
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of dispirooxindoles via [3+2] cycloaddition of azomethine ylides to 3-phenacylideneoxindoles and evaluation of their cytotoxicity†
Ying Huangab,
Yi-Xin Huangb,
Jing Suna and
Chao-Guo Yan *a
*a
aCollege of Chemistry & Chemical Engineering, Yangzhou University, Yangzhou 225002, China. E-mail: cgyan@yzu.edu.cn
bCollege of Medicine, Yangzhou University, Yangzhou 225001, China
First published on 2nd July 2018
Abstract
The three-component reaction of 1,2,3,4-tetrahydroisoquinoline, isatins and 3-phenacylideneoxindoles in refluxing ethanol afforded dispiro[indoline-3,1′-pyrrolo[2,1-a]isoquinoline-3′,3′-indolines] (4a–4x) in good yields via 1,3-dipolar cycloaddition of in situ generated azomethine ylide with the exocyclic double bond of 3-phenacylideneoxindoles. 1H NMR spectra and single crystal structures indicated the reaction has high regioselectivity and diastereoselectivity. Furthermore, their biological activities have been preliminarily demonstrated by in vitro evaluation against mouse breast cancer cells 4T1 and human liver cancer cells HepG2 by MTT assay. The results demonstrated that some of the compounds showed cytotoxicities to cell lines of 4T1 and HepG2, and indicated that novel spirooxindoles may become potential lead compounds for further biological screenings of their medicinal applications.
1. Introduction
The spirooxindole core is one of the most privileged heterocyclic rings, which not only exists in a number of naturally occurring substances, but also is featured in many medicinally relevant compounds with wide biological applications.1,2 Among various carbocyclic and heterocyclic spirooxindole systems, spiropyrrolidinyl oxindoles are frequently encountered in natural alkaloids and are often considered as attractive templates for drug discovery.3 Spiropyrrolidinyl oxindole is the spiro ring fusioning at the 3-position of the oxindole core with various substitutions around the pyrrolidine and oxindole rings. Some of them exhibit significant bioactivities such as horsfiline, strychnofoline, spirotryprostatin A and palmirine4 (Fig. 1). Apart from natural oxindole alkaloids, synthesized spiropyrrolidinyl oxindoles have been widely studied for their antiviral, antibacterial and anti-cancer activities.5,6 Their remarkable pharmacological activity and unique molecular architecture have made spiropyrrolidinyl oxindoles and their derivatives attractive synthetic targets. As a consequence, many efficient synthetic procedures have been developed for the preparation of the diversely structural spiropyrrolidinyl oxindoles.7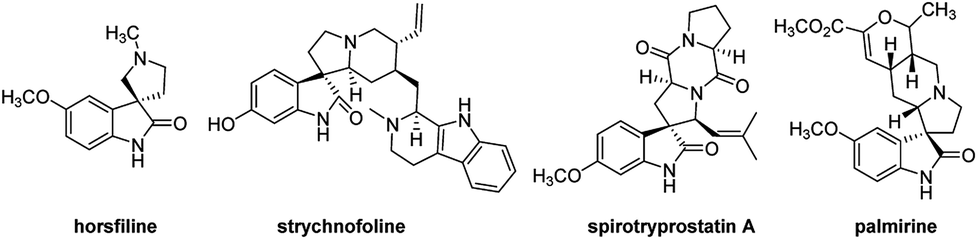 | ||
| Fig. 1 Representative spiropyrrolidinyl oxindole alkaloids. | ||
1,3-Dipolar cycloaddition reaction is an efficient and high-yielding, regio- and stereo controlled method for the synthesis of heterocyclic compounds. For the preparation of five-membered nitrogen-containing cyclic compounds, in particular pyrrolidines, dihydropyrroles, and pyrroles, [3+2] cycloaddition of azomethine ylides with alkenes is very effective and has been studied widely.8,9 If the azomethine ylides are generated from isatin derived compounds and α-amino acids through the thermal decarboxylation, pyrrolidine-containing spirooxindoles with high regioselectivity and stereoselectivity will be obtained.10,11
Heteroaromatic N-ylides such as pyridinium, thiazolium, quinolinium, isoquinolinium methylides which are readily available from the alkylation of azaaromatic heterocycles and sequential deprotonation reaction have also been used as one kind of reactive azomethine ylides extensively in cycloadditions for the synthesis of the fused heterocycles with a nitrogen at the point of fusion.12 Our group had reported the green synthetic methods for complex heterocyclic compounds, such as efficient synthesis of spiro[indoline-3,1′-pyrrolo[2,1-a]isoquinolines via 1,3-cycloaddition reactions of 3-phenacylideneoxindoles with aza-aromatic N-ylides generated from isoquinolinium salts.13 Wang's group synthesized some analogues by using 3,4-dihydroisoquinolinium salts.14 In addition, the 1,3-dipolar cycloaddition reactions of isatin, benzylamine and chalcone derivatives or benzylideneacetones are reported, in which azomethine ylides are in situ generated from isatin and benzylamine.15 Using 1,2,3,4-tetrahydroisoquinoline instead of benzylamine, the spiro compounds with key structure of spiro[indoline-3,1′-pyrrolo[2,1-a]isoquinolines could be obtained.16 In this paper, we wish to report an efficient 1,3-dipolar cycloaddition reaction of isatins, 1,2,3,4-tetrahydroisoquinoline and 3-phenacylideneoxindoles for regioselective and diastereoselective synthesis of novel functionalized spirooxindoles. Additionally, their biological activities have been preliminarily demonstrated by in vitro evaluation against mouse breast cancer cells 4T1 and human liver cancer cells HepG2 by MTT assay.
2. Results and discussions
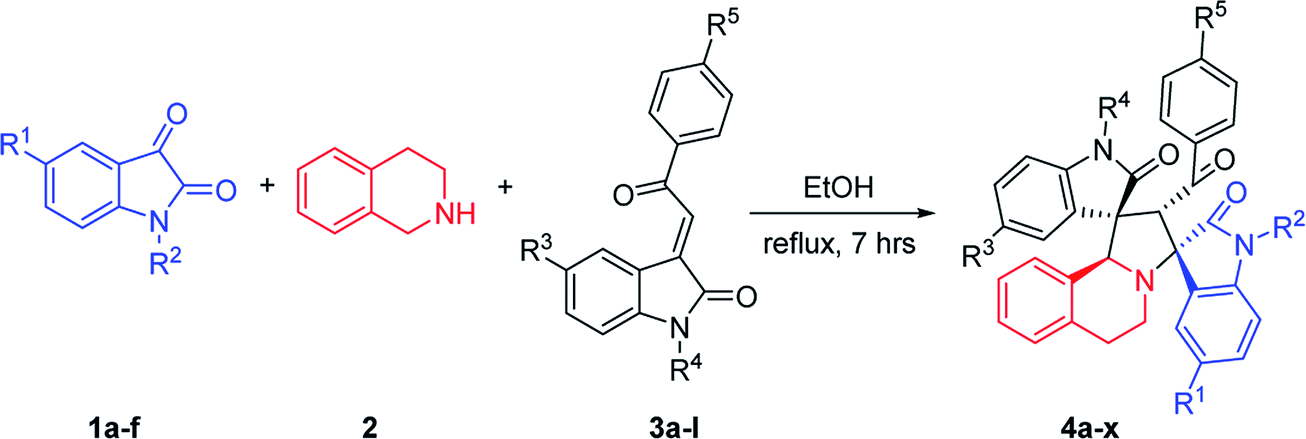
2.1. Synthesis of dispirooxindole derivatives
According to previously established reaction conditions for the domino reactions for the efficient synthesis of the functionalized spiro[indoline-3,1′-pyrrolo[2,1-a]isoquinolines,13 we have started the reaction of 5-methylisatin (1a), 1,2,3,4-tetrahydroisoquinoline (2) and 5-methyl-3-p-methylphenacylideneoxindole (3a) in a one-pot, three-component procedure depicted in Scheme 1. After refluxing the mixture of substrates in ethanol for about seven hours, it was pleased to find that the resulting precipitates were collected by filtration and characterized as the expected dispiro[indoline-3,1′-pyrrolo[2,1-a]isoquinoline-3′,3′-indoline] (4a) in 70% yield, in which the two scaffold of oxindolines exist on the 1,3-positions in the newly formed pyrrolidinyl ring. It should be pointed out that the isomeric product 5a, in which the two scaffold of oxindolines were connected together, was not formed in the reaction. This results also indicated this three-component reaction has high regioselectivity. Therefore, the expected three-component reaction successfully proceeded in very simple reaction conditions without adding other catalyst. When other solvent such as methanol, acetonitrile and toluene was used, the yield of the product (4a) was decreased.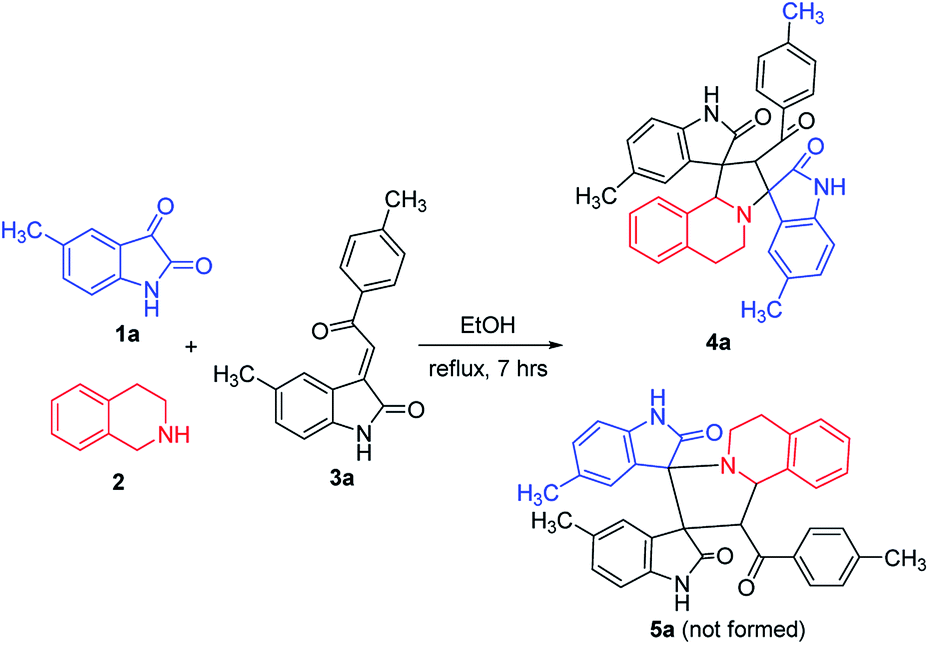 | ||
| Scheme 1 Three-component reaction for dispirooxindole 4a. | ||
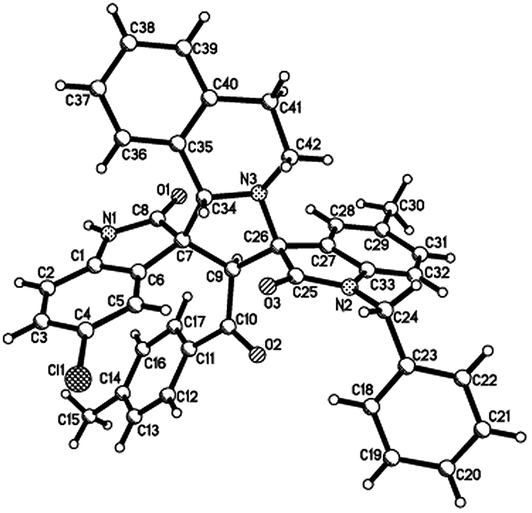
Then, we extended the scope to the reaction under this simple reaction conditions. Various isatins (1a–f) and 3-phenacylideneoxindoles (3a–l) with both electron-withdrawing and electron releasing-substituents were employed in the three-component reaction under same conditions. The corresponding novel dispirooxindoles (4b–x) were successfully synthesized in high yields. The substituents on the both oxindoles showed little effect on the yields of the products. The results are summarized in Table 1. It should be pointed out that the pure products were usually obtained by filtration of the formed precipitates. The products were fully characterized by the spectroscopic methods. For examples, the IR spectrum of 4a showed three peaks of the carbonyl groups at 1725, 1676, 1626 cm−1. Because there are four chiral carbon atoms in the newly formed ring of pyrrolidine, several diastereoisomers might be formed in the 1,3-dipolar cycloaddition reaction. In the 1H NMR spectrum of 4a, two singlets appeared at δ 5.59 and δ 4.89 ppm were the signs of two cyclic CH unit in newly-formed pyrrolidine, and the two characteristic signals appeared at δ 10.38 and δ 10.34 ppm were the corresponding signs of NH protons in two oxindole rings. This result clearly showed that only one diastereoisomer existed in the obtained product. In 13C NMR spectrum of 4a, the two carbonyl group of the oxindole ring showed signs at δ 179.1 and 177.4 ppm, and the carbonyl group in benzoyl group showed sign at δ 196.1 ppm. The mass spectrum displayed a distinguished peak at m/z 554.2447 which further supported the formation of cycloadduct 4a. Other spiro compounds also displayed similar spectroscopy. But the 1H and 13C NMR of the compounds 4t and 4u clearly indicated two diastereoisomers existed in the obtained samples. The major/minor ratios of 4t/4t′ (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) and 4u/4u′ (75:25) were determined by integral of signs in the 1H NMR spectra. For determining the relative configuration of the spiro compounds 4a–4x, the single crystal structures of three compounds 4a, 4u and 4v (Fig. 2, 3 and 4) were successfully determined by X-ray diffraction. From the figures, it is clearly seen that the three single crystal structures have same relative configuration, in which two oxindole units existed at trans-position. The two protons in the ring of pyrrolidine also existed in trans-configuration. On the basis of NMR spectra and single crystal structures, we can concluded that this three-component reaction predominately give this kind of the diastereoisomer as major product and the other diastereoisomers were formed as minor product in few cases.
:10) and 4u/4u′ (75:25) were determined by integral of signs in the 1H NMR spectra. For determining the relative configuration of the spiro compounds 4a–4x, the single crystal structures of three compounds 4a, 4u and 4v (Fig. 2, 3 and 4) were successfully determined by X-ray diffraction. From the figures, it is clearly seen that the three single crystal structures have same relative configuration, in which two oxindole units existed at trans-position. The two protons in the ring of pyrrolidine also existed in trans-configuration. On the basis of NMR spectra and single crystal structures, we can concluded that this three-component reaction predominately give this kind of the diastereoisomer as major product and the other diastereoisomers were formed as minor product in few cases.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Compd | R1 | R2 | R3 | R4 | R5 | Yieldb (%) |
| a Reaction conditions: isatin (0.30 mmol), tetrahydroisoquinoline (0.30 mmol), 3-phenacylideneoxindole (0.25 mmol); reflux, 7 h.b Isolated yields.c Ratio of 4t:4′t = 90:10.d Ratio of 4u:4′u = 75:25. |
|||||||
| 1 | 4a | CH3 | H | CH3 | H | CH3 | 70 |
| 2 | 4b | CH3 | H | Cl | H | OCH3 | 76 |
| 3 | 4c | Cl | H | CH3 | H | CH3 | 81 |
| 4 | 4d | CH3 | H | H | H | H | 90 |
| 5 | 4e | CH3 | H | CH3 | H | Cl | 80 |
| 6 | 4f | CH3 | H | Cl | H | CH3 | 50 |
| 7 | 4g | Cl | H | CH3 | H | Cl | 82 |
| 8 | 4h | Cl | H | Cl | H | OCH3 | 67 |
| 9 | 4i | Cl | H | F | H | CH3 | 57 |
| 10 | 4j | CH3 | H | Cl | CH2Ph | Cl | 57 |
| 11 | 4k | CH3 | H | H | CH2Ph | H | 58 |
| 12 | 4l | CH3 | H | CH3 | CH2Ph | CH3 | 62 |
| 13 | 4m | CH3 | H | CH3 | CH2Ph | Cl | 66 |
| 14 | 4n | CH3 | H | CH3 | C4H9 | CH3 | 90 |
| 15 | 4o | Cl | H | Cl | C4H9 | CH3 | 72 |
| 16 | 4p | H | CH3 | CH3 | H | CH3 | 85 |
| 17 | 4q | H | CH3 | Cl | H | CH3 | 63 |
| 18 | 4r | H | CH3 | CH3 | CH2Ph | Cl | 58 |
| 19 | 4s | CH3 | CH2Ph | CH3 | H | CH3 | 79 |
| 20 | 4t | CH3 | CH2Ph | Cl | H | OCH3 | 80c |
| 21 | 4u | CH3 | CH2Ph | Cl | H | CH3 | 62d |
| 22 | 4v | Cl | CH2Ph | Cl | H | CH3 | 76 |
| 23 | 4w | CH3 | CH2Ph | CH3 | CH2Ph | Cl | 60 |
| 24 | 4x | Cl | C4H9 | CH3 | H | CH3 | 50 |
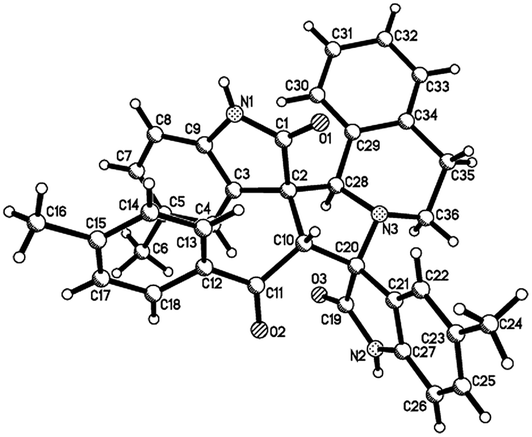 | ||
| Fig. 2 Single crystal structure of compound 4a. | ||
 | ||
| Fig. 3 Single crystal structure of compound 4u. | ||
 | ||
| Fig. 4 Single crystal structure of compound 4v. | ||
2.2. Viability of two cancer cells of compounds 4a–4x
All newly synthesized compounds were subjected to a preliminary evaluation on their in vitro cytotoxic activity, which was represented as the inhibition rate of the tested compounds to mouse breast cancer cells 4T1 and human liver cancer cells HepG2 at the concentration of 200 μg mL−1. The cytotoxic activity to 4T1 cells was not good as we expected. Among the tested compounds 4, compound 4i (R3 = F) displayed highest cytotoxic activity to HepG2 cells with the inhibition rate of 73.50%, followed by 4h (R3 = Cl) and 4d (R3 = H) (Table 2). Then the inhibition rate of human liver cancer cells line HepG2 of these three compounds were tested at three concentrations of 50, 100 and 200 μg mL−1 (Table 3).| Compound | 4T1 cell death | HepG2 cell death | ||||||
|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |||||
| % | Mean ± S.D. | % | Mean ± S.D. | % | Mean ± S.D. | % | Mean ± S.D. | |
| a /: no activity. | ||||||||
| 4a | 2.31 | 0.915 ± 0.011 | 25.80 | 0.870 ± 0.015 | 2.84 | 0.868 ± 0.024 | 32.86 | 0.506 ± 0.004 |
| 4b | 2.23 | 0.916 ± 0.016 | 15.05 | 0.984 ± 0.014 | 3.99 | 0.858 ± 0.023 | 19.34 | 0.591 ± 0.110 |
| 4c | 6.99 | 0.871 ± 0.015 | 20.36 | 0.928 ± 0.010 | 5.67 | 0.843 ± 0.016 | 19.71 | 0.589 ± 0.060 |
| 4d | 6.35 | 0.877 ± 0.040 | 20.82 | 0.923 ± 0.014 | 7.16 | 0.830 ± 0.016 | 66.72 | 0.145 ± 0.046 |
| 4e | 7.88 | 0.863 ± 0.022 | 20.64 | 0.925 ± 0.060 | 2.97 | 0.867 ± 0.014 | 13.27 | 0.629 ± 0.058 |
| 4f | 6.45 | 0.876 ± 0.012 | 19.43 | 0.937 ± 0.098 | 4.59 | 0.853 ± 0.013 | 14.22 | 0.623 ± 0.045 |
| 4g | / | 0.940 ± 0.061 | 27.64 | 0.851 ± 0.034 | 3.71 | 0.861 ± 0.012 | 23.16 | 0.567 ± 0.056 |
| 4h | 5.23 | 0.887 ± 0.045 | 23.25 | 0.896 ± 0.137 | 6.12 | 0.839 ± 0.013 | 70.11 | 0.098 ± 0.006 |
| 4i | 10.38 | 0.839 ± 0.025 | 33.31 | 0.791 ± 0.068 | 5.34 | 0.846 ± 0.028 | 73.50 | 0.088 ± 0.004 |
| 4j | 3.42 | 0.904 ± 0.020 | 17.87 | 0.954 ± 0.051 | 5.02 | 0.849 ± 0.019 | 25.25 | 0.554 ± 0.066 |
| 4k | 3.43 | 1.329 ± 0.112 | 10.88 | 1.237 ± 0.033 | / | 1.193 ± 0.024 | 2.36 | 1.211 ± 0.106 |
| 4l | / | 1.423 ± 0.013 | 10.54 | 1.241 ± 0.017 | / | 1.208 ± 0.012 | / | 1.563 ± 0.177 |
| 4m | 8.16 | 1.268 ± 0.032 | 20.71 | 1.099 ± 0.084 | / | 1.184 ± 0.003 | 4.22 | 1.188 ± 0.012 |
| 4n | 4.28 | 1.318 ± 0.018 | 15.68 | 1.169 ± 0.057 | 0.66 | 1.170 ± 0.019 | 11.82 | 1.094 ± 0.026 |
| 4o | 0.23 | 1.370 ± 0.021 | 12.69 | 1.211 ± 0.020 | / | 1.208 ± 0.030 | / | 1.441 ± 0.229 |
| 4p | / | 1.377 ± 0.011 | 22.86 | 1.070 ± 0.040 | / | 1.180 ± 0.028 | / | 1.393 ± 0.424 |
| 4q | / | 1.514 ± 0.009 | 4.65 | 1.322 ± 0.018 | 23.9 | 0.897 ± 0.022 | 9.88 | 1.118 ± 0.137 |
| 4r | / | 1.402 ± 0.027 | 5.11 | 1.316 ± 0.021 | / | 1.217 ± 0.018 | / | 1.285 ± 0.113 |
| 4s | / | 1.376 ± 0.016 | 8.20 | 1.273 ± 0.007 | / | 1.292 ± 0.025 | 7.23 | 1.151 ± 0.048 |
| 4t | / | 1.481 ± 0.016 | / | 1.465 ± 0.028 | / | 1.385 ± 0.020 | 10.09 | 1.115 ± 0.069 |
| 4u | 2.75 | 1.108 ± 0.039 | / | 1.131 ± 0.001 | 2.75 | 1.117 ± 0.154 | 1.053 ± 0.019 | |
| 4v | / | 1.125 ± 0.065 | / | 1.211 ± 0.021 | / | 1.172 ± 0.018 | / | 1.036 ± 0.010 |
| 4w | 2.40 | 1.187 ± 0.060 | 5.249 | 1.197 ± 0.005 | 2.40 | 1.121 ± 0.038 | 5.25 | 0.947 ± 0.026 |
| 4x | / | 1.185 ± 0.033 | / | 1.201 ± 0.002 | / | 1.194 ± 0.042 | / | 1.314 ± 0.110 |
| Compound | 200 μg mL−1 | 100 μg mL−1 | 50 μg mL−1 | |||
|---|---|---|---|---|---|---|
| Cell death | Cell death | Cell death | ||||
| % | Mean ± S.D. | % | Mean ± S.D. | % | Mean ± S.D. | |
| 4d | 67.40 | 0.325 ± 0.018 | 30.93 | 0.691 ± 0.062 | 16.12 | 0.838 ± 0.040 |
| 4h | 59.74 | 0.402 ± 0.014 | 30.64 | 0.693 ± 0.028 | 20.57 | 0.794 ± 0.028 |
| 4i | 79.30 | 0.209 ± 0.040 | 70.40 | 0.474 ± 0.056 | 44.08 | 0.559 ± 0.003 |
In detail, compounds 4a–4i which have no substituents on N atom of isatin (R2 = H) and 3-phenacylideneoxindole (R4 = H) inhibited the growth of HepG2 cells at the concentration of 200 μg mL−1, displayed promising cytotoxicity to HepG2 cells with inhibition rates varying from 13.27% to 73.50%. These results were much better than the other compounds (4j–4x) which have N-substituents such as CH3, C4H9 and CH2Ph. Compounds 4j–4x tend to precipitate out during the dilution process. These results would suggest that the polarity or lipo-hydro partition coefficient (logP) of the compound has a significant effect on its activity (Table 2).
As shown in Fig. 5, untreated cells exhibit regular blue colour. In contrast, cells treated with 4i at the concentration of 200 μg mL−1 showed clear red colour. It indicated that large amount of HepG2 cells died after treatment with 4i. Compounds 4d, 4h and 4i showed a tendency of concentration-dependent cytotoxicity because their inhibition rates were greatly enhanced with increasing concentration. Among them, compound 4i (R3 = F) was the most powerful to inhibit the growth of HepG2 cells to 44.08% at the concentration of 50 μg mL−1 (Table 3). The cytotoxicity of compounds 4d, 4h and 4i was tested on 3T3 cells at the concentration of 200 μg mL−1, the results showed that their cytocompatibility were good, which meant that they have cytotoxicity to cancer cells and were not toxic to normal cells (Fig. 6).
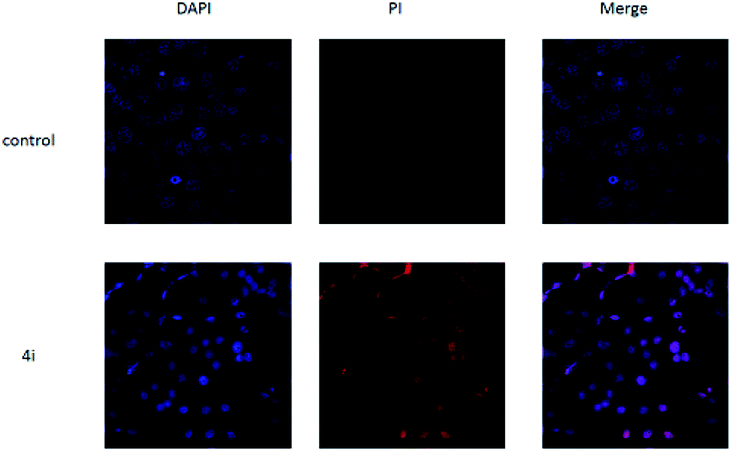 | ||
| Fig. 5 The morphological features of survival status were monitored by fluorescence microscopy after staining with DAPI. | ||
 | ||
| Fig. 6 Effect of compound 4d, 4h and 4i on 3T3 cell viabilities (200 μg mL−1). | ||
3. Conclusions
In summary, we have developed an efficient strategy for the synthesis of complex spiro compounds by a 1,3-dipolar cycloaddition of 3-methyleneoxindoles with 1,2,3,4-tetrahydroisoquinoline and isatin derivatives. This three-component reaction afforded the anticipated spirooxindoles in excellent yields with high regio- and stereoselectivity. This synthetic protocol has advantages of mild reaction conditions, easily accessible starting materials and easy purification of the products, which makes it a useful and attractive method for the synthesis of the complex spiro heterocycles in synthetic and medicinal chemistry. Furthermore, the bioactivity detection of these spirooxindoles has led to the discovery of three compounds with promising cytotoxicity to HepG2 cells. The preliminary bioassay of these compounds will enlighten further structural modification and bioactivity studies on spirooxindoles for their medicinal applications.4. Experimental
4.1. Chemistry
All reactions were performed in open atmosphere unless stated. All reagents, unless otherwise indicated, were obtained from commercial sources. 1H and 13C NMR spectra were recorded on a Bruker AV-600 instrument or Variance 400 spectrometer with DMSO-d6 or CDCl3 as solvent and tetramethylsailane (TMS) was used as internal standard (δ in ppm). IR spectra were obtained on a Bruker Tensor 27 spectrometer (KBr disc). HRMS were recorded on a Bruker UHR-TOF maXis spectrometer. X-ray data were collected on a Bruker Smart APEX-2 diffractometer. Melting points were taken on a hot-plate microscope apparatus and were uncorrected.4.2. General procedure for the preparation of 4a–4x
A mixture of isatin (0.3 mmol), 1,2,3,4-tetrahydroisoquinoline (0.3 mmol), and 3-phenacylideneoxindole (0.25 mmol) in ethanol (10 mL) was stirred at reflux for seven hours. After completion of the reaction, as indicated by TLC, the solid was separated by filtration, washed with cold ethanol and vacuum dried. In some cases, additional purification by TLC was necessary.5,5′′-Dimethyl-2′-(4-methylbenzoyl)-6′,10b′-dihydro-2′H, 5′H-dispiro[indoline-3,1′-pyrrolo[2,1-a]isoquinoline-3′,3′-indoline]-2,2′′-dione (4a). White solid, 70%, mp. 244–245 °C; 1H NMR (400 MHz, DMSO-d6) δ: 10.38 (s, 1H, NH), 10.34 (s, 1H, NH), 7.63 (s, 1H, ArH), 7.23 (s, 1H, ArH), 7.10–7.00 (m, 7H, ArH), 6.88 (brs, 1H, ArH), 6.82–6.77 (m, 2H, ArH), 6.36 (d, J = 7.6 Hz, 1H, ArH), 6.24 (d, J = 7.2 Hz, 1H, ArH), 5.59 (s, 1H, CH), 4.89 (s, 1H, CH), 2.86–2.78 (m, 2H, CH), 2.64–2.60 (m, 2H, CH), 2.29 (s, 3H, CH3), 2.21 (s, 6H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 196.1, 179.1, 177.4, 142.7, 141.1, 139.1, 135.2, 134.5, 134.2, 130.4, 130.3, 130.2, 129.9, 128.7, 128.5, 127.2, 126.9, 126.3, 126.1, 125.4, 124.8, 123.1, 109.1, 108.6, 70.6, 68.7, 66.1, 58.2, 41.4,29.3, 21.0, 20.9, 20.6; IR (KBr) ν: 3355, 3170, 3029, 2916, 2832, 1725, 1676, 1626, 1607, 1574, 1494, 1428, 1373, 1341, 1296, 1248, 1202, 1166, 1042, 1008, 947, 905, 810, 756, 730 cm−1; MS (m/z): HRMS (ESI) calcd for C36H32N3O3 ([M + H]+): 554.2438, found: 554.2447.
4.3. Biology
Three cancer cell lines are obtained from experimental cell resource centre of Shanghai Institutes for Biological Sciences. The relative cell viability was recorded by a microplate reader (SYenergy 2). The cell death stain was imaged by laser scanning confocal microscopy (LSCM, TLS SP8 STED).Cell viability assay. The cell viability was measured by MTT assay. Mouse breast cancer cells 4T1, Human liver cancer cells HepG2 and Mouse fibroblasts cells 3T3 were separately cultured in DMEM medium containing 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C and 5% CO2. For in vitro cytotoxicity assay, 4T1, HepG2 and 3T3 cells were seeded into 96-well plates at 8 × 103/well until adherent and then incubated with various concentrations of 4a–4x for 24 and 48 h. Ten microliters of 5 mg mL−1 MTT solution was then added into each well, followed by incubation for 24 and 48 h at 37 °C in the presence of 5% CO2. At the end of the incubated time, 100 μL of DMSO was added to dissolve the formazan crystals. Finally, the absorbance at 490 nm of each well was recorded by a microplate reader (SYenergy 2). The final cell viabilities were calculated by [OD(tested compounds group)/OD(control group)] × 100%.
Cell death stain. To investigate the cell survival status, HepG2 cells (1 × 105/well) suspended in 1 mL of DMEM medium were seeded in a 24-well plate with cell climbing slices on the bottom of the plate for 12 h, and then incubated with 4i (4h, 4d) (the final concentration was 200 μg mL−1) at 37 °C for 48 h. The cells were then rinsed twice with 0.01% PBS, fixed with 4% paraformaldehyde for 15 min, 0.4% Triton for 5 min and stained with DAPI and PI for 10 min. After rinsing with PBS three times, cell climbing slices were placed on the glass slides and imaged by laser scanning confocal microscopy (LSCM, TLS SP8 STED).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21572196) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We also thank Analysis and Test Centre of Yangzhou University and Translational Medicine Research Institute of Yangzhou University providing instruments for analysis.Notes and references
- (a) R. M. Williams and R. Cox, Acc. Chem. Res., 2003, 36, 127–139 CrossRef PubMed; (b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2963 CrossRef PubMed; (c) M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef; (d) D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef PubMed; (e) N. Ye, H. Y. Chen, E. A. Wold, P. Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef PubMed.
- (a) S. B. Kotha, A. C. Deb, K. Lahiri and E. Manivannan, Synthesis, 2009, 2, 165–193 CrossRef; (b) B. M. Trost and M. K. Brennan, Synthesis, 2009, 18, 3003–3025 CrossRef; (c) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165–5181 RSC; (d) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef PubMed.
- (a) Y. Y. Liu, H. Wang and J. P. Wan, Asian J. Org. Chem., 2013, 2, 374–386 CrossRef; (b) Z. Y. Cao, Y. H. Wang, X. P. Zeng and J. Zhou, Tetrahedron, 2014, 70, 2406–2415 CrossRef; (c) D. Q. Cheng, Y. Ishihara, B. Tan and C. B. Barbas, III, ACS Catal., 2014, 4, 743–762 CrossRef; (d) R. G. Redkin, V. V. Lipson and D. V. Atamanuk, Mol. Diversity, 2016, 20, 299–344 CrossRef PubMed.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef PubMed; (b) Q. Z. Yu, P. Guo, J. Jian, Y. Y. Chen and J. Xu, Chem. Commun., 2018, 54, 1125–1128 RSC.
- (a) M. M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef; (b) S. M. Wang, K. Ding, Y. P. Lu, N. C. Zaneta, S. Qiu, G. P. Wang, D. G. Qin and S. Sangjeev, WO 2006091646, 2006; (c) P. Ramesh, K. S. Rao, R. Trivedi and B. S. Kumar, RSC Adv., 2016, 6, 26546–26552 RSC.
- (a) G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef PubMed; (b) B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef PubMed; (c) Y. Arun, K. Saranraj, C. Balachandran and P. T. Perumal, Eur. J. Med. Chem., 2014, 74, 50–64 CrossRef PubMed; (d) Y. J. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef PubMed.
- (a) T. Penaska, K. Ormandyova, M. Meciarova, J. Filob and R. Sebesta, New J. Chem., 2017, 41, 5506–5512 RSC; (b) X. Fang and C. J. Wang, Org. Biomol. Chem., 2018, 16, 2591–2601 RSC; (c) Y. C. Wang, J. L. Wang, K. S. Burgess, J. W. Zhang, Q. M. Zheng, Y. D. Pu, L. J. Yan and X. B. Chen, RSC Adv., 2018, 8, 5702–5713 RSC.
- (a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–909 CrossRef PubMed; (b) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765–2809 CrossRef PubMed; (c) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484–4517 CrossRef PubMed; (d) S. Lanka, S. Thennarasu and P. T. Perumal, Tetrahedron Lett., 2014, 55, 2585–2588 CrossRef.
- (a) G. M. Ziarani, R. Moradi and N. Lashgari, Tetrahedron, 2018, 74, 1323–1353 CrossRef; (b) M. Y. Han, J. Y. Jia and W. Wang, Tetrahedron Lett., 2014, 55, 784–794 CrossRef; (c) B. Bdiri, B. J. Zhao and Z. M. Zhou, Tetrahedron: Asymmetry, 2017, 28, 876–899 CrossRef; (d) H. Pellissier, Tetrahedron, 2012, 68, 2197–2232 CrossRef.
- (a) L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef; (b) Y. Arun, G. Bhaskar, C. Balachandran, S. Ignacimuthu and P. T. Perumal, Bioorg. Med. Chem. Lett., 2013, 23, 1839–1845 CrossRef PubMed; (c) G. Subramaniyan, R. Raghunathan and M. Nethaji, Tetrahedron, 2002, 58, 9075–9079 CrossRef; (d) R. Jain, K. Sharma and D. Kumar, Tetrahedron Lett., 2012, 53, 1993–1997 CrossRef.
- (a) A. I. Almansour, R. S. Kumar, N. Arumugam, A. Basiri, Y. Kia, M. A. Ali, M. Farooq and V. Murugaiyah, Molecules, 2015, 20, 2296–2309 CrossRef PubMed; (b) A. R. S. Babu and R. Raghunathan, Tetrahedron, 2007, 63, 8010–8016 CrossRef; (c) A. R. S. Babu and R. Raghunathan, Tetrahedron Lett., 2007, 48, 6809–6813 CrossRef; (d) M. Poornachandran, R. Muruganantham and R. Raghunathan, Synth. Commun., 2006, 36, 141–150 CrossRef; (e) M. Poornachandran and R. Raghunathan, Synth. Commun., 2007, 37, 2507–2517 CrossRef.
- (a) D. S. Allgäuer and H. Mayr, Eur. J. Org. Chem., 2014, 14, 2956–2963 CrossRef; (b) D. S. Allgäuer, P. Mayer and H. Mayr, J. Am. Chem. Soc., 2013, 135, 15216–15224 CrossRef PubMed; (c) A. S. Al-Bogami and A. S. El-Ahl, Lett. Org. Chem., 2015, 12, 2–12 CrossRef; (d) A. V. Velikorodov, N. M. Imasheva, A. K. Kuanchalieva and O. Y. Poddubnyi, Russ. J. Org. Chem., 2010, 46, 971–975 CrossRef.
- (a) L. Wu, J. Sun and C. G. Yan, Org. Biomol. Chem., 2012, 10, 9452–9463 RSC; (b) R. Z. Liu, R. G. Shi, J. Sun and C. G. Yan, Org. Chem. Front., 2017, 4, 354–357 RSC; (c) J. Sun, G. L. Shen, Y. Huang and C. G. Yan, Sci. Rep., 2017, 7, 41024 CrossRef PubMed.
- (a) I. Fejes, M. Nyerges, A. Szollosy, G. Blasko and L. Toke, Tetrahedron, 2001, 57, 1129–1137 CrossRef; (b) M. Nyerges, L. Gajdics, A. Szollosy and L. Toke, Synlett, 1999, 1, 111–113 CrossRef.
- (a) Y. Sarra, M. Hamzehloueian, K. Alimohammadi and S. Yeganegi, J. Mol. Struct., 2012, 1030, 168–176 CrossRef; (b) C. Q. Peng, J. W. Ren, J. A. Xiao, H. G. Zhang, H. Yang and Y. M. Luo, Beilstein J. Org. Chem., 2014, 10, 352–360 CrossRef PubMed.
- (a) P. J. Xia, Y. H. Sun, J. A. Xiao, Z. F. Zhou, S. S. Wen, Y. Xiong, G. C. Ou, X. Q. Chen and H. Yang, J. Org. Chem., 2015, 80, 11573–11579 CrossRef PubMed; (b) K. Alimohammadi, Y. Sarrafi and B. Rajabpour, C. R. Chim., 2014, 17, 156–163 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all new compounds. CCDC 1556207–1556209. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04375b |
| This journal is © The Royal Society of Chemistry 2018 |