initially undergoes isomerization reaction to form fructose as an intermediate carbohydrate compound and subsequently, it is transformed to HMF via dehydration. Therefore, it is considered that glucose to fructose transformation is highly important because thermodynamic equilibrium conversion exists between both sugar molecules at each specific reaction temperature.2 On the other hand, the glucose isomerization (GI) reaction is predominantly applied in the food manufacturing sectors, where fructose is used as the primary ingredient in the production of food and beverage items due to its sweeter taste than glucose. Popularly, biochemical (involving glucose isomerase) and chemical (involving NaOH) methods are commercially used for this purpose. However, these suffer from serious drawbacks, e.g., low tolerance in variations of feedstock quality, prolonged reaction, product yield (20–26% fructose yield, in the case of NaOH), undesired side products and odors, toxicity, etc.3 With the interest of production of HMF from glucose, the majority of studies have suggested using a multi-step protocol owing to the advantages that fructose could be more easily, quickly, and completely dehydrated to obtain the target compound than glucose due to its higher reactivity.2,4–6In this context, a heterogeneous catalytic approach is preferred owing to the higher selectivity and yield, operation under a wide range of conditions, minimal side product generation, facility of recovery and regeneration, and environmentally safe process.7 To date, the selective glucose to fructose conversion has been demonstrated using a different category of solid materials, including modified zeolites,8–10 metal oxides,11 hydrotalcites,12 and metallosilicates.13 Therefore, GI reaction is possible through Lewis acid and/or Lewis base catalysis, however, each of the protocols undergo dissimilar reaction pathways resulting in delivering varied final product concentrations. In perception, Lewis base catalysis is promising over the other due to better selectivity and high yield, and offers less chance of further transformation of formed fructose to HMF in aqueous media.2,14 Moreover, it can be handled in ambient conditions because the adsorption of carbon dioxide or water is not too strong that considerably reduces the product yield.15 In recent years, efforts are being made to develop ideal catalyst materials through a variety of modified approaches to overcome the drawbacks facing during large-scale processing, i.e., catalyst instability (activity loss) and less recycle ability, and higher expenses at catalyst preparation. For instance, Faria et al. developed a catalyst by growing carbon nanotubes on NaX faujasite zeolites with the interest of converting glucose to HMF. The Lewis base catalysis could achieve as high as 26 mol kg−1 h−1 fructose formation (with 85% selectivity when glucose conversion is <20%) in aqueous medium at 110 °C. Fascinatingly, the catalyst offered improved hydrothermal stability and resistance against leaching of the active sites.16 Indeed, the specific surface area of catalyst promotes the catalytic activity of reaction. At the same time, the enhancement of the external surface area harmfully contributes to the possible diffusion of molecules.17
Among the classified low-cost metal oxide catalysts, titania (or titanium dioxide, TiO2) is proven to be stable under rigorous reaction conditions, offer excellent surface area, good ion exchangeability, better recyclable performance, inert and safe material. Whereas, nanotubes possess high specific surface area and hollow morphology and therefore, it is widely researched in many areas, including chemical catalysis, photocatalysis, electrocatalysis, sensors, and lithium batteries compared to other regular forms (nanoribbon, nanowire, nanosheet).17,18 Moreover, the characteristic TiO2 exhibit a bifunctional property, i.e., Lewis acid and base attributed to the lattice O− and Ti+ ions, and therefore, it has been extensively applied for chemical reactions.19,20 For instance, Lanziano et al. demonstrated the catalytic performance of TiO2 (superficial area of 128 m2 g−1) on glucose to fructose conversion in aqueous medium, but only 12% wt fructose yield could be obtained at 120 °C claiming limited catalytic activity.21 Therefore, in the present study, titanate nanotube consisting Na+ as charge compensating cation exhibiting a Lewis base character was invested for the GI reaction in aqueous medium with the objective of demonstrating a feasible, cost-effective process method.18 For this purpose, Na+ is the most investigated candidate due to its greater affinity towards the support material, more active, easy availability and cheap compared to other common monovalent (commonly, Li+, Na+, K+, Cs+) and divalent (Ca++ and Ba++) cations.22 In hypothesis, the sodium titanate (Na2Ti3O7) offers large basic sites due to the presence of two lone pair electrons (O−) that allows more ion compensation is responsible for the basicity, thereby promoting the isomerization reaction.
Materials and methods
Chemicals
All chemicals and reagents (TiO2 anatase, NaOH, HCl, D-glucose, D-fructose, and D-mannose, ethanol, methanol, 1-propanol, LiNO3 of 99% purity, KNO3 of 99% purity, and CsNO3 of 99% purity) were purchased from Sigma Aldrich India as analytical grade and used without any modification.Sodium titanate nanotube synthesis
Sodium titanate nanotubes (Na-TNT) was prepared by conventional hydrothermal synthesis method.23,24 Briefly, 1.0 g TiO2 powder was added to 80 ml of 10 M NaOH solution in 100 ml beaker. The mixture was continuously stirred for 8 h at 200 rpm to achieve a homogeneous mix, transferred to a Teflon lined stainless steel autoclave, and heated for 5 days at 180 °C. The resultant solids were collected and washed using ethanol–water mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio) until the pH becomes neutral. The as-synthesized catalyst was dried at 80 °C overnight. Other titanate nanotube catalysts x-TNT, where x = Li+, K+, and Cs+ were prepared from Na-TNT through ion exchange. About 100 mg Na-TNT was added to the corresponding nitrate solution of 0.5 M conc. (prepared in milli Q water) and stirred continuously for 24 h (at 200 rpm) at 80 °C. The slurry mixture was washed repeatedly using milli Q water and the solids collected were dried overnight at 50 °C. Similarly, the H2Ti3O7 catalyst was prepared by using 1 M HCl solution, as reported elsewhere.17
:50 ratio) until the pH becomes neutral. The as-synthesized catalyst was dried at 80 °C overnight. Other titanate nanotube catalysts x-TNT, where x = Li+, K+, and Cs+ were prepared from Na-TNT through ion exchange. About 100 mg Na-TNT was added to the corresponding nitrate solution of 0.5 M conc. (prepared in milli Q water) and stirred continuously for 24 h (at 200 rpm) at 80 °C. The slurry mixture was washed repeatedly using milli Q water and the solids collected were dried overnight at 50 °C. Similarly, the H2Ti3O7 catalyst was prepared by using 1 M HCl solution, as reported elsewhere.17
Characterization techniques
Powder X-ray diffraction (XRD) analysis was performed to the as-synthesized Na-TNT on Philips X′ Pert Pro Super X-ray diffractometer Cu-Kα radiation (λ = 1.54056 Å) operated at 40 kV and 30 mA. The structural morphology and chemical composition were determined by SEM-EDX equipment (JEOL 5900 LV microscope). The size and morphology were measured by using a high-resolution transmission electron microscopy (HRTEM) images and selected-area electron diffraction (SAED) using JEOL 2010 microscope (resolving power 1.9 Å at 200 kV). The nitrogen adsorption–desorption isotherms were obtained using a Micromeritics ASAP 2020 automatic analyzer at (−80 °C). Before analysis, the samples were degassed at less than 10−1 Pa pressure at 250 °C for 6 h. BET method was used to calculate the specific surface area (SBET), and the total pore volume (Vp) was determined by nitrogen adsorption capacity at a relative pressure of 0.98. Similarly, the average pore diameter (DP) and pore size distributions were obtained from the adsorption isotherms by the BJH method. Temperature-programmed desorption of adsorbed CO2 (CO2-TPD) technique was used to measure the basic sites of Na-TNT. Briefly, 0.1 g of catalyst was placed in a fixed-bed U-shaped quartz reactor. The sample was first pretreated in He flow (30 ml min−1) at 350 °C for 2 h, cooled down at room temperature, and flushed with a CO2/He gas mixture (50% at 30 ml min−1 flow) for 30 min. Followed by, purging of the remaining CO2 with He flow. The TPD was run from temperature 100 to 600 °C at a constant heating rate (10 °C min−1 in He gas stream). The basicity of the catalyst was estimated as being the total amount of CO2 released through thermal programmed desorption per gram of the Na-TNT sample. Na to Ti atomic ratio of the catalyst was determined by ICP-AES analysis (Shimadzu, ICP-1000IV).Glucose isomerization to fructose
Glucose isomerization reaction was carried out in a 3 ml thick-walled glass reactor. In a typical reaction, glucose (1 ml of 1.5% stock solution) and the as-synthesized catalyst were loaded into the reactor (maximum 16.67% wt on glucose) and sealed tightly. The reactor was pre-warmed depending on the reaction temperature and heated up to 120 °C in an oil-bath using a hot plate stirrer at 200 rpm. Once the reaction is completed (maximum 5 h), the reactor was cooled down immediately in an ice bath. The catalyst particles were filtered off by passing through 0.45 μm membrane under vacuum condition (S1). The filtrate collected was analyzed by high-performance liquid chromatography (HPLC, Agilent Technologies 1200 infinity series) after requisite dilution. The HPLC system was equipped with Agilent Hi-Plex Ca column (300 mm length, 8 μm porosity), and maintained at 85 °C and 0.6 ml min−1 of HPLC grade water (mobile eluent). The carbohydrates were analyzed on UV detector operated at 50 °C. The concentration of sugars was estimated using their respective calibration charts prepared by commercial grade standards. The glucose and products yield (fructose and mannose) was calculated based on the difference in concentration of residual glucose and products to initial glucose concentration, respectively. Similarly, the products selectivity were calculated based on the difference in concentration of products to glucose reacted. All experiments were conducted in triplicate and the data reported is the average.Catalyst recovery and regeneration
The collected catalyst solid after the first run (S1) was appeared to be slightly yellowish in color was washed repeatedly using milli-Q water until it becomes natural white (S2). It was dried at room temperature and freeze-dried for up to 48 h. The same procedure was repeated for each recycle runs. The catalyst regeneration was performed to the solid materials collected after four recycle runs. It was calcined at 300 °C before treating with 2 M NaOH solution in 100 ml beaker. The mixture was heated for 12 h at 80 °C under continuous stirring condition (200 rpm). The resultant solid material was collected and washed repeatedly using an ethanol–water mixture (50:50 ratio) until the pH becomes neutral.
Results and discussion
Characterization of as-synthesized sodium titanate nanotubes
Of the classical preparation of methods of titanate nanotube (TNT) using TiO2 as a precursor, hydrothermal treatment with alkali is of particular interest because nanotube aggregates can be produced in an unsophisticated manner at low temperatures. Indeed, the characteristic TiO2 in the form of nanotubes have a relatively larger specific surface area (400 m2 g−1) and pore diameter (5–7 nm inner and 7–11 nm outer diameter) than that in the form of nanoparticles (128 m2 g−1).21,25 Results in providing larger availability of surface active sites and facilitate interactions with the support which then can lead to a higher activity of the catalyst particles.23,26 Moreover, the nanotubes have a distinctive ion-exchangeable ability due to their layered structure and permit mobility of Na+ and H+ in the interlayer of TNTs. Owing to these advantages, the synthesis of titanate nanotubes became a routine, and it is generally accepted that the nanotubes have a layered titanate structure. However, there is a discrepancy in the proposed models of formation of titanate nanotubes in the hydrothermal environment and also, between the crystal structure and chemical composition. Furthermore, the stability and other structural characteristics, including pore structure and crystallinity (the common crystalline habits are anatase, rutile, and brookite, which own different band gap energies and chemical activities) of the TiO2 nanotube are dependent on the preparation method and subsequent calcination temperatures.25,27 Therefore, the as-synthesized sodium titanate nanotube (Na-TNT) obtained using TiO2 anatase, as a precursor, through hydrothermal treatment in NaOH solution at 180 °C was subjected to various instrumental techniques to determine its structural characteristics. The initial chemical analysis of the ion-exchanged TNT revealed that it allowed ca. >60% ion exchange for Na+ ions. Further, the instrumental analysis of the nanotubes by ICP-OES verified the atomic ratio of Na to Ti as 0.65, which lies in the close range to the theoretical value of Na2Ti3O7 (0.67). Presumably, it is caused due to some of the Na+ ions were exchanged by H+ during the H2O washing treatment, which leads to the synthesis of hydrous sodium titanate of composition NaxH2–xTi3O7·nH2O, resulting in forms multi-walled scroll-type nanotubes. Also, traces of unreacted TiO2 precursor were presented in the synthesized material.28The textural characteristics, mainly BET specific surface area, pore volume, and pore diameter, were determined from N2 adsorption isotherms, as displayed in Fig. 1a. The type IV N2 isotherms with N2 hysteresis loop indicated that solid material is mesoporous and exhibited 155.3 m2 g−1, 0.58 cm3 g−1, and 7.2 nm, respectively. The XRD pattern (Fig. S1a in the ESI†) of the solids showed that typical peaks at 10.1, 24.3, 28.4, and 48.5° corresponding to the reflections from (200), (110), (211) and (020) planes, respectively, of Na-TNT (Na2Ti3O7). The composition was confirmed using JCPDS-ICDD card 31-1329. The distinct peak at 10.1° (2θ) is attributed to the interlayer distance of Na-TNT, which is equal to 0.80 nm.25 In understanding, this characteristic peak and its position confirm the presence of Na in the interlayer spaces of the sodium titanate structure. Moreover, the diffraction signals point out the poor crystalline nature of the synthesized nanotubes based on the broadness of all observed peaks. Most importantly, no signals attributable to any crystalline phase of TiO2 or to other types of Na-TNTs (general formula of Na2TinO2n+1, where n = 4,5,6 varies depending on the heat treatment and condition) were observed in the diffraction profile of the synthesized nanotubes. Thus, the analysis result confirms that the successful transformation of TiO2 anatase into Na-TNT through the alkaline hydrothermal synthesis. Representative HR-TEM imaging was acquired to the nanotubes (Fig. 1b), which characterized that solids are in tubular morphology (open-ended tubes with 3 to 5 layered walls and not symmetric) with outer and inner diameters being 9.8 and 4.8 nm, respectively, and of varying lengths ranging up to several tens nm. Though the synthesized material appeared to be tubes are joined together in bundles, the selected area electron diffraction (SAED) pattern was recorded using large selected area aperture (Fig. 1c). The analysis results are revealing that the innermost ring to a d-spacing of (110) planes, whereas the other two circles to (200) and (211) inter-planer distances, confirming the tube-shaped crystals of Na-TNT. Further, the obtained spectroscopic and structural data were correlated with the morphology on scanning electron microscopy (SEM) with EDX measurement. The imaging result indexed the evident transformation of spherical particles into the fibrous tubular material (Fig. S1b in the ESI†), whereas the EDX data also reproduced its maximum ion exchange degree (∼65%).
 | ||
| Fig. 1 (a) Typical N2 adsorption–desorption isotherm and BJH pore size distribution plot (inset) of as-synthesized Na-TNT, (b) HR-TEM, and (c) SAED characterization analyses result of the as-synthesized Na-TNT. | ||
With regard to the basicity, typical CO2 desorption profile was obtained for the as-synthesized Na-TNT sample through CO2-TDP desorption analysis technique. Results presented (Fig. S2a in the ESI†) that an intermediate signal with the maximum at ∼202 °C corresponds to desorption of CO2 from sites of medium basic strength (106 μmol g−1). Whereas, the other two intensity peaks obtained at temperatures 338 °C and 518 °C were related to the CO2 desorption from sites with medium (81 μmol g−1) and strong basicity (43 μmol g−1), respectively. The estimated total amount of basic sites was 231 μmol g−1. However, it is comparatively lower than other conventional Lewis base catalysts, for example, MgO with 346 μmol g−1.29 Results revealed that nanotube catalysts consisted predominantly weak and medium basic sites. The functional groups of Na-TNT were examined on FTIR spectroscopy between 400 and 4000 cm−1. It can be noticed that nanotubes exhibit a broad and intense band located at about 3300 cm−1 (Fig. S2b in the ESI†), which is attributed to the O–H stretching mode, indicating the presence of surface hydroxyl groups and water molecules adsorbed on the surface and in the interlayer space of the nanotubes. Water molecules were confirmed by the presence of the band at 1630 cm−1 that is assigned to the H–O–H deformation mode. The band observed at 897 cm−1 can be attributed to the Ti–O stretching mode which involves non-bridging oxygen atoms. The band at 470 cm−1 is related to Ti–O–Ti vibrations of the interconnected octahedra that are rigid units responsible for the formation of the nanotube walls. Overall, the analyses result authenticated the expected structural property of the as-synthesized Na-TNT and found to be consistent with the literature reports.25,30,31
Performance of Na-TNT on glucose to fructose isomerization
Typical base catalyzed GI reaction follows the classical Lobry de Bruyn–Alberda van Ekenstein (LdB–AvE) rearrangement mechanism.32 Accordingly, the postulated reaction mechanism scheme of the GI reaction in the presence of Na-TNT is depicted in Fig. 2. When the glucose is in contact with Na2Ti3O7, the ring opening is induced by the Na+ ion, which is the most important step in the isomerization pathway, based on the fact that glucose primarily exists in its ring-form in H2O to a greater extent. Further, the hydride ion shift is occurred by the basic sites on the catalyst, the glucose C-2 is deprotonated, resulting in the formation of 1,2-enediol intermediate (trans-enediol). Followed by, a proton is then transferred from C-2 to C-1 and O-2 to O-1 to form fructose, and also, the formation of mannose via epimerization. Alongside, more complex reaction pathways (non-reversible) involving the 2,3-enediol anion, aldolization/retro-aldolization, β-elimination, and benzylic rearrangement generates a variety of unwanted byproducts (such as p-saccharinic acid, glyceric acid, 3-hydroxypropanoic acid, 3-hydroxypropanol, glyceraldehyde and other compounds).33 In concurrence to the base catalyzed isomerization reactions in the presence of NaOH or organic amines that systematically Na+ ion helps in the glucose ring opening mechanism, whereas OH− ion is responsible for the isomerization.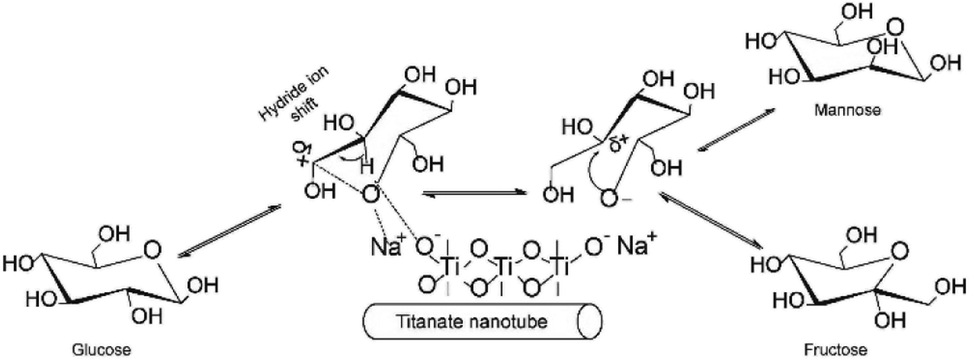 | ||
| Fig. 2 Proposed reaction scheme of glucose to fructose isomerization over Na2Ti3O7. | ||
The rate and extent of these undesired side reactions could be controlled by tuning of reaction parameters, including the type of base cation and its concentration, and temperature.34 Consequently, the as-synthesized Na-TNT was tested for its catalytic performance over aldose to ketose isomerization reaction under varying reaction conditions. Initially, the effect of catalyst dose to glucose was investigated under different loading levels ranging from 3.33 to 16.67% wt, while keeping the other parameters constant (at 90 °C for 30 min). Typical isomerization result was achieved during the incremented loadings up to the optimum, and further continuous supply of catalyst adversely affected the yield (Table 1). For example, a linearly increasing trend of fructose yield with 28% relative increments up to 10% wt catalyst load was achieved and started gradually decreasing as the load increases further and reaching as low as relatively 6.46% at the maximum load condition. Whereas, the fructose selectivity maintained a decreasing fashion from the start of the reaction (from 80.27 to 62.98%) even at low catalyst dosage mainly attributed to the further conversion of fructose. That is isomerization to glucose and undergo secondary transformations when it remains longer inside the nanotube structure because of partial blockage by the pores and interaction with the alkali ions present in the nanotubes.35 However, the glucose conversion showed a contrary trend with the corresponding incremented catalyst dose (increased from 28.34 to 46.71), this clearly indicates that additional catalyst load, in turn, provides higher basic conditions led to the formation of by-products under harsh conditions (90 °C for 30 min). Normally, the monosaccharides are unstable under strongly alkaline conditions and led to degrading them into numerous different byproducts at least 50 unwanted compounds.33,34 The evident color changes of the medium from pale yellow to dark brown as with the increased catalyst dose at 100 °C (Fig. S3 in the ESI†) indicates the progression of this thermally-induced nasty side reactions unconditionally.
| Catalyst loading% wt per wt Glu | YFru (%) | SFru (%) | YMan (%) | SMan (%) | TOF (×10−3) | Carbon balance (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 90 °C for 30 min in H2O medium. TOF – turn over frequency is calculated as moles of fructose formed per mole of Na charge compensating cation supply per second. Carbon balance is derived based on the difference between moles of carbon in products (fructose and mannose) and unreacted glucose, and to the mole of carbon in the initial glucose. N.D. – not detected. YFru-fructose yield; SFru-fructose selectivity; YMan-mannose yield; SMan-mannose selectivity. | ||||||
| 3.33 | 22.75 | 80.28 | 3.27 | 11.54 | 1.49 | 94.41 |
| 6.67 | 28.00 | 76.64 | 4.31 | 11.81 | 1.84 | 91.47 |
| 10.00 | 30.78 | 68.79 | 5.39 | 12.05 | 2.02 | 86.03 |
| 13.33 | 30.21 | 65.59 | 5.76 | 12.50 | 1.98 | 84.15 |
| 16.67 | 28.79 | 62.99 | 4.77 | 10.68 | 1.89 | 74.77 |
Mannose is the other main by product generated during typical GI reaction, formed in parallel to fructose by following the same trend throughout the incremented severities; it accounted for 5.76% wt (maximum) at the optimum (10.0% wt catalyst dose). While correlating the results with the estimated turnover frequency (TOF, moles of fructose produced per mole of charge compensating Na ion per s, assuming complete ionic compensation exist between Na+ and O−) of the reaction data, representing the same bell-shaped trend with maxima depending on the reaction condition (as summarized in Table 1). Furthermore, the results suggested that reaction was catalyzed only on Lewis basic sites without the aid of any of the other properties of support material except textural influence, based on the control run conducted in the presence of H2Ti3O7. In which, no fructose was formed due to compensation of ionic charges by H+ and O− exhibiting neither base nor acid sites (Fig. S4 in the ESI†). However, the decreased carbon balance (92%) attributed to the sugar degradation at 100 °C. In consistent with the hypothesis that the characteristic Na2Ti3O7 possess a Lewis basic site, which plays an important role in determining the catalytic activity of the reaction, suggesting the good ion exchange degree of Na+ to be responsible for the basic sites. The obvious decreasing value of carbon balance indicated that significant loss of carbohydrate molecules with the corresponding incremented severities (up to ∼33% with averagely 10% point differences). The reaction rate can also slow down at higher catalyst dose because of the approach to the thermodynamic equilibrium conversion for the glucose to fructose at the maintained temperature (100 °C). Therefore, the optimum Na-TNT/glucose ratio is considered to be 1:10, corresponding to a fructose yield and selectivity of 30.80% and 68.80%, respectively.
In a typical heterogeneous catalysis reaction, the catalytic activity of the reaction is greatly influenced by temperature. With regard to the texture of the support, the characteristic titanate nanotubes exhibit remarkable catalytic performance for the reaction even at room temperature condition compared to the other conventional catalysts. Overall literature studies have shown that relatively low reaction temperatures and times favor aldose isomerization activity under alkaline pH.33,34 In addition, the GI reaction is slightly endothermic.14 Therefore, it was expected to achieve a better glucose conversion accompanied with fructose selectivity under low-temperature conditions in the presence of Na-TNT. Consequently, the response of the glucose isomerization reaction was recorded with the correspondingly raised temperatures ranging from room temperature (25 °C) to 120 °C, while maintaining the catalyst to glucose ratio as 1:10 for 30 min constant (Fig. 3). No or less improvement in GI reaction was observed at 25 °C (<1% fructose yield with 28% glucose conversion), whereas the raised temperatures significantly influence products yield. For instance, ∼2-fold relative increase in the fructose yield was obtained when the temperature was raised from 60 to 80 °C and further increased temperatures showed a slight increment in the yield up to 100 °C, at which a maximum yield was achieved (30.94% fructose yield with 62.88% selectivity). A drastically decreased yield result was recorded beyond the optimum level, i.e., relatively ∼27% under higher temperature conditions, demonstrating higher temperatures favor more sugar degradation reactions.34 Whereas, the fructose selectivity was appeared to be the same pattern of decreasing value (relatively as low as ∼59%) and worse under harsh conditions (∼2-fold lower selectivity at 120 °C). Although the prolonged reaction times (>45 min) and at relatively higher temperatures (>60 °C) resulted in higher glucose conversion (ratio of glucose reacted to initial supply), lower fructose yield and selectivity was achieved attributed to the formation of unwanted byproducts through undesired reactions.11,34 This was supported by the color change indications of the reaction medium with respect to the temperatures (Fig. S5 in the ESI†). Mannose exhibited a nearly same production pattern of fructose concentration during the increased temperatures.
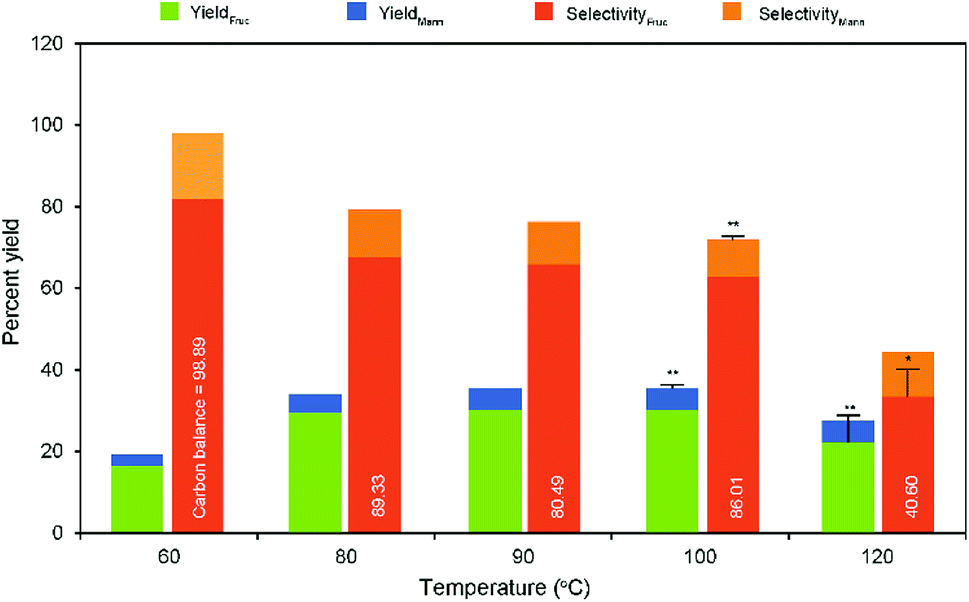 | ||
| Fig. 3 Effect of temperature on glucose isomerization to fructose in aqueous medium (10% wt catalyst dose on glucose). Error bar represents standard deviation, and asterisks (*) and (**) indicate significant and non-significant at p-value ≤ 0.05, respectively. | ||
Kinetics of glucose isomerization at different temperatures
Further investigation of the effect of time on GI reaction was studied at different temperatures viz. 60, 80 and 100 °C. The comparative results show that the reaction is time dependent and catalytic activity had a significant impact on the influence of the reaction (Fig. 4). For example, nearly 65% of the maximum yield (i.e., 31.26% wt after 15 min) could be reached within 2 min reaction at 100 °C, whereas the other reactions conducted at lower temperatures took 30 min (maximum) to reach the marked level. Further prolonged reaction attained the maximum fructose concentration at varied times depending on the temperature, e.g., 31.26% with 65.26% fructose selectivity within 15 min at 100 °C. Whereas, the low temperature reactions at 80 °C and 60 °C could yield the maximum after 30 min (30.31% with 67.60% fructose selectivity) and 150 min (28.80% with 76.75% fructose selectivity), respectively. This phenomenon explains that the reaction is controlled up to the specified times depending on the reaction severity and beyond, the activity practically reaches a plateau as displayed in Fig. 4. As mentioned earlier, the contrary responses achieved with the glucose conversion and fructose selectivity with all the temperatures are due to the carbohydrate degradation and reversible reactions.8 Mechanistically, the equilibrium constant for the glucose isomerization reaction lies <0.6, therefore, the reaction is reversible to form fructose and is unavoidable under the specified conditions.32 The temperature dependency of the GI reaction was verified using Arrhenius equation through the correlation plot obtained between lnK vs. 1/T (Fig. S6 in the ESI†), outlining the increased catalytic activity of the reaction under raised temperature conditions (R2 = 0.99).22 The kinetic reaction rates also pronounced remarkable differences with respect to the temperatures (60, 80 and 100 °C), i.e., 1.0, 6.0 and 19.0 × 10−4 s−1. The measured activation energy (Ea) of the reaction was 76 kJ mol−1, which is consistent with prior literature studies investigated under Lewis base condition in water system.36 The estimated carbon balance and TOF values were summarized in Table S1 in the ESI.† Although the increased productivity accomplished within the shorter residence time possibly ascribed to the enhanced surface reaction of nanotubes offering more basic sites and within mesopores, the stability of the catalyst was not satisfactory, resulting in a low glucose conversion with fructose yield after 15 min. This suggests that higher catalyst deactivation and proximity to the thermodynamic equilibrium conversion for this reaction at 100 °C and higher reaction times.8 At last, it can be noticed that the post-reaction catalysts color turned from white to yellow, suggesting it contained a considerable amount of organic matter, based on the spectroscopic analysis23 (Fig. S7 in the ESI†). At the same time, a proportionately increased mannose concentration with decreasing fructose yield with respect to time (after 15 min) (Fig. 4). Suggesting that progression of reversible reaction through cis-enediol intermediate formation under favourable conditions due to the shift of reaction equilibrium, which results in producing a mixture of stereoisomers (i.e., mannose and glucose),37,38 as scheme depicted in Fig. 2 and S8 in the ESI.† Nevertheless, the catalyst stimulated the maximum fructose productivity within relatively shorter residence time than the reported Lewis base catalysis, where slightly harsher conditions were employed (using more than 20% catalyst loading, 100 °C, and 30 min).8 Therefore, the plausible cause for this comparable yield achievement is due to leaching of Na+ ions, and surface passivation by strongly adsorbed unwanted organic compounds led to catalyst deactivation.16
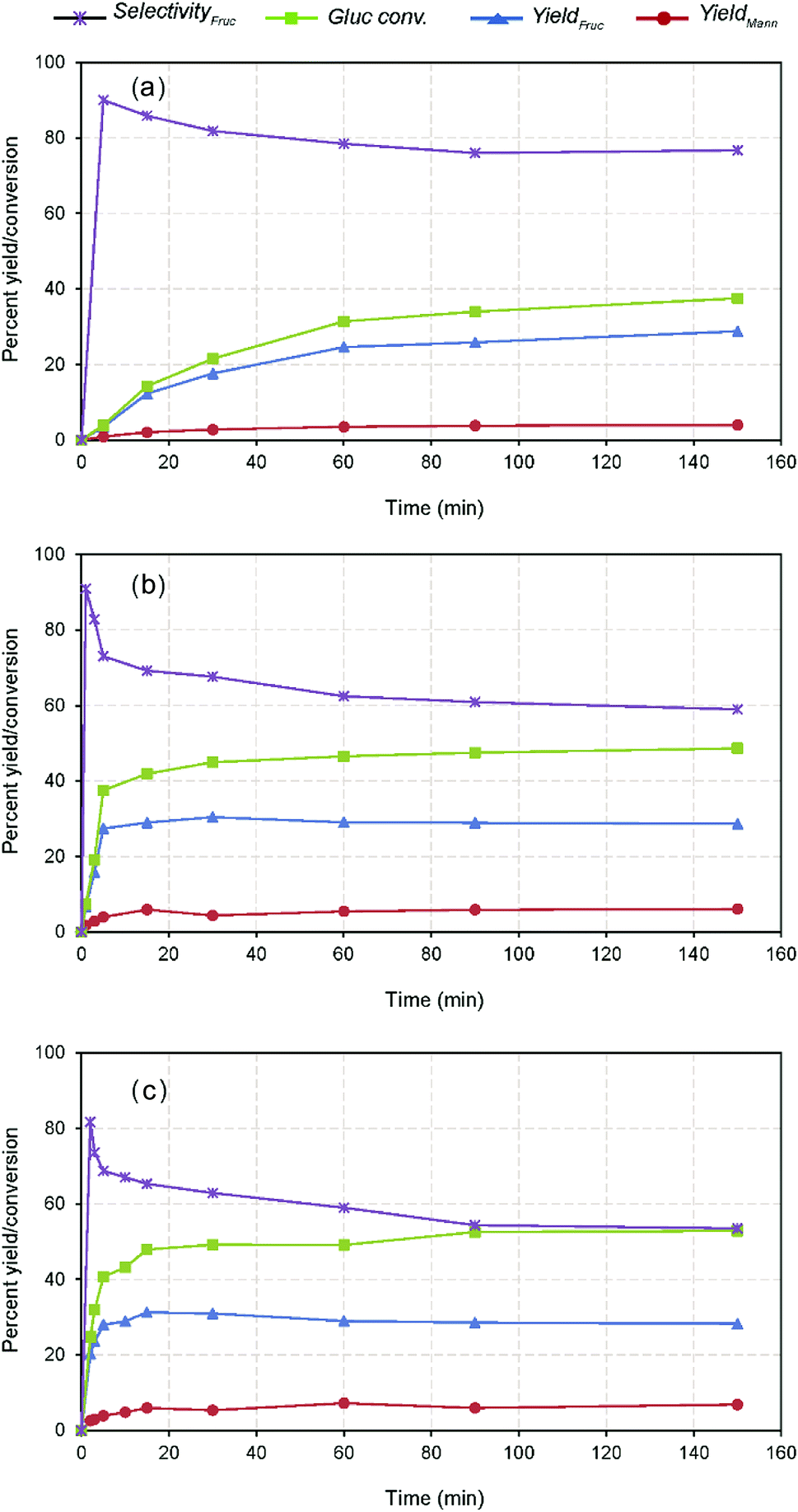 | ||
| Fig. 4 Kinetics of glucose isomerization at different temperature conditions, (a) 60 °C, (b) 80 °C, and (c) 100 °C (10% wt catalyst dose on glucose). | ||
Catalyst recycle-ability and regeneration
In complementary, the nanotubes allowed easy recovery through the formal filtration and washing after the finish of the reaction. Solid catalysts reuse in heterogeneous catalysis is one of the most important advantages. However, the activity decay is the only in subject to without loss of their catalytic activity.22 In perception, formation and retention of carbonaceous materials (coke) inside pore structure, and leaching of ions from the solid material to the liquid product results in the deactivation of the solid catalysts during the reaction.39 In order to evaluate the recyclability of the recovered catalyst, consecutive repeat studies were conducted under the modest reaction conditions. The comparative results show (Fig. 5) that an adverse effect on glucose conversion (16% relative lower to the fresh runs) and fructose yield (10%) during recycling runs up to 4 was achieved when using the recovered nanotubes thermal treatment. This is supposed to be leaching of Na+ ion, thereby reducing the basicity, blockage of pores due to the deposited unwanted carbonaceous matters, and agglomeration of catalyst particles.22 In addition, results of the SEM analysis of both pre- and post-reaction catalysts clearly show the increased agglomeration of catalyst particles during the reaction that in turn cause a reduction in catalytic activity (Fig. S9 in the ESI†). Consequently, a simple regeneration technique involving NaOH was employed to the post-reaction catalyst solids after calcination under the similar conditions. The result shows that comparable fructose yield (29.98% wt) with selectivity (67.86%) was obtained under the modest reaction conditions to the maximum yield. This conveys that it is possible to regain its total catalytic activity through the conventional ion exchange protocol, demonstrating a cost-effective isomerization process. The results proposed that Na-TNT could be a promising candidature for the large-scale isomerization of glucose to fructose reaction through continuous process operations (most suitably fixed bed reactors).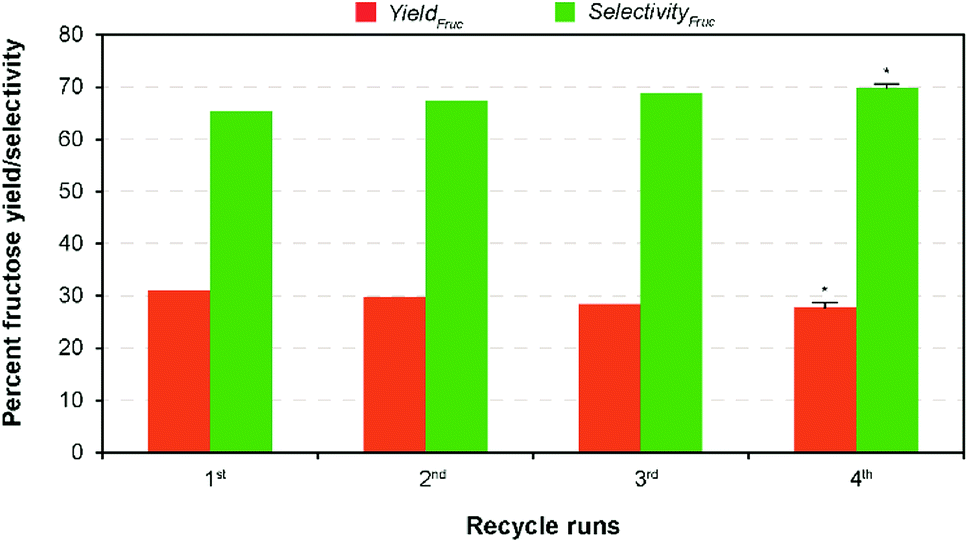 | ||
| Fig. 5 Performance of recovered catalyst on each recycle runs of glucose isomerization conducted under modest reaction conditions (10% wt catalyst dose at 100 °C for 15 min). Error bar represents standard deviation, and asterisk (*) indicates significant at p-value ≤ 0.05, respectively. | ||
In a typical GI reaction, it is postulated that stabilization of ions via hydroxylation of alkali metal oxide surface with the hydroxyl anions available in the solution (aqueous) initiates the reaction.34 In order to verify the mechanism, a series of comparative studies were conducted in alcohol systems, such as methanol (pKa = 15.5), ethanol (pKa = 15.9), and 1-propanol (pKa = 16.1) medium (both diluted and non-diluted levels) under the modest conditions (10% wt catalyst dose at 100 °C for 30 min) (Fig. 6). Since larger the substituents in alcohols are better electron donors, which destabilize the resulting OH− anions. The discouraging results show that effectiveness of the reaction was not up to the mark in alcohols, and no constructive correlation between the order of sequence of alcohols and yield results, predominantly, might be attributed to the combination of polarizability and solvation effects,40 and solubility of carbohydrate sugars.41 For instance, the systems achieved relatively 27–50% lower fructose yield (2:1 alcohol to water ratio) than water system, whereas the diluted systems remarkably increased the fructose concentrations (∼4-fold) accompanied by a better fructose selectivities (∼90%). Nevertheless, the alcohol systems minimized the formation of byproducts and degradation products (Fig. S9 in the ESI†). This suggests that Na-TNT is effective in the glucose to fructose conversion but only in H2O medium (pKa = 14.0).
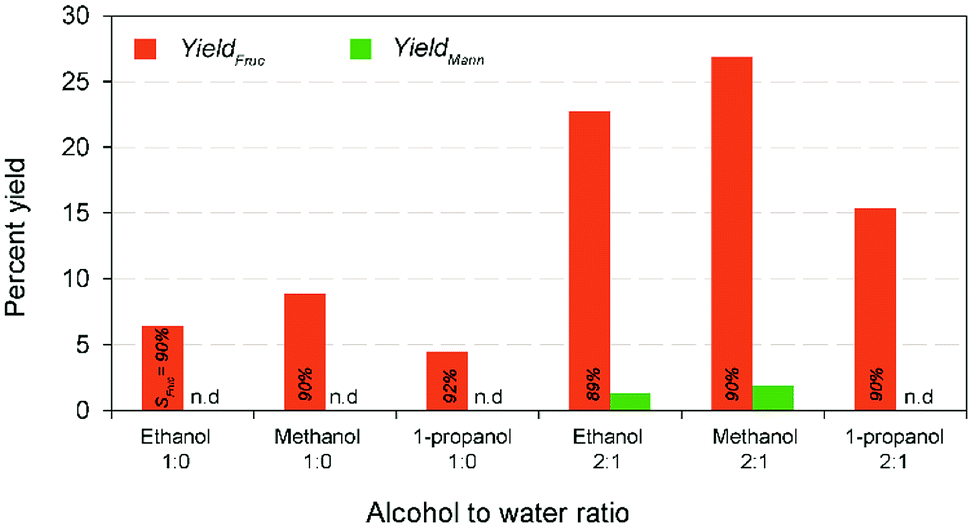 | ||
| Fig. 6 Response result of glucose isomerization in different alcohol mediums at various dilution levels (at 100 °C for 15 min using 10% wt catalyst dose). SFruc – fructose selectivity. N.D. – not detected. | ||
Evaluation of cation exchanged-TNTs on glucose isomerization
With regard to the catalyst basicity, the basic strength of the alkali-ion exchanged solid catalyst is rationalized by the negative charge of the oxygen atoms. Therefore, the base strength of TNT catalyst depends on the exchangeable alkali cations and it is reasonably increasing in the order of Li+ < Na+ < K+ < Rb+ < Cs+ within the monovalent alkali elements, according to the atomic properties (i.e., electrostatic interaction and hydrated radius).2,22 In order to evaluate the effectiveness of the other monovalent cation exchanged titanate nanotubes, namely Li-TNT, K-TNT and Cs-TNT on glucose to fructose isomerization, a comparative experiment was planned under the optimum processing conditions. The initial characterization of the as-synthesized catalysts revealed the maximum degree of ion exchange were ca. 56%, 48%, and 36%, respectively. Although Cs+ has the smallest hydrated radius within the group (226 pm), it can approach the surface the closest and be held the more tightly. These variations might be attributed to the relative affinity of a cation for a sorbent or the selectivity. In accordance with the literature studies that a reverse order is often observed with some hydrous oxides, for instance, the order of affinity of cations on titanium metal oxides is Li+ > Na+ > Cs+.42 The apparent isomerization results revealed (Fig. 7) that the fructose yield and selectivity varies depending on the basicity strength of the charge compensating ions. For example, Li+ exhibited a slightly lower fructose yield (29.97%) with higher selectivity (68.89%) when compared to the results achieved using Na+, whereas the others attained 16–22% relatively lower yields accompanied with a contrarily improved fructose selectivity (10–16%). This is consistent with the ion exchange capacity of the elements (K+ and Cs+) had comparatively lower degree value might be attributed to the increased hydrated radius in aqueous medium and other potential factors.43 Hence, even with the increase in basic nature with the ion-exchange of Na+ on titanate nanotubes the poor activity was observed. | ||
| Fig. 7 Comparative result of glucose isomerization in the presence of various monovalent alkaline cations of titanate nanotubes under the optimum conditions. Error bar represents standard deviation, and asterisks (*) and (**) indicate significant and non-significant at p-value ≤ 0.05, respectively. | ||
Conclusions
The present study demonstrated a single-step reaction of glucose isomerization to fructose over low-cost titanate material with Na+ ions in aqueous media. In a breakthrough, typical heterogeneous Lewis base catalysis expedites maximum fructose yield (31.26%) with higher selectivity (65.26%) within 15 min or less, establishing a cost-effective process methodology. Presumably, the increased basic sites due to the enhanced surface morphology of the catalyst (titanate nanotube) greatly influenced the catalytic reaction, thereby enabling higher glucose conversion. Na-TNTs exhibited a satisfactory performance over glucose isomerization to fructose reaction among the nanotubes tested consisting of Li+, K+, and Cs+, suggesting it could serve as a potential catalyst for large-scale with recyclable advantages. Though the fast leaching of Na+ ions during reaction impedes the fructose productivity, it is possible to regain its activity and durability through a simple regeneration protocol involving only NaOH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank Department of Biotechnology (Government of India), New Delhi, India for their consistent financial support. S. Elumalai acknowledges Department of Science and Technology (DST-SERB), New Delhi for the financial support through Grant No. YSS/2014/000031.Notes and references
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- I. Delidovich and R. Palkovits, ChemSusChem, 2016, 9, 547–561 CrossRef PubMed.
- H. Li, S. Yang, S. Saravanamurugan and A. Riisager, ACS Catal., 2017, 7, 3010–3029 CrossRef.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed.
- A. Norton, H. Nguyen, N. L. Xiao and D. G. Vlachos, RSC Adv., 2018, 8, 17101–17109 RSC.
- Z. Xue, M.-G. Ma, Z. Li and T. Mu, RSC Adv., 2016, 6, 98874–98892 RSC.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef PubMed.
- I. Graça, D. Iruretagoyena and D. Chadwick, Appl. Catal., B, 2017, 206, 434–443 CrossRef.
- S. Li, T. Josephson, D. G. Vlachos and S. Caratzoulas, J. Catal., 2017, 355, 11–16 CrossRef.
- Z. Zhu, H. Xu, J. Jiang, Y. Guan and P. Wu, J. Catal., 2017, 352, 1–12 CrossRef.
- M. Watanabe, Y. Aizawa, T. Iida, R. Nishimura and H. Inomata, Appl. Catal., A, 2005, 295, 150–156 CrossRef.
- S. Yu, E. Kim, S. Park, I. K. Song and J. C. Jung, Catal. Commun., 2012, 29, 63–67 CrossRef.
- S. Lima, A. S. Dias, Z. Lin, P. Brandão, P. Ferreira, M. Pillinger, J. Rocha, V. Calvino-Casilda and A. A. Valente, Appl. Catal., A, 2008, 339, 21–27 CrossRef.
- Q. Yang, S. Zhou and T. Runge, J. Catal., 2015, 330, 474–484 CrossRef.
- Y. Ono and T. Baba, Catal. Today, 1997, 38, 321–337 CrossRef.
- J. Faria, M. Pilar Ruiz and D. E. Resasco, ACS Catal., 2015, 5, 4761–4771 CrossRef.
- M. Kitano, K. Nakajima, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2010, 132, 6622–6623 CrossRef PubMed.
- L. L. Marciniuk, P. Hammer, H. O. Pastore, U. Schuchardt and D. Cardoso, Fuel, 2014, 118, 48–54 CrossRef.
- M. Watanabe, Y. Aizawa, T. Iida, T. M. Aida, C. Levy, K. Sue and H. Inomata, Carbohydr. Res., 2005, 340, 1925–1930 CrossRef PubMed.
- G. Martra, Appl. Catal., A, 2000, 200, 275–285 CrossRef.
- C. Lanziano, F. Rodriguez, S. Rabelo, R. Guirardello, V. Da Silva and C. Rodell, Chem. Eng. Trans., 2014, 37, 589–594 Search PubMed.
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193, 257–264 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef.
- D. Nepak and S. Darbha, Catal. Commun., 2015, 58, 149–153 CrossRef.
- A. Gajović, I. Friščić, M. Plodinec and D. Iveković, J. Mol. Struct., 2009, 924, 183–191 CrossRef.
- N. Liu, X. Chen, J. Zhang and J. W. Schwank, Catal. Today, 2014, 225, 34–51 CrossRef.
- Y. R. Smith, R. S. Ray, K. Carlson, B. Sarma and M. Misra, Materials, 2013, 6, 2892–2957 CrossRef PubMed.
- X. Sun, X. Chen and Y. Li, Inorg. Chem., 2002, 41, 4996–4998 CrossRef PubMed.
- F. Cavani, C. Felloni, D. Scagliarini, A. Tubertini, C. Flego and C. Perego, Stud. Surf. Sci. Catal., 2000, 143, 953–961 CrossRef.
- V. Štengl, S. Bakardjieva, J. Šubrt, E. Večerníková, L. Szatmary, M. Klementová and V. Balek, Appl. Catal., B, 2006, 63, 20–30 CrossRef.
- C. M. Rodrigues, O. P. Ferreira and O. L. Alves, J. Braz. Chem. Soc., 2010, 21, 1341–1348 CrossRef.
- J. M. Carraher, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2015, 5, 3162–3173 CrossRef.
- C. Liu, J. M. Carraher, J. L. Swedberg, C. R. Herndon, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2014, 4, 4295–4298 CrossRef.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, ChemCatChem, 2016, 8, 1100–1110 CrossRef.
- V. Galstyan, E. Comini, G. Faglia and G. Sberveglieri, Sensors, 2013, 13, 14813–14838 CrossRef PubMed.
- Q. Yang, M. Sherbahn and T. Runge, ACS Sustainable Chem. Eng., 2016, 4, 3526–3534 CrossRef.
- Y. J. Topper and D. Stetten, J. Biol. Chem., 1951, 189, 191–202 Search PubMed.
- S. J. Angyal, in Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates, ed. A. E. Stütz, Springer, Berlin, Heidelberg, 2001, pp. 1–14 Search PubMed.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef.
- Y. Zeng, X. Chen, D. Zhao, H. Li, Y. Zhang and X. Xiao, Fluid Phase Equilib., 2012, 313, 148–155 CrossRef.
- L. A. Alves, J. B. Almeida e Silva and M. Giulietti, J. Chem. Eng. Data, 2007, 52, 2166–2170 CrossRef.
- Y. Berube and P. L. de Bruyn, J. Colloid Interface Sci., 1968, 27, 305–318 CrossRef.
- S. Despax, B. Estrine, N. Hoffmann, J. Le Bras, S. Marinkovic and J. Muzart, Catal. Commun., 2013, 39, 35–38 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04353a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |