
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of a porous β-cyclodextrin-polymer-coated solid-phase microextraction fiber for the simultaneous determination of five contaminants in water using gas chromatography-mass spectrometry†
Jiongxiu Pan,
Shuming Li,
Fuquan Dang ,
Zhiqi Zhang and
Jing Zhang*
,
Zhiqi Zhang and
Jing Zhang*
Key Laboratory of Analytical Chemistry for Life Science of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: zhangjing8902@snnu.edu.cn
First published on 19th June 2018
Abstract
A novel solid-phase microextraction fiber coated with a porous β-cyclodextrin polymer was developed. The porous β-cyclodextrin polymer cross-linked using tetrafluoroterephthalonitrile, possessed well-distributed pores and the largest surface area among current β-cyclodextrin polymers. Scanning electron microscopy revealed that the coating had a continuous wrinkled and folded structure, which guarantees a sufficient loading capacity for contaminants. The properties of the developed fiber were evaluated using headspace solid-phase microextraction of five contaminants as model analytes coupled with gas chromatography-mass spectrometry. Owing to the advantages of a large surface area and three-dimensional cavities, the novel fiber exhibited excellent operational stability and extraction ability. After optimisation of the extraction conditions, including extraction temperature, extraction time, salt effect, and desorption time, validation of the method with water samples achieved good linearity over a wide range (0.01–120 μg L−1) and low detection limits (0.003–1.600 μg L−1). The single-fiber and fiber-to-fiber repeatabilities were 1.7–11.0% and 1.9–11.0%, respectively. The method was applied to the simultaneous analysis of five analytes with satisfactory recoveries (76.6–106.0% for pond water and 89.0–105.9% for rainwater).
1. Introduction
Solid-phase microextraction (SPME) is a simple, solvent-free, time-saving, and powerful pretreatment technique for various complex matrices at the trace level. Coating materials play a significant role in SPME;1 therefore, various coatings with excellent performance have been developed, including crown ester,2 fullerene,3 calixarene,4 and β-cyclodextrin (β-CD).5 Coated microextraction fibers can extract different analytes from various complex matrices, such as gaseous or aqueous, subsequently thermal desorption in the inlet of a gas chromatograph or solvent desorption in liquid chromatography equipment.With the development of SPME, a series of fibers with high selectivity and thermal stability have been prepared. For example, graphene-coated fiber prepared via layer-by-layer chemical bonding and a hydrofluoric-acid-etched bare stainless steel wire fiber have high affinities for polycyclic aromatic hydrocarbons owing to π–π and cation–π interactions, respectively.6,7 A solvent-evaporation-induced self-assembly approach combined with dip-coating technology has been used to fabricate an ordered mesoporous silica film coating supported on anodised titanium wires, which overcame the fragility of commonly used fused silica fibers and showed high thermal stability.8 Further, in combination with molecularly imprinted polymer (MIP) technology, SPME fibers have been prepared with high selectivity for target molecules.9
β-CD, a macrocycle composed of seven glucose units, forms host–guest complexes with thousands of organic compounds. Owing to its structural characteristics and nontoxic properties, β-CD has been explored as a coating material for SPME.10–14 Li et al. prepared polydimethylsiloxane (PDMS)-β-CD SPME membranes that exhibited high thermal stability and excellent selectivity for polar compounds.15 Zeng et al. used β-CD fibers and β-CD capillaries to extract drugs from human urine samples.16
Further, coatings based on β-CD composite materials or β-CD bonded to other substrates have been investigated. Such fibers have excellent performance. Song et al. developed a β-CD-modified carbon-nanotube-reinforced hollow fiber to determine trace 1-naphthaleneacetic acid and 2-naphthoxyacetic acid in tomatoes for food quality control.17 Jia et al. prepared a poly(butyl methacrylate-co-ethyleneglyceldimethacrylate) monolithic column modified with allylamine-β-CD and nano-cuprous oxide. The monolithic column exhibited superior performance for the preconcentration of polychlorinated biphenyls.18 Nojavan et al. prepared a water-insoluble β-CD–epichlorohydrin polymer, which exhibited satisfactory results for the micro-solid phase extraction of benzene, toluene, ethylbenzene, and xylenes from water samples.19 Li et al. prepared a β-CD-bonded silica particle coating to detect phenol compounds in environmental water samples.20 Liu et al. bonded β-CD and β-CD-co-poly(ethylenepropylene glycol) to the surface of nanostructured titanium wires and established analytical methods for ephedrine and methylephedrine determination in urine samples.21
β-CD and its commercial derivatives have been widely applied as coating materials in SPME. Zeng et al. used a permethylated-β-CD/hydroxyl-terminated silicone oil fiber to successfully detect polybrominated diphenyl ethers in soil samples.22 Feng et al. prepared a β-CD-derivative coating for the analysis of nonsteroid anti-inflammatory drugs.23 Xu et al. prepared a series of SPME coatings based on β-CD derivatives and MIP technology for the determination of polychlorophenols in water samples.24
Cross-linked β-CD polymers are likewise of interest for removing micropollutants from water by means of adsorption. Wang et al. synthesised β-CD-based electrospun nanofiber membranes for highly efficient adsorption and separation of methylene blue from methylene blue/methyl orange mixtures in solution.25 Meo et al. used a click chemistry approach based on a copper-catalyzed 1,3-dipolar cycloaddition reaction to prepare CD hyper-reticulated co-polymers, which exhibit highly efficient adsorption of nitroarenes and commercial dyes.26 Suri et al. developed a high-efficiency silica-coated β-CD polymeric adsorbent for the removal of 17-estradiol, perfluorooctanoic acid, and bisphenol A from water.27 Bikiaris et al. used β-CD and chitosan as polymeric matrix substrates to prepare two types of novel MIPs for the adsorption of toxic and carcinogenic dyes.28 Zhao et al. reported a green bifunctional adsorbent based on ethylenediaminetetraacetic-acid-cross-linked β-CD, which achieved simultaneous high-efficiency adsorption of metals and cationic dyes.29 These previous studies demonstrate the active development of β-CD as a coating and adsorption material. However, compared with broad-spectrum activated carbons, β-CD polymers still have lower surface areas and inferior removal performance.30,31
Among a series of β-CD-based polymers reported by Alsbaiee et al., a porous β-CD polymer (P-CDP) possessed the largest surface area and well-distributed pores. Compared with other adsorbent materials and nonporous β-CD, this polymer can rapidly adsorb a variety of organic micropollutants. In addition, P-CDP can be regenerated several times with no loss in performance.32,33 Compared with coconut-shell activated carbon (SBET = 1085 m2 g−1), P-CDP achieves high-efficiency adsorption of organic micropollutants.34 These findings demonstrate the potential of P-CDP for quick adsorption of water pollutants with high removal efficiencies. We predict that the performance of P-CDP as a new type of SPME coating will be better than those of existing β-CD and β-CD polymer coatings. Five micropollutants were used as model analytes to evaluate the extraction performance of the P-CDP coating.
2. Experimental
2.1. Materials
Geosmin (GSM), tetrafluoroterephthalonitrile, and hydroxyl-terminated PDMS (OH-PDMS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,3-Dichlorobenzene, indole (standard for GC, >99.5%), β-CD, tetraethoxysilane (TEOS) (>99%, GC), trifluoroacetic acid (TFA), and 3-glycidyloxypropyltrimethoxysilane (KH-560) were purchased from Aladdin Chemistry (Shanghai, China). Analytical-grade phenol, benzene, and organic solvents such as methanol and acetone were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). A Millipore water purification system provided ultrapure water for the experiments (18 MΩ cm; Milli-Q) (Millipore, Bedford, MA, USA).2.2. Apparatus and chromatography
An Agilent 7890B-5977B gas chromatography-mass spectrometry (GC-MS) system (Agilent Technologies, USA) with a split/splitless injector was used for sample analysis. An HP-5MS capillary column was used for the separations. MS (5977B): electron ionization mode at 70 eV; quadrupole, 150 °C; ion source temperature, 230 °C; carrier gas, helium (99.999% purity); gas flow rate, 2 mL min−1. GC (7900B): injector temperature, 270 °C; transfer line temperature, 300 °C. The initial oven temperature of 60 °C (maintained for 2 min) was increased at a rate of 6 °C min−1 to 150 °C and then increased to 300 °C at a rate of 20 °C min−1 (maintained for 12 min); running time, 36.5 min.Fourier transform infrared (FT-IR) spectra were recorded using a Tensor 27 instrument (Bruker Daltonics, Germany). Surface area measurements were performed using an accelerated surface area and porosimetry analyser (ASAP 2020, Micromeritics, USA). Scanning electron microscopy characterisation was conducted using an environmental scanning electron microscope (Quanta200, FEI, USA). Thermal analysis was performed using a differential scanning calorimeter coupled to a thermogravimetric analyser (Q1000DSC + LNCS + FACSQ600SDT, TA, USA). Commercial SPME assemblies divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS SPME fibers, 50/30 μm) were purchased from Supelco (Bellefonte, PA, USA) and used for the comparison study. SPME devices equipped with P-CDP were laboratory-made.
2.3. P-CDP synthesis
P-CDP was prepared based on the method described by Alsbaiee et al.33 Briefly, 0.205 g (0.181 mmol) of β-CD, 0.100 g (0.515 mmol) of tetrafluoroterephthalonitrile, and 0.320 g (2.32 mmol) of K2CO3 were added to 8 mL of anhydrous tetrahydrofuran (THF)/dimethylformamide (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and maintained for two days under a nitrogen atmosphere at 85 °C and 500 rpm. The obtained orange product was cooled to room temperature (25 °C) and the resulting solid was filtered. Remaining K2CO3 was wiped off using 1 mol L−1 HCl until no bubbles were generated. The resulting pale yellow product was isolated and then activated by soaking in H2O (2 × 10 mL) for 15 min, THF (2 × 10 mL) for 30 min, and CH2Cl2 (1 × 15 mL) for 15 min. Finally, the product was dried in a liquid nitrogen bath under high vacuum at 77 K for 10 min and subsequently at room temperature for 2–3 days.
:1, v/v) and maintained for two days under a nitrogen atmosphere at 85 °C and 500 rpm. The obtained orange product was cooled to room temperature (25 °C) and the resulting solid was filtered. Remaining K2CO3 was wiped off using 1 mol L−1 HCl until no bubbles were generated. The resulting pale yellow product was isolated and then activated by soaking in H2O (2 × 10 mL) for 15 min, THF (2 × 10 mL) for 30 min, and CH2Cl2 (1 × 15 mL) for 15 min. Finally, the product was dried in a liquid nitrogen bath under high vacuum at 77 K for 10 min and subsequently at room temperature for 2–3 days.
2.4. Preparation of P-CDP-coated stainless steel fibers
Stainless steel fibers were immersed in acetone for 30 min and subsequently treated with an alkaline oil-removing solution at 80 °C for 20 min. The alkaline oil-removing solution consists of 50 g L−1 sodium hydroxide, 20 g L−1 sodium carbonate, and 20 g L−1 sodium phosphate. Subsequently, fiber oxidation was performed in an oxidation solution at 60 °C for 30 min. The oxidation solution consists of 10 g L−1 sodium carbonate and 50 g L−1 sodium nitrite. Followed by drying in an oven at 150 °C for 120 min. Finally, the fibers were washed with ethanol three times and dried under a nitrogen flow.A schematic for the preparation of P-CDP-coated stainless steel SPME fibers is shown in Fig. 1. A sol–gel solution was prepared using a method similar to that previously described in literature.22 Briefly, 10 mg of P-CDP was dissolved in 300 μL of methylene chloride and then 70 mg of OH-PDMS was added. Subsequently, 100 μL of TEOS, 100 μL of KH-560, and 60 μL of TFA (95%) aqueous solution were added to the solution. The solution was ultrasonically mixed in a centrifuge tube and then centrifuged at 10000 rpm for 10 min. The solution collected at the top was used for fiber coating. The treated stainless steel fibers were inserted into the sol–gel solution for 50 min to coat the surfaces of the fibers. The coating process was repeated several times until a satisfactory thickness was obtained. Subsequently, the fibers were placed in a drying oven at room temperature for 24 h, and finally aged under a helium atmosphere at 60–280 °C for 1.5 h in the GC injector. For comparison, β-CD fibers were prepared in a similar way.
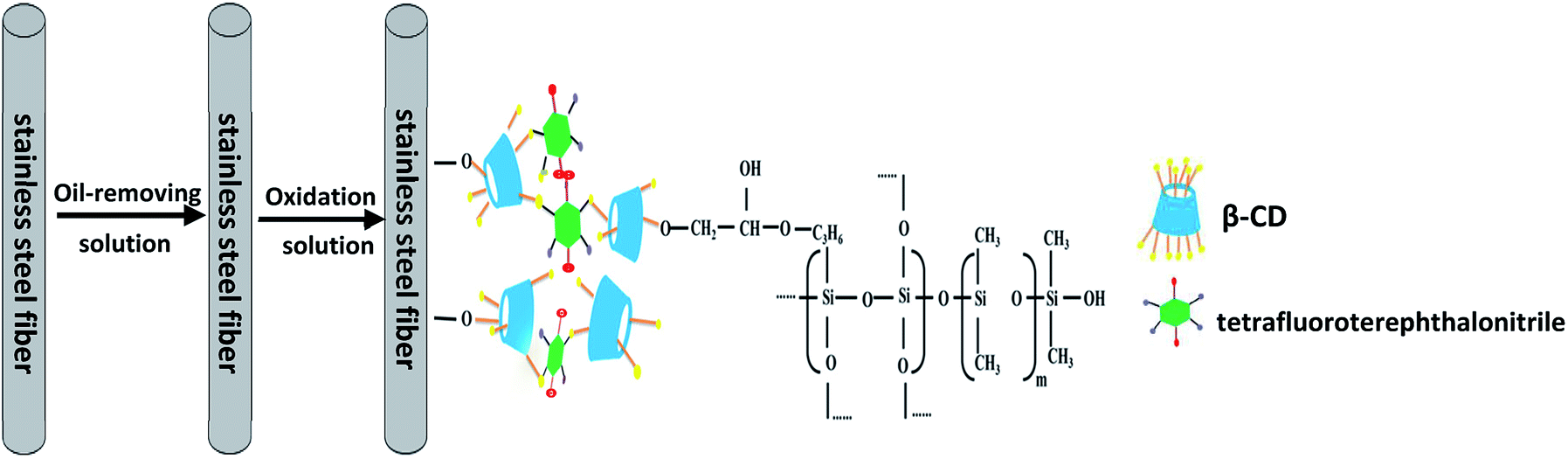 | ||
| Fig. 1 Schematic of the preparation of P-CDP-coated stainless steel SPME fibers. | ||
2.5. HS-SPME process
Standard volatile organic compounds (VOCs) and odours (1000 mg L−1) were prepared by dissolving appropriate concentrations of analytes in methanol as stock solutions. The stock solutions were diluted with methanol in a step-by-step manner to prepare the working solutions, added above solutions to 5 mL ultrapure water giving the final concentration ranging from 0.01–120 μg L−1. For SPME extraction experiments, 10 mL amber vials containing 5 mL of the obtained solutions. The solutions were stirred magnetically at 900 rpm and a water bath was used to control the extraction temperature. For the HS-SPME process, the fiber was inserted into the headspace of the sample analysis for some time. After extraction, the fiber was moved to the GC inlet for thermal desorption.2.6. Sample preparation
As real water samples, pond water was obtained from a local river (Xi'an, China) and rainwater was collected from (Xi'an, China). The samples were analysed without any pretreatment process.3. Results and discussion
3.1. Characterization of P-CDP and coating
P-CDP was obtained by nucleophilic aromatic substitution between β-CD and tetrafluoroterephthalonitrile. The obtained polymer was characterised using FT-IR, N2 adsorption–desorption, and environmental scanning electron microscopy (ESEM). The coating was characterised using ESEM and thermogravimetry.The FT-IR spectra of β-CD, P-CDP, and tetrafluoroterephthalonitrile are illustrated in Fig. S1.† The characteristic spectral features of β-CD, i.e. C–O stretches (1030 cm−1), aliphatic C–H stretches (2927 cm−1), and O–H stretches (3333 cm−1), were also observed in the spectrum of P-CDP. Furthermore, C–C aromatic stretches (1670 cm−1, 1480 cm−1), C–F stretches (1290 cm−1), and nitrile stretches (2235 cm−1), which are characteristic spectral features of tetrafluoroterephthalonitrile, were observed in the spectrum of P-CDP. Compared with β-CD and tetrafluoroterephthalonitrile, the intensities of the O–H and C–F of P-CDP were decreased. This observation is consistent with our prediction that the –F groups of tetrafluoroterephthalonitrile are substituted by the –OH of β-CD when a reaction occurs between β-CD and the cross-linker.
Nitrogen adsorption–desorption is the most commonly used technique to characterise the surface area and pore size distribution of solid samples.35,36 Therefore, we used this technique to characterise the properties of P-CDP. The results are shown in Fig. S2.† The N2 adsorption and desorption isotherms of P-CDP indicated that P-CDP has an SBET value of 208 m2 g−1. The pore diameter distribution revealed pore diameters in the range of 1.9–3.5 nm. The N2 adsorption and desorption isotherms and pore diameter distribution were similar to those reported in the literature.33 The above results demonstrated that P-CDP was successfully prepared.
An electron micrograph of the surface of P-CDP is shown in Fig. S3a.† As illustrated under ×1200 and ×5000 magnification, P-CDP has a porous structure, which could improve the kinetics of the SPME process and thus enhance the extraction performance. The ×5000 magnified image of the fiber surface showed a continuous wrinkled and folded structure (Fig. S3b†). Such a structure could enhance the effective surface area of the coating, which would be beneficial for the extraction process.
The thermal stability of the coating plays an important role in the SPME-GC process. The results of the thermal stability analysis of the P-CDP coating are shown in Fig. S4.† The weight loss near 100 °C was attributed to the loss of physically adsorbed water. Between 100 and 320 °C, the P-CDP coating exhibited no obvious weight loss, indicating that this coating could be used in the GC inlet at high temperatures.
3.2. Extraction procedure optimization
The extraction efficiency is influenced by various experimental parameters. In this work, the influences of extraction temperature, extraction time, ion strength, and desorption time were investigated, and the results are shown in Fig. S5.† All optimisation experiments were conducted using a solution consisting of 50 μg L−1 benzene, 200 ng L−1 1,3-dichlorobenzene, 2.5 ng L−1 indole, 120 μg L−1 phenol, and 100 ng L−1 GSM.3.3. Enrichment factor determination
The adsorption affinity between the P-CDP coating and analytes was evaluated using the enrichment factor (EF). The EF is defined as the ratio between the chromatographic peak areas of analytes after HS-SPME and those obtained by direct injection of different concentrations of standard solution. The results presented in Table S1† reveal that P-CDP exhibited good extraction efficiency for all the tested compounds. However, P-CDP showed different affinities for the analytes, ranging from 116.1 to 17.2, and this phenomenon is worth further investigation. β-CD is a natural molecule originating from starch that can form inclusion complexes with various compounds, especially aromatic compounds.31 The various binding forces that can contribute to β-CD inclusion include hydrophobic interactions, hydrogen bonding, electrostatic interactions, and other weak intermolecular interactions. In addition to these interactions, we consider that solubility of the analytes in water may have an influence on the EFs. 1,3-Dichlorobenzene (0.123 g L−1) and phenol (50 g L−1) have the lowest and highest solubilities, respectively. The EFs of all analytes are consistent with the solubility trend, which supports the above consideration.3.4. Comparison extraction efficiencies of various fibers
The extraction efficiencies of β-CD fiber, commercial DVB/CAR/PDMS fiber, and P-CDP-coated fiber were evaluated (Fig. 2). The P-CDP coating provided an enrichment of approximately two times compared with the β-CD fiber and three times compared with the commercial fiber; for example, for the P-CDP-coated fiber the enrichment amount was 2.8 for indole compared with the β-CD fiber and 3.5 for benzene compared with the commercial fiber. This phenomenon indicated that the porosity of P-CDP assisted the extraction process. From previous reports, it is known that pore structure and surface area play important roles in sorption processes. The adsorption ability of a polymer increases with increasing surface area, as significantly more adsorbates can be adsorbed onto the surface.37 If the three-dimensional cavities generated by the polymerisation reaction are similar to the interior cavity of CDs, the guest molecules can further insert themselves into the cavities, thereby increasing the adsorption amount. The driving forces for host–guest complexation include van der Waals forces, electrostatic interactions, hydrophobic interactions, and hydrogen bonds, which are considered the basis for separation by the CD polymer material.32 We concluded that the P-CDP-coated fiber exhibited good extraction ability for VOCs and odours owing to the synergistic effect of the β-CD rings, secondary cavities, and surface physical sorption.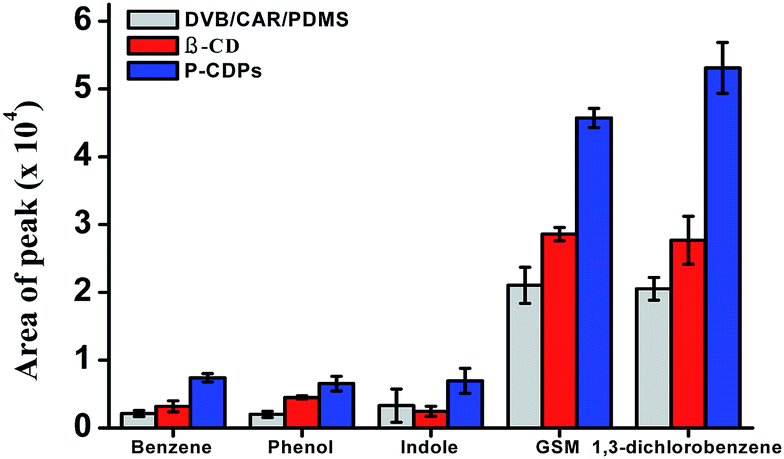 | ||
| Fig. 2 Comparison of the extraction efficiencies of P-CDP-coated fibers, β-CD fibers, and commercial DVB/CAR/PDMS fibers. All experiments were conducted under optimal conditions using 1 μg L−1 benzene, 5 μg L−1 phenol, 0.3 μg L−1 indole, 0.1 μg L−1 GSM, and 0.05 μg L−1 1,3-dichlorobenzene. | ||
3.5. Quantitative analysis
Calibration curves, limits of detection (LODs), and repeatability experiments were used to validate the proposed method. The results are presented in Table 1. The linear correlation coefficients for the analytes reached 0.9946. The LODs for both target analytes were determined according to a signal-to-noise-ratio (S/N) of three ranged from 0.003 to 1.6 μg L−1. The quality of the obtained coating allowed it to be applied in real samples at the trace level. The repeatability of the method was investigating by using each fiber five times to extract the standard solutions under the same conditions. Fiber-to-fiber reproducibility was determined by using three different fibers, fabricated using the same method to extract the standard solutions. The RSDs of both the single-fiber and fiber-to-fiber repeatabilities were less than 11%.| Analyte | Regression equation | Linear range (μg L−1) | R2 | LOD (μg L−1) | RSD single fiber (%, n = 5) | RSD fiber-to-fiber (%, n = 3) |
|---|---|---|---|---|---|---|
| GSM | y = 356x − 1988 | 0.01–0.2 | 0.9943 | 0.003 | 4.5 | 4.6 |
| Indole | y = 68536x − 15746 |
0.3–5.0 | 0.9946 | 0.100 | 3.9 | 5.1 |
| 1,3-Dichlorobenzene | y = 833x − 2934 | 0.01–0.3 | 0.9874 | 0.003 | 8.1 | 7.0 |
| Benzene | y = 2652x + 2668 | 1.0–100.0 | 0.9937 | 0.300 | 1.7 | 1.9 |
| Phenol | y = 916x − 1638 | 5.0–120.0 | 0.9779 | 1.600 | 11.0 | 11.0 |
3.6. Lifetime of P-CDP coating
A reusability experiment was conducted to determine the lifetime of the P-CDP-coated fiber (Fig. 3). A P-CDP-coated fiber was used for repeated HS-SPME of 100 ng L−1 GSM standard solutions. Comparisons of the obtained averages chromatographic peak areas for the batches 1–39, 40–69, 70–99, 100–130 experiments. The fiber could be used more than 130 times for repeated extractions without significant performance degradation. The experimental results demonstrated that the P-CDP coating has a long lifetime.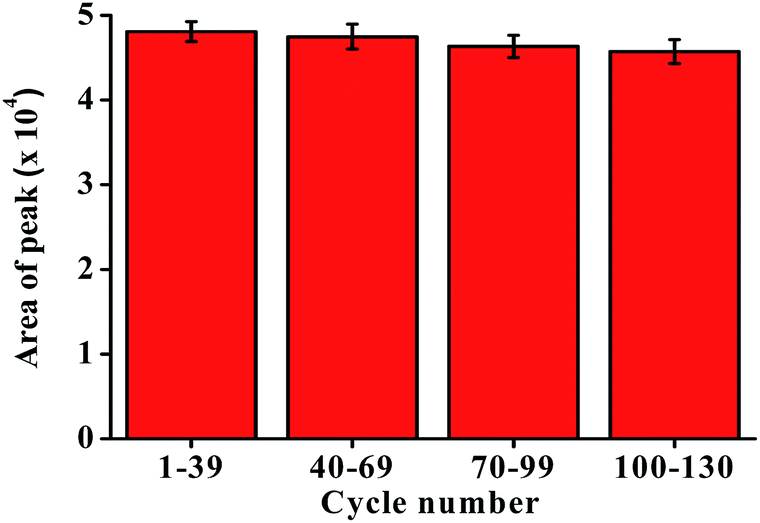 | ||
| Fig. 3 Replicate extraction of 100 ng L−1 GSM solutions using a P-CDP-coated fiber. | ||
3.7. Sample analysis
The developed P-CDP-coated fiber was used to analyse VOCs and odours in real water samples, as summarised in Table 2. Benzene and 1,3-dichlorobenzene were detected in pond water, whereas benzene and GSM were detected in rainwater. Solutions were spiked with different concentrations of the analytes to determine the recovery of the method. The recoveries ranged from 76.6% to 106.0% for all the analytes. Typical chromatograms for the analysis of real sample are illustrated in Fig. S6.†| Real samples | Analyte | Found (μg L−1) | Spiked concentration (μg L−1) | Detected concentration (μg L−1) | Recovery (%) | RSD (%, n = 3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Nd = not detected. | ||||||||||||||
| Pond water | Benzene | 1.8 | 12.0 | 50.0 | 70.0 | 12.2 | 49.6 | 71.1 | 86.6 | 95.6 | 99.0 | 6.9 | 10.9 | 5.6 |
| 1,3-Dichlorobenzene | 0.041 | 0.01 | 0.15 | 0.25 | 0.049 | 0.156 | 0.240 | 88.2 | 76.6 | 79.6 | 10.0 | 2.6 | 6.2 | |
| Indole | Nd | 0.3 | 2.5 | 4.0 | 0.28 | 2.16 | 3.95 | 93.3 | 86.4 | 98.7 | 8.7 | 10.3 | 4.5 | |
| Phenol | Nd | 30.0 | 70.0 | 110.0 | 25.8 | 65.4 | 111.8 | 86.0 | 93.4 | 101.6 | 7.8 | 2.6 | 4.6 | |
| GSM | Nd | 0.05 | 0.1 | 0.15 | 0.053 | 0.105 | 0.155 | 106.0 | 105.6 | 103.3 | 7.5 | 10.9 | 5.6 | |
| Rainwater | Benzene | 2.23 | 12.0 | 50.0 | 70.0 | 13.0 | 52.9 | 73.2 | 90.1 | 101.5 | 101.4 | 2.0 | 5.8 | 3.7 |
| 1,3-Dichlorobenzene | Nd | 0.01 | 0.15 | 0.25 | 0.009 | 0.152 | 0.235 | 99.8 | 101.5 | 94.1 | 5.0 | 2.6 | 7.3 | |
| Indole | Nd | 0.3 | 2.5 | 4.0 | 0.28 | 2.25 | 3.84 | 92.5 | 89.8 | 95.8 | 10.8 | 10.4 | 6.2 | |
| Phenol | Nd | 30.0 | 70.0 | 110.0 | 26.7 | 62.7 | 107.0 | 89.0 | 89.6 | 97.3 | 9.4 | 5.9 | 5.6 | |
| GSM | 0.012 | 0.05 | 0.1 | 0.15 | 0.058 | 0.11 | 0.17 | 92.6 | 98.8 | 105.9 | 6.4 | 3.1 | 1.8 | |
4. Conclusions
In this work, we developed a novel P-CDP-coated fiber. This fiber was prepared using sol–gel technology and applied to SPME processes. For HS-SPME, the fiber exhibited high operational stability and thermal stability. The satisfactory extraction efficiencies obtained for VOCs and odours were mainly due to the synergistic effect of the cavity of β-CD, the secondary cavities of P-CDP, and physical adsorption on the surface of P-CDP. The prepared fibers exhibited good linearity (R2 > 0.9946), precision (RSD < 11%), and lower detection limits (0.003–1.600 μg L−1), when used for the analysis of real water samples obtained satisfactory recoveries (76.6–106.0% for pond water and 89.0–105.9% for rainwater).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 885025).References
- Y. l. Fu, Y. l. Hu, Y. J. Zheng and G. K. Li, J. Sep. Sci., 2006, 29, 2684–2691 CrossRef PubMed.
- J. X. Yu, C. Y. Wu and J. Xing, J. Chromatogr. A, 2004, 1036, 101–115 CrossRef PubMed.
- J. X. Yu, D. Li, C. Y. Wu, L. Wu and J. Xing, J. Chromatogr. A, 2002, 978, 37–48 CrossRef PubMed.
- X. J. Li, Z. R. Zeng, S. Z. Gao and H. B. Li, J. Chromatogr. A, 2004, 1023, 15–25 CrossRef PubMed.
- J. J. Zhou and Z. R. Zeng, Anal. Chim. Acta, 2006, 556, 400–406 CrossRef.
- S. L. Zhang, Z. Du and G. K. Li, Anal. Chem., 2011, 83, 7531–7541 CrossRef PubMed.
- H. L. Xu, Y. Li, D. Q Jiang and X. P. Yan, Anal. Chem., 2009, 81, 4971–4977 CrossRef PubMed.
- H. Jiang, J. S. Li, X. R. Hu, J. Y. Shen, X. Y. Sun, W. Q. Han and L. J. Wang, Talanta, 2017, 174, 307–313 CrossRef PubMed.
- X. G. Hu, G. M. Dai, J. J. Huang and T. T. Ye, J. Chromatogr. A, 2010, 1217, 5875–5882 CrossRef PubMed.
- Y. F. Zhou, Y. N. Song, W. J. Zhen and W. T. Wang, Macromol. Res., 2015, 23, 1103–1111 CrossRef.
- A. M. Rubim, J. B. Rubenick, M. Maurer, L. V. Laporta and C. M. B. Braz, J. Pharm. Sci., 2017, 5, 1–11 Search PubMed.
- Y. Xiao, T. T. Y. Tan and Y. Wang, J. Chromatogr. A, 2012, 1269, 52–68 CrossRef PubMed.
- A. Ammala, Int. J. Cosmet. Sci., 2013, 35, 113–124 CrossRef PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef PubMed.
- Y. L. Hu, Y. Y. Yang, J. X. Huang and G. K. Li, Anal. Chim. Acta, 2005, 543, 17–24 CrossRef.
- J. J. Zhou and Z. R. Zeng, Anal. Chim. Acta, 2006, 556, 400–406 CrossRef.
- X. Y. Song, W. Ha, J. Chen and Y. P. Shi, J. Chromatogr. A, 2014, 1374, 23–30 CrossRef PubMed.
- H. Zheng, Q. Liu and Q. Jia, J. Chromatogr. A, 2014, 1343, 47–54 CrossRef PubMed.
- S. Nojavan and M. Yazdanpanah, J. Chromatogr. A, 2017, 1525, 51–59 CrossRef PubMed.
- Y. L. Hu, Y. X. Zheng and G. K. Li, Anal. Sci., 2004, 20, 667–671 CrossRef PubMed.
- X. Liu, L. L. Xu and S. Wang, Anal. Bioanal. Chem., 2014, 406, 3209–3217 CrossRef PubMed.
- J. J. Zhou, F. X. Yang, D. M. Cha, Z. R. Zeng and Y. Xu, Talanta, 2007, 73, 870–877 CrossRef PubMed.
- Y. Fan, Y. Q. Feng, S. L. Da and Z. H. Wang, Talanta, 2005, 65, 111–117 Search PubMed.
- Y. C. Liu, Y. J. Liu and Z. G. Xu, Anal. Bioanal. Chem., 2018, 410, 509–519 CrossRef PubMed.
- R. Zhao, Y. Wang and X. Li, ACS Appl. Mater. Interfaces, 2015, 7, 26649–26657 CrossRef PubMed.
- P. L. Meo, G. Lazzara and L. Liotta, Polym. Chem., 2014, 5, 4499–4510 RSC.
- B. Bikash, M. Muruganandham and R. P. S. Suri, J. Hazard. Mater., 2014, 273, 146–154 CrossRef PubMed.
- G. Z. Kyzas, N. K. Lazaridis and D. N. Bikiaris, Carbohydr. Polym., 2013, 91, 198–208 CrossRef PubMed.
- F. P. Zhao, E. Repo, D. L. Yin, Y. Meng, S. Jafari and M. Sillanp, Environ. Sci. Technol., 2015, 49, 10570–10580 CrossRef PubMed.
- M. C. Nadia and G. Crini, Prog. Polym. Sci., 2013, 38, 344–368 CrossRef.
- L. M. Paolo, G. Lazzara, L. Liotta, S. Riela and R. Noto, Polym. Chem., 2014, 5, 4499–4510 RSC.
- A. Alsbaiee, B. J. Smith, L. L. Xiao, Y. H. Ling, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 7689–7692 CrossRef PubMed.
- A. Alsbaiee, B. J. Smith, L. L. Xiao, Y. H. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef PubMed.
- Y. H. Ling, M. J. Klemes, L. L. Xiao, A. Alsbaiee, W. R. Dichtel and D. E. Helbling, Environ. Sci. Technol., 2017, 51, 7590–7598 CrossRef PubMed.
- A. H. Karoyo and L. D. Wilson, J. Colloid Interface Sci., 2013, 402, 215–222 CrossRef PubMed.
- G. Mamba, X. Y. Mbianda and P. P. Govender, Carbohydr. Polym., 2013, 98, 470–476 CrossRef PubMed.
- H. Mohamed, L. D. Wilson, R. Guo, C. Wu and H. John, Carbohydr. Polym., 2012, 87, 1241–1248 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of P-CDP fiber, analysis conditions, extraction procedure optimization, FT-IR and N2 adsorption–desorption of P-CDP, schematic and thermogravimetric analysis of coating, typical chromatograms, EFs of P-CDP coating. See DOI: 10.1039/c8ra04394a |
| This journal is © The Royal Society of Chemistry 2018 |