
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electroactive Co(III) salen metal complexes and the electrophoretic deposition of their porous organic polymers onto glassy carbon†
Marcello B. Solomon a,
Aditya Rawalb,
James M. Hookb,
Seth M. Cohenc,
Clifford P. Kubiakc,
Katrina A. Jolliffe*a and
Deanna M. D'Alessandro*a
a,
Aditya Rawalb,
James M. Hookb,
Seth M. Cohenc,
Clifford P. Kubiakc,
Katrina A. Jolliffe*a and
Deanna M. D'Alessandro*a
aSchool of Chemistry, The University of Sydney, New South Wales 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au; kate.jolliffe@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 3777
bNMR Facility, Mark Wainwright Analytical Centre, The University of New South Wales, 2052, Australia
cDepartment of Chemistry and Biochemistry, University of California, San Diego, California 92093, USA
First published on 3rd July 2018
Abstract
This paper reports the CO2 electroreduction properties of three bis-bromo Co(III) salen metal complexes and their Porous Organic Polymers (POPs) as a platform for using the salen core as a multi-electron reducing agent. Although Co(III) salen metal complexes have been studied extensively for their chemical catalysis with CO2, their electrochemical behaviour, particularly their reduction, in the presence of CO2 is much less explored. The discrete Co(III) complexes enabled the reduction of CO2 to CO in faradaic efficiencies of up to 20%. The reductive electrochemical processes of Co(III) salen complexes are relatively unknown; therefore, the mechanism of reduction for the complexes was investigated using IR and UV-Vis-NIR spectroelectrochemical (SEC) techniques. The discrete bis-bromo salen complexes were incorporated into POPs with tris-(p-ethynyl)-triphenylamine as a co-ligand and were characterised using solid state NMR, IR, UV-Vis-NIR and Field Emission Scanning Electron Microscopy (FE-SEM). The POP materials were electrophoretically deposited onto glassy carbon under milder conditions than those previously reported in the literature. Direct attachment of the POP materials to glassy carbon enabled improved solid state electrochemical analysis of the samples. The POP materials were also analysed via SEC techniques, where a Co(II/I) process could be observed, but further reductions associated with the imine reduction compromised the stability of the POPs.
Introduction
Since the industrial revolution, the consumption of fossil fuels has been rapidly increasing to cater for the needs of an ever growing world population. Industrialisation has been heavily dependent on the use of coal, which today is responsible for over 40% of the production of electricity worldwide.1 In excess of 12 million tonnes of oil and 8 million cubic tonnes of natural gas are consumed daily to provide energy, which has resulted in excess carbon dioxide (CO2) emissions into the atmosphere at a rate faster than the current carbon cycle can mitigate.2–10 Carbon capture and sequestration (CCS) has long been seen as a viable option as it allows for the retrofitting of existing power plants to separate CO2 from the flue stream prior to its release into the atmosphere.11 A concern arising from the storage of CO2 is its effective transport, which poses financial, logistical and environmental challenges. Other chief concerns for CCS are the safety and environmental aspects that arise from the high pressures and large concentrations of CO2. Processes that can combine the capture of CO2 with its use as a feedstock may assist in the handling of emissions.The benefit of using CO2 as a feedstock lies in its abundance and low cost; however, its use as a cheap feedstock is limited by the presence of C in its most oxidised form. Investing energy to convert CO2 into a more reactive intermediate could enable the generation of commodity chemicals, which could eventually provide a cleaner fuel source for the human population to use. From a thermodynamic viewpoint, CO2 can accept an electron to form a CO2˙− radical anion, but this is only observed directly at high reduction potentials (Epc = −1.90 V vs. NHE).12,13 The proton-coupled reduction of CO2 provides a feasible alternative to direct reduction and can lead to the generation of useful products (carbon monoxide or formic acid) at lower reduction potentials.13,14 It is important that there is a kinetic preference for CO2 reduction over H2 due to the required presence of a proton source to facilitate the process.
Porous Organic Polymers (POPs) are considered viable candidates as materials that can both capture and convert CO2 into commodity chemicals. They are generally cheap to produce, simple to synthesise on a larger scale, possess high thermal and chemical stability,15 are lightweight, contain hydrophobic pores from their extended aromaticity and are chemically resistant. Hupp and co-workers have previously shown that the porosity and stability of POPs can be exploited by generating a polymer based on cheap and abundant amine- and anhydride-bearing monomers.16 The activated polymer demonstrated stability up to 500 °C and retained porosity after exposure to HCl. Patel and co-workers explored N2-phobicity within POPs by generating a class of nitrogen-rich azo POPs, which possessed high point selectivity for CO2/N2 (S = 288 at 323 K) and were chemically stable in boiling water.17 Zhou and co-workers synthesised POPs containing tetratopic adamantane cages which exhibited BET surface areas up to 5400 m2 g−1 and were suitable for CO2 and CH4 separation.18 It was found through ideal adsorbed solution theory (IAST) that several high surface area POPs demonstrated selectivity for CO2 at 295 K, suggesting their suitability for CO2 capture. Many early studies of POPs exploited porosity as their most important property; however, subsequent studies identify that these materials can also have applications in catalysis.19 Bare metal sites can also be incorporated into POPs, which may promote their use in catalysis.20,21
Creating a class of solid state materials that is capable of both the capture of CO2 and its catalytic conversion requires the choice of multifunctional building blocks. The ubiquity of salen metal complexes over the past eighty years has shown that they are capable of diverse chemistry. Since the first reported preparation of the vibrantly coloured salen-based ligands in 1933 by Pfeiffer,22 this class of ligand has found extensive use in chemical catalysts,23 electrochemical agents,24 and charge transfer complexes,25 among other applications. The variability of substituents on the salen core enables the preparation of diverse libraries of compounds that may be generated by systematic variation.26 There is scope to modify substituents along the backbone of the salen ligand to study their steric and electronic influence on catalysis (Fig. 1). The inherently redox-active nature of the salen metal complex allows for its properties to be modulated.
 | ||
| Fig. 1 The structure of the salen metal complexes used here. The backbone can be systematically modified by varying Y to produce the analogs Co1, Co2 and Co3. | ||
The readily functionalisable core of the salen metal complex allows for its covalent immobilisation into a POP material. A class of salen-based POPs were reported by Chun and co-workers, who varied the bridging moiety in the salen to incorporate the diaminocyclohexane functionality, and introduced Al(III), Co(III) and Cr(III) metals into the chelating site. Not only did these polymers exhibit similar BET surface areas of 522–650 m2 g−1, but they also converted CO2 to cyclic carbonates with yields of up to 94%.20 Xie and co-workers also used POPs to examine the capture of CO2 and its conversion to propylene carbonate under mild conditions.27 Surface areas up to 965 m2 g−1 were achieved and the catalyst could be recycled up to 22 times without any significant decrease in activity. To the best of our knowledge, work has not yet appeared on the electrochemical reduction of salen-based POPs.
The electrochemical examination of POP materials requires their effective attachment to a conductive surface.28 A large proportion of the literature addressing the surface attachment of solid state materials (in particular Metal Organic Frameworks (MOFs)) onto a conductive surface has reported Indium-doped Tin Oxide (ITO) or Fluorine-doped Tin Oxide (FTO) on glass as the substrate of choice.29–33 These two substrates are both transparent, and therefore allow for the spectroscopic study of the surface attached material prior to, during and after reduction experiments using transmission measurements;34 however, there are limitations to their use. For example, ITO readily corrodes from glass substrates upon the application of highly anodic and cathodic potentials, and indium is a scarce and expensive element.35 Although FTO coated glass has a larger electrochemical window and uses more abundant fluorine, it has a high resistance, which affects the reversibility of the ferrocene/ferrocenium ion (Fc0/Fc+) couple.36 Glassy carbon is more conductive than FTO and ITO coated glass, and is therefore is a strong candidate as the substrate for solid state electrochemistry. The major limitation for glassy carbon is that mechanical immobilisation of materials such as POPs is difficult. Electrophoretic deposition (EPD) has been successfully applied to synthesise homogeneous, continuous films of UiO, MIL, NU,30 ZIF37 and porphyrin-based38 MOFs. The limitations of this particular surface attachment method lie in the high voltages required for deposition to occur. It is likely that immobilising of a POP onto glassy carbon using EPD could be achieved at lower voltages.39
Herein, we report the synthesis of three Co(III) salen metal complexes to explore their use as electrochemical reducing agents for CO2. The electrochemical properties of the discrete complexes are analysed to examine the effect of modulating the bridging diamine functionality of the complexes on their electrochemical properties. We perform a detailed electrochemical study on the conversion of CO2 using these complexes to address the gap in the literature regarding salen complexes as reducing agents. We also report the inclusion of the discrete complexes into novel POPs and examine the impact of salen immobilisation on their redox properties. We further report a method for the surface attachment of POPs onto glassy carbon via EPD which uses milder potentials than those previously reported for EPD onto an FTO substrate, thus providing a benign pathway for attaching solid-state materials to glassy carbon.
Experimental Methods
General
All chemicals used were purchased from Aldrich, Alfa Aesar and Merck, and were used without further purification unless stated otherwise. Solvents were obtained from a PureSolv system, or purchased and used without further purification. Low resolution electrospray ionisation mass spectra (ESI-MS) were acquired as a solution in MeCN, MeOH or DMF with a 100 μL min−1 flow rate on a Finnegan LCQ or amazon MS detector. Spectra were collected over the mass range m/z 100 to 1000. An ESI spray voltage of 5 kV was applied with a heated capillary temperature of 200 °C and a nitrogen sheath gas pressure of 60 psi. Melting points were measured using a Gallenkamp melting point apparatus with the sample placed in a glass capillary. The final melting points are uncorrected. Room temperature FT-IR spectra were obtained using a PerkinElmer UATR 2 infrared spectrometer over the range 400–4000 cm−1 with a resolution of 4 cm−1. Samples were mechanically compressed on the surface of a diamond crystal on which the background was collected. ICP-OES was performed at the Mark Wainwright Analytical Centre at The University of New South Wales.Nuclear magnetic resonance
Solution state 1H and 13C{1H} NMR spectra were recorded on a Bruker AVANCEIII 300, 400 or 500 spectrometer operating at 300, 400, 500 MHz for 1H and 75, 100, 125 MHz for 13C, respectively. 1H and 13C NMR chemical shifts were referenced internally to residual solvent resonances. Spectra were recorded at 298 K and chemical shifts (δ), with uncertainties of ±0.01 Hz for 1H and ±0.05 Hz for 13C, are quoted in ppm. Coupling constants (J) are quoted in Hz and have uncertainties of ±0.05 Hz for 1H–1H. Deuterated solvents were obtained from Cambridge Stable Isotopes and used as received. Chemical shifts for all ligands are reported relative to the deuterated solvents used.40The 13C cross polarisation magic angle spinning (CPMAS) solid state NMR experiments on the diamagnetic discrete complexes and their polymers were carried out at The Mark Wainwright Analytical Centre, The University of New South Wales, on a wide-bore Bruker Biospin AVANCEIII solids-300 MHz spectrometer operating at a frequency of 75 MHz for the 13C nucleus. The sample (∼80 mg) was placed into 4 mm zirconia rotors fitted with Kel-f® caps and spun in a double resonance H-X probehead at 8 kHz magic angle spinning (MAS). The 13C and 1H 90° radio frequency pulse lengths were optimised to 3.5 μs each. The 13C spectra were acquired with 1 ms cross polarisation contact time with a total suppression of spinning side bands (TOSS) scheme, followed by 1H decoupling at 75 kHz field strength using spinal-64 decoupling. The 13C non-quaternary suppression (NQS) spectra were recorded by turning off the 1H decoupling for 40 μs during the TOSS period. For sufficient signal-to-noise, ca. 10 K transients were acquired for each sample with recycle delays of 3.0 s in between to ensure sufficient relaxation of the 1H nuclei. The spectra were obtained at room temperature. The 13C chemical shifts were referenced to the glycine CO peak at 176 ppm.
Cyclic voltammetry
Solution and solid state electrochemical measurements were performed using a Bioanalytical Systems BASi Epsilon Electrochemical Analyser at 298 K. A single compartment cell was used, consisting of either a glassy carbon working electrode (3.0 mm diameter) for solution state or a glassy carbon plate (active area of 2 cm2) with immobilised polymer attached through a steel alligator clip to tinned copper wire for solid state, a platinum wire auxiliary electrode and an electrolysed Ag/AgCl wire reference electrode separated from the solution by a CoralPor tip. Cyclic and differential pulse voltammograms were performed using a 10 mL solution of the analyte (1 mM complex in 0.1 M [(n-C4H9)4N]PF6 in either MeCN or DMF). The solution was purged with dried N2, Ar, or CO2 prior to each experiment. Ferrocene (Fc) (1 mM) was added as an internal standard during each experiment. All potentials are quoted in V vs. Fc0/Fc+. Uncompensated resistance between the working and the reference electrodes was corrected by using iR compensation on the potentiostat. Scan rate dependence studies were carried out for each complex between 50–1600 mVs−1 to ensure the homogeneity of the system.Infrared spectroelectrochemistry (IR SEC)
Solution state IR SEC was performed on the discrete complexes using the IR-SEC cell previously reported by Kubiak et al.41 A Pine Instrument Co. Model AFCBP1 potentiostat was employed to control the cell potential, referenced to Ag/Ag+. Thin-layer bulk electrolysis was measured by reflectance IR off the electrode as the potential was scanned. All experiments were conducted in 0.1 M [(n-C4H9)4N]PF6/MeCN/DMF (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with known analyte loadings prepared under an inert atmosphere. FT-IR spectra were recorded on a Thermo Scientific Nicolet 6700, with resolution of 4 cm−1. Unlike the CV or UV-Vis-NIR SEC experiments, it was not possible to use pure DMF in the IR SEC experiments, since the strong νC
:1) with known analyte loadings prepared under an inert atmosphere. FT-IR spectra were recorded on a Thermo Scientific Nicolet 6700, with resolution of 4 cm−1. Unlike the CV or UV-Vis-NIR SEC experiments, it was not possible to use pure DMF in the IR SEC experiments, since the strong νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1740 cm−1 overwhelmed the comparatively weaker νCN stretch expected from the salen metal complex at ∼1600 cm−1.
O stretching vibration at 1740 cm−1 overwhelmed the comparatively weaker νCN stretch expected from the salen metal complex at ∼1600 cm−1.
Ultraviolet-visible-near infrared spectroelectrochemistry (UV-Vis-NIR SEC)
Solution state UV-Vis-NIR SEC was measured over the range 5000−35000 cm−1 using a CARY5000 spectrophotometer interfaced to Varian WinUV software. The absorption spectra of the electrogenerated species were obtained in situ by the use of an Optically Semi-Transparent Thin-Layer Electrosynthetic cell, path length 0.685 mm, mounted in the path of the spectrophotometer. Solutions for the spectroelectrochemical experiment contained 0.1 M [(n-C4H9)4N]PF6/MeCN or [(n-C4H9)4N]PF6/DMF supporting electrolyte and ca. 0.4 mM of the compound for analysis. Appropriate potentials were applied by using an eDAQ e-corder 410 potentiostat and the current was carefully monitored throughout the electrolysis. The electrogenerated species formed in situ, and their absorption spectra were recorded at regular intervals through the electrolysis. The attainment of a steady-state spectrum and the decay of the current to a constant minimum at a potential appropriately beyond E1/2 (for the redox process) was indicative of the complete conversion of the starting material.
Solid state diffuse reflectance UV-Vis-NIR spectra of the redox-active species were collected in situ using a CARY5000 UV-Vis-NIR spectrophotometer equipped with a Harrick Omni Diff Probe attachment interfaced to Varian WinUV software over the range 5000–25000 cm−1 in a custom-made cell previously reported by D'Alessandro et al.42 The cell consisted of a Pt wire counter electrode and a Ag/Ag+ quasi-reference electrode. The solid sample was immobilised by a thin strip of Teflon tape onto a 0.1 mm thick Indium-doped Tin Oxide (ITO) coated quartz slide, which functioned as the working electrode. The applied potential was controlled using an eDAQ potentiostat. Continuous scans of the sample were obtained and the potential increased gradually until a change in the spectrum was observed.
Bulk electrolysis
Solution and solid state bulk electrolysis were performed in a threaded 60 mL single compartment cell with a custom airtight Teflon top, as reported by Kubiak et al.43 The set-up consisted of a carbon rod (surface area = 7.4 cm2) for solution state or glassy carbon plate with immobilised POP for solid state as the working electrode (active area = 2 cm2), a coiled Pt wire counter electrode protected from the bulk solution by fritted glass and an electrolysed Ag/AgCl pseudo reference, separated from solution by a CoralPor tip. The analyte solution (∼40 mL) consisted of complex (1–2 mM) in 0.1 M [(n-C4H9)4N]PF6/MeCN/DMF (8:2). The solution was purged with CO2 for 20 min prior to each electrolysis experiment, with the optimal applied potential determined by the CV experiments.
Gas phase analysis was performed by sampling 1 mL of the headspace of the cell at 20 min intervals and injecting into a Hewlett-Packard 7890A series gas chromatograph with two molecular sieve columns (30 m × 0.53 mm i.d. × 25 μm film). The 1 mL injection was split between two columns, one with N2 carrier gas and one with He carrier gas to quantify both CO and H2 respectively. Instrument specific calibration curves were measured prior to analysis to determine the amount of each gas produced.
Solution phase analysis was performed on the bulk solution to quantify formic acid production by sampling the bulk electrolysis solution (5 mL) after electrolysis. D2O (1 mL) was added to the solution and this was well mixed, prior to its dilution with dichloromethane. The D2O layer was separated prior to the addition of concentrated hydrochloric acid (1 drop). Samples were analysed by 1H NMR and spectra were recorded on a Bruker AVANCEIII 300 spectrometer operating at 300 MHz for 1H. Spectra were recorded at 298 K.
Thermogravimetric analysis
TGA measurements were carried out on a TA Instruments Hi-Res 2950 Thermogravimetric Analyser or Discovery Thermogravimetric Analyser. Dry N2 (0.1 L min−1) flowed over the sample during data collection. The sample temperature was ramped at 1 °C min−1 from 25 to 600 °C. Samples were loaded dry after exposure to air.Gas adsorption
Adsorption isotherms were measured using the Accelerated Surface Area & Porosity (ASAP) 2020 or the 3-Flex, both supplied by Micromeritics Instruments Inc. The sample (∼50–100 mg) was loaded into a glass analysis tube and outgassed for 24 h under vacuum at 80 °C, prior to analysis.N2 adsorption and desorption isotherms were measured at 77 K and data were analysed using the Brunauer, Emmett and Teller (BET) models to determine the surface area.44 Pore size distributions were calculated using the Density Functional Theory (DFT) cylindrical model in the Micromeritics MicroActive Software Package Version 4.03.
Electrophoretic deposition (EPD) onto glassy carbon
Glassy carbon substrates were cut to 3 × 1 cm electrodes. The POP (20 mg) was suspended in toluene (20 mL) and sonicated for 30 s. Two identical glassy carbon substrates were dipped into the deposition solution (1 cm separation distance) and a constant DC voltage of 30 V was applied from an Extech 382270 High Precision Quad Output DC power supply. The deposition occurred over a period of 6 h, with a stirred suspension that was sonicated every 30 minutes to break up larger aggregates of POP. Caution: electrical sparking due to the accidental contact of electrodes and/or their leads can result in the spontaneous ignition of toluene. Prior to the undertaking of these experiments, ensure that the experiment is set up in an empty fume hood, clear of flammables and with a blast shield.Field emission scanning electron microscopy
FE-SEM measurements were obtained at either the Nano3 facility at The University of California, San Diego or at the Australian Centre for Microscopy & Microanalysis at The University of Sydney. POP materials were deposited onto a glassy carbon substrate, which was adhered to conductive carbon tape on a sample holder disk. The disk was coated using a Cr-sputter coating for 8 s. A Philips XL30 ESEM was used for acquiring images using a 10 kV energy source under vacuum at a working distance at 10 mm. ∼19000× magnification images were collected.
Results and discussion
Synthesis and structural characterisation
The synthesis of salen ligands was achieved by the Schiff-base condensation of 5-bromosalicylaldehyde with varying bridging diamines to afford the free-base salen in good yield. Co(III) metalation was achieved by the addition of Co(OAc)2·4H2O in the presence of LiCl as an oxidising agent to afford all Co(III) complexes in good yields (Scheme 1A). Salen metal complexes were successfully incorporated into POPs via a Sonogashira–Hagihara palladium cross coupling reaction between tris(p-ethynyl)triphenylamine (TPA) and the bis-bromo salen metal complexes under an inert N2 atmosphere in the presence of [Pd(PPh3)4] and CuI to form the series of POPs. In all reaction mixtures, the light yellow solution darkened and a brown precipitate formed (Scheme 1B).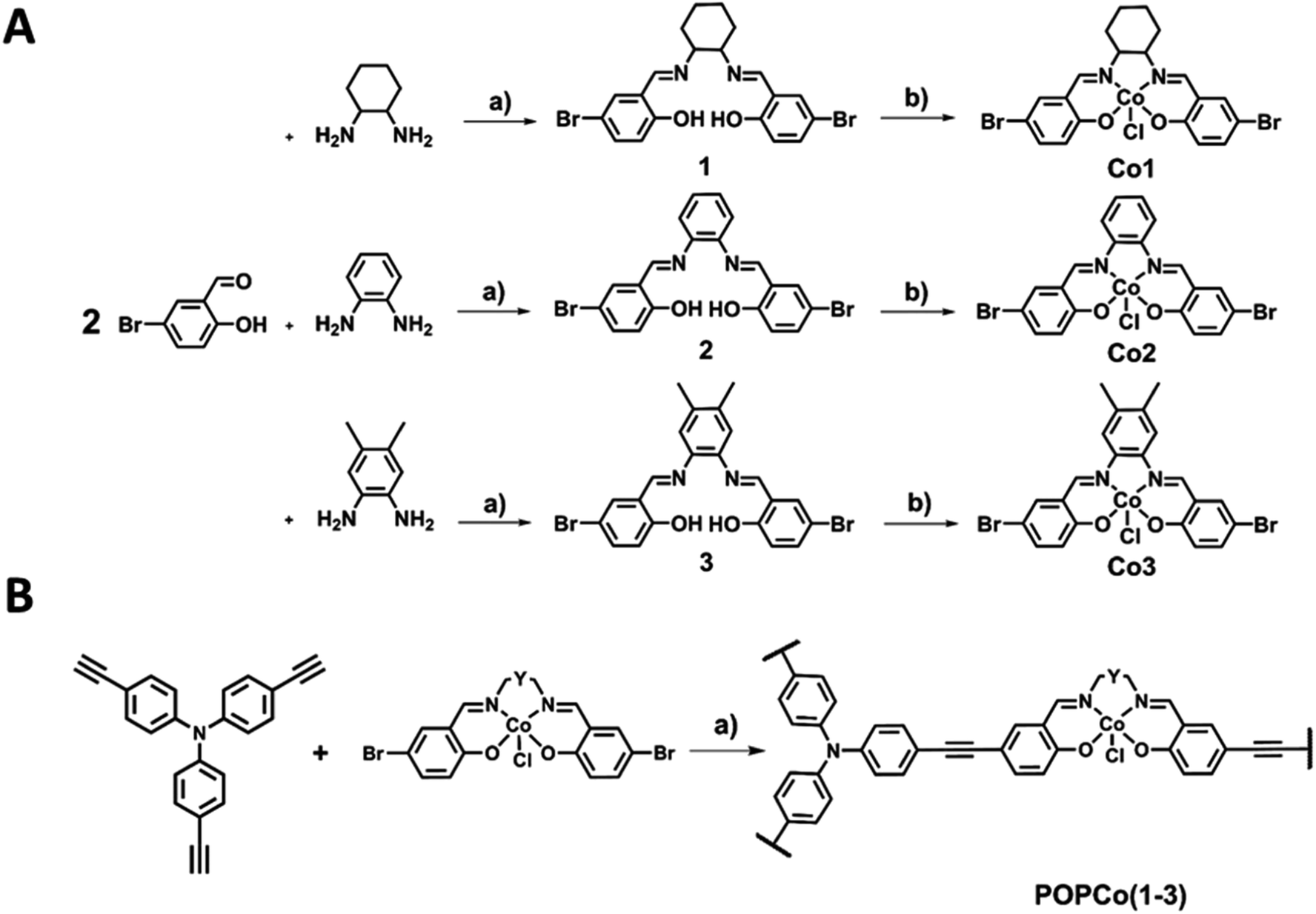 | ||
| Scheme 1 (A) Synthesis of bromo-terminated Co(III) salen metal complexes (a) solvent: MeOH, 80 °C, 2 h, (b) metal: Co(OAc)2·4H2O (1.1 eq.), LiCl (4 eq.), solvent: EtOH, r.t., 48 h. (B) Synthesis of the Co(III) salen polymer (a) catalysts: [Pd(PPh3)4] (30 mol%), CuI (30 mol%), solvent: toluene/EtOH (2:1), 85 °C, 72 h. | ||
Extensive characterisation was performed on the amorphous polymers to determine their composition. Despite their non-crystalline nature, TGA studies revealed that all POPs possessed thermal stability above 200 °C, with a number of polymers showing thermal stability of up to 400 °C, prior to partial thermal degradation (Fig. S1†). IR experiments indicated that the TPA co-ligand was covalently linked to the salen metal complex. The IR spectrum of TPA exhibited a νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) H stretch at 3264 cm−1, which disappeared upon polymerisation. The weak νCC stretch also shifted from 2102 cm−1 in the monomer to 2191 cm−1 in the polymer.45 The νCN stretch belonging to the imine in the salen metal complex was observed to shift upon incorporation into the salen POP compared with the polymer (Fig. S2†). Further evidence of the covalent linkage between co-ligand and salen was detected in the UV-Vis-NIR spectra of the monomers and polymers. The UV-Vis-NIR spectrum of TPA exhibited a sharp n–π* charge transfer at 26460 cm−1 and π–π* charge transfer bands at 31970 and 34260 cm−1. When compared to the discrete salen complexes, many of the characteristic charge transfer bands in the salen metal complexes were shifted to lower energies in the polymers, consistent with polymeric systems that are more delocalised. The UV-Vis-NIR spectra of the polymers all exhibited the n–π* charge transfer band, consistent with TPA incorporation into the polymer (Fig. S3†). ICP-OES indicated that the Co(III) salen was incorporated into the POP; however, the lower than calculated values for the Co(III) content suggests that homocoupling between the TPA may be occurring because of uncontrolled propagation, as well as the possible leaching of the Co(III) species (Table S1†).
H stretch at 3264 cm−1, which disappeared upon polymerisation. The weak νCC stretch also shifted from 2102 cm−1 in the monomer to 2191 cm−1 in the polymer.45 The νCN stretch belonging to the imine in the salen metal complex was observed to shift upon incorporation into the salen POP compared with the polymer (Fig. S2†). Further evidence of the covalent linkage between co-ligand and salen was detected in the UV-Vis-NIR spectra of the monomers and polymers. The UV-Vis-NIR spectrum of TPA exhibited a sharp n–π* charge transfer at 26460 cm−1 and π–π* charge transfer bands at 31970 and 34260 cm−1. When compared to the discrete salen complexes, many of the characteristic charge transfer bands in the salen metal complexes were shifted to lower energies in the polymers, consistent with polymeric systems that are more delocalised. The UV-Vis-NIR spectra of the polymers all exhibited the n–π* charge transfer band, consistent with TPA incorporation into the polymer (Fig. S3†). ICP-OES indicated that the Co(III) salen was incorporated into the POP; however, the lower than calculated values for the Co(III) content suggests that homocoupling between the TPA may be occurring because of uncontrolled propagation, as well as the possible leaching of the Co(III) species (Table S1†).
Solid state 13C NMR experiments were performed to further characterise the salen-based POPs. Solid state 13C NMR spectra could be obtained for POPCo1, POPCo2 and POPCo3, which all contain the Co(III) d6 species. This implies that the Co(III) salen metal complexes are low spin when incorporated into the POPs. The 13C CPMAS spectra of the polymers containing the diamagnetic salen metal complexes were collected and compared to the discrete complexes to show incorporation of the salen into the POP (Fig. 2, S4–S6†). A shift in the position of the alkynyl peaks from those observed in discrete TPA previously reported,46 as well as their broadening, suggests that a polymeric material has formed. Evidence for the incorporation of the salen metal complex came from the appearance of peaks from the bridging diamine around 160–165 ppm, corresponding to the imine carbon and phenolic carbon environments present in the salen. These varied dependent on the bridging moiety. The comparatively small signal intensity of the salen moiety suggests an incorporation of salen around 10%, which is consistent with the ICP-OES data (Table S1†). Variation in the incorporated salen metal complex could be deduced from the appearance of different carbon environments at lower field. For POPCo1, evidence for the diaminocyclohexane bridging moiety came from the appearance of peaks corresponding to the sp3 hybridised secondary and quaternary carbons on the bridging diamine between 20–30, and 70 ppm. The additional signals in the alkyl region at 15 and 50 ppm have been tentatively assigned to small quantities of residual triethylamine. For the aromatic POPCo2 and POPCo3, a larger integration under the aromatic signals from 110–160 ppm was detected, which implies additional carbon environments from the discrete complexes Co2 and Co3, respectively (Fig. S4 and S5†). Finally, the synthesis was repeated in the absence of salen metal complex, forming POPTPA. The 13C CPMAS spectrum for this deliberately homocoupled polymer POPTPA demonstrated the absence of peaks corresponding to the phenolic carbon and the imine of the salen moiety at 162–165 ppm. Additionally, a single ethynyl signal at 90 ppm was observed (Fig. S6†).
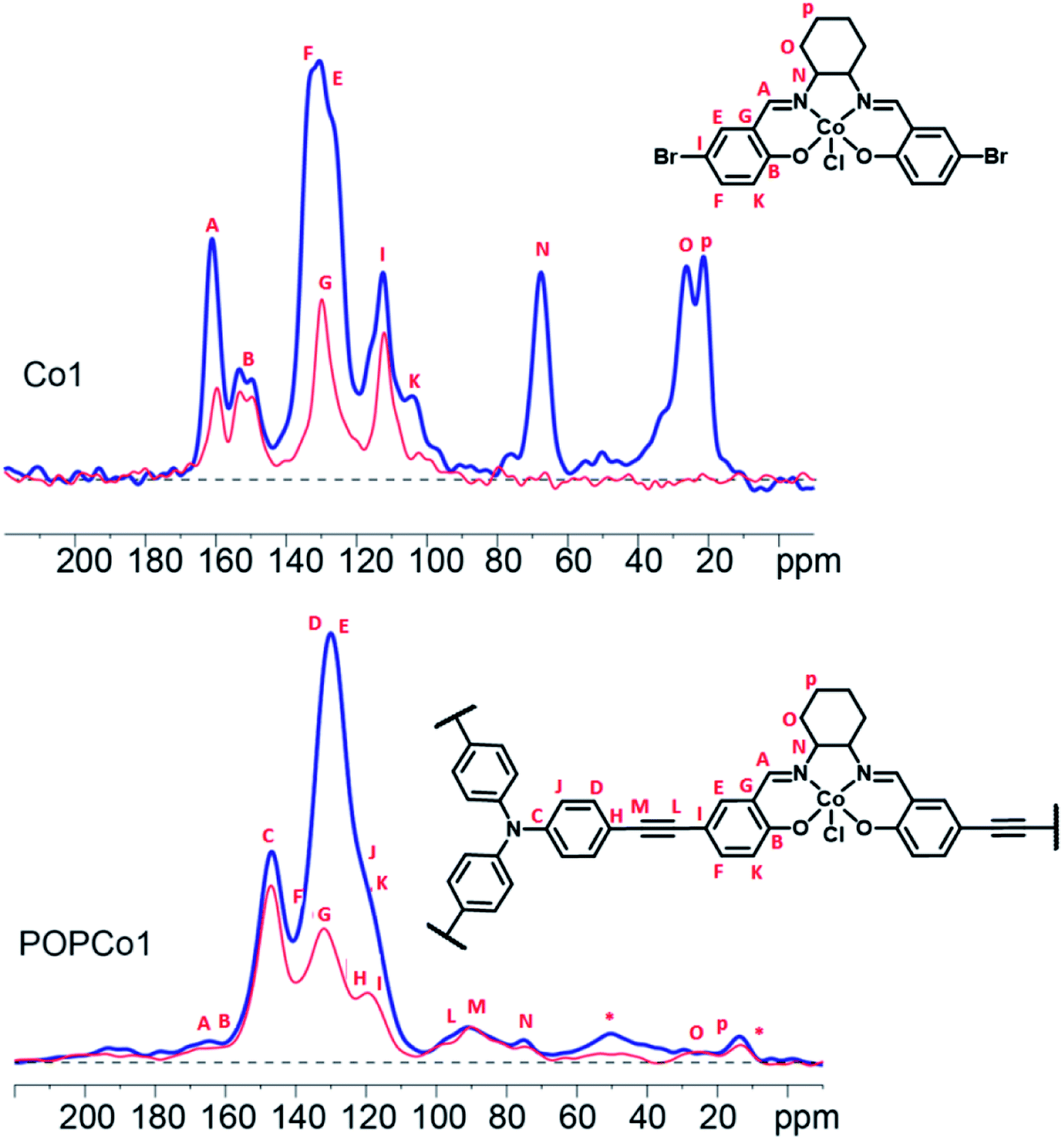 | ||
| Fig. 2 13C CPTOSS of Co1 (above) and POPCo1 (below). The full 13C NMR spectrum is plotted in blue, while the spectrum in red is the non-protonated methyl/carbon species detected after 40 μs of dipolar dephasing. The signals marked * are attributed to excess triethylamine, which was unable to be completely removed from the pores. | ||
Gas adsorption in polymers
Methods for the activation of POPs were initially probed as a step towards examining their gas sorption properties. The activation of the polymers was achieved by initially immersing the newly synthesised POP materials in DMF (10 mL) in an oven at 100 °C for 1 h, prior to filtration and repetition of the process three times. The polymer was subjected to washing via a Soxhlet washing process with methanol at 80 °C for 48 h.The porosities of the POPs were analysed using N2 gas sorption experiments at 77 K to determine their BET surface areas. All POPs demonstrated Type I BET isotherm behaviour, which is indicative of a microporous material (Fig. 3A).47 There is a significant amount of hysteresis, which is commonly observed within highly flexible porous materials. The amorphous nature of the POPs is reflected in the pore size distribution, where there is a wide variety of sizes, consistent with the uncontrolled propagation of the POP (Table 1, Fig. S7†).
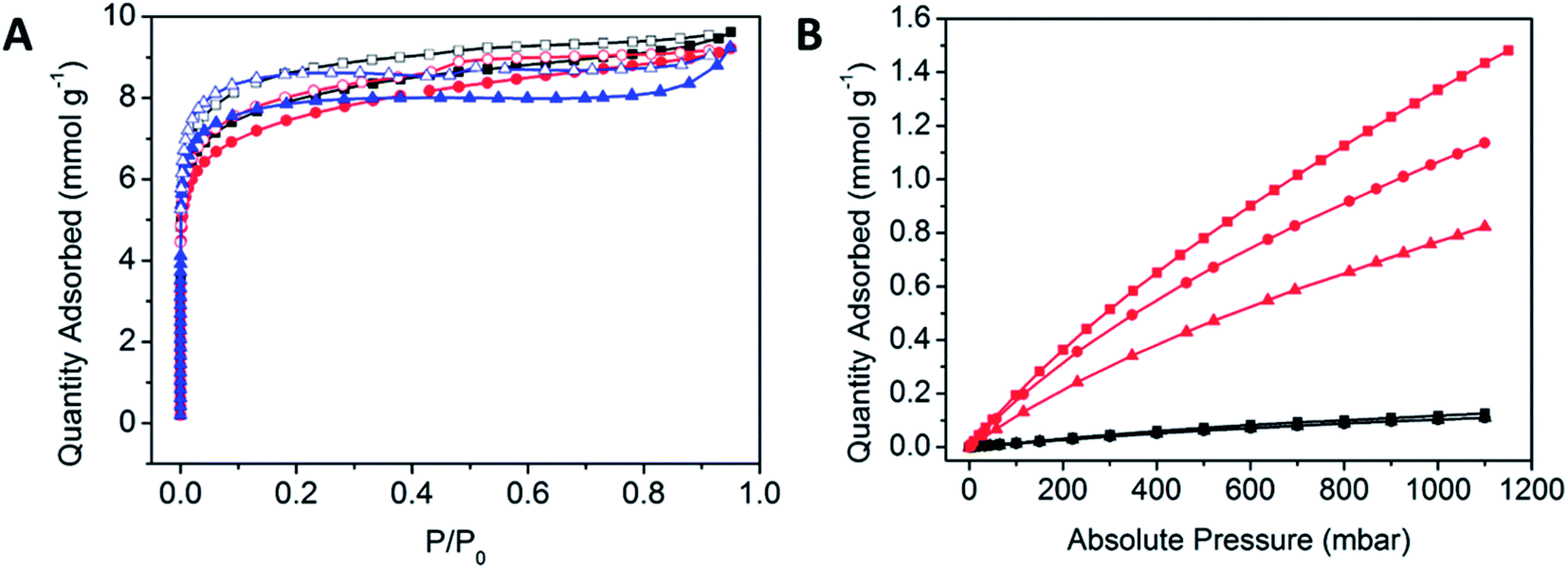 | ||
| Fig. 3 (A) N2 gas sorption isotherms at 77 K of POPCo1 (black), POPCo2 (red) and POPCo3 (blue). Closed squares represent N2 adsorption isotherms while open squares represent N2 desorption isotherms. (B) CO2 adsorption isotherms (red) and N2 gas adsorption isotherms (black) at 298 K of POPCo1 (circles), POPCo2 (triangles) and POPCo3 (squares). | ||
| Polymer | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size distribution (Å) | Point selectivity S | −Qst (kJ mol−1) |
|---|---|---|---|---|---|
| POPCo1 | 657 ± 1 | 0.36 | 4.9–7.7, 8.3–13 | 15.4 | 25–27 |
| POPCo2 | 669 ± 1 | 0.37 | 4.8–7.8, 8.4–13, 17−21 | 9.5 | 27–29 |
| POPCo3 | 688 ± 1 | 0.36 | 5.6–7.8, 8.8–12, 14−21 | 15.1 | 20–27 |
N2 and CO2 gas sorption experiments were performed at 298 K (Fig. 3B). The comparable N2 isotherms indicate that at 298 K, there is a limited uptake of N2 and a preference for CO2 uptake which could arise from differences in the salen backbone. Having to overcome the increased π-stacking from the aromatic POPCo2 may hinder the adsorption of CO2, while an increased bulk from the bridging diamine may disrupt these interactions, justifying why POPCo1 and POPCo2 have higher uptakes. The isosteric heats of adsorption indicate that there are physisorptive interactions between CO2 and the POP (Table 1, Fig. S8†). The slight variations in −Qst suggest that the backbone of the salen does not play a significant role in the physisorptive interactions between CO2 and the POP. Finally, the CO2/N2 selectivities (S) were taken with respect to the conditions of a post-combustion flue stream at 298 K, consisting of N2 (P/P0 = 0.75) and CO2 (P/P0 = 0.15) (Table 1).
Surface attachment of Co(III) salen POPs to glassy carbon
POPCo1, POPCo2 and POPCo3 were successfully immobilised onto glassy carbon via electrophoretic deposition by suspending the POP in toluene, with sonication and stirring while a constant DC voltage of 30 V was applied over a period of 6 h. The reaction was paused every hour to allow for the further sonication of the mixture to disrupt aggregation of the POP. The experiment resulted in coverage of POPCo1, POPCo2 and POPCo3, which could be imaged on the surface via FE-SEM (Fig. 4). POPCo1 and POPCo2 demonstrated better coverage of the glassy carbon plates than POPCo3. | ||
| Fig. 4 (A) Coverage of glassy carbon plates by Co(III) polymers. FE-SEM images of (B) POPCo1 (C) POPCo2 and (D) POPCo3. | ||
Electrochemistry
| Complex | IR | IIR | IIIR | IVR | Fc0/+ |
|---|---|---|---|---|---|
| Co1 | Epc = −0.49 V | E1/2 = −1.66 V | E1/2 = −2.36 V | E1/2 = 0 V | |
| ΔE = 64 mV | ΔE = 60 mV | ΔE = 60 mV | |||
| (ipa/ipc) = 0.642 | (ipa/ipc) = 0.837 | (ipa/ipc) = 0.914 | |||
| Co2 | Epc = −0.37 V | E1/2 = −1.56 V | E1/2 = −2.23 V | E1/2 = −2.43 V | E1/2 = 0 V |
| ΔE = 35 mV | ΔE = 63 mV | ΔE = 84 mV | ΔE = 61 mV | ||
| (ipa/ipc) = 0.448 | (ipa/ipc) = 0.597 | (ipa/ipc) = 0.684 | (ipa/ipc) = 0.944 | ||
| Co3 | Epc = −0.39 V | E1/2 = −1.57 V | E1/2 = −2.25 V | E1/2 = − 2.46 V | E1/2 = 0 V |
| ΔE = 33 mV | ΔE = 58 mV | ΔE = 59 mV | ΔE =62 mV | ||
| (ipa/ipc) = 0.540 | (ipa/ipc) = 0.705 | (ipa/ipc) = 0.859 | (ipa/ipc) = 0.918 |
| POP | IR | IIR | IIIR | IVR | Fc |
|---|---|---|---|---|---|
| POPCo1 | Epc = −0.12 V | E1/2 = −0.80 V | Epc = −1.10 V | Epc = −2.16 V | E1/2 = 0 V |
| POPCo2 | Epc = −1.42 V | Epc = −2.35 V | E1/2 = 0 V | ||
| POPCo3 | Epc = −0.86 V | Epc = −1.86 V | −2.26 V | E1/2 = 0 V |
Spectroelectrochemistry
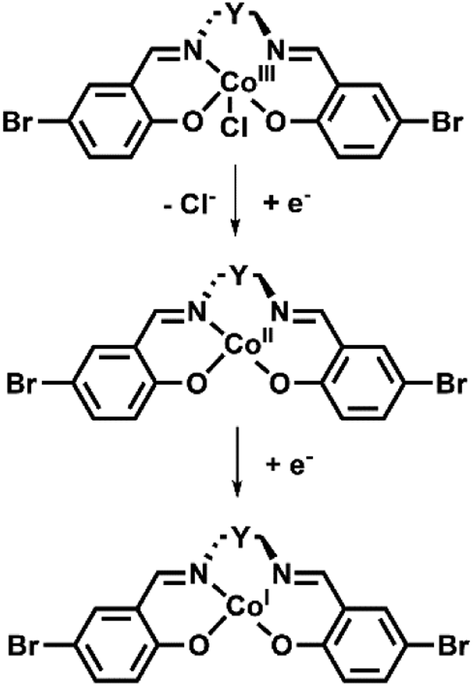 | ||
| Scheme 2 Proposed mechanism of the Co(III) complexes observed over the initial two reduction processes. | ||
The first reduction processes in the complexes were consistent with the reduction of the Co(III) centre to Co(I). In the SEC for all complexes, an increase in the intensity of the d–d bands (16280 and 19990 cm−1 for Co1, 18570 cm−1 for Co2 and 15040 cm−1 for Co3) was consistent with a transition from d6 to d7 cobalt species (Fig. S11†) and decreases in LMCT bands (24470 in Co1, 20980, 25550 and 29600 cm−1 in Co2 and 21720, 29550 and 30980 cm−1 for Co3), as seen in other Co(III) salen complexes.48,49 The maintenance of isosbestic points suggests that there is a clean conversion between the Co(III), Co(II) and Co(I) species.
Interesting electronic properties appear after the IIR reduction. Solution state differential pulse voltammetry (DPV) experiments on Co2 in 0.1 M [(n-C4H9)4N]PF6/DMF under N2 reveal a non-Gaussian peak, indicating that the redox couple has a number of underlying processes (Fig. 5A). Despite multiple processes observed, the integration of the area under the combined processes is the same as the integration of the Fc0/Fc+ signal, indicating that only one electron is being transferred. UV-Vis-NIR SEC shows not only evidence for the Co(II/I) couple (vide supra), but also an appearance of a radical band at 12690 cm−1. IR SEC experiments were performed on Co2 (4 mM) in 0.1 M [(n-C4H9)4N]PF6/MeCN/DMF (9:1), which indicated that ligand-based processes are also occurring. The appearance of a new IR stretch at ν = 1674 cm−1, corresponding to a change in the νC–O stretch on the phenol, implies that there may be evidence of charge transfer from the Co(I) metal centre to the ligand (Fig. 5B).
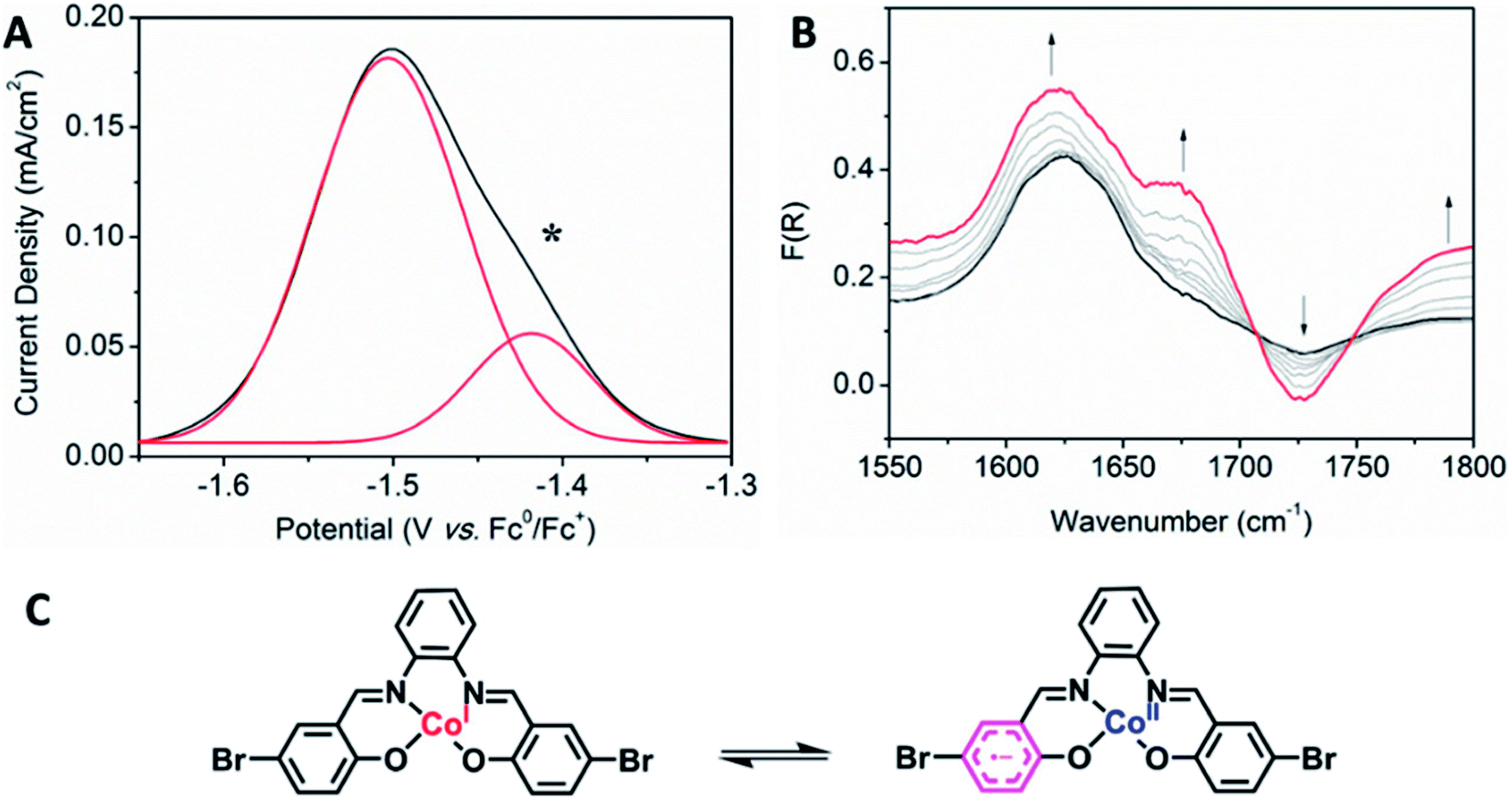 | ||
| Fig. 5 (A) Solution state DPV of Co2 (1 mM) (scan rate: 0.02 V s−1, Fc (1 mM) was used as an internal standard) and (B) solution state IR SEC of Co2 (4 mM) upon changing the potential from −0.8 to −1.5 V vs. Ag/Ag+. (0.1 M [(n-C4H9)4N]PF6/MeCN/DMF (9:1) as the supporting electrolyte). (C) The predicted mechanism of the IIR reduction process, showing possible charge transfer between the Co(I) metal and the salen backbone. | ||
Clear differences in the ligand based reductions are also apparent between the materials. In Co1, the third reduction results in the generation of a number of important bands, consistent with intervalence charge transfer (IVCT). Upon application of a potential at Epc = −1.66 V vs. Fc0/Fc+, the observation of a weak IVCT band at 10010 cm−1 and a radical band at 12860 cm−1 are suggestive of the formation of the salen radical anion. The lower energies associated with the radical anion band suggest that the Co(III) metal centre promotes delocalisation. To the best of our knowledge, such IVCT transitions have not been previously reported for Co(III) salen metal complexes. The appearance of the IVCT band is also accompanied by an increase in the π–π* transitions at 31080 cm−1, consistent with improved electron delocalisation in the conjugated parts of the salen backbone. In Co2, there is evidence for a weaker IVCT transition. Changes appear in the LMCT and n–π* transitions at 19590 and 23680 cm−1, respectively. This is accompanied by the appearance of a weak IVCT band at 8340 cm−1 and a weak radical band at 12670 cm−1. Similar processes in Co3 are observed. The dominant changes in the UV-Vis spectra are the decrease in the transitions at 20500, 25120, 29550 and 30980 cm−1, and the appearance of new transitions at 22490 and 23870 cm−1. This is also supported by the fact that the IIIR process in Co3 is more cathodic than its Co2 analogue, which is consistent with improved delocalisation and electron density increasing the voltage required for reduction of the Co3 backbone. The absence of an observable IVCT band suggests that the degree of aromaticity affects the nature of the IVCT transition (Fig. 6).
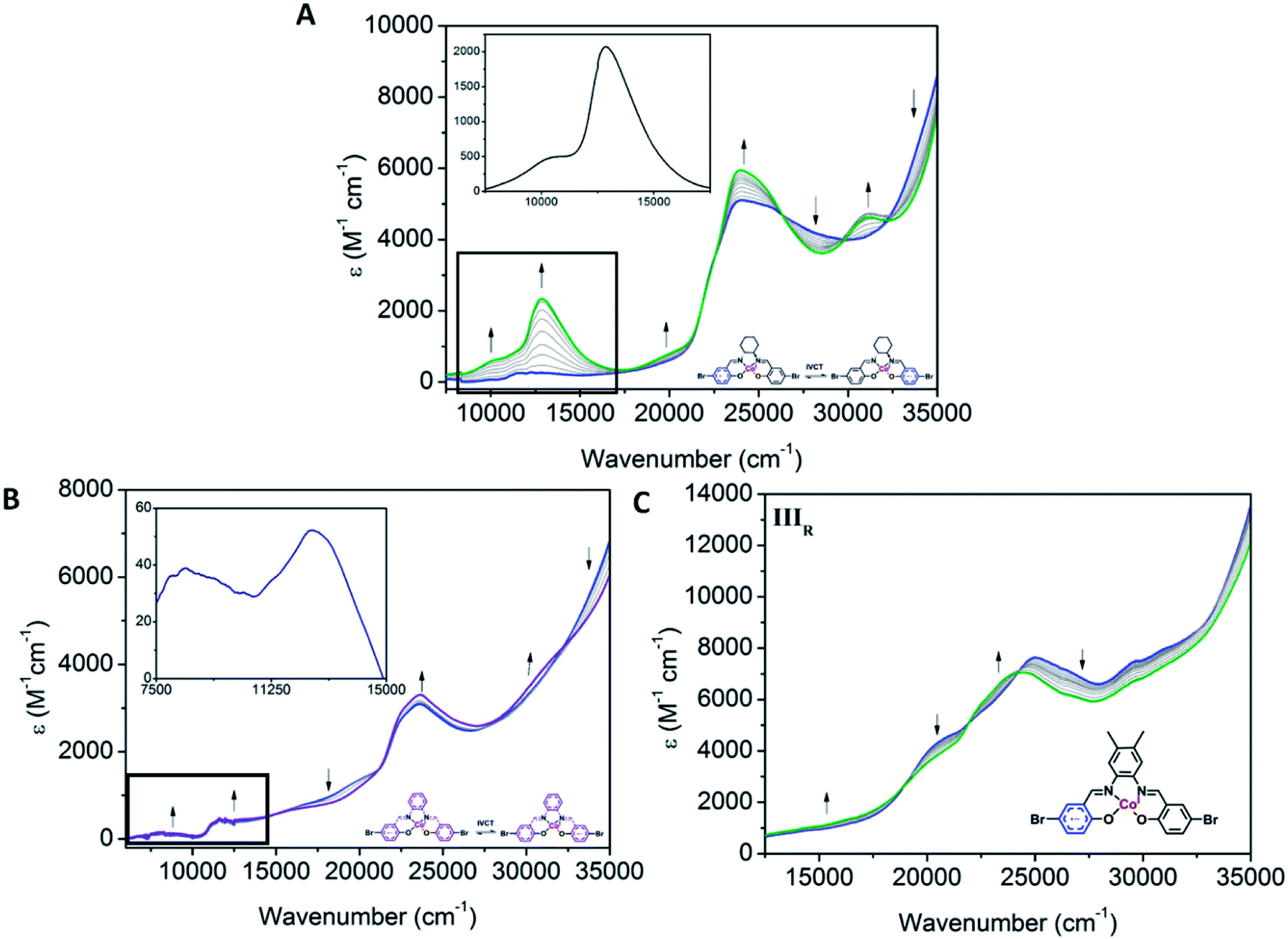 | ||
| Fig. 6 Solution state UV-Vis-NIR SEC of (A) Co1 (0.61 mM) upon changing the potential from −1.5 V to −1.8 V vs. Ag/Ag+, (B) Co2 (0.51 mM) upon changing the potential from −1.6 to −2.1 V vs. Ag/Ag+ and (C) Co3 (0.32 mM) upon changing the potential from −1.8 to −2.2 V vs. Ag/Ag+ (0.1 M [(n-C4H9)4N]PF6/DMF as the supporting electrolyte). | ||
410 and 19940 cm−1 (for POPCo1) (Fig. S12†) and 18170 cm−1 and 20510 cm−1 (for POPCo3) (Fig. 7). This change is consistent with a Co(II/I) process and is reflected by corresponding changes in discrete complexes. There were small changes in the intensity of the peaks for POPCo2. Upon reaching the IIIR potential, losses in intensity occurred in all of the bands in the spectra and the lack of stable isosbestic points indicated a degradation of the polymer. This is also evident from the change in the electrolyte solution from clear and colourless to green.
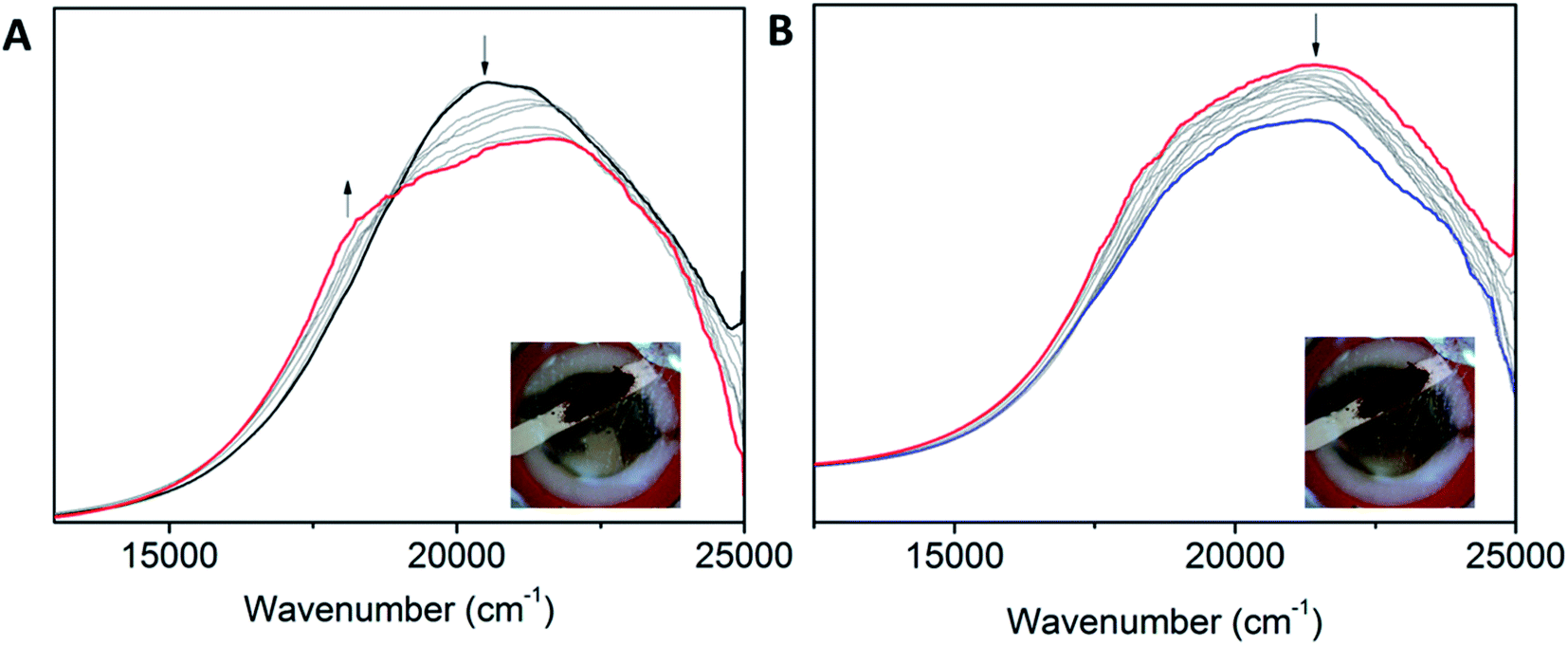 | ||
| Fig. 7 Solid state Vis-NIR SEC of POPCo3 upon changing the potential from (A) −1.0 to −1.5 V vs. Ag/Ag+, (B) −1.7 to −2.0 V vs. Ag/Ag+ (0.1 M [(n-C4H9)4N]PF6/MeCN as the supporting electrolyte). | ||
Electrochemical behaviour in the presence of CO2
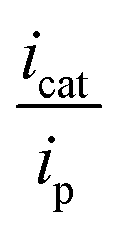 of 3.34, corresponding to a TOF k[Q] of 2.16 s−1 at 0.63 M TFE. Only a weak association of CO2 with Co2 was observed (Fig. 8).
of 3.34, corresponding to a TOF k[Q] of 2.16 s−1 at 0.63 M TFE. Only a weak association of CO2 with Co2 was observed (Fig. 8).
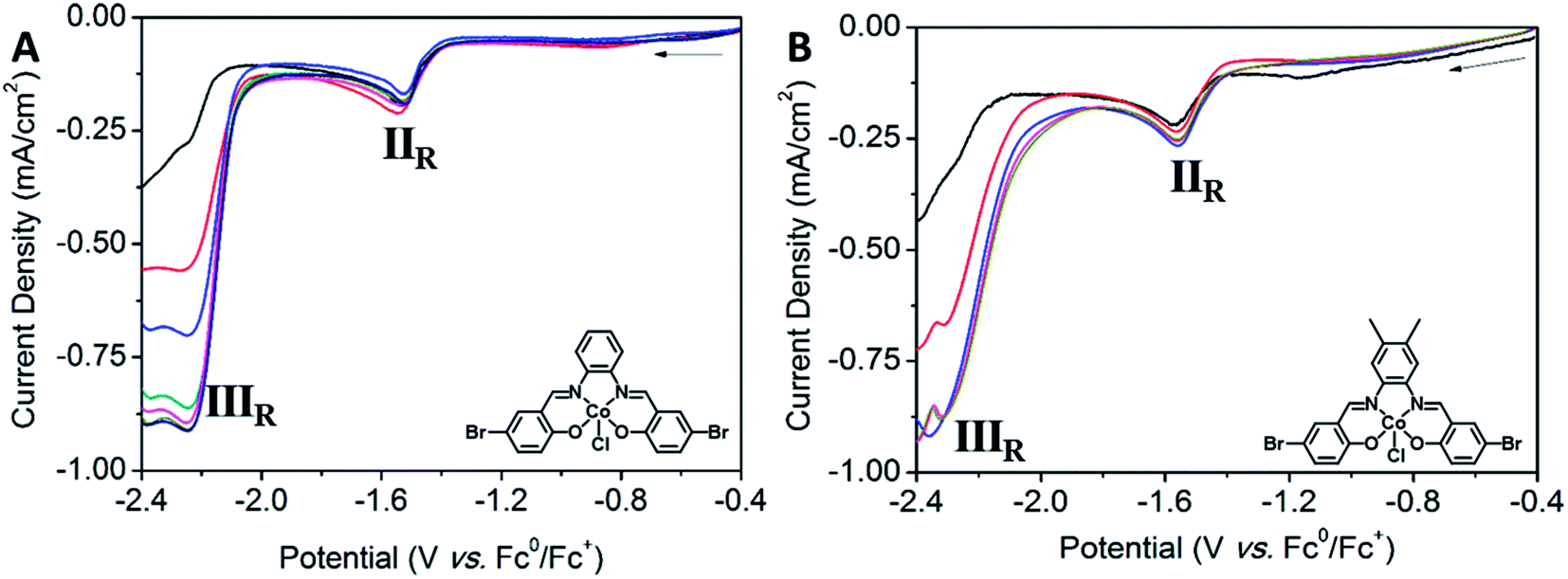 | ||
| Fig. 8 Solution state CV showing the electrochemical response of (A) Co2 (1 mM) under saturation of N2 (black), CO2 (red) and CO2 with TFE (0.7 mmol-blue, 1.4 mmol-green, 2.1 mmol-light pink, 2.8 mmol-pink, 3.5 mmol-brown) and (B) Co3 (1 mM) under saturation of N2 (black), CO2 (red) and CO2 with TFE (0.7 mmol-blue, 1.4 mmol-pink). Return sweeps are not shown for clarity (0.1 M [(n-C4H9)4N]PF6/DMF as the supporting electrolyte, scan rate: 0.1 V s−1, Fc (1 mM) was used as an internal standard). | ||
Solution state CV experiments performed on Co3 in the presence of CO2 resulted in a slight anodic shift in the IIR redox process at E1/2 = −1.57 V vs. Fc0/Fc+, corresponding to the Co(II/I) process and indicating a weak association of CO2 to the metal centre. The IIIR redox process at E1/2 = −2.25 V vs. Fc0/Fc+ increased under CO2 saturation, and was further enhanced to a maximum upon the addition of successive aliquots of TFE, indicating that a proton source is required to achieve optimal activity. The absence of this enhancement under N2 with the presence of TFE indicates that the process is not solely related to proton reduction, but is more likely to arise from the reduction of CO2. Like Co2, Co3 also follows pseudo first order kinetics. A plot of icat against the square root of [CO2] showed a linear relationship, suggesting first order kinetics in [CO2] (Fig. S15A†). The plot of icat vs. [H+] shows an initial second order dependence on TFE, prior to reaching saturation (Fig. S15B†). In Co3, saturation of the proton source occurs at lower concentrations than its Co2 analogue, implying that a lower concentration of a proton source facilitates the reduction of CO2. Plotting icat vs. [analyte] reveals a linear relationship, indicating a first order dependence of peak current on [analyte] (Fig. S15C†). CO2 (0.23 M in DMF) is well in excess of the analyte. Solution state CV experiments were performed on Co3 (1 mM) with varying concentrations of TFE titrated into the solution. The titration of TFE into the reaction mixture under an atmosphere of CO2 resulted in an increase in the current density of the IIIR process to a maximum at Epc = −2.25 V vs. Fc0/Fc+ after only two aliquots. Addition of excess TFE after this point resulted in no significant change. The addition of TFE into the reaction mixture yielded a peak ratio 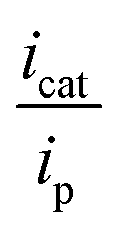 of 2.98, corresponding to a TOF k[Q] of 1.72 s−1 at 0.14 M TFE, which is a smaller turnover than the Co2 analogue. A negligible association with CO2 was observed.
of 2.98, corresponding to a TOF k[Q] of 1.72 s−1 at 0.14 M TFE, which is a smaller turnover than the Co2 analogue. A negligible association with CO2 was observed.
CPE experiments were performed on solutions of both Co2 (3.60 mM) with TFE (0.63 M) and Co3 (1.11 mM) with TFE (0.14 M) in 0.1 M [(n-C4H9)4N]PF6/DMF/MeCN (8:2) under CO2. The generation of CO was observed in the gas phase from both complexes, as measured from the first 3.73 turnovers over 3 h in Co2 and measured from the first 4.15 turnovers over the space of 4 h in Co3 (Table 4). A linear relationship in the production of gas was maintained throughout the duration of the CPE experiments for both complexes, indicating that the true lifetime of the catalyst may be slightly longer, although there is evidence for both H2 and CO production plateauing (Fig. S16†). The analyte sustained current densities of 2.57 mA cm−2 for Co2 and 0.4 mA cm−2 for Co3, with CPE experiments revealing a sustained current density over the period of the transformation. This suggests that the electrochemical transformation of CO2 is more likely to be stoichiometric rather than catalytic.
:2) with TFE as the supporting electrolyte under CO2, Epc = −1.85 V vs. Ag/Ag+)
| Experiment | FCO (%) | FH2 (%) | FHCOOH (%) |
|---|---|---|---|
| Solvent | 0 | 0 | 0 |
| Solvent + TFE (0.7 M) | 0 | 12 | 0 |
| Co2 +TFE (0.63 M) | 20 | 13 | 7 |
| Co3 + TFE (0.14 M) | 11 | 14 | 0 |
The solution of the electroanalyte was analysed via 1H NMR experiments for solution phase products post electrolysis. After aqueous work up into D2O, 1H NMR revealed a peak at δ = 8.00 ppm, corresponding to the generation of formic acid (Fig. S17†). An NMR time-course experiment was performed concurrently with CPE experiments on Co2, which demonstrated that the peak increased with the CPE. Broadening of the peak may be indicative of paramagnetic metal ions leaching into the solution. In the absence of CO2, the peak corresponding to the formation of formic acid did not appear. No formic acid was observed during experiments where CO2 was present but Co2 was absent, indicating that the transformation of CO2 into formic acid is due to both the presence of CO2 and Co2.
The prime difference between the analytes of Co2 and Co3 lies in the generation of both gaseous phase and solution phase products from the reduction of CO2. Although CO was formed from both complexes, it was formed in lower efficiencies from the more electron donating Co3. Following the CPE experiments on Co3, the electroanalyte was analysed via 1H NMR experiments after work up into D2O. No evidence for formic acid was found, which may imply that selectivity for the generation of CO over formic acid could be achieved by varying the electronics of the salen backbone. Future studies will require the analysis of more electron withdrawing species and their effect on the reduction of CO2.
Conclusions
This work sought to explore the use of Co(III) salen metal complexes and POPs as agents for the electrochemical reduction of CO2. A series of discrete Co(III) bis-bromo terminated salen metal complexes were synthesised and characterised using NMR, UV-Vis-NIR, IR, CV and various SEC techniques to understand their physical and electrochemical properties. The two salen metal complexes, Co2 and Co3, successfully reduced CO2 to CO in 20% and 11% Faradaic efficiencies, respectively, suggesting scope to use these complexes as multi-electron reducing agents. Formic acid was also observed as a product of CO2 reduction in Co2 but not in Co3, implying that a more electron poor salen metal complex improves the conversion of CO2 into commodity chemicals. Further functionalisation of the bridging diamine may allow for the selectivity of products to be tuned. This work has shown the potential for expanding the studies of Co(III) salen complexes as reducing agents for two-electron transfer reactions; however, additional work is required to improve the stability of the electrogenerated imine radical that forms.The discrete salen complexes were successfully incorporated into POP materials through the Sonogashira–Hagihara reaction with TPA to generate POPCo1, POPCo2 and POPCo3, and were characterised by UV-Vis-NIR, IR, elemental analysis, FE-SEM, solid state NMR and gas sorption. The polymers were found to be porous, with BET surface areas up to 690 m2 g−1 (comparable with those reported in the literature).21,27,49 The successful reduction of CO2 into CO and formic acid by the discrete complexes led to the examination of the electrochemical properties of the POPs through solid state CV, CPE and SEC experiments. It was possible to use mild EPD conditions to adhere the POP materials to the conductive glassy carbon surface as a thin film, which could be characterised through FE-SEM. Solid state CV experiments revealed a number of irreversible processes for these polymers, suggesting that the instability of the POP material for electrochemical purposes arises from the imine core. When exposed to CO2, the redox behaviour was turned off, suggesting that there is an irreversible chemical transformation of CO2 in the polymer, which could be confirmed from Vis-NIR SEC. There is further scope to address this shortcoming by reinforcing the stability of the imine.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Australian Research Council (ARC) and the Science and Industry Endowment Fund (SIEF) as part of the ‘Solving the Energy Waste Roadblock’ venture. We gratefully acknowledge Dr Danielle Kennedy at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) for her support in the provision of an OCE PhD top-up scholarship for M. B. S. We are also grateful to Dr Charles Machan from The University of Virginia and Dr Mark Reineke from The University of California, San Diego for their assistance with IR SEC and bulk electrolysis measurements, Dr Carol Hua for guidance on UV-Vis-NIR SEC and Dr Dorothy Yu for the analysis of the polymer samples via ICP-OES. S.M.C was supported by a grant from the office of Basic Energy Sciences, Department of Energy, under Award DE-FG02-08ER46519. C. P. K. gratefully acknowledges NSF (CHE-1145893) for support.References
- R. Carapellucci, L. Giordano and M. Vaccarelli, Analysis of CO2 Post-combustion Capture in Coal-fired Power Plants Integrated with Renewable Energies, J. Photosci., 2015, 82, 350–357, DOI:10.1016/j.egypro.2015.11.801.
- R. Barker, Y. Hua and A. Neville, Internal corrosion of carbon steel pipelines for dense-phase CO2 transport in carbon capture and storage (CCS) – a review, Int. Mater. Rev., 2016, 62(1), 1–31, DOI:10.1080/09506608.2016.1176306.
- D. M. D'Alessandro, B. Smit and J. R. Long, Carbon Dioxide Capture: Prospects for New Materials, Angew. Chem., Int. Ed., 2010, 49(35), 6058–6082, DOI:10.1002/anie.201000431.
- A. Das and D. M. D'Alessandro, Tuning the functional sites in metal–organic frameworks to modulate CO2 heats of adsorption, CrystEngComm, 2015, 17(4), 706–718, 10.1039/C4CE01341G.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Selective gas adsorption and separation in metal–organic frameworks, Chem. Soc. Rev., 2009, 38(5), 1477–1504, 10.1039/B802426J.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Carbon dioxide capture-related gas adsorption and separation in metal–organic frameworks, Coord. Chem. Rev., 2011, 255(15–16), 1791–1823, DOI:10.1016/j.ccr.2011.02.012.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Progress in adsorption-based CO2 capture by metal-organic frameworks, Chem. Soc. Rev., 2012, 41(6), 2308–2322, 10.1039/C1CS15221A.
- E. S. Rubin, J. E. Davison and H. J. Herzog, The cost of CO2 capture and storage, Int. J. Greenhouse Gas Control, 2015, 40, 378–400, DOI:10.1016/j.ijggc.2015.05.018.
- D. M. Thomas, J. Mechery and S. V. Paulose, Carbon dioxide capture strategies from flue gas using microalgae: a review, Environ. Sci. Pollut. Res., 2016, 23(17), 1–15, DOI:10.1007/s11356-016-7158-3.
- R. Wennersten, Q. Sun and H. Li, The future potential for Carbon Capture and Storage in climate change mitigation – an overview from perspectives of technology, economy and risk, J. Cleaner Prod., 2015, 103, 724–736, DOI:10.1016/j.jclepro.2014.09.023.
- R. Stanger and T. Wall, Sulphur impacts during pulverised coal combustion in oxy-fuel technology for carbon capture and storage, Prog. Energy Combust. Sci., 2011, 37(1), 69–88, DOI:10.1016/j.pecs.2010.04.001.
- J. Wang, Analytical Electrochemistry, John Wiley & Sons, New Jersey, 2006, p. 250 Search PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels, Chem. Soc. Rev., 2009, 38(1), 89–99, 10.1039/B804323J.
- C. Amatore and J. M. Saveant, Mechanism and kinetic characteristics of the electrochemical reduction of carbon dioxide in media of low proton availability, J. Am. Chem. Soc., 1981, 103(17), 5021–5023, DOI:10.1021/ja00407a008.
- V. A. Davankov and M. P. Tsyurupa, Structure and properties of hypercrosslinked polystyrene—the first representative of a new class of polymer networks, React. Polym., 1990, 13(1), 27–42, DOI:10.1016/0923-1137(90)90038-6.
- O. K. Farha, A. M. Spokoyny, B. G. Hauser, Y.-S. Bae, S. E. Brown, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Synthesis, Properties, and Gas Separation Studies of a Robust Diimide-Based Microporous Organic Polymer, Chem. Mater., 2009, 21(14), 3033–3035, DOI:10.1021/cm901280w.
- H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Unprecedented high-temperature CO2 selectivity in N2-phobic nanoporous covalent organic polymers, Nat. Commun., 2013, 4, 1357–1366, DOI:10.1038/ncomms2359.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Porous Polymer Networks: Synthesis, Porosity, and Applications in Gas Storage/Separation, Chem. Mater., 2010, 22(21), 5964–5972, DOI:10.1021/cm1021068.
- Y. Zhang and S. N. Riduan, Functional porous organic polymers for heterogeneous catalysis, Chem. Soc. Rev., 2012, 41(6), 2083–2094, 10.1039/C1CS15227K.
- J. Chun, S. Kang, N. Kang, S. M. Lee, H. J. Kim and S. U. Son, Microporous organic networks bearing metal-salen species for mild CO2 fixation to cyclic carbonates, J. Mater. Chem. A, 2013, 1(18), 5517–5523, 10.1039/C3TA10477J.
- H. Li, B. Xu, X. Liu, Sigen A, C. He, H. Xia and Y. Mu, A metallosalen-based microporous organic polymer as a heterogeneous carbon-carbon coupling catalyst, J. Mater. Chem. A, 2013, 1(45), 14108–14114, 10.1039/C3TA13128A.
- P. Pfeiffer, E. Breith, E. Lübbe and T. Tsumaki, Tricyclische orthokondensierte Nebenvalenzringe, Liebigs Ann. Chem., 1933, 503(1), 84–130, DOI:10.1002/jlac.19335030106.
- D. J. Darensbourg. Salen Metal Complexes as Catalysts for the Synthesis of Polycarbonates from Cyclic Ethers and Carbon Dioxide, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, W. G. Coates, R. Reichardt, E. Dinjus and A. T. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012; pp. 1–27 Search PubMed.
- H. Khoshro, H. R. Zare, A. Gorji, M. Namazian, A. A. Jafari and R. Vafazadeh, The effect of electronic structure on electrocatalytic behaviors of cobalt Schiff base complexes: Electrosynthesis of 2-phenylacetic acid using carbon dioxide, J. Electroanal. Chem., 2014, 732, 117–121, DOI:10.1016/j.jelechem.2014.07.039.
- T. Kurahashi and H. Fujii, One-Electron Oxidation of Electronically Diverse Manganese(III) and Nickel(II) Salen Complexes: Transition from Localized to Delocalized Mixed-Valence Ligand Radicals, J. Am. Chem. Soc., 2011, 133(21), 8307–8316, DOI:10.1021/ja2016813.
- T. P. Yoon and E. N. Jacobsen, Privileged Chiral Catalysts, Science, 2003, 299(5613), 1691–1693, DOI:10.1126/science.1083622.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Capture and conversion of CO2 at ambient conditions by a conjugated microporous polymer, Nat. Commun., 2013, 4, 2960, DOI:10.1038/ncomms2960.
- M. B. Solomon, T. L. Church and D. M. D'Alessandro, Perspectives on metal–organic frameworks with intrinsic electrocatalytic activity, CrystEngComm, 2017, 19(29), 4049–4065, 10.1039/c7ce00215g.
- M. C. So, S. Jin, H.-J. Son, G. P. Wiederrecht, O. K. Farha and J. T. Hupp, Layer-by-Layer Fabrication of Oriented Porous Thin Films Based on Porphyrin-Containing Metal–Organic Frameworks, J. Am. Chem. Soc., 2013, 135(42), 15698–15701, DOI:10.1021/ja4078705.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C.-W. Kung, M. J. Katz, M. So, B. Klahr, D. Jin, Y.-W. Chung, T. W. Odom, O. K. Farha and J. T. Hupp, Directed Growth of Electroactive Metal–Organic Framework Thin Films Using Electrophoretic Deposition, Adv. Mater., 2014, 26(36), 6295–6300, DOI:10.1002/adma.201401940.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Functionalization of robust Zr(IV)-based metal-organic framework films via a postsynthetic ligand exchange, Chem. Commun., 2015, 51(1), 66–69, 10.1039/C4CC08218D.
- C. R. Wade, M. Li and M. Dincă, Facile Deposition of Multicolored Electrochromic Metal–Organic Framework Thin Films, Angew. Chem., Int. Ed., 2013, 52(50), 13377–13381, DOI:10.1002/anie.201306162.
- C.-W. Kung, J. E. Mondloch, T. C. Wang, W. Bury, W. Hoffeditz, B. M. Klahr, R. C. Klet, M. J. Pellin, O. K. Farha and J. T. Hupp, Metal–Organic Framework Thin Films as Platforms for Atomic Layer Deposition of Cobalt Ions To Enable Electrocatalytic Water Oxidation, ACS Appl. Mater. Interfaces, 2015, 7(51), 28223–28230, DOI:10.1021/acsami.5b06901.
- L. Ye, J. Liu, Y. Gao, C. Gong, M. Addicoat, T. Heine, C. Woll and L. Sun, Highly oriented MOF thin film-based electrocatalytic device for the reduction of CO2 to CO exhibiting high faradic efficiency, J. Mater. Chem. A, 2016, 4(40), 15320–15326, 10.1039/C6TA04801C.
- R. Wilson, H. Akhavan-Tafti, R. DeSilva and A. P. Schaap, Comparison Between Acridan Ester, Luminol, and Ruthenium Chelate Electrochemiluminescence, Electroanal., 2001, 13(13), 1083–1092, DOI:10.1002/1521-4109(200109)13:13<1083::AID-ELAN1083>3.0.CO;2-D.
- G. Wang and Y. Lin, Novel counter electrodes based on NiP-plated glass and Ti plate substrate for dye-sensitized solar cells, J. Mater. Sci., 2007, 42(13), 5281–5285, DOI:10.1007/s10853-006-0395-y.
- H. Zhu, H. Liu, I. Zhitomirsky and S. Zhu, Preparation of metal–organic framework films by electrophoretic deposition method, Mater. Lett., 2015, 142, 19–22, DOI:10.1016/j.matlet.2014.11.113.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, Fe-Porphyrin-Based Metal–Organic Framework Films as High-Surface Concentration, Heterogeneous Catalysts for Electrochemical Reduction of CO2, ACS Catal., 2015, 5(11), 6302–6309, DOI:10.1021/acscatal.5b01767.
- Q.-G. Fu, X.-Y. Nan, X. Chen, W.-L. Wang, H.-J. Li, Y.-Y. Li and L.-T. Jia, Electrophoretic deposition of SiC nanowires onto carbon/carbon composites to improve the interface bonding of Ti–Ni–Si joint, Mater. Des., 2015, 80, 137–143, DOI:10.1016/j.matdes.2015.04.042.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities, J. Org. Chem., 1997, 62(21), 7512–7515, DOI:10.1021/jo971176v.
- I. S. Zavarine and C. P. Kubiak, A versatile variable temperature thin layer reflectance spectroelectrochemical cell, J. Electroanal. Chem., 2001, 495(2), 106–109, DOI:10.1016/s0022-0728(00)00394-6.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Rapid determination of the optical and redox properties of a metal–organic framework via in situ solid state spectroelectrochemistry, Chem. Commun., 2012, 48(33), 3945–3947, 10.1039/C2CC30568B.
- M. L. Clark, B. Rudshteyn, A. Ge, S. A. Chabolla, C. W. Machan, B. T. Psciuk, J. Song, G. Canzi, T. Lian, V. S. Batista and C. P. Kubiak, Orientation of Cyano-Substituted Bipyridine Re(I) fac-Tricarbonyl Electrocatalysts Bound to Conducting Au Surfaces, J. Phys. Chem. C, 2016, 120(3), 1657–1665, DOI:10.1021/acs.jpcc.5b10912.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60(2), 309–319, DOI:10.1021/ja01269a023.
- S. W. Kim, S. C. Shim, D. Y. Kim and C. Y. Kim, Synthesis and properties of novel triphenylamine polymers containing ethynyl and aromatic moieties, Synth. Met., 2001, 122(2), 363–368, DOI:10.1016/S0379-6779(00)00398-2.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D'Alessandro, Redox tunable viologen-based porous organic polymers, J. Mater. Chem. C, 2016, 4(13), 2535–2544, 10.1039/C6TC00132G.
- D. Dollimore, P. Spooner and A. Turner, The bet method of analysis of gas adsorption data and its relevance to the calculation of surface areas, Surf. Technol., 1976, 4(2), 121–160, DOI:10.1016/0376-4583(76)90024-8.
- A. R. Silva, T. Mourão and J. Rocha, Oxidation of cyclohexane by transition–metal complexes with biomimetic ligands, Catal. Today, 2013, 203, 81–86, DOI:10.1016/j.cattod.2012.07.043.
- M. H. Alkordi, L. J. Weselinski, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, R. G. AbdulHalim, J.-M. Basset and M. Eddaoudi, CO2 conversion: the potential of porous-organic polymers (POPs) for catalytic CO2-epoxide insertion, J. Mater. Chem. A, 2016, 4(19), 7453–7460, 10.1039/C5TA09321J.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04385j |
| This journal is © The Royal Society of Chemistry 2018 |